
Sandmeyer cyanation of arenediazonium tetrafluoroborate using acetonitrile as a cyanide source†
Wenbin
Xu
,
Qinghui
Xu
and
Jizhen
Li
*
Department of Organic Chemistry, College of Chemistry, Jilin University, 2519 Jiefang Road, Changchun, 130023, China. E-mail: ljz@jlu.edu.cn
First published on 14th January 2015
Abstract
Palladium-catalyzed cyanation of aryldiazonium tetrafluoroborate using acetonitrile as a non-metallic cyanide source was achieved in the presence of Ag2O under ambient air, eliminating the involvement of highly toxic CuCN used in the traditional Sandmeyer reaction, in which the CN group comes from metallic cyanides. The substrate scope and limitation of this protocol were investigated.
Introduction
The development of methods for the introduction of cyano groups into functionalized arenes is of great interest in organic synthesis because aromatic nitriles not only are present widely as a key functional group in various natural products, biologically active molecules and designed functional materials,1 but also are important precursors to a variety of functional groups, such as aldehydes, amines, amidines, tetrazoles, amides, and other carboxy derivatives.2 One group of the main methods available for the synthesis of aromatic nitriles is the traditional direct cyanation of Ar–X [X = N2+Y−, I, Br, Cl, OTf, B(OR)2, etc.] with the cyano-group originally from metal cyanides (MCN),3 for which the Sandmeyer reaction of aryldiazonium salts4 and the Rosenmund–Von Braun reaction of aryl halides5 are two representative methods (Scheme 1).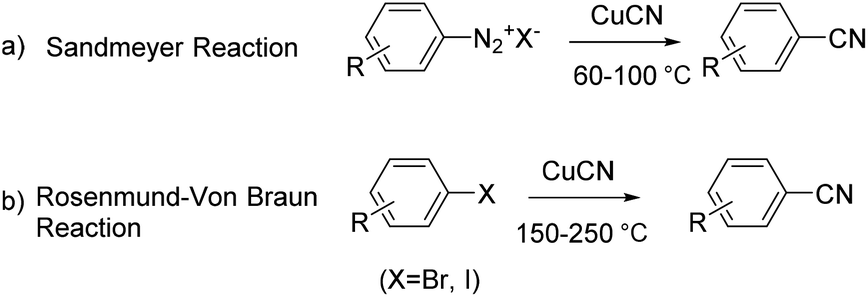 | ||
| Scheme 1 Traditional Sandmeyer reaction and Rosenmund–Von Braun reaction. | ||
In previous investigations on the Sandmeyer reaction and Rosenmund–Von Braun reaction, various kinds of CN sources have been widely explored.6 Conventional CN sources, metal cyanides (MCN), are limited by their toxicity and hazardous handling. Thus in recent years non-metallic CN sources such as acetocyanohydride, N-cyano-N-phenyl-p-toluenesulfonamide (NCTS), BnSCN, DDQ, tert-butylisocyanide, ethyl cyanoacetate, benzyl cyanide, malononitrile, N-cyanobenzimidazole etc.7 have been investigated as promising alternatives, particularly along with their contribution to the development of C–H functionalization as well.8 Special among various CN sources investigated, acetonitrile is very appealing as a potential economic candidate, even though it has rarely been explored as a cyanide precursor due to its strong C–CN energy.9 In 1998, Cheng demonstrated the activation and cleavage of the acetonitrile C–CN bond in the cyano-group transfer to aromatic bromides mediated by metal complexes of palladium and zinc species.10a This is the first example of the Rosenmund–Von Braun reaction that used acetonitrile as the CN source, even though it was limited to bromoarenes with methyl at the ortho position and the low yields reported depended strongly on the amount of the phosphine ligand. A few other reports appeared later in regard to C–CN bond activation and cleavage with a transition metal as the catalyst.10b–f Similar to the initial report, in 2012 Li developed a Cu/Ag system for the oxidative cyanation of aryl iodides with acetonitrile.11 Recently, the economical cyanation of the aromatic C–H bond mediated by Cu/Ag and Cu/Si systems with inert acetonitrile has been documented by Zhu and Shen respectively.12,13 These reports and mechanistic investigations open a new door for transition metal mediated inert alkylnitrile C–CN bond activation and cleavage for the introduction of a new aromatic C–CN bond. However there is no report on the utilization of Sandmeyer substrates, aryldiazonium salt, in this type of transformation.
