
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boronic esters of corannulene: potential building blocks toward icosahedral supramolecules†
Sara
Da Ros
,
Anthony
Linden
,
Kim K.
Baldridge
* and
Jay S.
Siegel
*
Department of Chemistry, University of Zurich, CH-8057 Zurich, Switzerland. E-mail: dean_spst@tju.edu.cn
First published on 24th March 2015
Abstract
Direct iridium-catalyzed multi-borylation provides a valuable tool for the symmetric functionalization of various polycyclic aromatic hydrocarbons, inter alia, regular fivefold derivatization of corannulene. In this paper, highly efficient microwave-assisted synthesis of 1,3,5,7,9-(Bpin)5-corannulene is reported, resulting in a significant decrease in reaction time compared to the routine bench-top preparation. In addition, conversion to more reactive boron species, such as the corresponding pentatrifluoroborate and pentaboronic acid, was realized under mild conditions in excellent yields. 1,3,5,7,9-corannulene pentaboronic acid gave further access to a series of boronic esters of corannulene via simple alcohol exchange. This convenient methodology to 1,3,5,7,9-corannulene pentaboronic acid portends its ability to serve as a key building block for formation of icosahedral supramolecules, alone or together with suitable bridging ligands.
Introduction
Significant effort has been invested in derivatization of polycyclic aromatic hydrocarbons, such as corannulene (1), in order to introduce functionality and provide building blocks for more complex architectures.1 Due to its fivefold symmetry, 1 can be used as a suitable core element for construction of higher-order supramolecules, such as host–guest complexes,2 dendrimers,3 and liquid crystals.4 The recently realized kilogram-scale production of 1 opened up unprecedented possibilities and created a solid foundation for further synthetic manipulations.5 This newly gained availability also led to an increased demand for convenient and efficient functionalization strategies of 1.The conventional method for five-fold symmetric functionalization of 1 has been by its conversion to 1,3,5,7,9-pentachlorocorannulene (2), which may further undergo alkylation6,7 or arylation8,9 reactions; however, this approach suffers from several challenges including poor solubility of 2, difficult purification, and low reactivity of aryl chlorides in Suzuki cross-coupling reactions.
Scott et al., developed a new synthetic strategy for the functionalization of 1, by using iridium-catalyzed poly-borylation under basic conditions.10 The reaction proceeds via direct C–H activation, affording sym-1,3,5,7,9-(Bpin)5-corannulene (3)11 in good yields. Addition of small amounts of base was found to promote equilibration via a postulated slow substituent-redistribution process, driving the reaction towards maximal borylation. Unlike 2, the resulting five-fold borylated species, 3, is highly soluble in dichloromethane and can be purified with ease by recrystallization (Fig. 1).
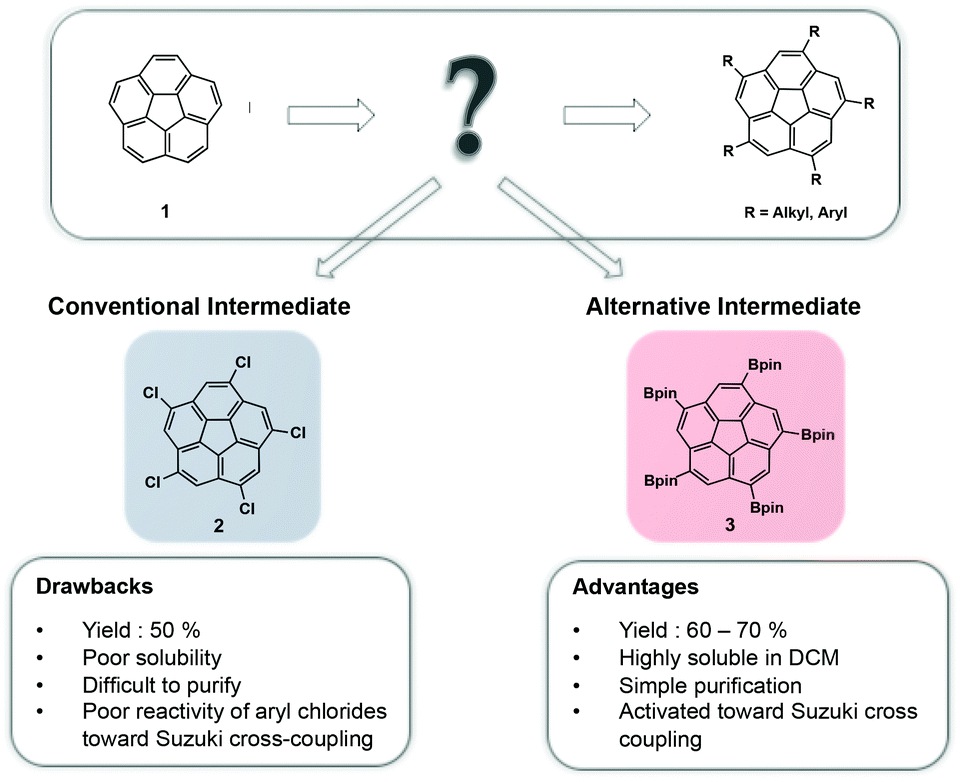 | ||
| Fig. 1 Comparison of 1,3,5,7,9-(Bpin)5 corannulene 3 and 1,3,5,7,9-pentachlorocorannulene 2 as key intermediates in the functionalization process of corannulene 1. | ||
Borylated substrates, such as 3, are activated towards Suzuki cross-coupling with various partners (e.g. halides or related leaving groups), and afford a coupling-partner tolerance that can include pyridines or bulkier ortho-substituted molecules. It was demonstrated by Scott and coworkers that coupling of a sterically demanding 1-bromo-2,6-dichlorobenzene can be achieved in good yields.10
One of the limitations of the iridium-catalyzed borylation of 1 is the long reaction time; also the subsequent Suzuki cross-couplings suffer from this restriction. Another drawback is that boronic esters, such as pinacol esters, tend to be less reactive than the corresponding acids or the recently emerging trifluoroborates.
In addition to the wide range of applications for use of boronic acids in organic synthesis,12 trifluoroborates have gained considerable attention in the past decade, not only associated with Suzuki cross-couplings,13 but also in other synthetic areas such as allylation,14 boronic Mannich,15 and ether coupling reactions.16 Both functionalities thus represent attractive synthetic targets.
In the present work, microwave-assisted synthesis of 3 is shown to result in substantially shorter reaction times compared to the bench-top procedure developed by Scott et al.10 Microwave irradiation also accelerates subsequent Suzuki cross-couplings of 3. The optimized borylation procedure enabled the synthesis of several grams of 3, which allowed the development of methods for conversion of 3 to more reactive boron species such as potassium 1,3,5,7,9-corannulene pentatrifluoroborate (6) and 1,3,5,7,9-corannulene pentaboronic acid (8) at room temperature in near quantitative yields. The boronic acid species 8 further gave access to a series of boronic acid esters of corannulene via simple alcohol exchange. The physical organic properties of the newly established series of borylated corannulene derivatives were extensively studied.
