
Enantioselective synthesis of bicyclo[2.2.2]octane-1-carboxylates under metal free conditions†
Ying-Zi
Li
a,
Jie
Wang
a,
Wang-Bin
Sun
ab,
Yi-Fan
Shan
a,
Bing-Feng
Sun
*a,
Guo-Qiang
Lin
*a and
Jian-Ping
Zou
b
aKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: bfsun@sioc.ac.cn; lingq@sioc.ac.cn
bKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry and Chemical Engineering, Soochow University, Suzhou, Jiangsu 215123, China
First published on 29th January 2015
Abstract
A new tandem reaction was realized that permits rapid access to a wide range of bicyclo[2.2.2]octane-1-carboxylates in good to excellent yields with excellent enantioselectivities under metal free, mild, and operationally simple conditions. An open transition state was deemed operative in this highly enantioselective process mediated by an organic base.
Bicyclo[2.2.2]octane represents a privileged structure found in a myriad of natural products such as atisanes and ent-atisanes, atisine-, denudatine-, as well as daphmanidin-type alkaloids,1,2 in addition to being the core unit of a type of molecular rotor.3 Some biologically significant molecules such as platencin,4 maoecrystal V,5 crotogoudin and crotobarin6 feature this bicyclic structure (Fig. 1). In particular, the antibiotic platencin (1) has become one of the most prominent natural products in recent years.7 Platencin blocks FabF and FabH enzymes and exhibits broad-spectrum antibacterial activity against Gram-positive pathogens that show resistance to current antibiotics.4,8,9 However, the poor in vivo efficacy has precluded platencin from being a clinical drug, thereby entailing the procurement of analogues for further biological evaluations.10 Tremendous efforts have been made to access platencin,11a–s synthetic analogues,11t–w as well as related natural products.12
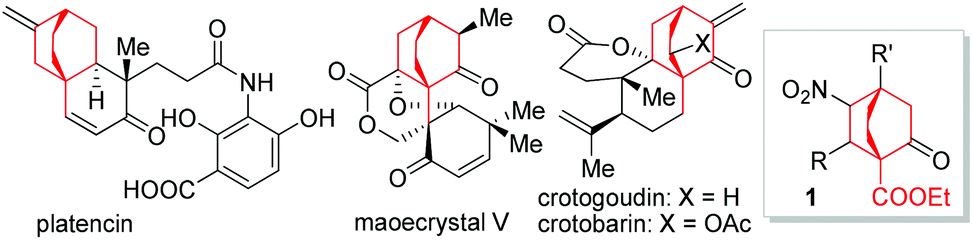 | ||
| Fig. 1 Platencin and the related natural products. | ||
In our recent efforts to realize a diversity-oriented synthesis of platencin and related natural products, bicyclo[2.2.2]octane-1-carboxylate 1 was selected as an early-stage key intermediate. Therefore, exploration of a method that allows gram-scale preparation of 1 under operationally simple conditions is highly desirable. The development of enantioselective approaches to bicyclo[2.2.2]octanes has remained an attractive yet challenging arena, and to date only a few methods have been successfully established.13 Among these, Córdova,13c Xu,13d and Chen13e developed catalytic asymmetric [4 + 2] cycloaddition reactions of cyclohexenones with electron-deficient alkenes based on enamine/iminium catalysis.14 However, only cyclohexenones devoid of α′-substituents were employed as substrates in these reactions, leading to [2.2.2] bicyclic products without substitution on the C1 bridgehead (Scheme 1A). Based on our experiences in natural product synthesis,15 an enantioselective approach to bicyclo[2.2.2]octane-1-carboxylate 1 could fill a significant gap in the current repertoire. We envisioned 1 to be assembled from α′-ethoxycarbonyl cyclohexenone 2 and nitroolefin 3 through a formal [4 + 2] cycloaddition reaction (Scheme 1B). To the best of our knowledge, to date this formal [4 + 2] cycloaddition reaction has not been reported. Actually, under relevant circumstances, formation of the formal [4 + 2] products was not documented.12d,16 In this paper, we delineate the highly enantioselective synthesis of 1 by employing metal free, mild, and operationally simple conditions.