On the other hand, while a variety of transition-metal catalyzed procedures were developed successfully in the Rosenmund von Braun reaction for aryl halides,14 a limited number of investigations have been reported on the Sandmeyer reaction for catalytic cyanation of aryldiazonium salt.15 In recent years, using aryldiazonium salt as the substrate, very useful transition metal (TM) catalyzed protocols for the formation of C–C, C–F, C–CF3, and C–S bonds through the cross-coupling process have been described.16 These investigations proved the high versatility of aryldiazonium salt. Furthermore, aryldiazonium salts are readily accessible from various aromatic amines through diazotized reaction with NaNO2 or other organic nitriles. Taking into consideration the above mentioned points, we envisioned that TM catalyzed Sandmeyer cyanation of aryldiazonium salt with non-metallic CN sources would be of great interest. Herein, we would like to report the cyanation of aryldiazonium tetrafluoroborates with CH3CN as the CN source under TM conditions.
To examine the feasibility of the proposed reaction, we started with p-ethoxybenzenediazonium fluoroborate 1a as the model substrate. The combination of one equivalent of Cu(OAc)2 and Ag2O has been shown to be effective in catalyzing cyanation of arylhalide with CH3CN as the CN source,11,12 we started with this combination in our initial exploration of the conditions. As shown in Table 1, the Cu(OAc)2/Ag2O combination turned out to be ineffective in promoting the cyanation of p-ethoxybenzenediazonium salts by CH3CN (entry 1). With arylhalide, the reference reported elevated temperature to drive the reaction; in our case the arenediazonium fluoroborate substrates are not stable at elevated temperature. However it was pleasing to find out that switching to catalytic amount of Pd(OAc)2 coupled with one equivalent of Ag2O at 55 °C for 24 h resulted in a fair amount of the expected product (entry 2). Replacing Pd(OAc)2 with PdCl2 further improved the yield modestly (entry 3). In an attempt to determine the role of each component of this combination, we tested with either catalytic amount of PdCl2 or one equivalent of Ag2O alone, no cyanation product was observed in both cases (entries 4 and 5). Therefore PdCl2 was utilized as the catalyst of choice in the current study, and a series of experiments were then carried out to screen the other additives. The results indicated that PdCl2 in combination with Cu(II) species such as Cu(OAc)2, CuO or Cu(OTf)2 was mostly ineffective in this reaction (entries 6–8). However Cu2O did promote the formation of a cyanation product, although the yield was inferior to that of Ag2O (entry 9 vs. 3), which suggested that the presence of Ag2O was important to the transformation. Other silver salts such as Ag2CO3, AgOAc and AgNO3 were also examined and they demonstrated similar effect on the product yield compared with Ag2O (entries 13–15). Surprisingly AgOTf did not exhibit similar reactivity as the other silver salts did in this reaction (entry 10). Slightly increasing either the amount of PdCl2 or Ag2O did not significantly affect the yields (entries 11 and 12) and thus the equivalence of PdCl2 or Ag2O was kept to be 0.1 or 1.0 in relation to the starting aryldiazonium fluoroborate.
