
Oxidative C(sp3)–H functionalization of acetonitrile and alkanes with allylic alcohols under metal-free conditions†
Xue-Qiang
Chu
,
Hua
Meng
,
You
Zi
,
Xiao-Ping
Xu
* and
Shun-Jun
Ji
*
Key Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou, 215123, People's Republic of China. E-mail: xuxp@suda.edu.cn; shunjun@suda.edu.cn; Fax: +86 512 65880307; Tel: +86-512-65880307
First published on 6th January 2015
Abstract
An efficient method for the construction of ketonitriles and α-aryl ketones by tert-butyl peroxybenzoate (TBPB) mediated direct alkylation of acetonitrile and alkanes with α,α-diaryl allylic alcohols has been developed. The reactions involve C(sp3)–H functionalization and radical addition/1,2-aryl migration processes under metal-free conditions.
Nitriles are versatile intermediates for the synthesis of amides, primary amines, amidines, ketones and carboxylic acids.1 In particular, ketonitriles are featured in the synthesis of various heterocycles and biologically active compounds,2 such as pyridinone derivatives,2a–e α-hydroxyketones,2f–h and piperidines.2i As a result, extensive research effort has been made to develop practical approaches for their facile construction.3–12
Among them, conjugate addition of a nucleophile to unsaturated nitriles represents one of the most important methods, which is problematic in the substrate compatibility and basic conditions.3 Various precursors, such as α-silylnitrile,4 α-bromonitrile,5 diazoacetonitrile,6 α-aminoacetonitrile,7 cyanoacetate salt8etc., have been applied to the C–C bond formation by introducing the cyanomethyl group but they are limited by prefunctionalization. In addition, strategies involving transition metal-catalyzed C–H activation of nitrile compounds have been well documented over the past few decades.9,10 However, catalytic C–H functionalization of simple alkylnitriles (such as acetonitrile, low acidity, pKa 31.1 in DMSO) remains rare.11 Recently, Liu,11b Li,11e Zhu,11g You11hetc. have reported elegant direct oxidative cyanomethylation reactions by incorporating the acetonitrile with alkene. From an eco-sustainable and atom economical standpoint, the direct utilization of simple substrates and transition metal-free processes would be more attractive, and further development of new type-reactions toward nitrile derivatives is still highly desirable.12
Recently, much attention has been paid to new methods for the C–C bond formation through free-radical-initiated C–H bond functionalization.13 Especially the selective activation of C(sp3)–H bonds in ethers, alcohols, amines, alkanes, and other activated compounds has achieved significant success.14,15 Since 2013, radical-mediated 1,2-migration reactions16 of α,α-diaryl allylic alcohols have been expanded to CF3,17 N3,18 POPh2,19 and alkyl20 radicals by Wu, Tu, Sodeoka, Han, and our group, and new Csp3–X (X = C, P, N) and Csp3–Csp2 bonds have been concomitantly constructed by this type rearrangement (Scheme 1a). Herein, we communicate our recent efforts to present the metal-free oxidative C(sp3)–H functionalization of acetonitrile with α,α-diaryl allylic alcohols, which is initiated with radical addition and terminated by a 1,2-aryl migration (Scheme 1b). The protocol is also efficient when simple alkanes were used as the raw materials instead of acetonitrile.
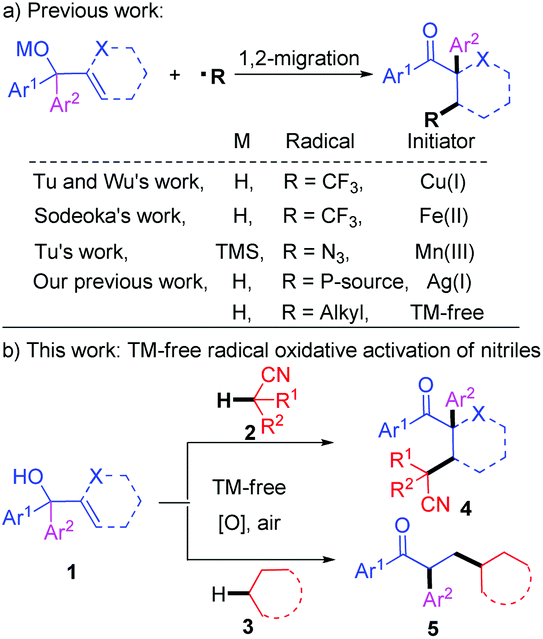 | ||
| Scheme 1 Radical-initiated tandem addition/1,2-migration reactions. | ||