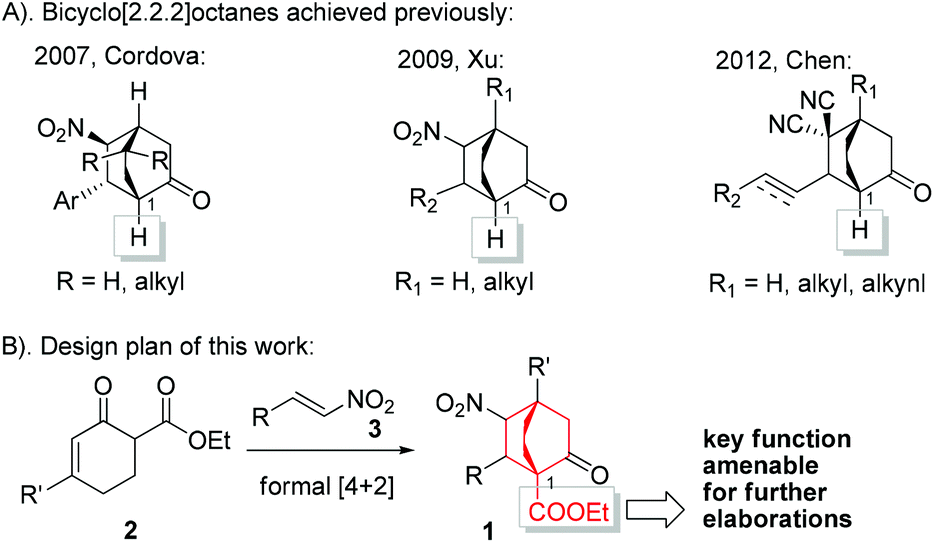 | ||
| Scheme 1 Conceptual formal [4 + 2] reaction leading to 1. | ||
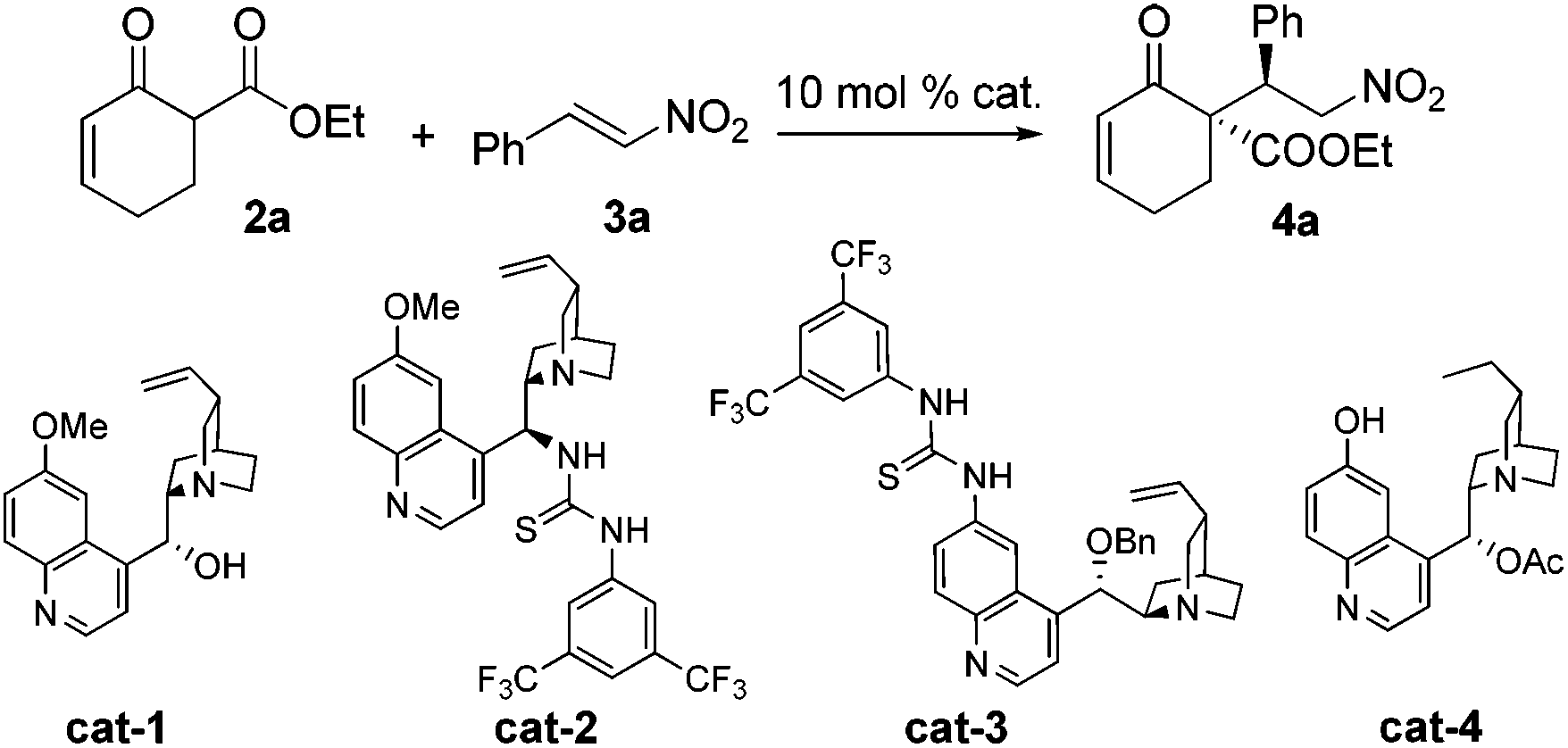
We began our studies by investigating the reaction of 2a and 3a. Surprisingly, under various examined conditions, 4a was validated as the sole product. Therefore, optimization of this conjugate addition reaction17,13c–i was first conducted and the selected results are summarized in Table 1. The best results were obtained with cat-4, delivering 4a in a quantitative yield with excellent stereoselectivity (entry 4). Importantly, when (Z)-3a instead of 3a was employed as the electrophile, the same product 4a was obtained with excellent selectivity (entry 5). This result could be attributed to the propensity of (Z)-3a undergoing isomerization to 3a in the presence of the catalyst, as evidenced by our 1H NMR studies. Accordingly, the use of geometrically pure nitroolefin was not necessary.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Cat. | Conv.b [%] | drb | eec [%] |
| a The reaction was conducted with 2a (0.40 mmol), 3a (0.20 mmol), catalyst (10 mol%), in DCM (1.0 mL) at RT. b Determined by 1H NMR analysis of the crude products. c Determined by chiral HPLC analysis. d The isolated yield. | |||||
| 1 | 3a | cat-1 | 81 | >20/1 | −31 |
| 2 | 3a | cat-2 | 94 | >20/1 | 87 |
| 3 | 3a | cat-3 | 88 | >20/1 | −90 |
| 4 | 3a | cat-4 | 99 | >20/1 | 99 |
| 5 | (Z)-3a | cat-4 | 84d | 19/1 | 97 |
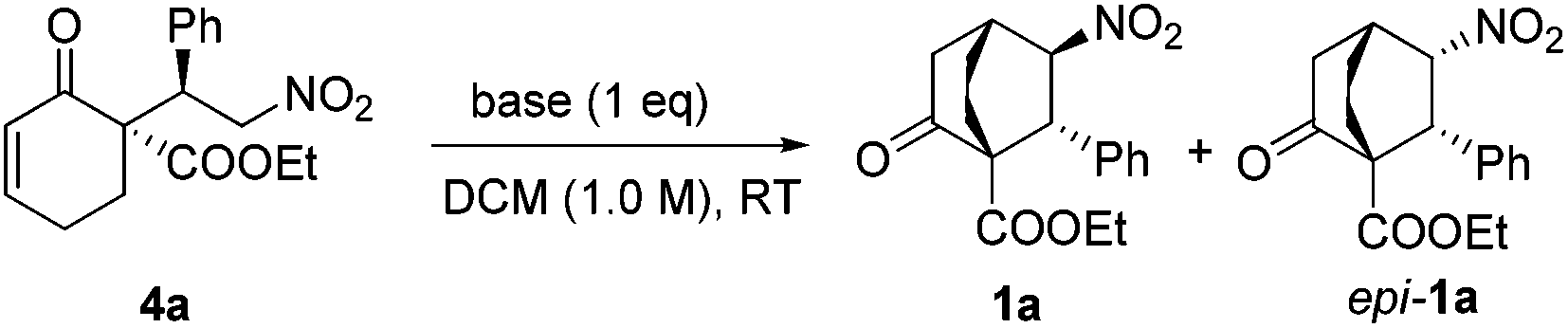
After obtaining enantiopure 4a, we focused on the crucial intramolecular conjugate addition. Due to the reversibility of the previous step, it was critical to define suitable conditions for this step to proceed without invoking racemization. Various bases were scrutinized (Table 2). While K2CO3 gave no reaction, by switching to Cs2CO3, 1a and epi-1a in a combined yield of 26% were obtained with poor selectivity (entry 2). Interestingly, with LiOH employed as the base, a high diastereoselectivity favoring epi-1a was observed, although the product was completely racemic (entry 5). This compelling evidence of racemization indicated that 4a did revert to 2a and 3a to certain levels under these reaction conditions. To our delight, after the failure of several conventional amine bases (entries 6–8), DBU and TBAF were found to be efficient promoters for this reaction (entries 9, 10). Thus, by treatment of 4a with 1.0 equiv. TBAF, 1a was isolated in 78% yield with >20/1 dr and 96% ee (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Yieldb [%] | drc [1a/epi-1a] | eed [%] |