|
|
||||
|---|---|---|---|---|
| No | Catalyst (equiv.) | Additives (equiv.) | Temp. (°C) | Yieldb (%) |
| a Reactions were carried out with 1a (0.5 mmol) in CH3CN (3.0 mL) for 24 h, under air, unless otherwise indicated. b Isolated yield. n.r., no reaction. c 48 h. d 2,9-Dimethyl-1,10-phenanthroline (1.0 equiv.) was added. | ||||
| 1 | Cu(OAc)2 (1.0) | Ag2O (1.0) | 55 | n.r. |
| 2 | Pd(OAc)2 (0.1) | Ag2O (1.0) | 55 | 39 |
| 3 | PdCl 2 (0.1) | Ag 2 O (1.0) | 55 | 64 |
| 4 | — | Ag2O (1.0) | 55 | n.r. |
| 5 | PdCl2 (0.1) | — | 55 | n.r. |
| 6 | PdCl2 (0.1) | Cu(OAc)2 (1.0) | 55 | Trace |
| 7 | PdCl2 (0.1) | CuO (1.0) | 55 | n.r. |
| 8 | PdCl2 (0.1) | Cu(OTf)2 (0.1) | 55 | Trace |
| 9 | PdCl2 (0.1) | Cu2O (1.0) | 55 | 29 |
| 10 | PdCl2 (0.1) | AgOTf (1.0) | 55 | n.r. |
| 11 | PdCl2 (0.1) | Ag2O (1.5) | 55 | 65 |
| 12 | PdCl2 (0.15) | Ag2O (1.0) | 55 | 65 |
| 13 | PdCl2 (0.10) | Ag2CO3 (1.0) | 55 | 64 |
| 14 | PdCl2 (0.10) | AgOAc | 55 | 59 |
| 15 | PdCl2 (0.10) | AgNO3 | 55 | 56 |
| 16 | PdCl2 (0.1) | Ag2O (1.0) | Reflux | 59 |
| 17 | PdCl2 (0.1) | Ag2O (1.0) | RT | 10 |
| 18 | PdCl2 (0.1) | Ag2O (1.0) | Sealed tube | 59 |
| 19c | PdCl2 (0.1) | Ag2O (1.0) | 55 | 59 |
| 20 | PdCl2 (0.1) | Ag2O (1.0) | 55 | 62 |
| PPh3 (1.0) | ||||
| 21 | PdCl2 (0.1) | Ag2O (1.0) | 55 | 34 |
| TEA(1.0) | ||||
| 22 | PdCl2 (0.1) | Ag2O (1.0)d | 55 | 62 |
| 23 | PdCl2 (0.1) | Ag2O (1.0) | 55 | 60 |
| O2 | ||||
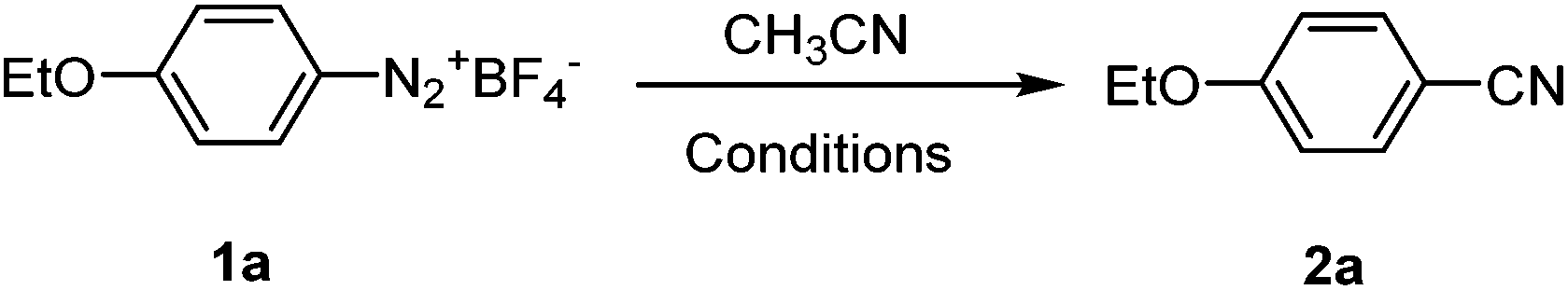
Further studies on the reaction temperature and time suggested high temperature or long reaction time did not improve the cyanation of arenediazonium salts (entries 16–23). Other experiments were also carried out with PPh3, Et3N and 2,9-dimethyl-1,10-phenanthroline as additives or under an oxygen atmosphere (entries 20–22). The presence of pure oxygen and addition of ligands did not seem to affect the formation of the cyanation product (entry 23). Similar phenomenon was observed previously in palladium-catalyzed coupling reactions with diazonium salts.16a,17 Therefore, the optimal reaction conditions were chosen as the following in further experiments with other substrates: PdCl2 0.1 equiv., Ag2O 1.0 equiv., 55 °C, the reaction time is 24 h, under ambient air.