The initial study was carried out with allylic alcohol 1a and acetonitrile 2a as the model substrates (Table 1). Fortunately, using TBPB (tert-butylperoxybenzoate) as the radical initiator without any redox catalyst21 at 120 °C for 12 h, the direct oxidative alkylation product 4aa could be obtained in 90% GC yield, along with an isolated yield of 87% (Table 1, entry 1). It was found that other oxidants such as DTBP, TBHP or CHP showed low reaction efficiency (Table 1, entries 2–4). Meanwhile, PhI(OAc)2 and K2S2O8 resulted in no product (Table 1, entries 5 and 6). Lower equivalent of acetonitrile in 1,2-dichloroethane or toluene reduced the yield dramatically (Table 1, entry 7). Further screening of the amount of oxidant (Table 1, entries 8–10), the reaction temperature (Table 1, entry 11) and the reaction time (Table 1, entry 12) revealed that allylic alcohol 1a with 2 equiv. TBPB in 3 mL of acetonitrile at 120 °C for 12 h would be the optimal conditions (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Time (h) | Temp. (°C) | GC-yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (3 mL), and oxidant (1–3 equiv.) (TBPB = tert-butylperoxybenzoate, DTBP = di-tert-butyl peroxide, TBHP = tert-butyl hydroperoxide (70% in aqueous solution), CHP = cumyl hydroperoxide) at 120 °C. b Yields were determined by GC with an internal standard. c Isolated yields. d 2a (10 equiv.) in 1,2-dichloroethane or toluene (3 mL). | ||||
| 1 | TBPB (2) | 12 | 120 | 90(87)c |
| 2 | DTBP (2) | 12 | 120 | 20 |
| 3 | TBHP (2) | 12 | 120 | <5 |
| 4 | CHP (2) | 12 | 120 | 0 |
| 5 | K2S2O8 (2) | 12 | 120 | 0 |
| 6 | PhI(OAc)2 (2) | 12 | 120 | <5 |
| 7 | TBPB (2) | 12 | 120 | Traced |
| 8 | TBPB (1.5) | 12 | 120 | 71 |
| 9 | TBPB (2.5) | 12 | 120 | 89 |
| 10 | — | 12 | 120 | 0 |
| 11 | TBPB (2) | 12 | 80 | Trace |
| 12 | TBPB (2) | 6 | 120 | 84 |
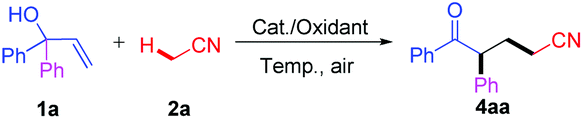
Encouraged by the above results, we then proceeded to explore the generality of this novel cascade oxidative addition/migration reaction to various α,α-diaryl allylic alcohols. As shown in Table 2, all of the symmetrical substrates with electron-withdrawing groups (F, Cl, Br, and CF3) were effectively converted to the corresponding ketonitriles 4 in moderate to excellent yields (Table 2, 4ba–da and 4ga). Although the electronic effect on the aromatic rings reduced the reactivity to some extent, the expected products 4ea (Me) and 4fa (OMe) were still furnished in 69% and 58% yields, respectively (Table 2, 4ea–fa). Subsequently, allylic alcohols bearing two different aryl groups were studied in order to obtain information on this type rearrangement. The application of asymmetric alcohol 1h provided an inseparable mixture in 72% total yield with a ratio of 3.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 determined by 1H NMR (Table 2, 4ha). In addition, preferential migration of the more electron-poor aryl groups was detected for substrates 1i–k and heterocycle-containing compound 1l (Table 2, 4ia–la). ortho-Substituted aryl rings 1m–1n migrated less effectively with the carbonyl derivatives 4ma–na being isolated in moderate yields (Table 2, 4ma–na). This chemoselective manner suggested that both the electronic character and the steric demand of the substituents affected the migration procedure. Based on this, the reaction is more likely to be a radical (“neophyl”) rearrangement route to the products.17–20 Furthermore, pentafluorophenyl 1o was almost inert under our conditions (Table 2, 4oa).
:1 determined by 1H NMR (Table 2, 4ha). In addition, preferential migration of the more electron-poor aryl groups was detected for substrates 1i–k and heterocycle-containing compound 1l (Table 2, 4ia–la). ortho-Substituted aryl rings 1m–1n migrated less effectively with the carbonyl derivatives 4ma–na being isolated in moderate yields (Table 2, 4ma–na). This chemoselective manner suggested that both the electronic character and the steric demand of the substituents affected the migration procedure. Based on this, the reaction is more likely to be a radical (“neophyl”) rearrangement route to the products.17–20 Furthermore, pentafluorophenyl 1o was almost inert under our conditions (Table 2, 4oa).