Results and discussion
Because microwave irradiation is known to accelerate significantly various reaction processes,17 detailed investigations were carried out to perform the fivefold borylation of 1 in a reduced time frame with the aid of a microwave reactor. Modified borylation conditions, which were originally developed for the bench-top procedure established by Scott and coworkers,10 enabled successful synthesis of 3 in a microwave reactor under constant irradiation of 300 W for 4 hours (Fig. 2). The reaction conditions included: [Ir(OMe)COD]2 (0.2 eq.) as catalyst, 4,4′-dimethyl-2,2′-bipyridine (0.4 eq.) as ligand, B2pin2 (5.5 eq.) as borylating agent and NaOMe as base (0.1 eq.). The yields of the reaction were typically found to be 51%, with yields up to 62% detected. Suitable solvent choice was found to be crucial for the performance of the reaction in a microwave reactor; only with cyclohexane were satisfying yields afforded. Use of dioxane or THF showed a significant drop in yields, to 29% and 34%, respectively. Milder reaction conditions were also investigated using microwave-assisted borylations at 150 W and 230 W, however in both cases a decrease in yield was detected (45%). | ||
| Fig. 2 Comparison of two different synthetic routes for synthesis of 3 starting from 1: (a) routine bench-top preparation, and (b) newly proposed microwave-assisted synthesis. | ||
This relatively powerful microwave procedure for 3 motivated a microwave-assisted synthesis of monoborylated corannulene. The current preparation of 2-corannulyl-4,4,5,5-tetramethyl-1,2,3-dioxaborolane (4) established by Scott and de Meijere involves a two-step synthesis starting from 1, which proceeds via bromination to the mono(bromo)-corannulene intermediate followed by palladium cross-coupling to yield the pinacol boronic ester 4.18 Using the microwave-assisted borylation procedure together with slight modifications to equivalencies of reagents, 4 was generated in a one-step synthesis (yields 55–60%) (Fig. 3). In addition to unreacted 1 (24–26%), diborylated corannulene (13–15%) was also observed; however, tri- and tetra-borylated products were not detected.
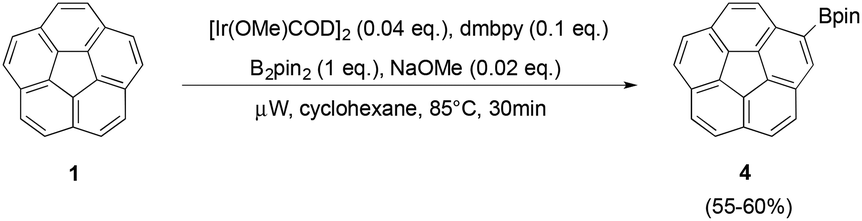 | ||
| Fig. 3 One-step synthesis of 4via direct Ir-catalyzed borylation of 1. | ||
By increasing the amounts of [Ir(OMe)COD]2, 4,4′-dimethyl-2,2′-bipyridine, [Bpin]2, and NaOMe, respectively, elevated conversion rates of 1 to 4 can be detected, however, higher amounts of the diborylated side product also occur. The proposed synthesis in Fig. 3, therefore, represents a good balance between these two extremes, and results in a suitable product to side product ratio, when considering that 1 can be recovered from the reaction mixture. In contrast to the fivefold borylation, base is not essential for this monoborylation procedure; the reaction conditions of Fig. 3, run without addition of base, still affords 4 in comparable yields.
The diborylated side product can be efficiently deborylated at high temperatures under acidic conditions via the procedure described by Scott,10 which increases the return of 1. The drawback of this synthesis remains in the separation of the various mono- and di-borylated isomers, similar to the Scott and de Meijere's procedure. Nevertheless, pure product can be obtained by column chromatography.
Suzuki cross-coupling of 3 was additionally carried out in a microwave-reactor to test whether a similar acceleration as with the fivefold-borylation could be observed. This reaction resulted in successful conversion of 3 in the presence of 2-bromofluorobenzene (10 eq.), Pd(dppf)Cl2 (0.2 eq.) and Cs2CO3 (12 eq.) to 1,3,5,7,9-pentakis(2-fluorophenyl)-corannulene 5 under constant irradiation of 300 W in only 2 h (Fig. 4).
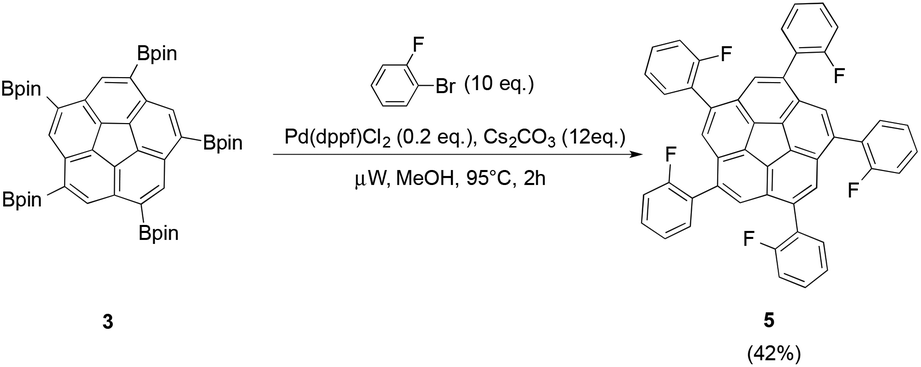 | ||
| Fig. 4 Microwave-assisted Suzuki cross-coupling of pinacol boronic ester 3 resulted in the desired ortho-fluorobenzene derivative 5. | ||
The optimized microwave-assisted poly-borylation procedure of 1 led to increased availability of 3, which allowed study of the conversion of 3 to other more reactive boron species. For conversion of boronic esters to trifluoroborates, two common synthetic strategies are available in the literature, via C–B bond formation by reaction of R-SnMe3 with BF3/KF,19 or via B–F bond formation with HF/KOH,20 or KHF2.21 The latter procedure, introduced by Vedejs et al., has become the standard method for conversion of boronic acids or esters to the corresponding trifluoroborates.
Treating pinacol boronic ester 3 with excess aqueous KHF2, yielded successful conversion to the corresponding trifluoroborate, 6, in nearly quantitative yields (Fig. 5a). The drawback of the synthesis was the difficulty in separating the pentapodal aryl trifluoroborate, 6, from the formally liberated potassium hydroxide, leaving the product as a mixture of salts.
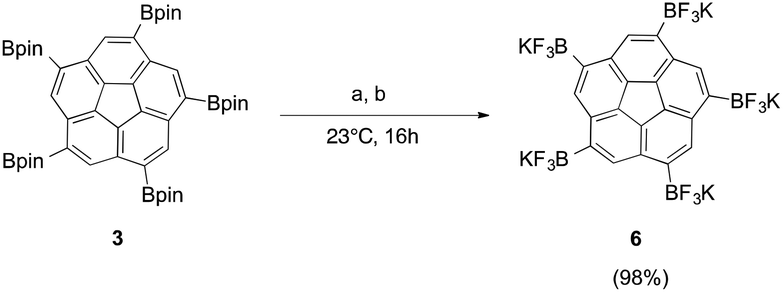 | ||
Fig. 5 Conversion of boronic ester 3 to the corresponding potassium trifluoroborate 6 by (a) excess 4.5 eq. KHF2, MeOH. (b) KF, L-(+)-tartaric acid, MeOH–MeCN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1). | ||
An improved synthetic method, proposed by Lloyd-Jones et al.,22 involves trifluoroborate formation via pre-equilibration by KF followed by consumption of the in situ generated potassium hydroxide via addition of mild organic acid. They determined that stoichiometric amounts of L-(+)-tartaric acid results in neutralization of the in situ generated potassium hydroxide and precipitation of potassium bitartrate, which shifts the equilibrium towards product formation and drives the reaction to completion.