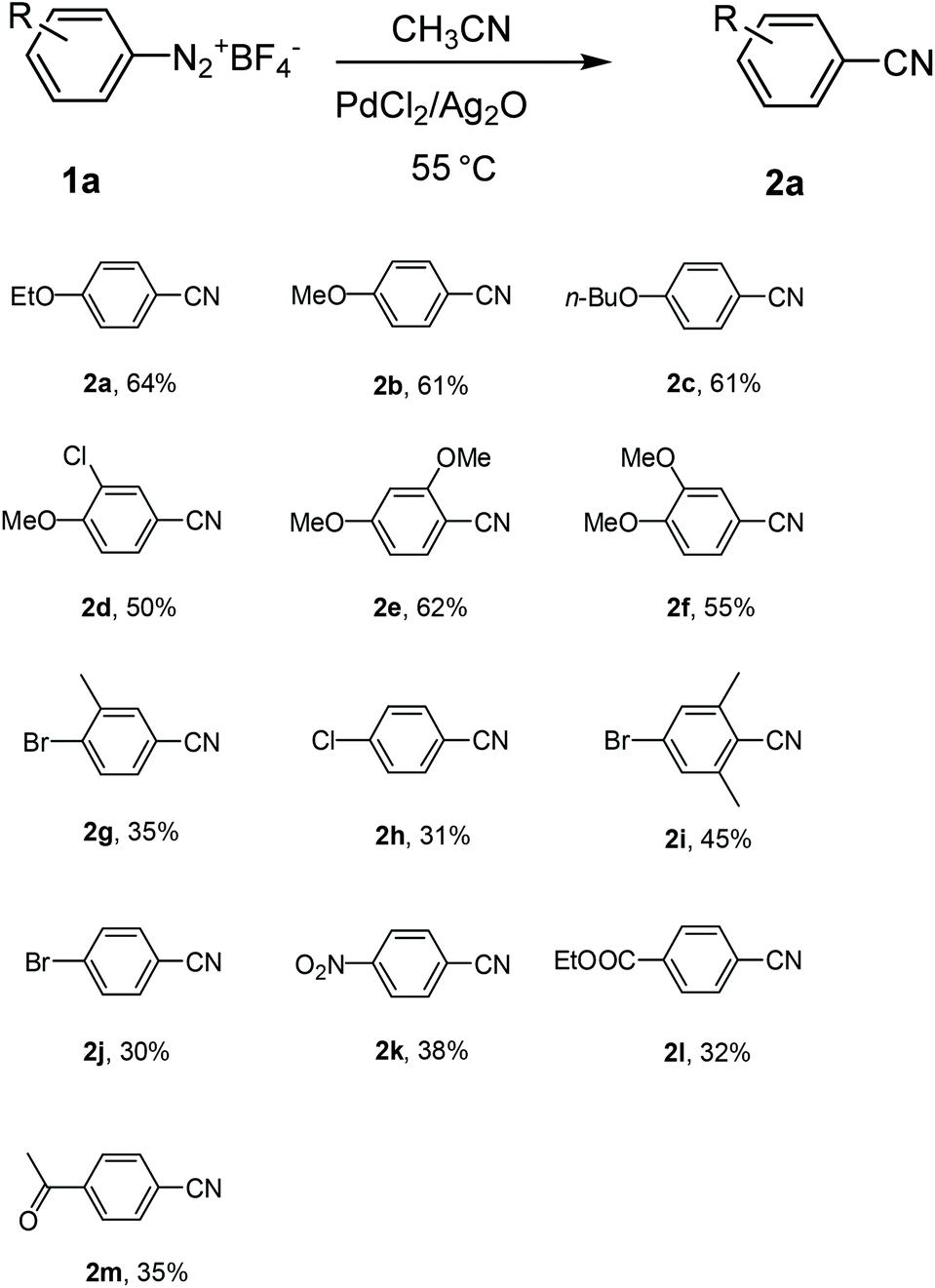
With the optimized reaction conditions in hand, the substrate scope and limitations of the protocol were examined subsequently (Table 2). In general, there are some noticeable trends and observations. Among those substrates that afforded aromatic nitriles in the product mixture, those bearing a strong para-electro-donating group proceeded most efficiently to give aromatic nitriles in moderate yields 50%–64% (2a–2f). When the starting aryldiazonium fluoroborates contain halide substitutions (2g–2j), the yield of the corresponding aromatic nitriles were usually lower, likely due to the interference of halides, which may also undergo oxidative addition with the palladium catalyst, facilitating a coupling process at the position of halides to complicate the product matrix. Other electron-withdrawing groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NO2, CO2Et, etc.) decreased the yields of aromatic nitriles even lower after column chromatography (2k–2m). It is worth mentioning that for all those substrates that gave moderate or low yield of cyanation products from the starting aryldiazonium salts in acetonitrile, there is either a lone pair p electron or π electrons conjugating with the phenyl ring (2a–2m). On the other hand, substrates without such a structural feature did not afford the corresponding aromatic nitriles and the products were acetanilides in excellent yield (Scheme 2). Due to the high reactivity of ArN2+BF4− to CH3CN, likely the nitrilium ions were formed and subsequently hydrolyzed to give acetanilides 3n–3r.18 Moreover, aryldiazonium fluoroborate with substituents at the meta and ortho positions could be smoothly transformed into the corresponding aromatic nitriles, which indicated that the steric hindrance did not significantly affect the reactivity (2d–2g, 2i). In some cases the ortho substitution may even have beneficiary effect that increased the yields of the corresponding aromatic nitriles (2ivs.2g and 2j), however such effect was not obvious when the ortho substitution is a methoxy group (2evs.2b and 2f).
