
Synthesis of silafluorenes and silaindenes via silyl radicals from arylhydrosilanes: intramolecular cyclization and intermolecular annulation with alkynes†
Liang
Xu‡
a,
Shuai
Zhang‡
ab and
Pengfei
Li
*a
aCenter for Organic Chemistry, Frontier Institute of Science and Technology (FIST) and Frontier Institute of Chemistry, Xi'an Jiaotong University, 99 Yanxiang Road, Xi'an, Shaanxi 710054, China. E-mail: lipengfei@mail.xjtu.edu.cn
bDepartment of Chemistry, School of Science and MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an 710049, China
First published on 2nd March 2015
Abstract
Intramolecular homolytic aromatic silylation of biphenyl-2-hydrosilanes via silyl radicals has been developed for the synthesis of silafluorenes. Moreover, silaindenes are synthesized via a silyl radical cascade process, assembling internal alkynes with readily available arylhydrosilanes by C–H and Si–H cleavage and C–C and C–Si bond formation in one step. These represent a new simple and efficient approach towards the synthesis of benzosilole derivatives.
Siloles (silacyclopentadienes) and their π-extended derivatives, such as silaindenes and silafluorenes, have attracted considerable interest because of their application as building blocks or structural moieties in organic electronic materials.1,2 Conventional synthetic approaches towards silole derivatives (Scheme 1) employed the reactions of dichlorosilane with highly reactive organometallic reagents3 and intramolecular reductive cyclization of alkynyl silanes.4 Recently, transition metal-catalyzed approaches have also been developed to synthesize diverse silole,5 silaindene6 and silafluorene6b,7 derivatives under mild conditions. While these synthetic methods have greatly improved the accessibility to silaindenes and silafluorenes, most of them start from difunctionalized precursors, such as 2,2′-dihalobiaryls,3a–e,7e silicon-bridged diynes,7a,g 2-(arylsilyl)aryl triflate,7b,c (2′-bromobiphen-2-yl)silane,7f 2-trimethylsilylarylboronic acid,6b,f and 2-trimethylsilyl arylbromide.6e,h To the best of our knowledge, no precedent synthetic method towards silaindenes from ortho-unsubstituted arylsilanes through direct C–H bond functionalization has been reported. As for silafluorenes, two existing approaches involving a direct intramolecular aromatic C–H silylation process were based on Rh-catalyzed double activation of Si–H and C–H bonds,7d,h and sila-Friedel–Crafts reaction8 respectively. In continuation of our research on main group element-centered radicals,9 we wanted to devise more general and efficient methods to synthesize these molecules. In this report, we describe a silyl radical-based strategy for silafluorenes and silaindenes via direct C–H bond cleavage of readily available arylsilanes without the aid of transition metals, acids or bases. During our preparation of this manuscript, such an approach was also disclosed by Leifert and Studer for the synthesis of silafluorenes.10