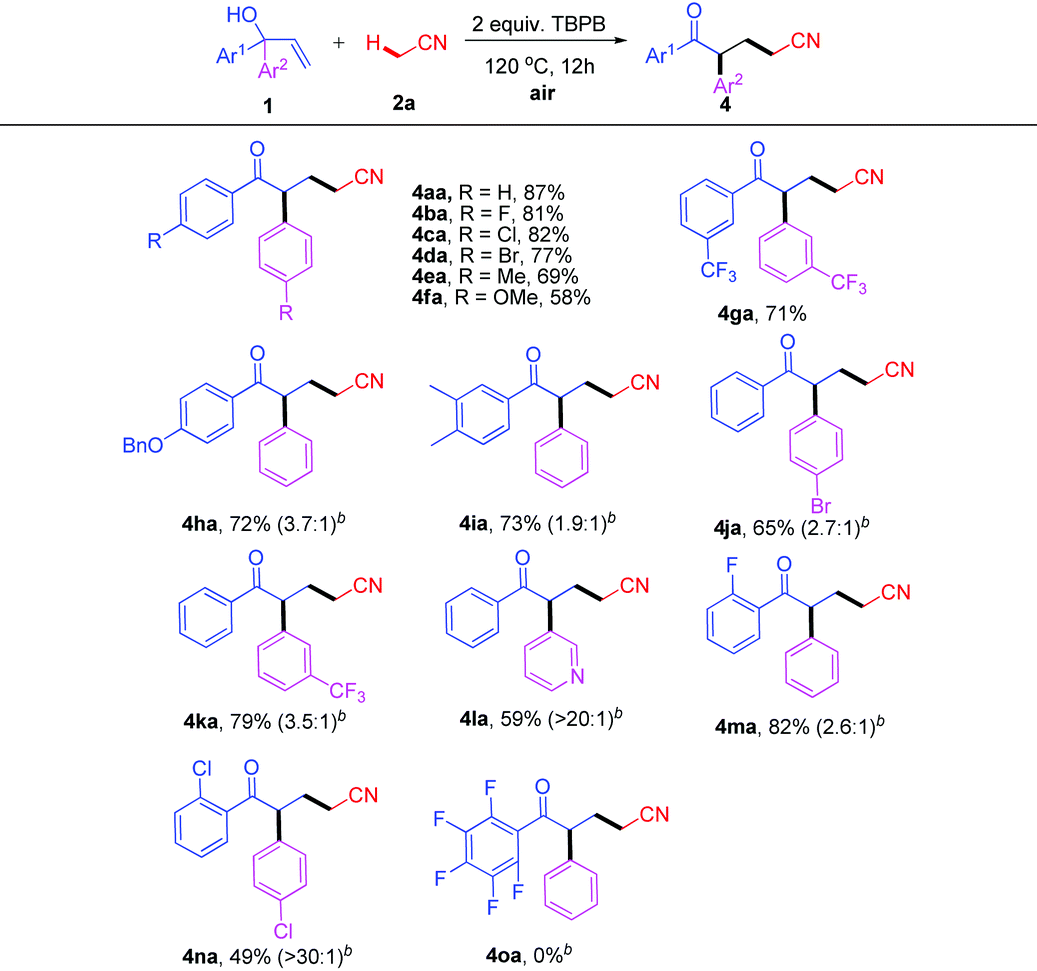
We also extended the direct oxidative cyanoalkylation to other types of allylic alcohols (Scheme 2). To our delight, when 1p–r with a 3,4-dihydro-2H-pyran motif were introduced to react with 2a, the reaction went smoothly under the stated conditions to afford products 4pa–ra, albeit in lower yields (29–33%). Regretably, this method was not suitable for the ring expansion of 9-vinyl-9H-fluoren-9-ol 1s.
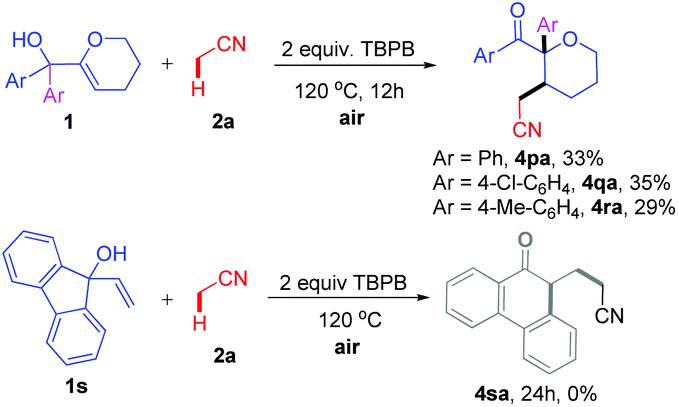 | ||
| Scheme 2 Reaction of allylic alcohols 1p–s with 2a. | ||
Next, other nitriles were also tested (Table 3). Cyclopropanecarbonitrile 2b underwent the oxidative C–H activation at the α-position to give the ketonitrile 4ab in a lower yield, probably due to the significant steric effect (Table 3, 4ab). Interestingly, α-substituted alkylnitriles 2c–e (Cl, Br, Ph) failed to participate in the TBPB-promoted addition reaction (Table 3, 4ac–ae). Additionally, activated nitriles were well tolerated, providing the corresponding products 4af–ah in 53–72% yields (Table 3, 4af–ah, the reactivity decreased as follows: X = CN > COOMe > PhCO).