O, NO2, CO2Et, etc.) decreased the yields of aromatic nitriles even lower after column chromatography (2k–2m). It is worth mentioning that for all those substrates that gave moderate or low yield of cyanation products from the starting aryldiazonium salts in acetonitrile, there is either a lone pair p electron or π electrons conjugating with the phenyl ring (2a–2m). On the other hand, substrates without such a structural feature did not afford the corresponding aromatic nitriles and the products were acetanilides in excellent yield (Scheme 2). Due to the high reactivity of ArN2+BF4− to CH3CN, likely the nitrilium ions were formed and subsequently hydrolyzed to give acetanilides 3n–3r.18 Moreover, aryldiazonium fluoroborate with substituents at the meta and ortho positions could be smoothly transformed into the corresponding aromatic nitriles, which indicated that the steric hindrance did not significantly affect the reactivity (2d–2g, 2i). In some cases the ortho substitution may even have beneficiary effect that increased the yields of the corresponding aromatic nitriles (2ivs.2g and 2j), however such effect was not obvious when the ortho substitution is a methoxy group (2evs.2b and 2f).
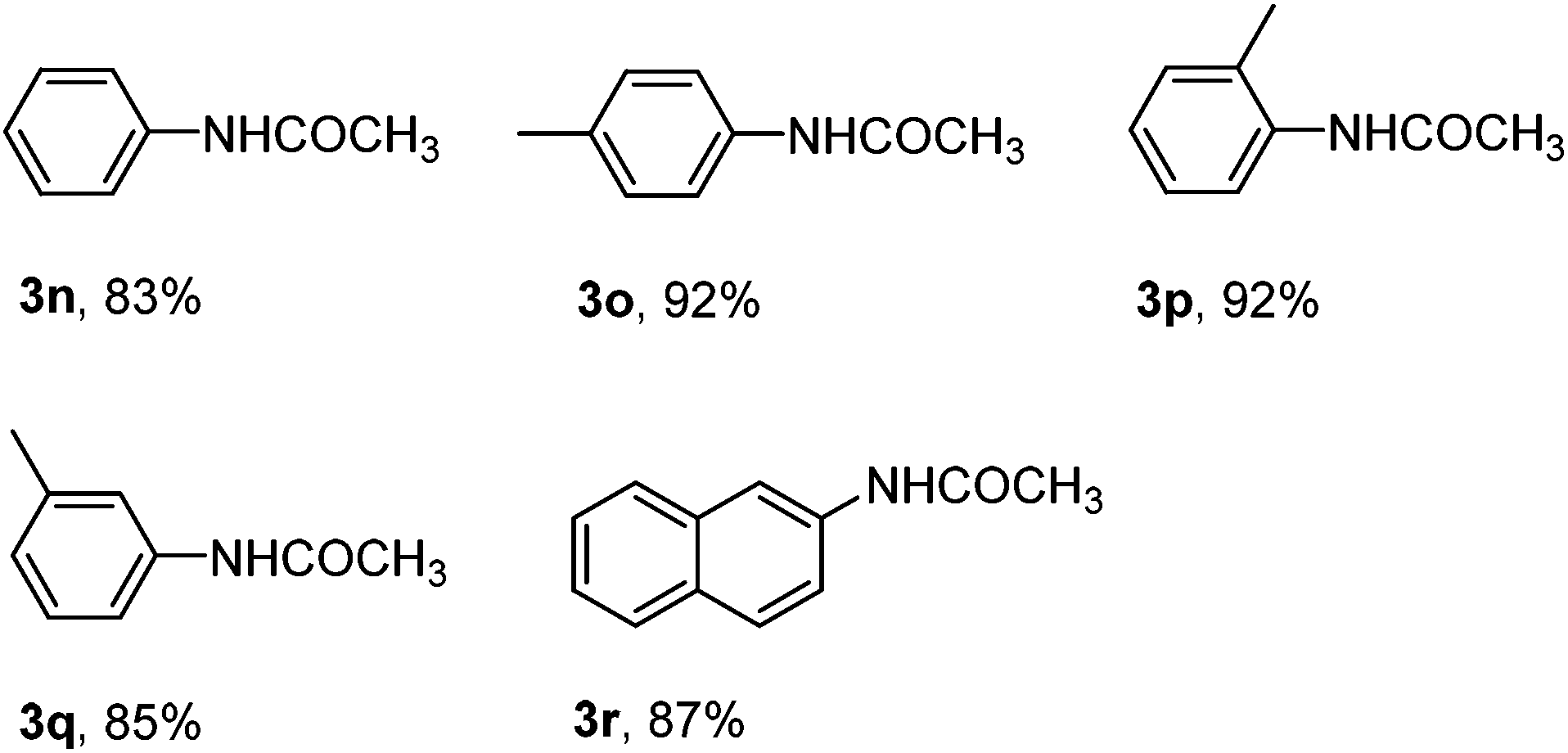 | ||
| Scheme 2 Acetanilides formed in excellent yields from arenediazonium salts under the standard conditions. | ||
|
It is worth noting that, the formation of bisaryl byproducts derived from PdCl2-catalyzed homocoupling of arenediazonium salts were obvious (e.g. 28% yield of 4,4′-dibromobiphenyl starting with 1j and 12% yield of 4,4′-dimethoxy-biphenyl starting with 1b). Such products have also been reported in other cases as the major products in refluxing methanol catalyzed by Pd(OAc)2.19 Expectedly in this protocol, the homocoupling aryldiazonium salts and cyanation with CH3CN are two competitive reactions, probably accompanied by simple reduction as well. As a result, a different catalysis system such as Pd/Ag species in CH3CN or Pd species alone in CH3OH19 affected the formation of different major products from the same arenediazonium fluoroborates. So, it is concluded that the relative yields of aromatic nitriles, byproducts of biaryls and acetanilides, depended on and varied with the type of substitution in aryldiazonium tetrafluoroborates in our protocol here. The complete product distribution of each substrate is listed in ESI;† we tabulated the yields for those products that can be isolated pure and characterized appropriately.
