
Deuterative cyclization of sulfanyl 1,6-diynes: complete and monodeuteration of functional groups on heterocycles†‡
Yukiteru
Ito
and
Mitsuhiro
Yoshimatsu
*
Department of Chemistry, Faculty of Education, Gifu University, Yanagido 1-1, Gifu 501-1193, Japan. E-mail: yoshimae@gifu-u.ac.jp
First published on 20th January 2015
Abstract
Regioselective H/D exchange reaction of functional groups on heterocycles proceeded via a transition metal-free reductive cyclization of sulfanyl 1,6-diynes using sodium borodeuteride/ethanol-D1. Both alkoxide- and aryloxide-mediated cyclizations and amination–cyclization resulted in the deuteration of functional groups with high deuterium incorporation. Reductive cyclization using sodium borodeuteride/ethanol exclusively afforded the monodeuterated furans and pyrroles in good yields.
1. Introduction
The H/D exchange reaction of N- and O-containing heterocycles is of considerable interest in the preparation of isotopically labeled compounds for investigating reaction mechanisms in organic chemistry and for pharmacokinetics and metabolism studies in drug development.1 Two conceptually different methodologies have been developed for H/D exchange reactions. One methodology is pH-dependent H/D exchange reactions (classical methods) mediated or catalyzed by acids or bases.2 As shown in Fig. 1, the acid- or base-catalyzed reactions deuterated through a simple autoprotic equilibrium when the reaction mixtures are heated using a conventional method (i.e., thermal and/or microwave methods); however, pH-dependent H/D exchange reactions require a large excess of the deuterium source (e.g., D2O, D2 or alcohol-D), long reaction times, and high reaction temperatures.3 In many cases, procedures must be repeated to achieve a high degree of deuteration (%DD).4 Another methodology is a metal-catalyzed H/D exchange reaction, which involves comparably mild reaction conditions and is highly tolerant toward numerous functional groups.5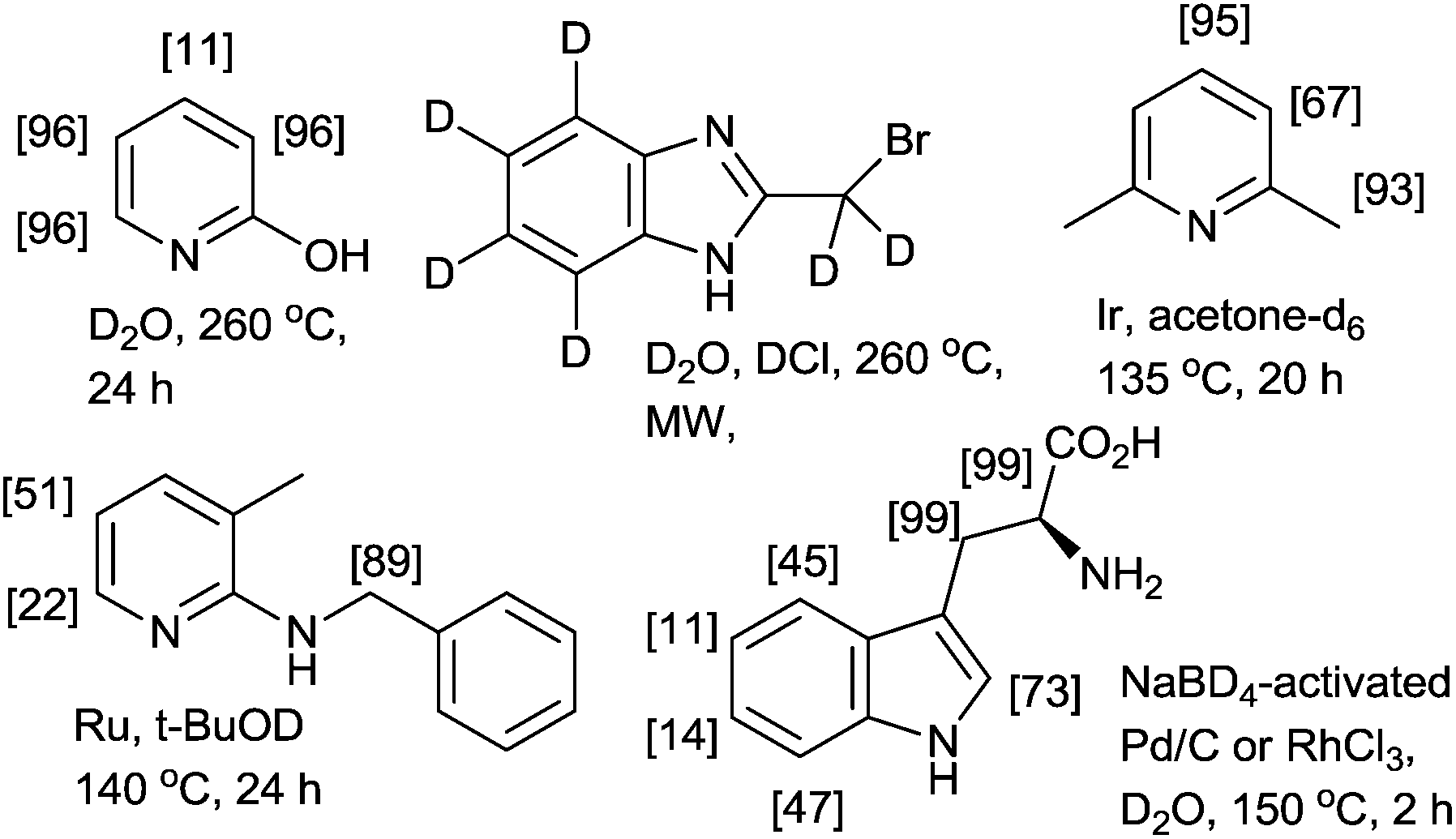 | ||
| Fig. 1 Deuteration of N-containing hetaryl compounds. | ||