| a The reaction was conducted with 4a (0.10 mmol) in DCM (0.1 mL) at RT. b Isolated yields. c Determined by 1H NMR analysis of the crude products. d Determined by chiral HPLC analysis. | ||||
| 1 | K2CO3 | Trace | — | — |
| 2 | Cs2CO3 | 26 | 1/2.2 | 71|15 |
| 3 | CsOH | 30 | 1.2/1 | 31|02 |
| 4 | CsF | 22 | 1/1.1 | 38|19 |
| 5 | LiOH | 35 | 1/12 | 0 |
| 6 | NEt3 | Trace | — | — |
| 7 | DIPEA | Trace | — | — |
| 8 | DMAP | Trace | — | — |
| 9 | DBU | 61 | >20/1 | 94 |
| 10 | TBAF·3H2O | 78 | >20/1 | 96 |
With the two separate steps successfully established, the stage was now set for development of a one-pot tandem reaction. To this end, after the first conjugate addition catalyzed by cat-4 was complete, TBAF was introduced into the reaction mixture to promote the second conjugate addition. The use of 0.3 equiv. TBAF (1.0 M in THF) was determined to be optimal, wherein 1a could be isolated in 84% yield with >20/1 dr and 97% ee (Table 3, entry 1). Under the optimized conditions, a broad substrate scope of nitroolefins was found for this tandem reaction, providing bicyclo[2.2.2]octane-1-carboxylates in good yields and mostly with excellent stereoselectivities. For nitrostyrene substrates, both electron donating and electron withdrawing substituents on the benzene ring are well tolerated (entries 2–11). In general, substrates with electron withdrawing groups gave better results than those with electron donating groups (entries 5–11 versus 2–4), presumably as a result of the higher reactivity of the former than that of the latter. Of particular interest were nitrostyrenes with an ortho substituent wherein surprisingly high yields were obtained (entries 10 and 11 versus 5). This may be accounted for by resorting to the strain release effect in the reaction, considering that there is a repulsive interaction between the vinyl proton and the ortho substituent coplanar in the substrates. This rationale is in agreement with the fact that 1k was obtained in a higher yield than 1j since the bromine atom, albeit with a less electronegativity, is more sterically demanding than the chlorine atom (entry 11 versus 10). Gratifyingly, other aryl, heteroaryl, as well as alkenyl and alkyl nitroolefins also proved to be suitable substrates for this reaction (entries 12–17). The relative as well as the absolute stereochemistry of the products were established unambiguously by the X-ray crystallographic analysis of enantiopure 1e (Fig. 2).21