Even though the mechanism is not clear in detail at the current stage, based on the experiments and previous reports, we proposed a possible general mechanism outlined in Scheme 3.10–13,16a,18 Initially, divalent palladium is reduced to zero-valent palladium II. Oxidative addition of II to ArN2+BF4− gives Ar–Pd species III upon the release of nitrogen and complexing with CH3CN. Subsequently the cleavage of CH3–CN bond likely occurred in the presence of Ag2O to provide the intermediate IV. Finally, reductive elimination of intermediate IV resulted in aromatic nitriles along with the regeneration of Pd species II to complete the catalytic cycle.
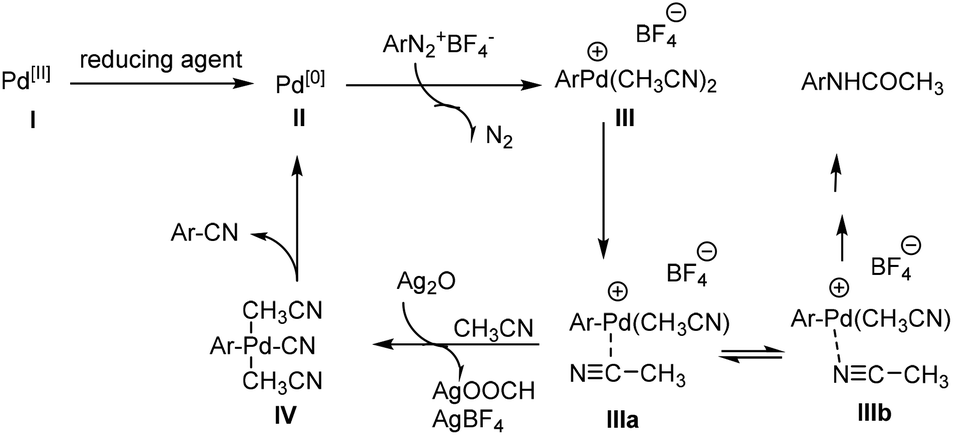 | ||
| Scheme 3 Proposed possible mechanism for Pd-catalyzed cyanation of arenediazonium fluoroborate with CH3CN as the cyanide source. | ||
Conclusions
In summary, we have discovered a novel approach to form aromatic nitriles from aryldiazonium tetrafluoroborates with a cheap organic solvent acetonitrile as the non-metal cyanide source.20 In such a case, the “CN” group could be installed regiospecifically in the position of aryldiazonium. This strategy involves PdCl2 catalyzed cyanation process via the cleavage of inert acetonitrile C–CN bond in the presence of Ag2O under ambient air. Even though this approach has a limited substrate scope with moderate yield at its current stage, it eliminates the involvement of toxic CuCN, in comparison with the traditional Sandmeyer reaction and it is an example of preparative cyanation of arenediazonium tetrafluoroborates with simple acetonitrile. Further studies are ongoing to expand the reaction scope, simplify the reaction procedure, increase the yield, and study the reaction mechanism in detail in our lab.Acknowledgements
Financial support from the Natural Science Foundation of Jilin Province, P. R. China (grant no. 20130101004JC, 3D513P491412) is gratefully acknowledged. We thank Dr Zhenfa Zhang for his help in this manuscript preparation.Notes and references
- For examples, see: (a) A. Kleemann, J. Engel, B. Kutschner and D. Reichert, Pharmaceutical substances: Syntheses, Patents, Applications, Thieme, Stuttgart, New York, 4th edn, 2001 Search PubMed; (b) Review: J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS PubMed.