Bergman and co-workers demonstrated iridium-catalyzed H/D exchange reactions in acetone-D6, which are suitable for the specific deuteration of aliphatic and nonfunctionalized aromatic compounds.6 As is evident in recently reported methods using transition-metal catalysts, regioselective deuteration of the aliphatic C–H bonds on heterocycles are difficult, even when excellent C–H-activating metals, such as ruthenium,1c,7 palladium,8 platinum9 and rhodium are used (Fig. 1).10 Therefore, the development of facile, rapid, and efficient protocols for the deuteration of N-containing heterocycles would represent a substantial advancement.
We recently reported the transition-metal-free reductive cyclizations of sulfanyl 1,6-diynes using sodium borohydride in ethanol.11 According to the reaction mechanism, the unique metal-free cyclization comprises three processes: (i) the base-promoted alkyne–allene isomerization–protonation of diyne (i.e., autoprotic equilibrium); (ii) the intramolecular cyclization of a bis-allene intermediate; and (iii) the addition of a hydride (i.e., the nucleophilic addition of hydride). Further studies have demonstrated that the use of alcohol-D1 instead of solvents and sodium borodeuteride instead of reducing agents lead to the protocol for complete deuteration–cyclization of heterocycles through the base-promoted H/D exchange reactions. Furthermore, the use of a non-deuterated alcohol led to alkyne–allene isomerization and protonation to afford the mono-deuterated heterocycles by the nucleophilic addition of a deuteride. Here, we report the regioselective deuteration–cyclization of sulfanyl 1,6-diynes leading to the completely deuterated- and mono-deuterated pyrroles and furans.
2. Results and discussion
Using our previously reported reductive cyclization conditions,11 we examined the reductive cyclization of 1,6-diyne 1 using sodium borodeuteride (3 equiv.) and DBU (3 equiv.) in ethanol-D1 at 78 °C for 15 min (Scheme 1). Through quenching with H2O, we obtained product 2a in 75% yield (Scheme 1). The 1H NMR spectral data of 2a revealed two very small peaks assigned to methylene at δ 3.80 ppm and methyl protons at δ 1.97 ppm, which should also be observed in the 1H NMR spectrum of the corresponding non-deuterated pyrrole (Fig. 2). The