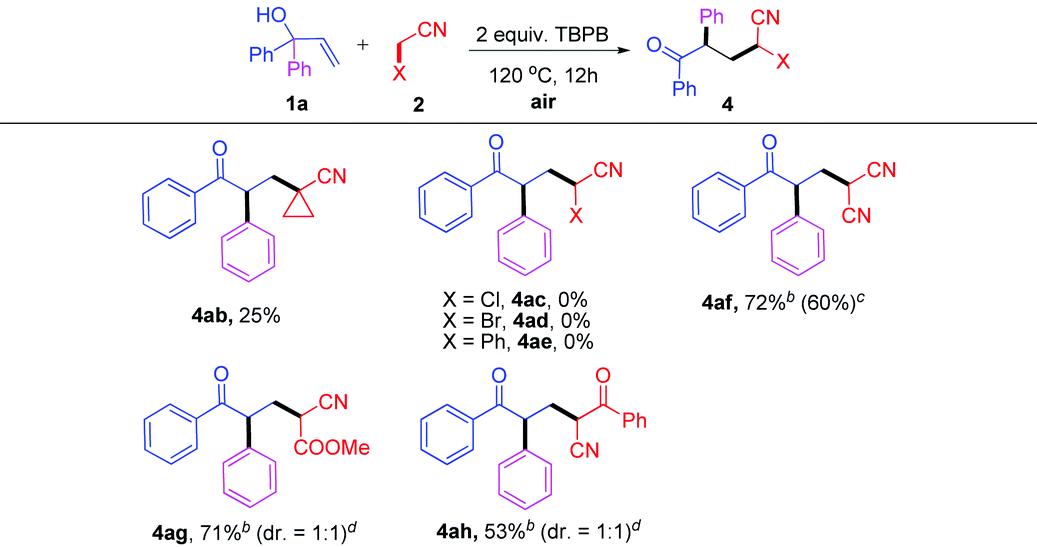
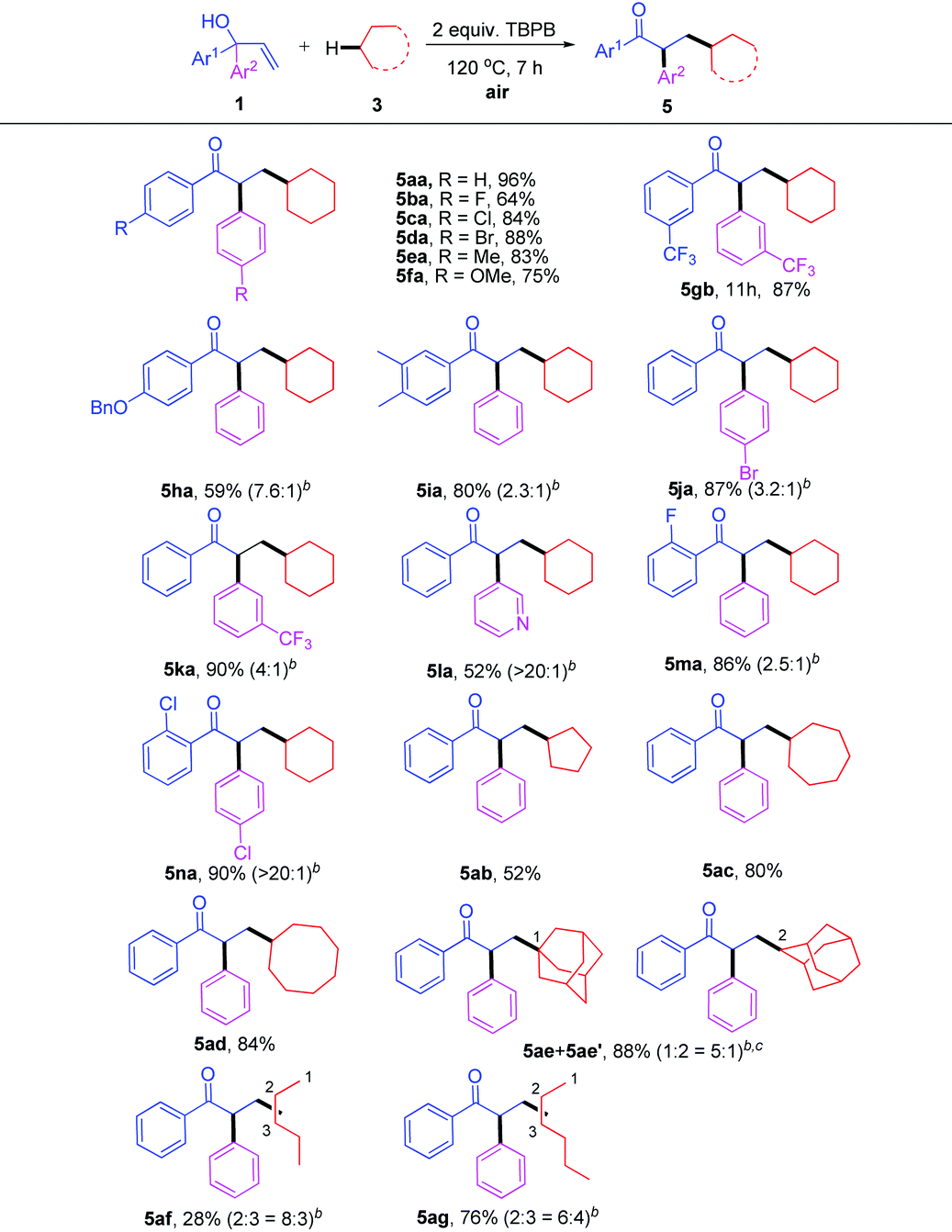
As a continuation on the free radical-initiated C(sp3)–H bond functionalization, we next turned our attention to the reaction of α,α-diaryl allylic alcohols with simple alkanes. The results are summarized in Table 4. A series of highly functionalized ketones was synthesized in moderate to excellent yields under the typical conditions (Table 4, 5aa–na). Apart from cyclic alkanes 3b–d, open-chain alkanes such as pentane 3f and hexane 3g could also act as effective substrates in this system (Table 4, 5ab–5ag).
The presented methodology can be conveniently used in the synthesis of complicated heterocycles 6 and 7 (Scheme 3). Dihydropyridone derivative 6 was obtained in 50% yield by intramolecular annulation of ketonitrile 4aa, which was an important class of biologically active relevant products.2a–c Moreover, 4aa could be reduced by LiAlH4 to give 2,3-diphenylpiperidine 7 in 79% yield.
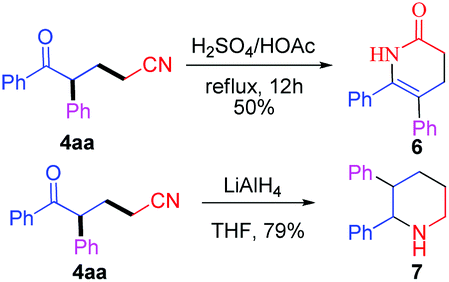 | ||
| Scheme 3 Further transformations of the ketonitrile. | ||
To define the possible reaction pathway, 2.0 equiv. of the radical-trapping reagent (TEMPO) was added, the reaction was completely suppressed, and only TEMPO-acetonitrile adduct was observed under the standard conditions (determined by LC-MS analysis), including that an acetonitrile radical was involved in the reaction process (Scheme 4). On the basis of previous studies19,20 and the results above, a possible mechanism was proposed in Scheme 