Application of this method to the fivefold substituted polycyclic aromatic hydrocarbon, 3, shows that, in addition to potassium bitartrate, 6 also precipitates out of solution (Fig. 5b); however, this problem could be overcome by redissolving the reaction mixture in DMF, which brings 6 in solution and precipitates out the other salts. Subsequent filtration affords trifluoroborate, 6, in near quantitative yields and high purity.
Mono(BF3K)corannulene (7) can be synthesized analogously to its fivefold substituted analogue 6. In this case, the trifluoroborate formation strategies of both Vedejs and Lyoyd-Jones were shown to be suitable, and the final compound purified via column chromatography without additional salt impurities (Fig. 6).
 | ||
| Fig. 6 Conversion of boronic ester 4 to the corresponding potassium trifluoroborate 7 by (a) excess 4.5 eq. KHF2, MeOH. (b) KF, L-(+)-tartaric acid, MeOH–MeCN (1:1). | ||
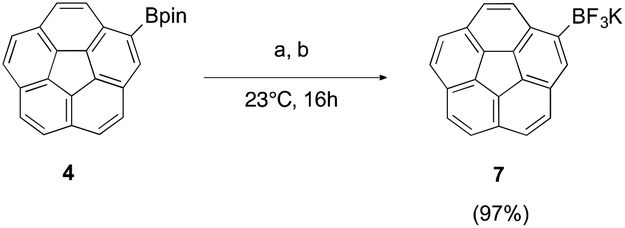
Further challenges were exposed in the mass spectroscopic characterization of pentapodal trifluoroborate, 6. Whereas the mono-substituted derivative, 7, could be characterized by mass spectrometry without any difficulties, the fivefold analogue failed to be detected by any mass spectrometry method employed. On the other hand, NMR spectroscopy revealed the proposed fivefold trifluoroborate, 6, with 1H-NMR clearly showing a singlet at 8.08 ppm for the corannulyl protons of 6, shifted upfield (∼1 ppm) compared to the corannulyl protons of the pinacol ester derivative 3 (Fig. 7). Moreover, 11B-NMR illustrates a nicely resolved quadruplet resulting from B–F coupling in the trifluoroborate substituents.
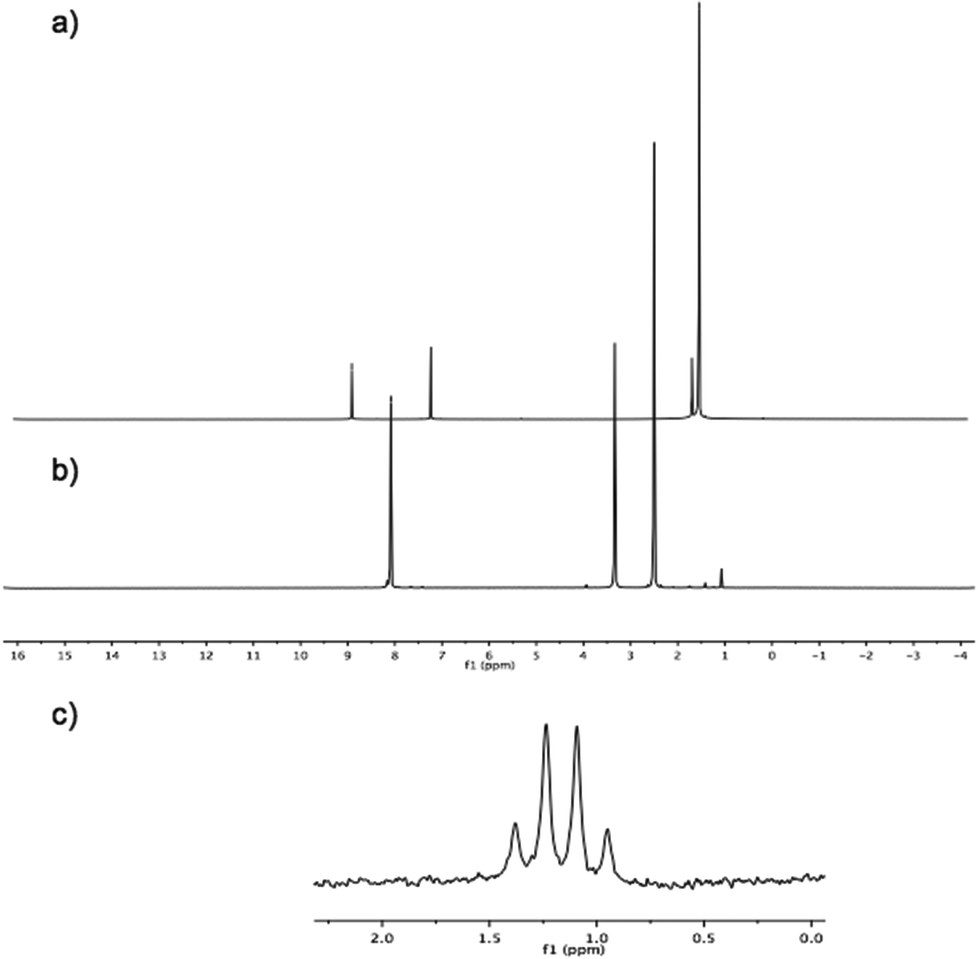 | ||
| Fig. 7 NMR Spectra: (a) 1H-NMR of 3; (b) 1H-NMR of 6; and, (c) 11B-NMR of 6. | ||
To provide further evidence for the presence of the proposed compound 6, Suzuki cross-coupling reactions starting from 6 were carried out according to the procedure developed by Lloyd-Jones and coworkers (Fig. 8).23 Even though the reaction suffered from low yields, the desired product 5 could be obtained successfully.
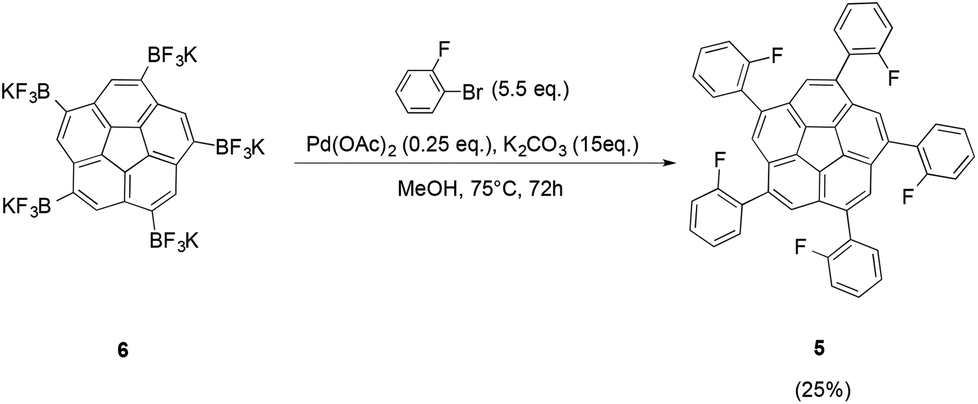 | ||
| Fig. 8 Suzuki cross-coupling of trifluoroborate 6 with 2-bromofluorobenzene yields the desired ortho-fluorobenzene derivative 5. | ||
Conversion of boronic esters to boronic acids have been carried out in the literature using various hydrolysis conditions, including acid hydrolysis,24 oxidative hydrolysis,25 reduction,26 and transesterification.27 With such a broad range of synthetic procedures, a screening was carried out. Unfortunately, standard conditions, such as sodium periodate in the presence of ammonium acetate or palladium on carbon (10 wt%) under hydrogen atmosphere did not result in satisfying product formation (34% and traces, respectively). Reaction with diethanolamine under acidic conditions also did not lead to detectable conversion.