deuterium incorporation of each proton was determined as CD2 (92%) and CD3 (93%) on the basis of the intensities of an external standard (1,4-dioxane). The 2H NMR spectrum also showed two corresponding peaks at δ 1.98 and 3.82 ppm that were assigned to methyl and methylene groups, respectively; however, it did not show two peaks due to 2-H and 5-H on pyrrole. In the mass spectrum, five deuterium atoms were observed for 2a obtained using this method. Furthermore, the regioselectivity of deuteration was excellent, and deuterium was selectively introduced only onto the functional groups of the pyrrole. | ||
| Scheme 1 Initial study for deuteration–cyclization. | ||
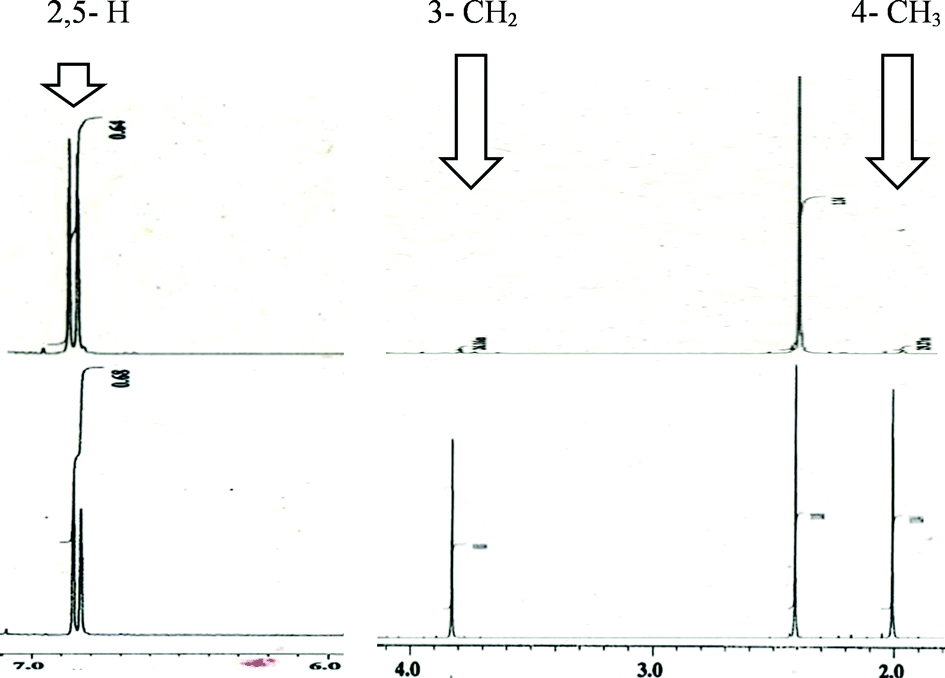 | ||
| Fig. 2 NMR spectral data of the compound 2a and that of the corresponding no-deuterated compound. | ||
After obtaining the initial results of the unique deuterative cyclization of 1,6-diynes, we investigated the substrate scope of this reaction. Although the chemical yields of products 2b and 2c were moderate to low, deuterium was successfully incorporated into their functional groups. In addition, their regioselectivity was completely controlled under the experimental conditions. We also performed the deuteration–cyclization of O-tethered 1,6-diynes to afford the deuterated furans in 85–95%DD (Fig. 3).
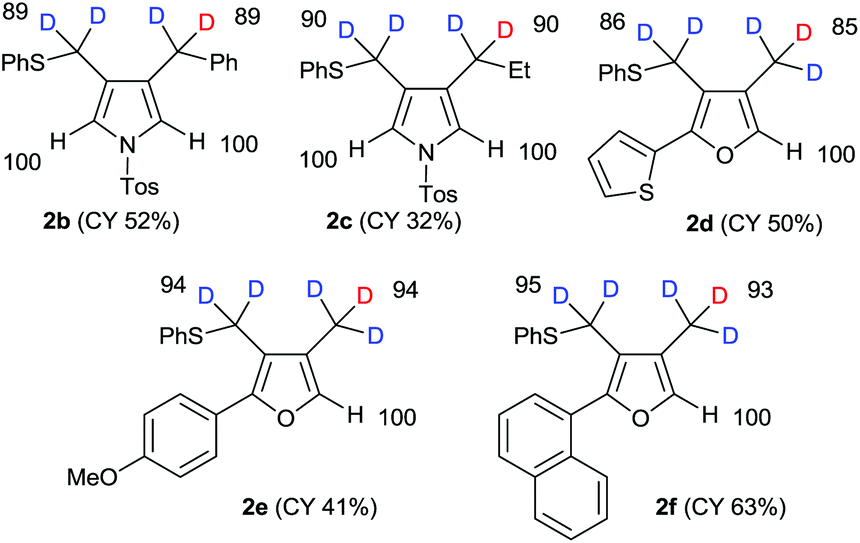 | ||
| Fig. 3 Scope and limitations of reductive cyclization of 1 using NaBD4/EtOD. | ||
Next, we focussed our attention on the deuterative cyclization mediated by other nucleophiles such as alkoxides and aryloxides, which were reported a few years ago.12 Treatment of 1 with sodium ethoxide in ethanol-D1 at room temperature produced 4-ethoxymethylpyrrole 3a at a 56% yield. We observed the deuterium incorporation of each proton in the 1H NMR spectrum, as shown in Scheme 2. We observed the 96% deuterium incorporation of both the 3- and 4-methylene groups and found that the acidic pyrrole protons were not deuterated at all. In addition, we conducted a DBU-mediated cyclization reaction in ethanol-D1. Deuterium incorporation of this product had almost the same result; however, the chemical yield was very low.