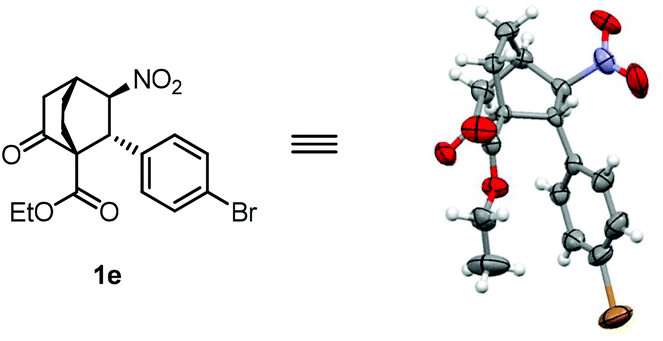 | ||
| Fig. 2 X-ray structure of 1e. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Prod. | Yieldb [%] | drc | eed [%] |
| a Conditions: 2a (0.40 mmol), 3 (0.20 mmol), cat-4 (3 mol%), DCM (0.2 mL), TBAF (1.0 M in THF, 60 μL), RT. b Isolated yields based on 3. c Determined by 1H NMR analysis of the crude products. d Determined by chiral HPLC analysis. | |||||
| 1 | Ph | 1a | 85 | >20/1 | 97 |
| 2 | 4-Me-C6H4 | 1b | 80 | 19/1 | 95 |
| 3 | 4-MeO-C6H4 | 1c | 70 | 13/1 | 95 |
| 4 | 3,4-(MeO)2-C6H3 | 1d | 72 | 13/1 | 96 |
| 5 | 4-Br-C6H4 | 1e | 86 | 19/1 | 98 |
| 6 | 4-F-C6H4 | 1f | 76 | >20/1 | 99 |
| 7 | 4-CF3-C6H4 | 1g | 87 | >20/1 | 98 |
| 8 | 4-NO2-C6H4 | 1h | 85 | >20/1 | 99 |
| 9 | 3-NO2-C6H4 | 1i | 96 | >20/1 | 97 |
| 10 | 2-Cl-C6H4 | 1j | 93 | >20/1 | 97 |
| 11 | 2-Br-C6H4 | 1k | 97 | >20/1 | 98 |
| 12 | 1-Naphthyl | 1l | 88 | 7.3/1 | 96 |
| 13 | 2-Furanyl | 1m | 69 | 12/1 | 90 |
| 14 | Styryl | 1n | 60 | 13/1 | 94 |
| 15 | Isobutyl | 1o | 93 | 19/1 | 97 |
| 16 | Phenethyl | 1p | 62 | >20/1 | 97 |
| 17 | Cyclohexyl | 1q | 53 | 19/1 | 99 |
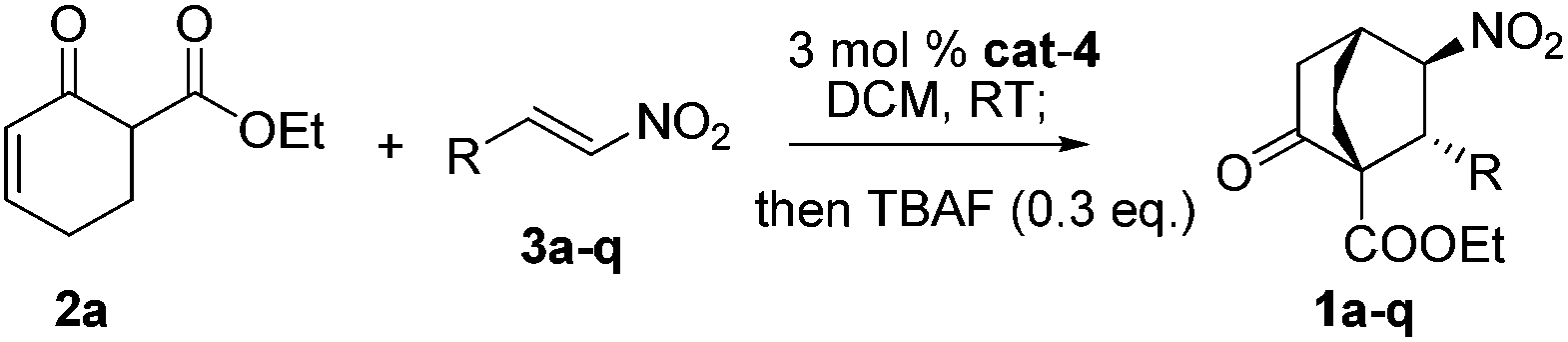
Gratifyingly, variations on the enone component proved viable (Scheme 2). Bicyclo[2.2.2]octane functions as the bridge in some molecular rotors,18 and the synthetic challenge for such rotors resides in the construction of the bridgehead quaternary carbons.19 As a proof of concept, the reaction of 2b with 3a was examined, and 1r was isolated in 82% yield with 19/1 dr and 99% ee (Scheme 2A). Another result of particular interest was obtained in the reaction of 2c with 3a, delivering 1s harboring five contiguous stereocenters with >20/1 dr and 96% ee, probably through the intermediacy of 5 (Scheme 2B). Notably, 1s contains a tricyclo[3.2.1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2,7]octane core characteristic of trachylobanes.20 Remarkably, this reaction could be readily scaled up, as exemplified by the five-gram scale preparation of 1a (Scheme 3).