5. Firstly, TBPB decomposed to give tert-butoxyl radical and benzoate radical at high temperature. Subsequently, hydrogen abstraction of 2a or 3a with tert-butoxyl radical (or benzoate radical) provided a cyanomethyl or alkyl radical I, which was added to allylic alcohol 1, thus affording an alkyl intermediate II. Next, intramolecular 5-ipso cyclization led to generate the spiro[2,5]octadienyl III,17a followed by 1,2-aryl migration to give radical species IV. Finally, the desired product 4 or 5 was delivered via further oxidation and deprotonation steps.17–20
 | ||
| Scheme 4 Mechanism studies. | ||
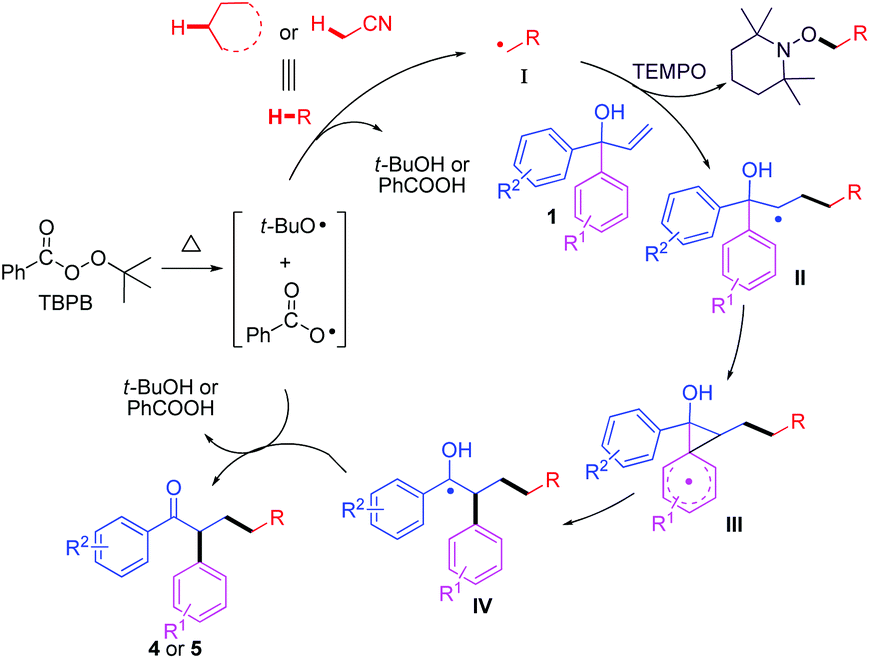 | ||
| Scheme 5 Possible mechanism. | ||
In conclusion, we have developed a novel and convergent alkylation reaction of acetonitrile and alkanes with allylic alcohols that involves C(sp3)–H functionalization and radical addition/1,2-aryl migration. The direct utilization of commercially available nitrile derivatives and simple alkanes under metal-free conditions represents a practical and straightforward route for access to a variety of highly functionalized ketones.
We gratefully acknowledge the Natural Science Foundation of China (no. 21172162 and 21372174), the Young National Natural Science Foundation of China (no. 21202113), the Ph.D. Programs Foundation of the Ministry of Education of China (2013201130004), Key Laboratory of Organic Synthesis of Jiangsu Province (KJS1211), PAPD, and Soochow University for financial support.
Notes and references