As a solution to this problem, a polymer-supported transesterification procedure was applied.28 The use of solid phase reagents in this synthesis is advantageous since a large excess of reagent can be used and simply filtered off after completion of the reaction process. Using polystyrene-boronic acid as a solid phase reagent in the presence of hydrochloric acid at room temperature, pinacol boronic ester 3 was efficiently converted to 1,3,5,7,9-corannulene pentaboronic acid (8) (Fig. 9). With subsequent filtration, the desired product could be afforded in quantitative yields and good purity. Only minor amounts of tetra-substituted isomers are observed as side products. IR-spectroscopy clearly establishes formation of product 8, by a band at 3400 cm−1 characteristic of OH stretching with hydrogen bonding in the free boronic acid, (Fig. 10). Monoboronic acid corannulene can be synthesized analogously.
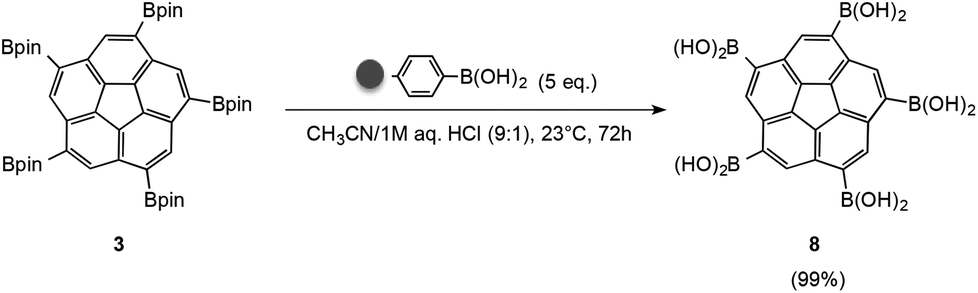 | ||
| Fig. 9 Polymer-supported transesterification of 3via polystyrene-boronic acid under acidic conditions yields the boronic acid 8. | ||
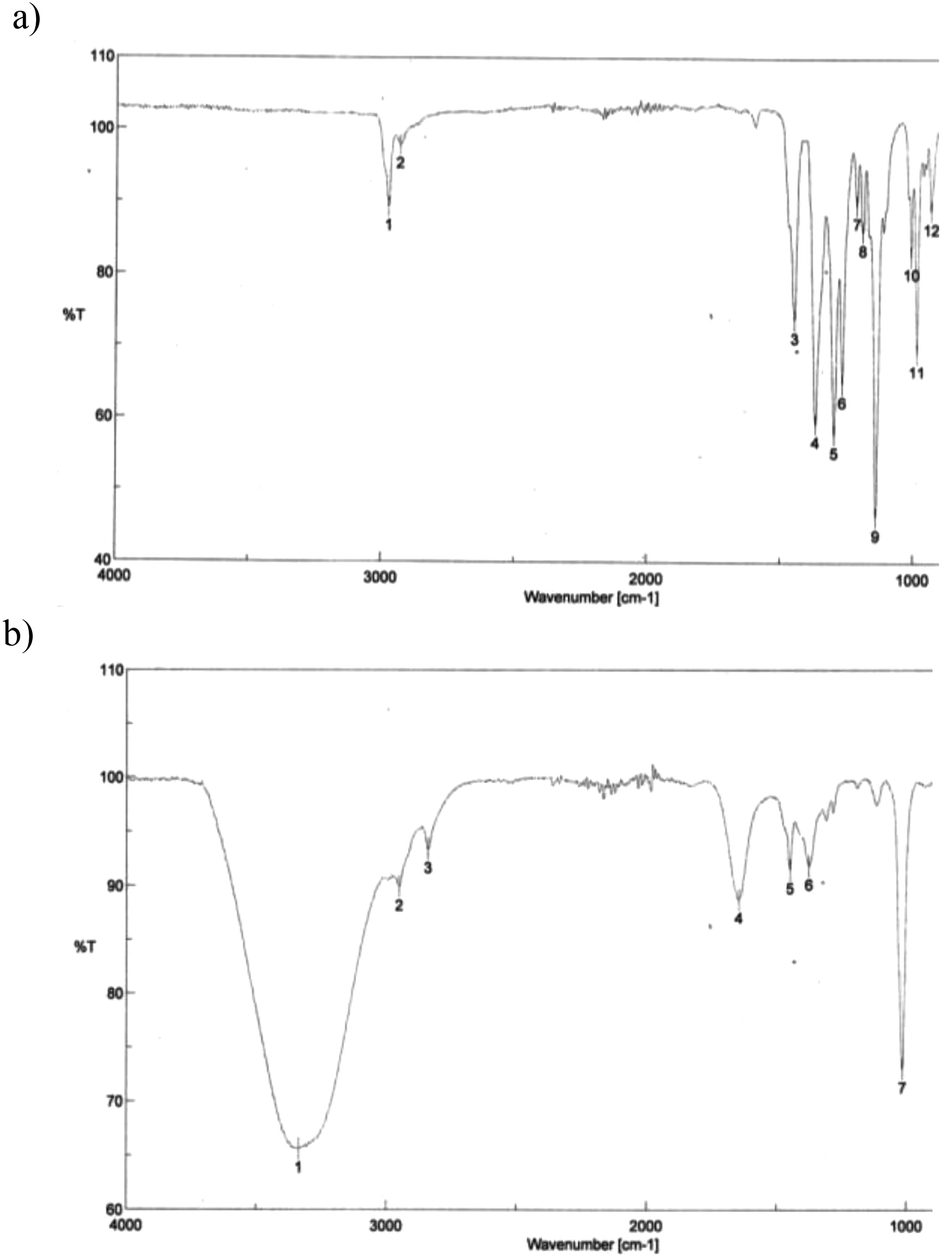 | ||
| Fig. 10 Experimental IR spectra of (a) 3 and (b) 8, showing evidence for hydrolysis of the pinacol ester to boronic acid 8, with H-bond interactions of the boronic acid moieties appearing at 3400 cm−1. | ||
Simple boronic acids are prone to undergo alcohol exchange in the presence of cis-diols, e.g., with monosaccharide moieties such as glucose. Pentaboronic acid 8 undergoes fivefold alcohol exchange in the presence of simple organic cis-diols, and strongly favors alcohol exchange at room temperature with various cis-diols either neat or as excess in MeOH. This methodology allowed synthesis of a set of novel boronic esters of corannulene, 9–13, in high yields. The only difficulties occurred during synthesis of 13, which required performing the reaction under inert conditions (Fig. 11).