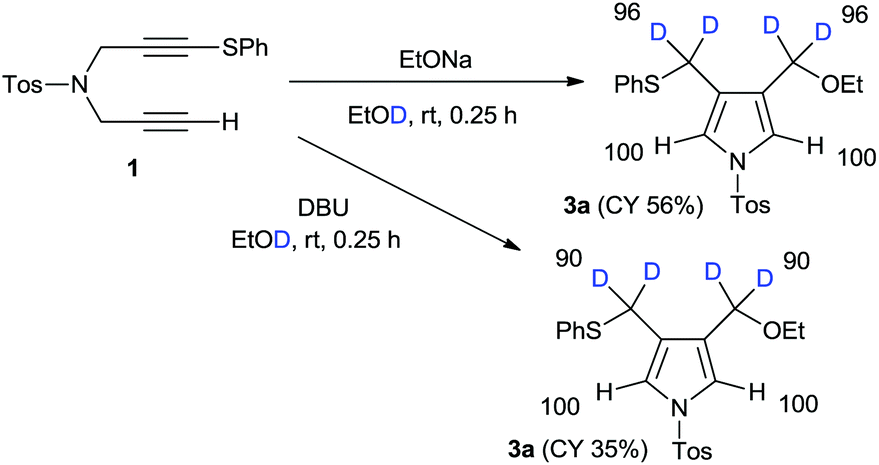 | ||
| Scheme 2 Alkoxide-mediated cyclization using RONa/ROD. | ||
The results of the substrate scope of alkoxide-mediated deuterative cyclizations of 1,6-diynes are summarized in Fig. 4. Sodium methoxide and ethoxide reacted with similar substrates to give pyrrole 3b and furans 3c–d with excellent deuterium incorporation. The reactions with aryloxide, generated in situ by treatment with sodium hydride in ethanol-D1, also afforded aryloxymethylpyrrole 3e–g with highly regioselective deuteration. The classical pH-dependent methods require repetitive cycles, under high thermal conditions, and long reaction times to achieve high deuterium incorporation; however, our practical protocol was observed to proceed in a single cycle, at low temperature, within 10–15 min. Furthermore, the deuteration of each substrate was exclusively regioselective.
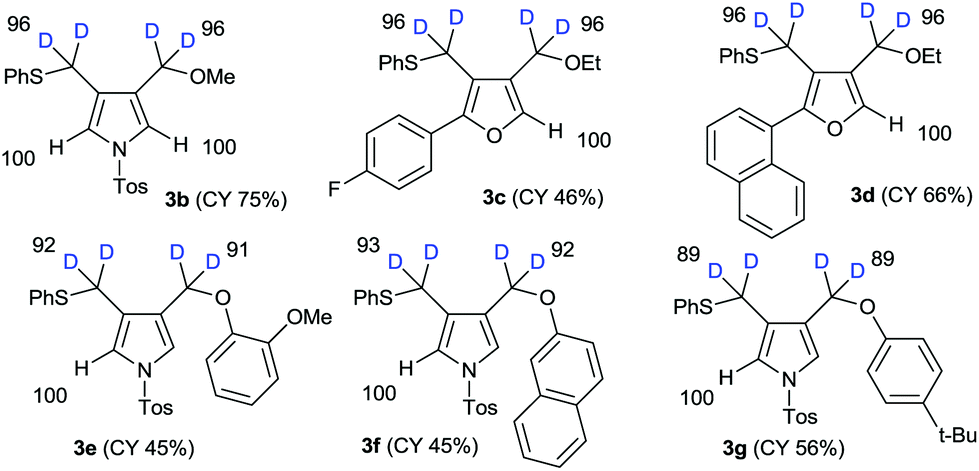 | ||
| Fig. 4 Scope and limitation of alkoxide-mediated cyclization. | ||
With our successful results for high deuterium incorporation into both alkoxymethyl- and aryloxymethylpyrroles and furans, we attempted the amination–cyclization of 1,6-diynes in the presence of deuterium sources.13 The introduction of amine functional groups is an important process in drug development. On the basis of our previous work on the nickel–palladium-catalyzed amination–cyclization of 1,6-diynes, we conducted the reactions in this study using the secondary amine-D1, which were prepared via the usual method involving R1R2NH/NaH/D2O,14 and a DMSO–D2O mixed solvent. The reaction of 1 with diethylamine-D1 in the presence of mixed catalysts (i.e., hexafluoroacetylacetonate nickel(II) hydrate, bis(triphenylphosphine)palladium(II) chloride) in DMSO–D2O was examined. The regioselectivity of deuteration in the case of product 4a was observed to be relatively high; however, the deuterium incorporation was relatively low because of water contamination of the nickel-based-catalyst. In the case of 1,4-benzothiazin-3-one, the H/D exchange reaction on the active methylene group adjacent to the sulfur atom in 55%DD was observed. In the reaction with imidazole, the H/D exchange on imidazole was not observed. The different reactivity between imidazole and benzimidazole is not clear. Similar results were obtained for furan syntheses (Scheme 3).