- (a) Z. Rappoport, The Chemistry of the Cyano Group, Interscience Publishers, New York, 1970 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- For examples, see: (a) P. Y. Yeung, C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2010, 122, 9102 CrossRef; (b) T. Schareina, A. Zapf and M. Beller, J. Organomet. Chem., 2004, 689, 4576 CrossRef CAS PubMed; (c) G. Ishii, K. Moriyama and H. Togo, Tetrahedron Lett., 2011, 52, 2404 CrossRef CAS PubMed; (d) S. Ushijima, K. Moriyama and H. Togo, Tetrahedron, 2011, 67, 958 CrossRef CAS PubMed; (e) Z. H. Zhang and L. S. Liebeskind, Org. Lett., 2006, 8, 4331 CrossRef CAS PubMed; (f) G. Y. Zhang, L. L. Zhang, M. L. Hu and J. Cheng, Adv. Synth. Catal., 2011, 353, 291 CrossRef CAS.
- (a) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633 CrossRef; (b) J. K. Kochi, J. Am. Chem. Soc., 1957, 79, 2942 CrossRef CAS; (c) J. Lindley, Tetrahedron, 1984, 40, 1433 CrossRef CAS; (d) See review: H. H. Hodgson, Chem. Rev., 1947, 40, 251 CrossRef CAS.
- (a) K. W. Rosenmund and E. Struck, Chem. Ber., 1919, 52, 1749 CrossRef; (b) C. F. Koelsch and A. G. Whitney, J. Org. Chem., 1941, 6, 795 CrossRef CAS; (c) Review: C. Galli, Chem. Rev., 1988, 88, 765 CrossRef CAS; (d) Review: E. B. Merkushev, Synthesis, 1988, 923 CrossRef CAS PubMed.
- For recent examples, see: (a) J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS PubMed; (b) H. J. Cristau, A. Ouali, J. F. Spindler and M. Taillefer, Chem. – Eur. J., 2005, 11, 2483 CrossRef CAS PubMed.
- For recent examples using “non-metallic” CN sources for the cyanation reaction, see: (a) Review: J. kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed, and references therein; (b) T. J. Gong, B. Xiao, W. M. Cheng, W. Su, J. Xu, Z. J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630 CrossRef CAS PubMed; (c) Y. Yang and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 8677 CrossRef CAS PubMed.
- For examples, see: (a) Review: T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137 CrossRef CAS PubMed, and references therein; (b) H. Xu, P. T. Liu, Y. H. Li and F. S. Han, Org. Lett., 2013, 15, 3354 CrossRef CAS PubMed.
- (a) See recent minireview: C. Najera and J. M. Sansano, Angew. Chem., Int. Ed., 2009, 48, 2452 CrossRef CAS PubMed, and references therein; (b) P. Hanson, A. B. Taylor, P. H. Walton and A. W. Timms, Org. Biomol. Chem., 2007, 5, 679 RSC.