- (a) K. Friedrich and K. Wallenfels, The Chemistry of the Cyano Group, Wiley-Interscience, New York, 1970 Search PubMed; (b) R. J. H. Gregory, Chem. Rev., 1999, 99, 3649 CrossRef CAS PubMed.
- (a) Q.-S. Yu and A. Brossi, Heterocycles, 1988, 27, 1709 CrossRef CAS PubMed; (b) S. Wang, Y. Dong, X. Wang, X. Hu, J. O. Liu and Y. Hu, Org. Biomol. Chem., 2005, 3, 911 RSC; (c) L. Carles, K. Narkunan, S. Penlou, L. Rousset, D. Bouchu and M. A. Ciufolini, J. Org. Chem., 2002, 67, 4304 CrossRef CAS PubMed; (d) K. U. Krishnaraj and K. S. Devaky, Tetrahedron, 2014, 70, 6450 CrossRef CAS PubMed; (e) H. Asahrara, K. Muto and N. Nishiwaki, Tetrahedron, 2014, 70, 6522 CrossRef CAS PubMed; (f) Y. Yamamoto and D. Matsumi, Chem. Commun., 1998, 875 RSC; (g) G. Frey, H.-T. Luu, P. Bichovski, M. Feurer and J. Streuff, Angew. Chem., Int. Ed., 2013, 52, 7131 CrossRef CAS PubMed; (h) J. Streuff, M. Feurer, P. Bichovski, G. Frey and U. Gellrich, Angew. Chem., Int. Ed., 2012, 51, 8661 CrossRef CAS PubMed; (i) C. M. Pedersen and M. Bols, Tetrahedron, 2005, 61, 115 CrossRef CAS PubMed; (j) S. Dei, M. N. Romanelli, S. Scapecchi, E. Teodori, A. Chiarini and F. Gualtieri, J. Med. Chem., 1991, 34, 2219 CrossRef CAS; (k) A. S. Girgis, N. Mishriky, A. M. Farag, W. I. El-Eraky and H. Farag, Eur. J. Med. Chem., 2008, 43, 1818 CrossRef CAS PubMed.
- F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed.
- (a) L. Wu and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15824 CrossRef CAS PubMed; (b) A. Park and S. Lee, Org. Lett., 2012, 14, 1118 CrossRef CAS PubMed.