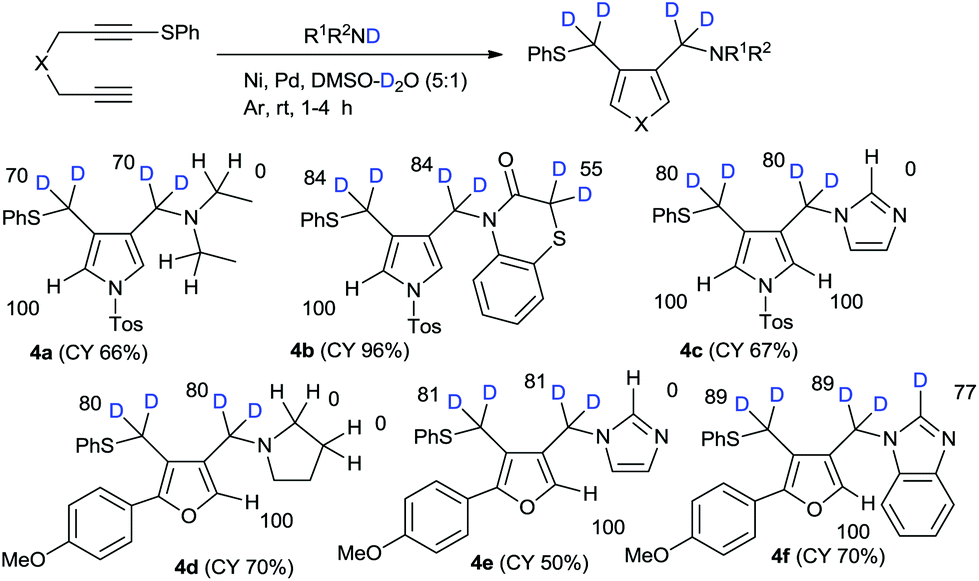 | ||
| Scheme 3 Amination–cyclization of 1,6-diynes with R1R2ND/DMSO/D2O. | ||
A plausible mechanism for the base-promoted deuterative cyclization of 1,6-diynes is shown in Scheme 4. The classical H/D exchange reaction occurs through an autoprotic equilibrium pathway under thermal or microwave conditions; however, in our system, the reaction was completed at low temperature and with a short reaction time to give the deuterated heterocycles in a regioselective manner and with high deuterium incorporation because of the strong substituent effect of the sulfur functional group, which facilitated alkyne–allene isomerization–deuteration. On the basis of our previous work, the sulfur-substituted 1,6-diynes should easily undergo cyclization during isomerization–protonation under basic conditions via anionic 6, monodeuterated 7,15 alleneyne 8 and bis(allene) 9 to give the key cationic intermediate 10. In the reactions with sodium borodeuteride, a deuteride would attack the exo-methylene carbon of 10 to give the product-D4 (R = Et or Ph) or -D5 (R = D) 11. On the other hand, the adduct-D412 (Nu = OR, OAr, NR1R2) was obtained in the presence of nucleophiles such as alkoxides, aryloxides and amines. Given these results, our interest turned towards the mono-deuteration–cyclization of 1,6-diynes using sodium borodeuteride/ethanol.