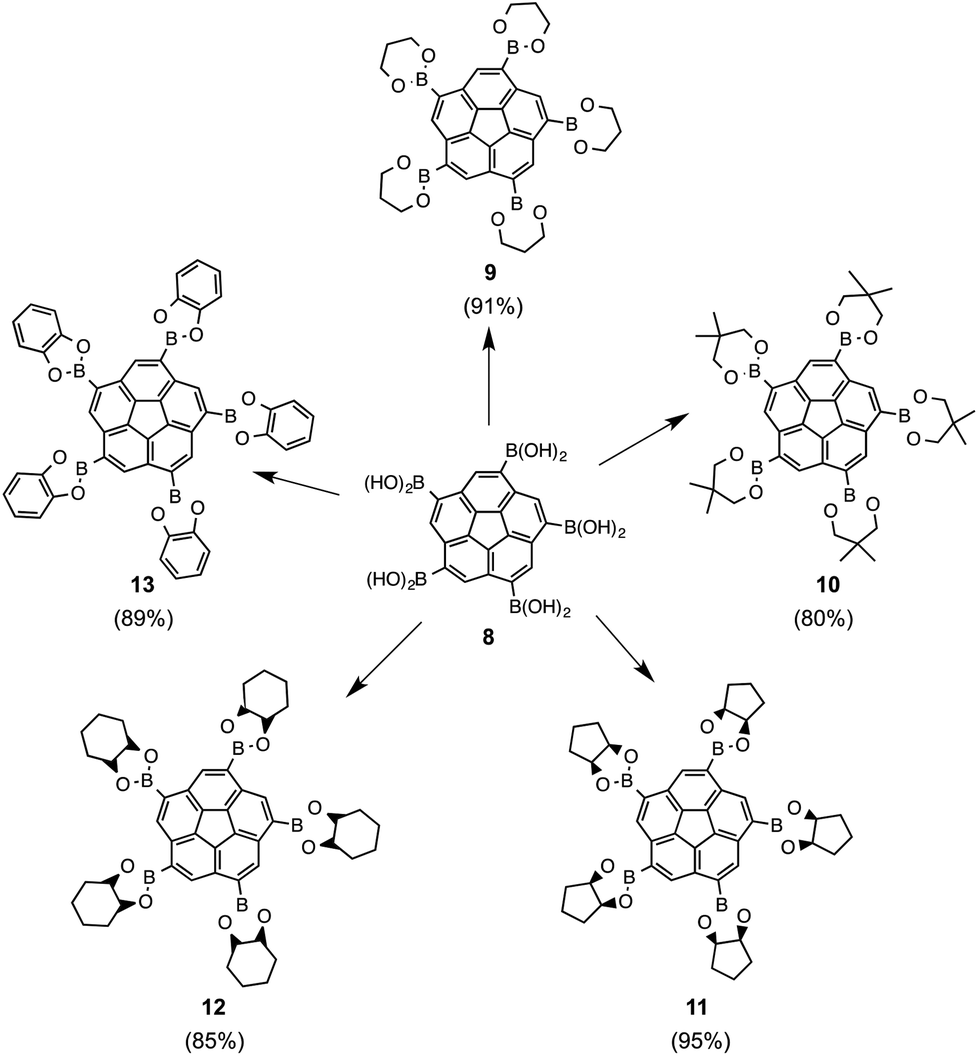 | ||
| Fig. 11 Boronic esters, 9–13, synthesized via alcohol exchange with 8 in the presence of the corresponding cis-diols. | ||
Physical organic properties
Crystals of 3 (Fig. 12) and 11 (Fig. 13) suitable for X-ray diffraction were obtained by slow evaporation of a mixture of CH2Cl2–n-hexane and CDCl3, respectively. Both structures reveal disorder of the entire molecules over two positions related by a mirror plane, with the mirror being crystallographic only in the case of 11. The mirror plane arises from superposition of the enantiomorphs of the chiral corannulene molecule on the same crystallographic site. In 11, the crystal is necessarily a racemic conglomerate, while in 3, the 69% occupancy of the major configuration suggests an unequal distribution of enantiomers in the chosen crystal. However, the presence of inversion twinning cannot be excluded in the absence of a reliable absolute structure parameter, and inversion twinning would result in a fully racemic mixture.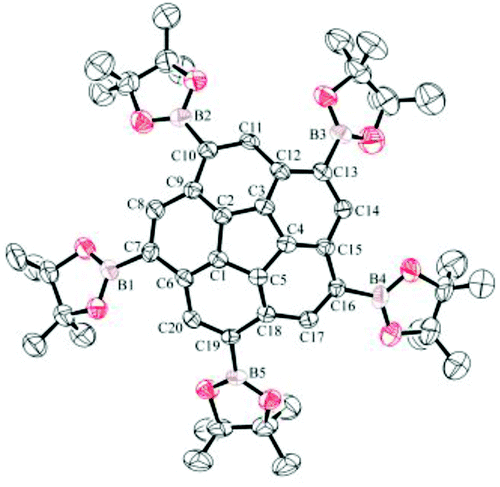 | ||
| Fig. 12 Displacement ellipsoid (50% probability) representation of 3 showing only one of the disordered arrangements. | ||
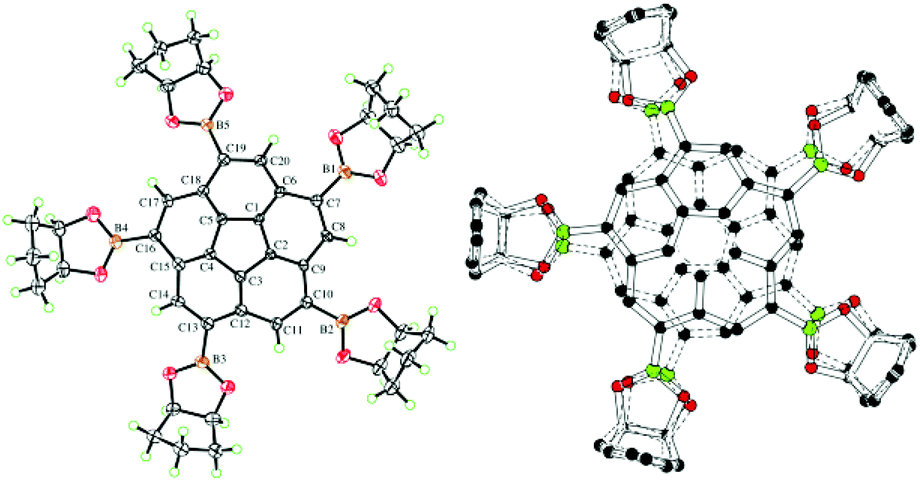 | ||
| Fig. 13 Displacement ellipsoid (50% probability) representation of 11 showing only one (left) and the superposition of both (right) disordered arrangements. | ||
Computations predict a slightly deeper bowl for 3 (0.90 Å) compared to the parent corannulene, 1 (0.87 Å); however, experimental estimates for the bowl depth in the major and minor disordered forms in the crystal structure of 3 (0.918(5) Å and 0.90(1) Å, respectively) have a standard uncertainty too large to establish a conclusive bowl depth difference. For comparison, the experimental bowl depth estimated from the crystal structure of 11 is 0.86(1) Å.