2,7]octane core characteristic of trachylobanes.20 Remarkably, this reaction could be readily scaled up, as exemplified by the five-gram scale preparation of 1a (Scheme 3).
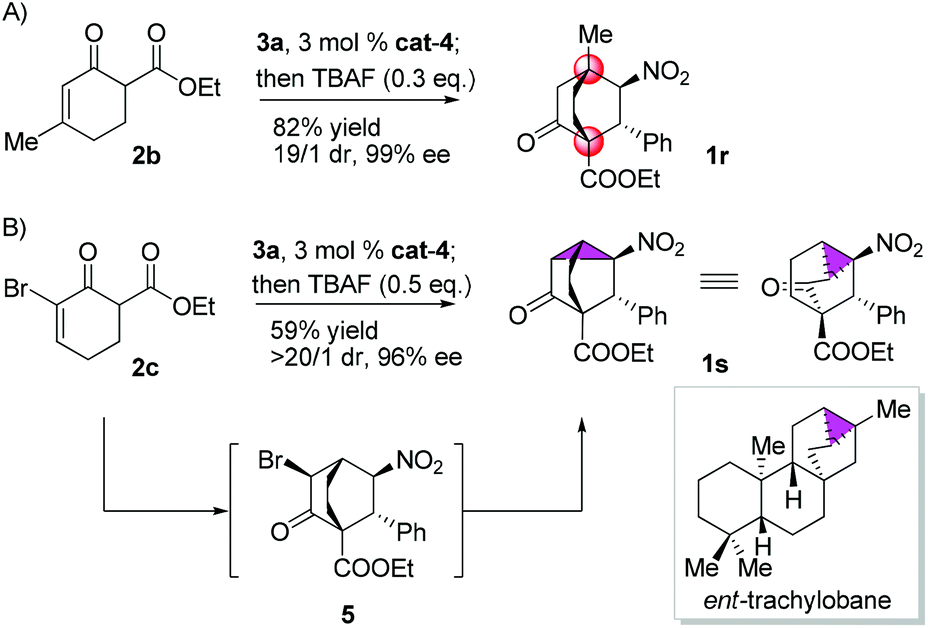 | ||
| Scheme 2 Reaction of 3a with 2b (A) and 2c (B). | ||
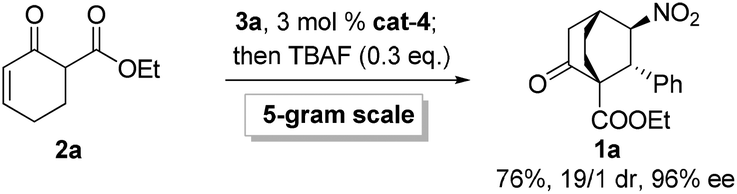 | ||
| Scheme 3 Five-gram-scale preparation of 1a. | ||
On the basis of the preceding results, the stereochemical outcome of the tandem reaction could be rationalized, as exemplified by the reaction of 2a with 3a (Scheme 4). In the first step, 2a may form a complex with cat-4via hydrogen-bonding interactions, thereby discriminating the two faces of the reactive enol. The nitroolefin approaches the enol selectively from the less hindered α-face with the substituents of the two bond-forming carbons positioned in a staggered arrangement (6), delivering 4a. In the second step, when DBU or TBAF is used as the base, the open transition state 7 might be operative to minimize the steric repulsion between the nitro and the phenyl groups, leading to the formation of 1a as the major product (path A). In contrast, when metal-containing bases are employed, the closed transition state 8 could be operative owing to the coordinating ability of metal ions. Framework 8 may break down to 2a-M and 3a which recombine to afford 8 in the racemic form before further proceeding to racemic epi-1a (path B). This rationale is in agreement with the fact that metal-containing bases are detrimental for this enantioselective reaction.