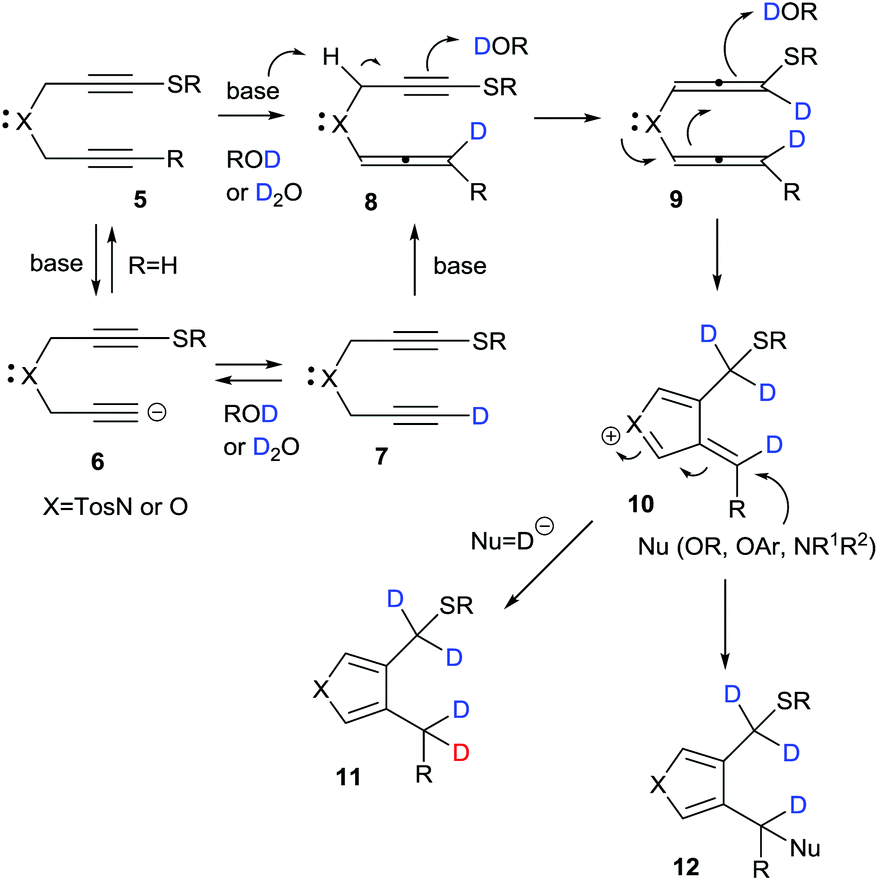 | ||
| Scheme 4 Plausible reaction mechanism for deuteration–cyclization of 1,6-diynes. | ||
In fact, the reaction of 1 with sodium borodeuteride in ethanol afforded the mono-deuterated pyrrole 13a in good yield (Scheme 5). All the spectral data show that the product was regioselectively deuterated at the methyl group. Furthermore, only one deuteride was introduced and its deuterium incorporation was as high as 88%DD. The chemical yields of the reactions of both the phenyl- and ethyl-substituted 1,6-diynes were low; however, both the regioselectivities and the deuterium incorporations were excellent. Similarly, the mono-deuterated furan derivatives were obtained in good yields.
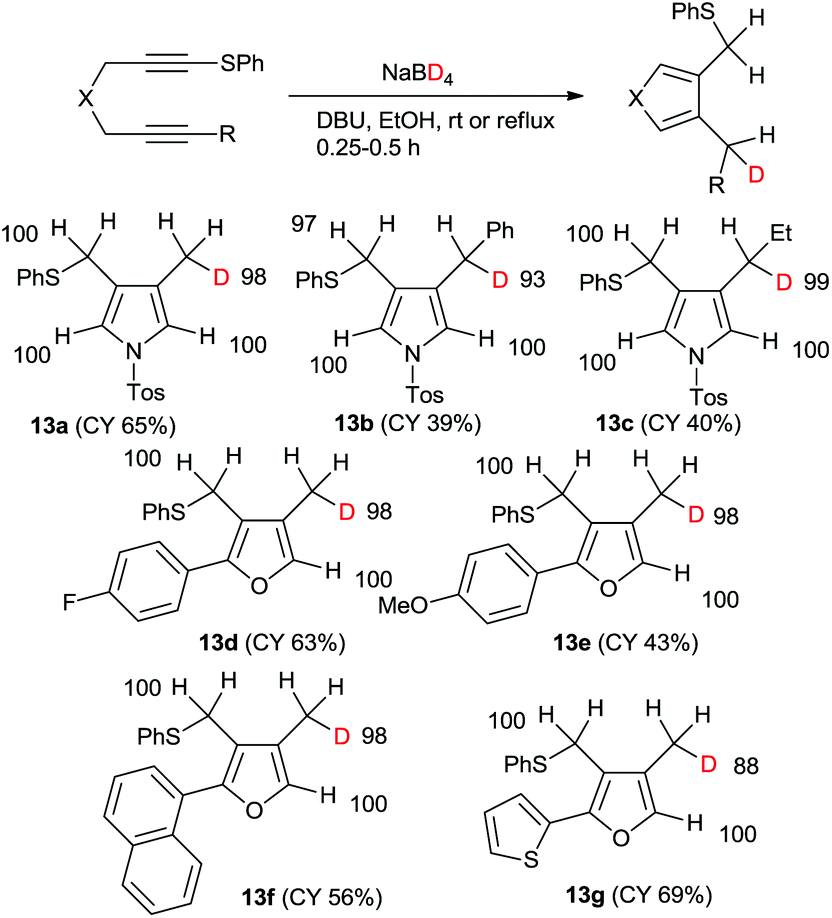 | ||
| Scheme 5 Mono-deuteration–cyclization of sulfanyl 1,6-diynes with NaBD4–EtOH. | ||
3. Conclusion
In summary, we have developed practical and efficient deuterative cyclization reactions of sulfanyl 1,6-diynes, leading to N- and O-containing heterocycles. Distinct from other common deuteration methods that involve autoprotic equilibrium processes or transition-metal-catalyzed C–H activations, our complete deuteration during the formation of heterocycles proceeded regioselectively at the functional groups of heterocycles to give the deuterated products with excellent deuterium incorporation in moderate to high chemical yields. Furthermore, the deuteration of heterocycles was not observed. Our useful protocol is also applicable to mono-deuteration–cyclization to give pyrroles-D1 and furans-D1, exclusively.4. Experimental
4.1 Typical procedure for complete deuterative cyclization of 1,6-diynes
To an EtOD (1.0 mL) solution of sodium borodeuteride (17.7 mg, 0.42 mmol) and DBU (64.2 mg, 0.42 mmol) was added 4-methyl-N-[3-(phenylthio)-2-propyn-1-yl]-N-(2-propyn-1-yl)benzenesulfonamide (1) (50 mg, 0.14 mmol) under an Ar atmosphere. The reaction mixture was refluxed for 15 min. The cooled mixture was poured into water (50 mL). The organic layer was separated and the aqueous layer was extracted with AcOEt. The combined organic layer was dried over MgSO4. The solvent was removed under reduced pressure. The residue was purified by preparative TLC on silica gel eluting with AcOEt–n-hexane (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 4-(methyl-d3)-3-(phenylsulfanylmethyl-d2)-1-(4-methylphenylsulfonyl)-1H-pyrrole (2a) (38.0 mg, 75%) as white powders.