- (a) B. W. Yoo, S. K. Hwang, D. Y. Kim, J. W. Choi, J. J. Ko, K. I. Choi and J. H. Kim, Tetrahedron Lett., 2002, 43, 4813 CrossRef CAS; (b) Y. Yang, S. Tang, C. Liu, H. Zhang, Z. Sun and A. Lei, Org. Biomol. Chem., 2011, 9, 5343 RSC.
- Z. Yang, K. Son, S. Li, B. Zhou and J. Xu, Eur. J. Org. Chem., 2014, 6380 CrossRef CAS.
- G. Wu, Y. Deng, C. Wu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 10510 CrossRef CAS PubMed.
- R. Shang, D.-S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470 CrossRef CAS PubMed.
- For reviews, see: (a) G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2002, 41, 953 CrossRef CAS; (b) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS PubMed; (c) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676 CrossRef CAS PubMed; (d) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS PubMed.
- (a) D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9330 CrossRef CAS PubMed; (b) J. You and J. G. Verkade, Angew. Chem., Int. Ed., 2003, 42, 5051 CrossRef CAS PubMed; (c) J. You and J. G. Verkade, J. Org. Chem., 2003, 68, 8003 CrossRef CAS PubMed; (d) R. Shetty and K. K. Moffett, Tetrahedron Lett., 2006, 47, 8021 CrossRef CAS PubMed.
- (a) N. Kumagai, S. Matsunage and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 13632 CrossRef CAS PubMed; (b) T. Wu, X. Mu and G. Liu, Angew. Chem., Int. Ed., 2011, 50, 12578 CrossRef CAS PubMed; (c) Y. Kawato, N. Kumagai and M. Shibasaki, Chem. Commun., 2013, 49, 11227 RSC; (d) J. Shen, D. Yang, Y. Liu, S. Qin, J. Zhang, J. Sun, C. Liu, C. Liu, X. Zhao, C. Chu and R. Liu, Org. Lett., 2014, 16, 350 CrossRef CAS PubMed; (e) J.-L. Zhang, Y. Liu, R.-J. Song, G.-F. Jiang and J.-H. Li, Synlett, 2014, 1031 Search PubMed; (f) S. B. Lang, T. M. Locascio and J. A. Tunge, Org. Lett., 2014, 16, 4308 CrossRef CAS PubMed; (g) A. Bunescu, Q. Wang and J. Zhu, Chem. – Eur. J., 2014, 20, 14633 CrossRef CAS PubMed; (h) J. Li, Z. Wang, N. Wu, G. Gao and J. You, Chem. Commun., 2014, 50, 15049 RSC.
- For selected examples, see: (a) M. Jeganmohan and C.-H. Cheng, Chem. Commun., 2006, 2454 RSC; (b) Z. Jiang, Q. Huang, S. Chen, L. Long and X. Zhou, Adv. Synth. Catal., 2012, 354, 589 CrossRef CAS; (c) S. Lin, Y. Wei and F. Liang, Chem. Commun., 2012, 48, 9879 RSC; (d) D. Stephens, Y. Zhang, M. Cormier, G. Chavez, H. Arman and O. V. Larionov, Chem. Commun., 2013, 49, 6558 RSC; (e) S. Lin, L. Li, F. Liang and Q. Liu, Chem. Commun., 2014, 50, 10491 RSC; (f) N. R. Rondla, J. M. Ogilvie, Z. Pan and C. J. Douglas, Chem. Commun., 2014, 50, 8974 RSC; (g) X. Huang and N. Jiao, Org. Biomol. Chem., 2014, 12, 4324 RSC; (h) Z. Shu, Y. Zhou, Y. Zhang and J. Wang, Org. Chem. Front., 2014, 1, 1123 RSC; (i) T. Maji and J. A. Tunge, Org. Lett., 2014, 16, 5072 CrossRef CAS PubMed.
- For selected reviews, see: (a) K. C. Majumdar, P. K. Basu and P. P. Mukhopadhyay, Tetrahedron, 2004, 60, 6239 CrossRef CAS PubMed; (b) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed; (c) S. Chiba and H. Chen, Org. Biomol. Chem., 2014, 12, 4051 RSC; (d) F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194 RSC.
- For selected recent reviews, see: (a) S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937 RSC; (b) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (c) X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807 RSC.