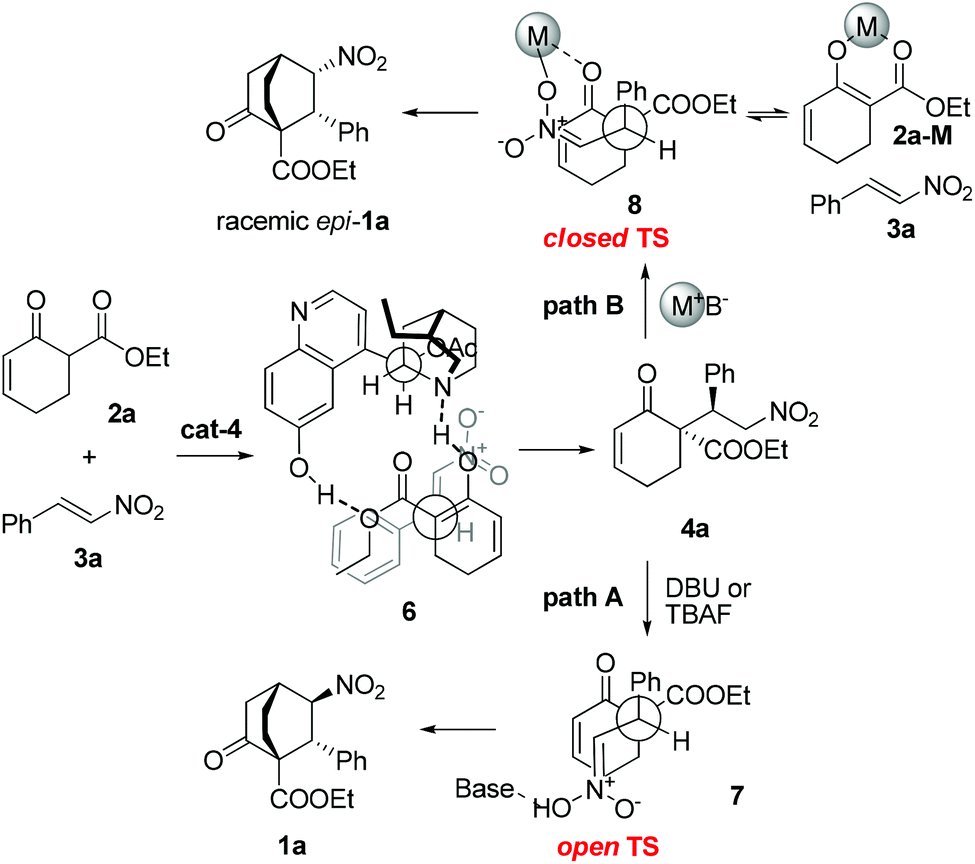 | ||
| Scheme 4 Proposed stereochemical course of the reaction in the presence of organic bases (path A) or metal-containing bases (path B). | ||
Conclusions
We have realized a formal [4 + 2] reaction that permits rapid access to a wide range of functionalized [2.2.2] bicyclic octanes in a highly enantioselective manner from simple starting materials. The reaction features metal free, mild, and operationally simple conditions, providing synthetically useful bicyclo[2.2.2]octane-1-carboxylates in good yields with excellent enantioselectivity. Notably, only 3 mol% of the organocatalyst is used for the enantioselective reaction. Importantly, this method is amenable to large scale preparation, thus facilitating relevant synthetic studies of natural products. Moreover, this work has also laid the basis for access to bicyclo[2.2.1]heptane-1-carboxylates. Endeavors along these lines are currently underway and will be reported in due course.Acknowledgements
Financial support from the National Natural Science Foundation of China (grant no. 21172246, 21290180, 21472210), the National Basic Research Program of China (973 Program, grant no. 2010CB833206), the Collaborative Innovation Center of Chemical Science and Engineering (Tianjin) and the State Key Laboratory of Bioorganic and Natural Products Chemistry is gratefully appreciated.Notes and references
- (a) P.-J. Zhao, S. Gao, L.-M. Fan, J.-L. Nie, H.-P. He, Y. Zeng, Y.-M. Shen and X.-J. Hao, J. Nat. Prod., 2009, 72, 645 CrossRef CAS PubMed; (b) F.-P. Wang, Q.-H. Chen and X.-Y. Liu, Nat. Prod. Rep., 2010, 27, 529 RSC.
- (a) J. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936 RSC; (b) M. E. Weiss and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11501 CrossRef CAS PubMed.
- B. Rodríguez-Molina, S. Pérez-Estrada and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2013, 135, 10388 CrossRef PubMed.
- J. Wang, S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. Colletti, K. Herath, R. Cummings, O. Salazar, I. Gonzalez, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. Cully and S. B. Singh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7612 CrossRef CAS PubMed.
- (a) S. H. Li, J. Wang, X. M. Niu, Y. H. Shen, H. J. Zhang, H. D. Sun, M. L. Li, Q. E. Tian, Y. Lu, P. Cao and Q. T. Zheng, Org. Lett., 2004, 6, 4327 CrossRef CAS PubMed; (b) J. Gong, G. Lin, W. Sun, C.-C. Li and Z. Yang, J. Am. Chem. Soc., 2010, 132, 16745 CrossRef CAS PubMed; (c) F. Peng and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 18860 CrossRef CAS PubMed; (d) P. Lu, Z. Gu and A. Zakarian, J. Am. Chem. Soc., 2013, 135, 14552 CrossRef CAS PubMed.