Corannulene and its derivatives undergo rapid bowl-to-bowl interconversion at room temperature; the bowl-inversion barrier was determined by VT-NMR measurements and computational analysis. Experiments were carried out with 3 in deuterated dichlorofluoromethane (Freon-21).29 This particular solvent was chosen because it has a very low melting point, −135 °C, which enables measurement over a larger temperature window. Furthermore, it exhibits low viscosity at low temperatures as well as good solubilizing properties for most compounds. Using this solvent, VT-NMR measurements could be performed from a temperature range of −50 °C to up to −135 °C.
Two dynamic processes are possible in structure 3, bowl inversion and rotation around the C–B bond. Provided both processes are slow enough to be resolved on the NMR time scale, NMR signals for all four diastereotopic methyl groups of the pinacol ester should be observable. Experimentally, only one dynamic process could be detected by NMR, namely the bowl-to-bowl inversion process (Fig. 14), leading to a splitting of the pinacol ester methyl peak. In contrast, rotation around the C–B bond is too fast to be resolved on the NMR time scale and could not be observed.
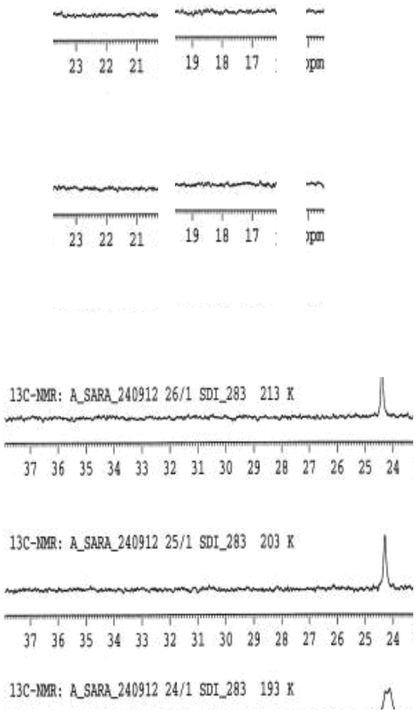 | ||
| Fig. 14 VT-NMR of 3 in dichlorofluoromethane-d1 (138–223 K). | ||
The coalescence temperature of the bowl-to-bowl inversion process was found to be 195 K, and the barrier to interconversion determined with the aid of the Gutowsky–Holm approximation and Eyring equation to be 9.43 kcal mol−1. This can be compared to the B97D/Def2-TZVPP(THF) calculated value of 8.82 kcal mol−1 (Table 1). Since the pinacol ester substituents of 3 are more sterically demanding than unsubstituted 1, 3 is expected to have a slightly more shallow bowl than 1. The consequence of a more shallow bowl is a lower bowl-to-bowl inversion barrier in 3 compared to 1 (11.5 kcal mol−1). The barrier for 3 is determined to be ∼2 kcal mol−1 lower than in 1 experimentally as well as computationally (Table 1).
| Cmpd | Bowl depth [Å] | Bowl inversion barrier [kcal mol−1] | Bowl inversion frequency [cm−1] | |
|---|---|---|---|---|
| Expt | 3 | 0.90 | 9.43 | — |
| 1 | 0.87 | 11.5 | — | |
| Calc | 3 | 0.87 | 9.7 | −71.4 |
| 1 | 0.92 | 11.2 | −116.2 |
Cyclic voltammetry data was collected for the three boronic esters, 3, 4, and 11. The cyclic voltammograms of the two different pentapodal boronic ester corannulenes, 3 and 11, appear similar in their electrochemical behavior, with three nearly identical reduction potential values (Table 2). In the case of the mono-trifluoroborate derivative 4 however, the first and third reduction potentials are more negative, whereas the first reduction potential is nearly identical to that of 1. Values calculated at the B97D/Def2-TZVPP level of theory are in reasonably good agreement with the experimental data for 3 and 1. Comparison of cyclic voltammograms showed that the reduction rate of 4 is significantly faster compared to the fivefold derivatives 3 and 11. This could be explained by the lesser steric bulk of monosubstituted corannulene 4 compared to pentasubstituted corannulenes 3 and 11.
| Compounds | Electrochemical reduction potentials (E1/2) [V] | ||
|---|---|---|---|
| 1 E 1/2 | 2 E 1/2 | 3 E 1/2 | |
| a B97D/Def2-TZVPP calculated values in THF solvent environment vs. Ag/AgCl (4.53) reference electrode. The potentials were referenced against the ferrocenium/ferrocene standard potential. | |||
| 1 | −2.45 (−2.50)a | — | — |
| 3 | −2.25 (−2.15)a | −2.38 | −2.70 |
| 4 | −2.42 | −2.41 | −2.84 |
| 11 | −2.26 | −2.40 | −2.72 |
In all borylated species, the inductive electron-donor effect along the B–C sigma bond and the electron-withdrawing π-resonance effect of the pinacol ester moieties lead to a slight shift in reduction potential toward more positive values. This affect on the redox potential of corannulene derivatives with a single boronic ester substituent, such as in derivative 4, is a small impact; however, when five electron withdrawing boronic ester substituents are present, such as in 3 and 11, the overall electrochemical properties change significantly. The HOMO/LUMO structure and electrophilic HOMO plot for 3, support this analysis (Fig. 15).
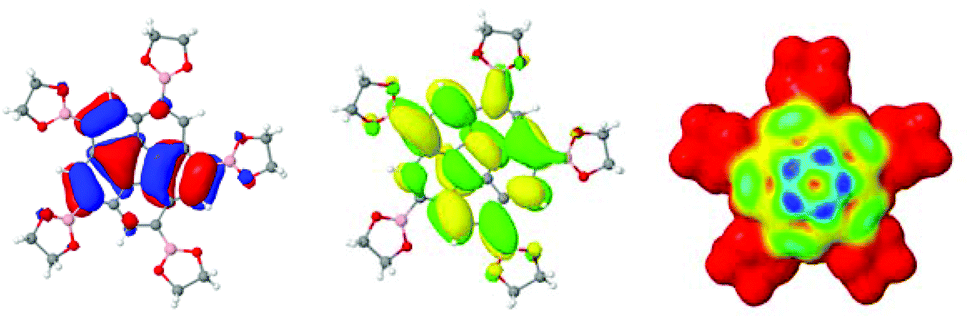 | ||
| Fig. 15 B97D/Def2-TZVPP optimized structure of 3. Depiction of (a) HOMO, (b) LUMO, and (c) electrophilic HOMO map (blue: highest probability for electrophilic attack). | ||
UV/Vis and emission data revealed for all boronic esters, 9–13, in addition to 3, similar absorption (λmax: 275 nm, 307 nm) and emission (λmax: ca. 440 nm) behavior (Fig. 16). All boronic esters show a large red shift of ∼20–24 nm for the π–π* absorption band in both the UV/Vis and emission spectra. The large Stokes shift of these boronic ester derivatives could be due to the significant geometrical distortion in the S1-state, as well as contribution from the intramolecular charge transfer.