- For selected recent examples of sp3 C–X (X = C, O, N, S) bond formation via sp3 C–H bond functionalization, see: (a) D. Liu, C. Liu, H. Li and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 4453 CrossRef CAS PubMed; (b) W.-T. Wei, M.-B. Zhou, J.-H. Fan, W. Liu, R.-J. Song, Y. Liu, M. Hu, P. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2013, 52, 3638 CrossRef CAS PubMed; (c) H. Wang, L.-N. Guo and X.-H. Duan, Chem. Commun., 2013, 49, 10370 RSC; (d) S.-L. Zhou, L.-N. Guo, S. Wang and X.-H. Duan, Chem. Commun., 2014, 50, 3589 RSC; (e) J.-J. Cao, X. Wang, S.-Y. Wang and S.-J. Ji, Chem. Commun., 2014, 50, 12892 RSC; (f) Z. Li, Y. Zhang, L. Zhang and Z.-Q. Liu, Org. Lett., 2014, 16, 382 CrossRef CAS PubMed; (g) J. Zhao, H. Fang, J. Han and Y. Pan, Org. Lett., 2014, 16, 2530 CrossRef CAS PubMed; (h) Y. Zhu and Y. Wei, Chem. Sci., 2014, 5, 2379 RSC; (i) B. D. Barve, Y.-C. Wu, M. El-Shazly, M. Korinek, Y.-B. Cheng, J.-J. Wang and F.-R. Chang, Org. Lett., 2014, 16, 1912 CrossRef CAS PubMed; (j) X. Zhang, M. Wang, P. Li and L. Wang, Chem. Commun., 2014, 50, 8006 RSC; (k) S. K. Rout, S. Guin, W. Ali, A. Gogoi and B. K. Patel, Org. Lett., 2014, 16, 3086 CrossRef CAS PubMed.
- (a) C. Rüchardt and R. Hecht, Chem. Ber., 1965, 98, 2471 CrossRef; (b) M. Tada, K. Inoue and M. Okabe, Bull. Chem. Soc. Jpn., 1983, 56, 1420 CrossRef CAS; (c) A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649 CrossRef CAS.
- (a) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962 CrossRef CAS PubMed; (b) H. Egami, R. Shimizu, Y. Usui and M. Sodeoka, Chem. Commun., 2013, 49, 7346 RSC; (c) Z.-M. Chen, W. Bai, S.-H. Wang, B.-M. Yang, Y.-Q. Tu and F.-M. Zhang, Angew. Chem., Int. Ed., 2013, 52, 9781 CrossRef CAS PubMed; (d) X. Liu and X. Wu, Synlett, 2013, 1882 CAS.
- Z.-M. Chen, Z. Zhang, Y.-Q. Tu, M.-H. Xu, F.-M. Zhang, C.-C. Li and S.-H. Wang, Chem. Commun., 2014, 50, 10805 RSC.
- X.-Q. Chu, Y. Zi, H. Meng, X.-P. Xu and S.-J. Ji, Chem. Commun., 2014, 50, 7642 RSC.
- (a) X.-Q. Chu, H. Meng, Y. Zi, X.-P. Xu and S.-J. Ji, Chem. Commun., 2014, 50, 9718 RSC; (b) X.-Q. Chu, H. Meng, Y. Zi, X.-P. Xu and S.-J. Ji, Chem. – Eur. J., 2014, 20, 17198 CrossRef CAS PubMed; (c) R.-J. Song, Y.-Q. Tu, D.-Y. Zhu, F.-M. Zhang and S.-H. Wang, Chem. Commun., 2015, 51, 749 RSC. During the manuscript preparation, two free-radical-promoted 1,2-aryl migration reactions of allylic alcohols with acetonitrile and alkane were reported: (d) J. Zhao, H. Fang, R. Song, J. Zhou, J. Han and Y. Pan, Chem. Commun., 2015, 51, 599 RSC; (e) Y. Li, B. Liu, H.-B. Li, Q. Wang and J.-H. Li, Chem. Commun., 2015, 51, 1024 RSC.
- (a) Y.-M. Li, X.-H. Wei, X.-A. Li and S.-D. Yang, Chem. Commun., 2013, 49, 11701 RSC; (b) J.-W. Zeng, Y.-C. Liu, P.-A. Hsieh, Y.-T. Huang, C.-L. Yi, S. S. Badsara and C.-F. Lee, Green Chem., 2014, 16, 2644 RSC; (c) T. Shen, Y. Yuan and N. Jiao, Chem. Commun., 2014, 50, 554 RSC; (d) J.-J. Cao, T.-H. Zhu, S.-Y. Wang, Z.-Y. Gu, X. Wang and S.-J. Ji, Chem. Commun., 2014, 50, 6439 RSC; (e) W. Sha, J.-T. Yu, Y. Jiang, H. Yang and J. Cheng, Chem. Commun., 2014, 50, 9179 RSC; (f) J. Zhao, H. Fang, J. Han, Y. Pan and G. Li, Adv. Synth. Catal., 2014, 356, 2719 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00314d |
| This journal is © the Partner Organisations 2015 |