- (a) O. L. Rakotonandrasana, F. H. Raharinjato, M. Rajaonarivelo, V. Dumontet, M.-T. Martin, J. Bignon and P. Rasoanaivo, J. Nat. Prod., 2010, 73, 1730 CrossRef CAS PubMed; (b) S. Breitler and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 11168 CrossRef CAS PubMed.
- (a) E. Martens and A. L. Demain, J. Antibiot., 2011, 64, 705 CrossRef CAS PubMed; (b) G. I. Moustafa, S. Nojima, Y. Yamano, A. Aono, M. Arai, S. Mitarai, T. Tanaka and T. Yoshimitsu, Med. Chem. Commun., 2013, 4, 720 RSC; (c) M. J. Smanski, R. M. Peterson, S. R. Rajski and B. Shen, Antimicrob. Agents Chemother., 2009, 53, 1299 CrossRef CAS PubMed.
- R. M. Peterson, T. Huang, J. D. Rudolf, M. J. Smanski and B. Shen, Chem. Biol., 2014, 21, 389 CrossRef CAS PubMed.
- G. A. I. Moustafa, S. Nojima, Y. Yamano, A. Aono, M. Arai, S. Mitarai, T. Tanaka and T. Yoshimitsu, Med. Chem. Commun., 2013, 4, 720 RSC.
- (a) S. Brinster, G. Lamberet, B. Staels, P. Trieu-Cuot, A. Gruss and C. Poyart, Nature, 2009, 458, 83 CrossRef CAS PubMed; (b) E. Martens and A. L. Demain, J. Antibiot., 2011, 64, 705 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, G. S. Tria and D. J. Edmonds, Angew. Chem., Int. Ed., 2008, 47, 1780 CrossRef CAS PubMed; (b) J. Hayashida and V. H. Rawal, Angew. Chem., Int. Ed., 2008, 47, 4373 CrossRef CAS PubMed; (c) K. Tiefenbacher and J. Mulzer, Angew. Chem., Int. Ed., 2008, 47, 6199 CrossRef CAS PubMed; (d) S. Y. Yun, J.-C. Zheng and D. Lee, Angew. Chem., Int. Ed., 2008, 47, 6201 CrossRef CAS PubMed; (e) D. C. J. Waalboer, M. C. Schaapman, F. L. van Delft and F. P. J. T. Rutjes, Angew. Chem., Int. Ed., 2008, 47, 6576 CrossRef CAS PubMed; (f) K. C. Nicolaou, Q.-Y. Toh and D. Y.-K. Chen, J. Am. Chem. Soc., 2008, 130, 11292 CrossRef CAS PubMed; (g) K. A. B. Austin, M. G. Banwell and A. C. Willis, Org. Lett., 2008, 10, 4465 CrossRef CAS PubMed; (h) K. Tiefenbacher and J. Mulzer, J. Org. Chem., 2009, 74, 2937 CrossRef CAS PubMed; (i) G. N. Varseev and M. E. Maier, Angew. Chem., Int. Ed., 2009, 48, 3685 CrossRef CAS PubMed; (j) A. K. Ghosh and K. Xi, Angew. Chem., Int. Ed., 2009, 48, 5372 CrossRef CAS PubMed; (k) K. C. Nicolaou, G. S. Tria, D. J. Edmonds and M. Kar, J. Am. Chem. Soc., 2009, 131, 15909 CrossRef CAS PubMed; (l) P. Li and H. Yamamoto, Chem. Commun., 2010, 46, 6294 RSC; (m) V. Singh, B. C. Sahu, V. Bansal and S. M. Mobin, Org. Biomol. Chem., 2010, 8, 4472 RSC; (n) S. Hirai and M. Nakada, Tetrahedron Lett., 2010, 51, 5076 CrossRef CAS PubMed; (o) K. Palanichamy, A. V. Subrahmanyam and K. P. Kaliappan, Org. Biomol. Chem., 2011, 9, 7877 RSC; (p) T. Yoshimitsu, S. Nojima, M. Hashimoto and T. Tanaka, Org. Lett., 2011, 13, 3698 CrossRef CAS PubMed; (q) S. Hirai and M. Nakada, Tetrahedron, 2011, 67, 518 CrossRef CAS PubMed; (r) L. Zhu, C. Zhou, W. Yang, S. He, G.-J. Cheng, X. Zhang and C.-S. Lee, J. Org. Chem., 2013, 78, 7912 CrossRef CAS PubMed; (s) J. S. Yadav, R. Goreti, S. Pabbaraja and B. Sridhar, Org. Lett., 2013, 15, 3782 CrossRef CAS PubMed; (t) O. V. Barykina, K. Rossi, L. M. J. Rybak and B. B. Snider, Org. Lett., 2009, 11, 5334 CrossRef CAS PubMed; (u) K. Tiefenbacher, A. Gollner and J. Mulzer, Chem. – Eur. J., 2010, 16, 9616 CrossRef CAS PubMed; (v) D. C. J. Waalboer, S. H. A. M. Leenders, T. Schuelin-Casonato, F. L. van Delft and F. P. J. T. Rutjes, Chem. – Eur. J., 2010, 16, 11233 CrossRef CAS PubMed; (w) G. Y. C. Leung, H. Li, Q.-Y. Toh, A. M.-Y. Ng, R. J. Sum, J. E. Bandow and D. Y.-K. Chen, Eur. J. Org. Chem., 2011, 183 CrossRef CAS.