:1) to give 4-(methyl-d3)-3-(phenylsulfanylmethyl-d2)-1-(4-methylphenylsulfonyl)-1H-pyrrole (2a) (38.0 mg, 75%) as white powders.
4.2 Typical procedure for monodeuteration–cyclization of 1,6-diynes
To an EtOH (1.0 mL) solution of DBU (64.2 mg, 0.42 mmol) and sodium borodeuteride (17.7 mg, 0.42 mmol) was added 4-methyl-N-[3-(phenylthio)-2-propyn-1-yl]-N-(2-propyn-1-yl)benzenesulfonamide (1) (50 mg, 0.14 mmol) under an Ar atmosphere. The reaction mixture was stirred under reflux conditions for 15 min. The work-up procedure gave 4-(methyl-d1)-3-(phenylsulfanylmethyl)-1-(4-methylphenylsulfonyl)-1H-pyrrole (13a) (33.0 mg, 65%) as white powder.Acknowledgements
We thank Prof. Hironao Sajiki and Prof. Yasunari Monguchi (Gifu Pharmaceutical University) for their kind advice to measure the 2H NMR spectrum.Notes and references
- (a) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 CrossRef CAS PubMed; (b) B. A. Arndtsen and R. G. Bergman, Science, 1995, 270, 1970 CAS; (c) G. Pieters, C. Taglang, E. Bonnefille, T. Gutmann, C. Puente, J.-C. Berthet, C. Dugave, B. Chaudret and B. Rousseau, Angew. Chem., Int. Ed., 2014, 53, 230 CrossRef CAS PubMed.
- Acids or bases: (a) N. H. Werstiuk and C. Ju, Can. J. Chem., 1989, 67, 5 CrossRef CAS; (b) E. Leppälä and K. Wähälä, J. Labelled Compd. Radiopharm., 2004, 47, 25 CrossRef; (c) S. Vaidyanathan and B. W. Surber, Tetrahedron Lett., 2005, 46, 5195 CrossRef CAS; (d) U. Schöllkopf, W. Hartwig and U. Groth, Angew. Chem., Int. Ed. Engl., 1979, 18, 863 CrossRef; (e) U. Schöllkopf, U. Groth and C. Deng, Angew. Chem., Int. Ed. Engl., 1981, 20, 798 CrossRef; (f) Y. Hirono, K. Kobayashi, M. Yonemoto and Y. Kondo, Chem. Commun., 2010, 46, 7623 RSC.
- (a) K. Wähälä and S. Rasku, Tetrahedron Lett., 1997, 38, 7287 CrossRef; (b) S. Rasku and K. Wähälä, Tetrahedron, 2000, 56, 913 CrossRef CAS; (c) K. Liu, J. Williams, H. Lee, M. M. Fitzgerald, G. M. Jensen, D. B. Goodin and A. E. McDermott, J. Am. Chem. Soc., 1998, 120, 10199 CrossRef CAS.