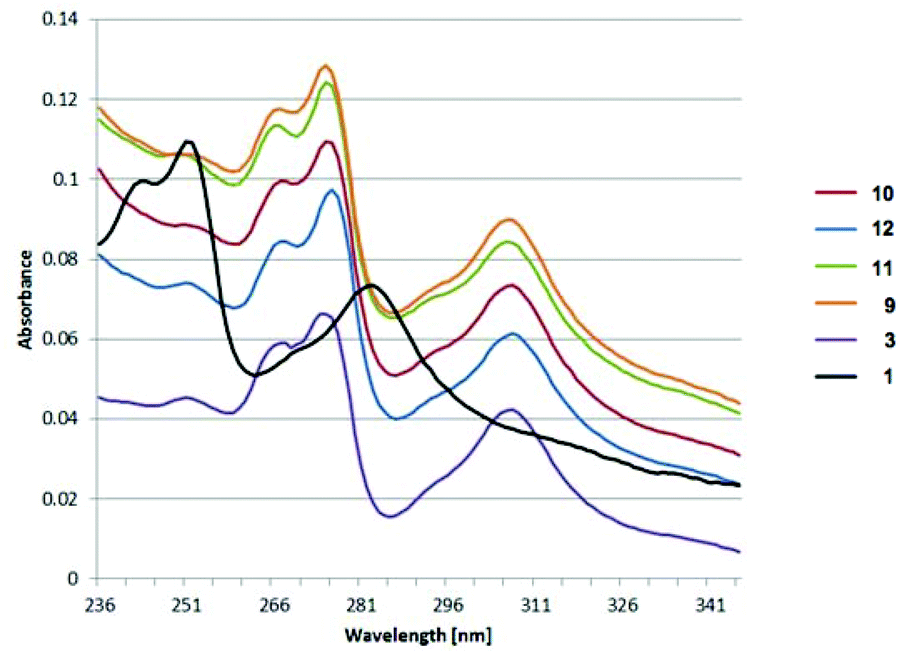 | ||
| Fig. 16 UV/Vis spectra of 1 and boronic ester derivatives 3 and 9–12. | ||
Targeting icosahedral constructs
Boronic acids are commonly used templates for supramolecular chemistry30 due to their ability to complementarily bind 1,2- and 1,3-diols, such as saccharides.31 This feature has lead to successful application in a range of saccharide sensors,32e.g., for monitoring of diabetic blood glucose. Moreover, boronic acids are prone to form boroxines (cyclized six-membered rings) upon dehydration. Yaghi et al. disclosed that, depending on choice of adequate building blocks, two- and three-dimensional supramolecular geometries can be realized.33 As such, symmetrical corannulene pentaboronic acid was considered here to be a potential building block for formation of icosahedral supramolecules with unique virus-like properties.1,3,5,7,9-Corannulene pentaboronic acid, 8, could serve as a potential template towards formation of icosahedral supramolecules either via boroxine formation, or by ligand-mediated assembly (Fig. 17). The stoichiometric ratio of the linkers has to be adjusted; divalent linkers require 30 eq. per 12 eq. of 8, while 20 eq. are sufficient for trivalent linkers. This is due to the fact that divalent linkers connect the core molecules via edges (30 edges in an icosahedron), whereas trivalent linkers bind via faces (20 faces in an icosahedron). Formation of such an icosahedral supramolecule is expected to be more effective with trivalent ligands, since they exhibit more rigidity and are therefore more directing towards the closure of the bowl, whereas divalent ligands are rather flexible.
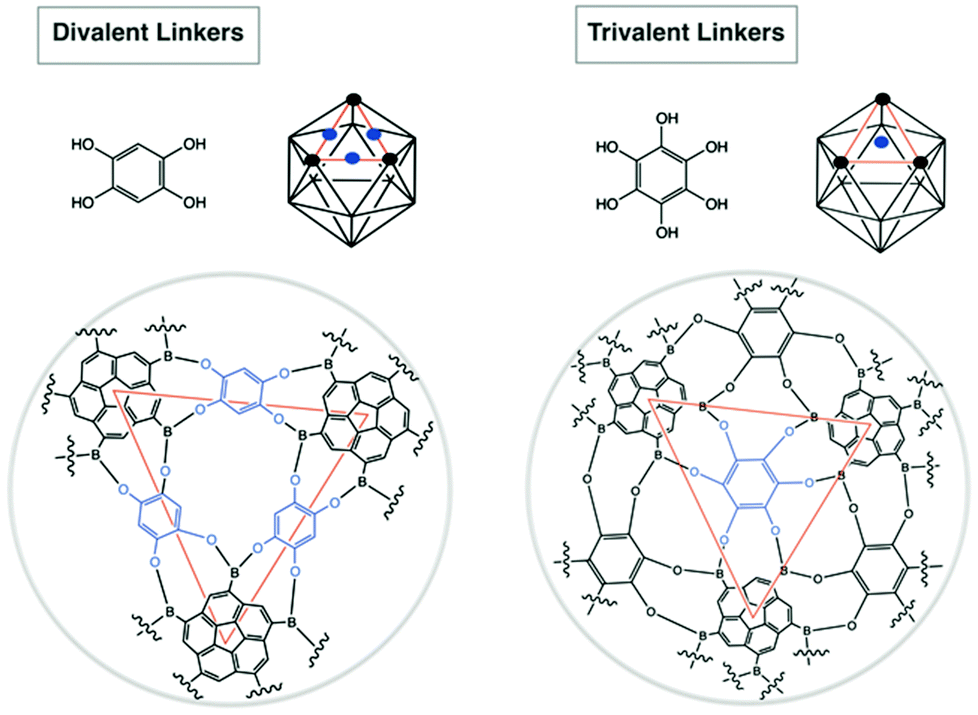 | ||
| Fig. 17 Potential coordination of divalent and trivalent linkers to 8 in the formation of icosahedron supramolecules. | ||
Computational modelling of the desired icosahedral supramolecule derived from boroxine formation of precursor 8 shows the possibility for such structures (Fig. 18). The resulting icosahedral geometry shows similarities with virus-capsids, such as present for example in human papilloma viruses and might therefore exhibit interesting properties as host–guest complexes.