- (a) F. H. Luo, C. I. Chu and C. H. Cheng, Organometallics, 1998, 17, 1025 CrossRef CAS; (b) T. A. Ateşin, T. Li, S. Lachaize, W. W. Brennessel, J. J. García and W. D. Jones, J. Am. Chem. Soc., 2007, 129, 7562 CrossRef PubMed; (c) F. L. Taw, P. S. White, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2002, 124, 4192 CrossRef CAS PubMed; (d) T. Lu, X. Zhuang, Y. Li and S. Chen, J. Am. Chem. Soc., 2004, 126, 4760 CrossRef CAS PubMed; (e) D. S. Marlin, M. M. Olmstead and P. K. Mascharak, Angew. Chem., Int. Ed., 2001, 40, 4752 CrossRef CAS; (f) H. Nakazawa, T. Kawasaki, K. Miyoshi, C. H. Suresh and N. Koga, Organometallics, 2004, 23, 117 CrossRef CAS.
- R. J. Song, J. C. Wu, Y. Liu, G. B. Deng, C. Y. Wu, W. T. Wei and J. H. Li, Synlett, 2012, 2491 CAS.
- C. D. Pan, H. M. Jin, P. Xu, X. Liu, Y. X. Cheng and C. J. Zhu, J. Org. Chem., 2013, 78, 9494 CrossRef CAS PubMed.
- X. Z. Kou, M. D. Zhao, X. X. Qiao, Y. M. Zhu, X. F. Tong and Z. M. Shen, Chem. – Eur. J., 2013, 19, 16880 CrossRef CAS PubMed.
- (a) Review: P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC, and references therein; (b) S. Zheng, C. Yu and Z. Shen, Org. Lett., 2012, 14, 3644 CrossRef CAS PubMed.
- (a) W. B. Xu, Q. H. Xu, Z. F. Zhang and J. Z. Li, Asian J. Org. Chem., 2014, 3, 1062 CAS; (b) I. P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, J. Organomet. Chem., 2004, 689, 3810 CrossRef CAS PubMed; (c) A. S. Sigeev, I. P. Beletskaya, P. V. Petrovskii and A. S. Peregudov, Russ. J. Org. Chem., 2012, 48, 1055 CrossRef CAS; (d) P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, Synthesis, 2007, 2534 CrossRef PubMed.
- (a) See review: A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622 CrossRef CAS PubMed; (b) See review: F. Y. Mo, G. B. Dong, Y. Zhang and J. B. Wang, Org. Biomol. Chem., 2013, 11, 1582 RSC; (c) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 7972 CrossRef CAS PubMed.
- S. Yasui, M. Fujii, C. Kawano, Y. Nishimura and A. Ohno, Tetrahedron Lett., 1991, 32, 5601 CrossRef CAS.
- (a) S. Milanesi, M. Fagnoni and A. Albini, J. Org. Chem., 2005, 70, 603 CrossRef CAS PubMed; (b) S. Milanesi, M. Fagnoni and A. Albini, Chem. Commun., 2003, 216 RSC; (c) V. Cepanec, M. Litvic, J. Udikovic, I. Pogorelic and M. Lovric, Tetrahedron, 2007, 63, 5614 CrossRef PubMed; (d) P. C. Buxton, M. Fensome and H. Heaney, Tetrahedron, 1995, 51, 2959 CrossRef CAS; (e) R. Kikukawa, T. Totok, F. Wada and T. Matsuda, J. Organomet. Chem., 1984, 270, 283 CrossRef.
- M. K. Robinsin, V. S. Kochurina and J. M. Hanna, Tetrahedron Lett., 2007, 48, 7687 CrossRef PubMed.
- General procedure. To a solution of aryldiazonium tetrafluoroborate (0.5 mmol) in CH3CN (3.0 mL) was added PdCl2 (8.8 mg, 0.05 mmol) and Ag2O (116 mg, 0.5 mmol). The mixture was stirred at 55 °C for 24 h under air. Then the reaction mixture was cooled to room temperature and filtered through a pad of celite (1.0 g) and rinsed with CH2Cl2 (10 mL). The resulting organic solution was concentrated under reduced pressure and further purified by flash chromatography (SiO2, petroleum ether–ethyl acetate gradient), yielding the corresponding aryl nitriles.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure, 1H NMR, 13C NMR, IR and MS. See DOI: 10.1039/c4qo00301b |
| This journal is © the Partner Organisations 2015 |