- (a) W. Nagata, T. Sugasawa, M. Narisada, T. Wakabayashi and Y. Hayase, J. Am. Chem. Soc., 1963, 85, 2342 CrossRef CAS; (b) W. A. Ayer, C. E. McDonald and G. G. Iverach, Tetrahedron Lett., 1963, 4, 1095 CrossRef; (c) R. A. Bell and R. E. Ireland, Tetrahedron Lett., 1963, 4, 269 CrossRef; (d) A. Abad, C. Agulló, A. C. Cuñat, I. de Alfonso Marzal, I. Navarro and A. Gris, Tetrahedron, 2006, 62, 3266 CrossRef CAS PubMed; (e) X.-Y. Liu, H. Cheng, X.-H. Li, Q.-H. Chen, L. Xu and F.-P. Wang, Org. Biomol. Chem., 2012, 10, 1411 RSC.
- (a) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS; (b) B. M. Trost, K. L. Sacchi, G. M. Schroeder and N. Asakawa, Org. Lett., 2002, 4, 3427 CrossRef CAS PubMed; (c) H. Sundén, R. Rios, Y. Xu, L. Eriksson and A. Córdova, Adv. Synth. Catal., 2007, 349, 2549 CrossRef; (d) D.-Q. Xu, A.-B. Xia, S.-P. Luo, J. Tang, S. Zhang, J.-R. Jiang and Z.-Y. Xu, Angew. Chem., Int. Ed., 2009, 48, 3821 CrossRef CAS PubMed; (e) X. Feng, Z. Zhou, R. Zhou, Q.-Q. Zhou, L. Dong and Y.-C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (f) H. Yang and R. G. Carter, J. Org. Chem., 2009, 74, 5151 CrossRef CAS; (g) H. Yang and R. G. Carter, Tetrahedron, 2010, 66, 4854 CrossRef CAS PubMed; (h) M. Bella, D. M. S. Schietroma, P. P. Cusella, T. Gasperi and V. Visca, Chem. Commun., 2009, 597 RSC; (i) D. M. S. Schietroma, M. R. Monaco, V. Visca, S. Insogna, J. Overgaard and M. Bella, Adv. Synth. Catal., 2011, 353, 2648 CrossRef CAS.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed.
- (a) J. Wang, S.-G. Chen, B.-F. Sun, G.-Q. Lin and Y.-J. Shang, Chem. – Eur. J., 2013, 19, 2539 CrossRef CAS PubMed; (b) R. Ding, B.-F. Sun and G.-Q. Lin, Org. Lett., 2012, 14, 4446 CrossRef CAS PubMed; (c) R. Ding, J.-G. Fu, G.-Q. Xu, B.-F. Sun and G.-Q. Lin, J. Org. Chem., 2014, 79, 240 CrossRef CAS PubMed; (d) X.-L. Wang, Y.-Y. Lu, J. Wang, X. Wang, H.-Q. Yao, G.-Q. Lin and B.-F. Sun, Org. Biomol. Chem., 2014, 12, 3562 RSC.
- (a) L. Duroure, T. Jousseaume, R. Araoz, E. Barre, P. Retailleau, L. Chabaud, J. Molgo and C. Guillou, Org. Biomol. Chem., 2011, 9, 8112 RSC; (b) P. Elsner, L. Bernardi, G. D. Salla, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2008, 130, 4897 CrossRef CAS PubMed.
- For selected examples of catalytic enantioselective conjugate addition reactions, see: (a) M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 5672 CrossRef CAS PubMed; (b) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119 CrossRef CAS PubMed; (c) H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS PubMed; (d) H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105 CrossRef CAS PubMed; (e) Y. Chi, N. A. Kopf and S. H. Gellman, J. Am. Chem. Soc., 2008, 130, 5608 CrossRef CAS PubMed.
- (a) G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281 CrossRef CAS PubMed; (b) T. M. Neubauer, T. van Leeuvan, D. Zhao, A. S. Lubbe, J. C. M. Kistemaker and B. L. Feringa, Org. Lett., 2014, 16, 4220 CrossRef CAS PubMed.
- (a) J. D. Roberts, W. T. Moreland and W. Frazer, J. Am. Chem. Soc., 1953, 75, 637 CrossRef CAS; (b) H. D. Holtz and L. M. Stock, J. Am. Chem. Soc., 1964, 86, 5183 CrossRef CAS; (c) K. Kumar, S. S. Wang and C. N. Sukenik, J. Org. Chem., 1984, 49, 665 CrossRef CAS.
- (a) Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 5375 CrossRef CAS PubMed; (b) L.-S. Shi, M. V. B. Reddy, H.-J. Yan, W.-L. Chang and H.-C. Lin, J. Chin. Chem. Soc., 2006, 53, 751 CAS.
- CCDC-1024512 (1e) contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1024512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00311j |
| This journal is © the Partner Organisations 2015 |