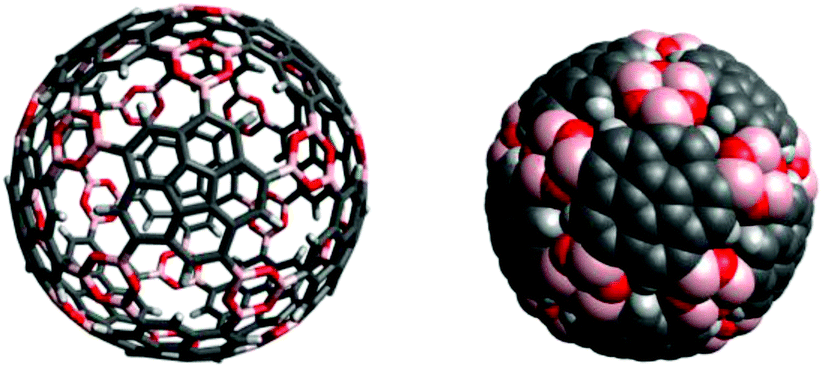 | ||
| Fig. 18 Computational model of a target icosahedral construct derived by boroxine formation from twelve molecules of precursor 8. | ||
Conclusions and outlook
A straightforward microwave-assisted synthetic strategy is shown to be a valuable tool for functionalization of corannulene. In particular, the method provides a new route towards preparation of 1,3,5,7,9-(Bpin)5-corannulene, 3, in a time-effective four hours, which is 24 times faster than the commonly used bench-top procedure developed by Scott and coworkers.10 However, loss in yield from the usual 65–70% to 51% is observed. Suzuki cross-coupling reactions with 3 are also enhanced by microwave irradiation, reducing the reaction time of 3 with 2-bromofluorobenzene from 2 days to 2 hours. In summary, the efficient microwave procedures enable for the first time the synthesis of arylated species 5 starting from 1 in only one day. Furthermore, investigations towards conversion to more reactive boron species, namely the corresponding pentatrifluoroborate species 6 and pentaboronic acid 8 were also facilitated and successfully realized. Moreover, 8 led to a series of novel pentapodal boronic ester analogues, 9–13, due to its reactivity with 1,2-diols. The 1,3,5,7,9-corannulene pentaboronic acid, 8, is therefore thought to be a suitable building block in the formation of supramolecule frameworks, using adequate divalent or trivalent linkers such as tetra- and hexa-hydroxybenzene, respectively. In the presence of 8, both types of linkers are potentially able to result in icosahedral geometry.Experimental procedure
Synthesis and characterization as well as measurement data are available in detail in the ESI.†Computational methods
The conformational analyses of the molecular systems described in this study, including structural and orbital arrangements as well as property calculations, were carried out using the GAMESS34 software package. Structures were fully optimized with the B97D dispersion enabled density functional35,36 using an ultrafine grid, in combination with the Def2-TZVPP basis set.37 Optimized geometries were uniquely characterized via second derivatives (Hessian) analysis. Effects of solvation were taken into account using the COSab method,38,39 with the dielectric for THF and solvent radii from Klamt.40 Calculations of reduction potential were carried out as E° = −ΔH/nF + Eref, where n = 1, F = 1 eV, with correction to the Ag/AgCl electrode, 4.53. Visualization and analysis of structural and property results, including electrophilic (HOMO) and nucleophilic (LUMO) frontier density plots, were obtained using Avogadro41 and WEBMO.42Notes and references
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- A. J. Olson, J. H. E. Hu and E. Keinan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20731–20736 CrossRef CAS PubMed.
- D. Pappo, T. Mejuch, O. Reany, E. Solel, M. Gurram and E. Keinan, Org. Lett., 2009, 11, 1063–1066 CrossRef CAS PubMed.
- D. Miyajima, K. Tashiro, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2009, 131, 44–45 CrossRef CAS PubMed.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
- T. Hayama, in Organic Chemistry Institute, University of Zürich, Zürich, 2008 Search PubMed.
- M. Mattarella and J. S. Siegel, Org. Biomol. Chem., 2012, 10, 5799–5802 CAS.
- G. H. Grube, E. L. Elliott, R. J. Steffens, C. S. Jones, K. K. Baldridge and J. S. Siegel, Org. Lett., 2003, 5, 713–716 CrossRef CAS PubMed.
- A. F. Littke and G. F. Fu, Angew. Chem., Int. Ed., 1998, 37, 3387–3388 CrossRef CAS.
- M. N. Eliseeva and L. T. Scott, J. Am. Chem. Soc., 2012, 134, 15169–15172 CrossRef CAS PubMed.
- Abbreviation for 1,3,5,7,9-pentakis(4,4,5,5-tetramethyl-1,3,2-dioxa-borolan-2-yl)corannulene.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- G. A. Molander and B. Biolatto, J. Org. Chem., 2003, 68, 4302–4314 CrossRef CAS PubMed.
- R. A. Batey, A. N. Thadani and D. V. Smil, Tetrahedron Lett., 1999, 40, 4289–4292 CrossRef CAS.
- N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583–586 CrossRef CAS.
- C.-V. T. Vo, T. A. Mitchell and J. W. Bode, J. Am. Chem. Soc., 2011, 133, 14082–14089 CrossRef CAS PubMed.
- P. Harrisson, J. Morris, T. B. Marder and P. G. Steel, Org. Lett., 2009, 11, 3586–3589 CrossRef CAS PubMed.
- H. A. Wegener, L. T. Scott and A. d. Meijere, J. Org. Chem., 2003, 68, 883–887 CrossRef PubMed.
- R. D. Chambers, H. C. Clark and C. J. Willis, J. Am. Chem. Soc., 1960, 82, 5298–5301 CrossRef CAS.
- R. A. Battey and T. D. Quach, Tetrahedron Lett., 2001, 42, 9099–9103 CrossRef.
- E. Vedejs, R. W. Chapman, S. C. Fields, S. Lin and M. R. Schrimpf, J. Org. Chem., 1995, 60, 3020–3027 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2012, 134, 7431–7441 CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431–7441 CrossRef CAS PubMed.
- M. E. Jung and T. I. Lazarova, J. Org. Chem., 1999, 64, 2976–2977 CrossRef CAS.
- H. Nakamura, M. Fujiwara and Y. Yamamoto, J. Org. Chem., 1998, 63, 7529–7530 CrossRef CAS.
- W. Yang, H. He and G. Druechkhammer, Angew. Chem., Int. Ed., 2001, 40, 1714–1718 CrossRef CAS.
- J. Wityak, R. A. Earl, M. M. Abelman, Y. B. Bethel, B. N. Fisher, G. S. Kauffman, C. A. Kettner, P. Ma, J. L. MecMillan, L. J. Mersinger, J. Pesti, M. E. Peierce, F. W. Rankin, R. J. Chorval and P. N. Confalone, J. Org. Chem., 1995, 60, 3717–3722 CrossRef CAS.
- T. E. Pennington, C. Kardiman and C. A. Hutton, Tetrahedron Lett., 2004, 45, 6657–6660 CrossRef CAS PubMed.
- F. A. L. Anet and J. S. Siegel, J. Org. Chem., 1988, 53, 2629–2630 CrossRef.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Foxxy, Chem. Commun., 2011, 47, 1124–1150 RSC.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- T. D. James, M. D. Phillips and S. Shink, Boronic Acids in Saccharide Recognition, Royal Society of Chemistry, 2006 Search PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- M. Schmidt, K. K. Baldridge, J. A. Boatz, S. Elbert, M. Gordon, J. H. Jenson, S. Koeski, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108–034115 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- K. K. Baldridge and A. Klamt, J. Chem. Phys., 1997, 106, 6622–6633 CrossRef PubMed.
- A. Klamt, V. Jonas, T. Bürger and C. W. Lohrenz, J. Phys. Chem., 1997, 102, 5074–5085 CrossRef.
- M. Hanwell, D. Curtis, D. Lonie, T. Vandermeersch, E. Zurek and G. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- WEBMO: T. Cundari and J. R. Schmidt, http://www.webmo.net.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1042202–1042203. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00009b |
| This journal is © the Partner Organisations 2015 |