- (a) T. Junk and W. J. Catallo, Tetrahedron Lett., 1996, 37, 3445 CrossRef CAS.
- Recent articles for C–H activation of heteroaromatic compounds: (a) S. Ma, G. Villa, P. S. Thuy-Boun, A. Homs and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 734 CrossRef CAS PubMed; (b) D. R. Armstrong, J. A. Garden, A. R. Kennedy, S. M. Leenhouts, R. E. Mulvey, P. O'Keefe, C. T. O'Hara and A. Steven, Chem. – Eur. J., 2013, 19, 13492 CrossRef CAS PubMed; (c) L. Wang and L. Ackermann, Chem. Commun., 2014, 50, 1083 RSC; (d) Y. Yang and S. L. Buchwald, Angew. Chem., Int. Ed., 2014, 53, 8677 CrossRef CAS PubMed; (e) M. Zhan, H. Jiang, X. Pang, T. Zhang, R. Xu, L. Zhao, Y. Liu, Y. Gong and Y. Chen, Tetrahedron Lett., 2014, 55, 5070 CrossRef CAS; (f) M. Pohjoispää, R. Mera-Adsme, D. Sundholm, S. Heikkinen, T. Hase and K. Wähälä, J. Org. Chem., 2014, 79, 10636 CrossRef PubMed; (g) R. Salter, J. Labelled Compd. Radiopharm., 2010, 53, 645 CrossRef CAS.
- (a) J. M. Buchanan, J. M. Stryker and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 1537 CrossRef CAS; (b) J. T. Golden, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 5837 CrossRef CAS PubMed; (c) S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092 CrossRef CAS PubMedM. B. Skaddan, C. M. Yung and R. G. Bergman, Org. Lett., 2004, 6, 11 CrossRef CAS PubMed; C. M. Yung, M. B. Skaddan and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 13033 CrossRef PubMed.
- Ru: (a) M. H. G. Prechtl, M. Holscher, Y. Ben-David, N. Theyssen, R. Loschen, D. Milstein and W. Leitner, Angew. Chem., Int. Ed., 2007, 46, 2269 CrossRef CAS PubMed; (b) B. Grobell, M. Schnurch and M. D. Mihovilovic, J. Org. Chem., 2012, 77, 4432 CrossRef PubMed.
- Pd: (a) H. Sajiki, H. Esaki, F. Aoki, T. Maegawa and K. Hirota, Synlett, 2005, 1385 CrossRef CAS; (b) H. Esaki, F. Aoki, T. Maegawa, K. Hirota and H. Sajiki, Heterocycles, 2005, 66, 361 CrossRef CAS; (c) V. Derdau and J. Atzrodt, Synlett, 2006, 1918 CrossRef CAS; (d) Y. Fujiwara, H. Iwata, Y. Sawama, Y. Monguchi and H. Sajiki, Chem. Commun., 2010, 46, 4977 RSC.
- M. Yamamoto, K. Oshima and S. Matsubara, Heterocycles, 2006, 67, 353 CrossRef CAS.
- Rh: (a) V. Derdau, J. Atzrodt, J. Zimmermann, C. Kroll and F. Bruckner, Chem. – Eur. J., 2009, 15, 10397 CrossRef CAS PubMed.
- Y. Ito and M. Yoshimatsu, Chem. Lett., 2014, 43, 1758 CrossRef.
- (a) M. Yoshimatsu, H. Watanabe and E. Koketsu, Org. Lett., 2010, 12, 4192 CrossRef CAS PubMed; (b) N. Takahashi, Y. Nagase, G. Tanabe, O. Muraoka and M. Yoshimatsu, Tetrahedron, 2012, 68, 1566 CrossRef CAS.
- H. Nagata, Y. Sugimoto, Y. Ito, M. Tanaka and M. Yoshimatsu, Tetrahedron, 2014, 70, 1306 CrossRef CAS.
- (a) M. Rubina and V. Gevorgyan, J. Am. Chem. Soc., 2001, 123, 11107 CrossRef CAS PubMed; (b) G. Wallerberg and P. Haake, J. Org. Chem., 1981, 46, 43 CrossRef CAS; (c) A. Bruha and J. R. Kincaid, J. Am. Chem. Soc., 1988, 110, 6006 CrossRef CAS PubMed.
- (a) S. P. Bew, G. D. Hiatt-Gipson, J. A. Lovell and C. Poullain, Org. Lett., 2012, 14, 456 CrossRef CAS PubMed; (b) Y. Yabe, Y. Sawama, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2013, 19, 484 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization data and 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c4qo00333k |
| This journal is © the Partner Organisations 2015 |