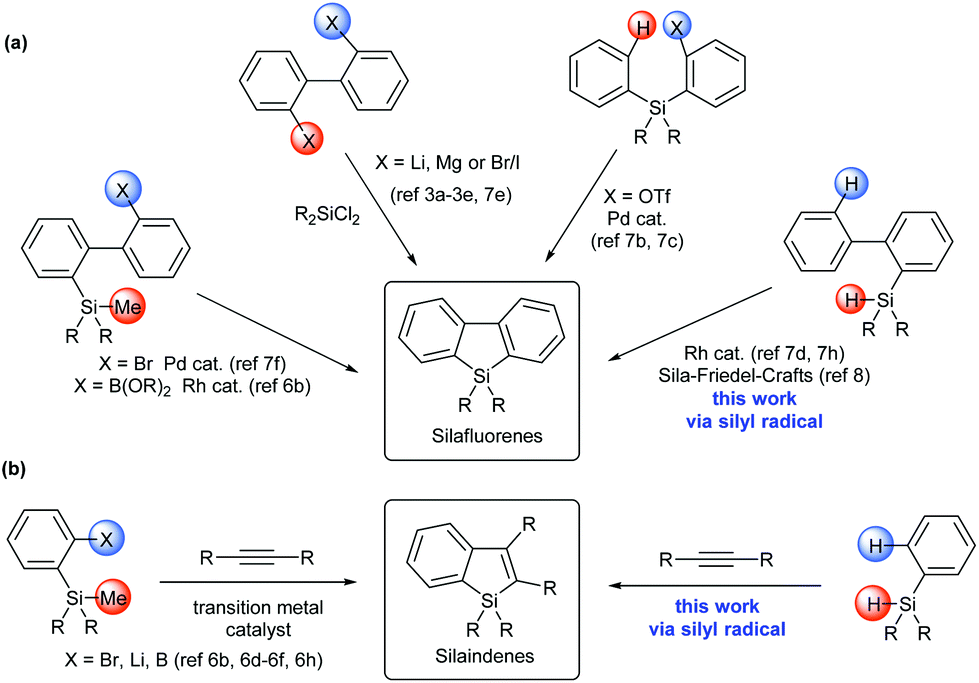 | ||
| Scheme 1 General approaches to access silafluorenes and silaindenes. | ||
Our hypothesis applying silyl radicals in the synthesis of silafluorenes was inspired by the recent development of intramolecular aromatic homolytic substitution reactions, especially the intramolecular radical cyclization reactions of biaryl frameworks.11 For example, radical phosphination12 and acylation13 on the aryl group were induced by simple radical generators to afford dibenzophospholes and fluorenones respectively.
We initiated our studies by examining the reaction between biphenyl-2-hydrosilane 1a as the silyl radical precursor and DTBP (di-tert-butyl peroxide) as a radical initiator in various solvents (Table 1). To our delight, the desired intramolecular homolytic aromatic silylation product, 2a, could be obtained in 79% yield based on 1H NMR analysis of the crude product when 1a was stirred with 3.0 equivalents of DTBP in benzene at 130 °C for 24 hours (entry 5). Using other solvents (e.g., MeCN or toluene) resulted in lower yields (entries 1–4). To further improve the yield, various radical initiators were examined, however, all the tested radical initiators including BPO (benzoyl peroxide), AIBN (azodiisobutyronitrile) and TBHP (tert-butyl hydroperoxide) gave very low yields (entries 7–9). The reaction was also performed under an air atmosphere and a moderate yield (66%, entry 14) was obtained. Because some examples of the base-promoted homolytic aromatic substitution (BHAS) process have been reported,14 the effect of additive bases was briefly explored. In the present reaction, however, the added bases did not show significant beneficial effect (entries 10–13). Finally, an extensive screening of various solvents revealed trifluorotoluene to be the optimal solvent because of its higher boiling point, potentially lower toxicity and the resulting higher yield than benzene (entry 9).
|
|
||||
|---|---|---|---|---|
| Entry | Initiator | Solvent | Additive | Yieldb (%) |
| a Reaction conditions: biphenyl-2-yl-dimethylsilane (0.1 mmol), radical initiator (0.3 mmol), additive (if necessary), solvent (0.5 ml) under an N2 atmosphere. b Yields based on 1H NMR analysis of the crude products with 1,3,5-trimethoxybenzene added as an internal standard. c 70% solvent in H2O. d Isolated yield shown in parenthesis. e The reaction was performed under air. | ||||
| 1 | DTBP | MeCN | — | 46 |
| 2 | DTBP | Toluene | — | 39 |
| 3 | DTBP | DCE | — | 36 |
| 4 | DTBP | 1,4-Dioxane | — | 57 |
| 5 | DTBP | Benzene | — | 79 |
| 6 | BPO | Benzene | — | 19 |
| 7 | AIBN | Benzene | — | Trace |
| 8 | TBHPc | Benzene | — | 26 |
| 9 | DTBP | PhCF 3 | — | 84 (77) |
| 10 | DTBP | Benzene | K2CO3 (3 eq.) | 75 |
| 11 | DTBP | Benzene | tBuOLi (3 eq.) | 64 |
| 12 | DTBP | Benzene | KOAc (3 eq.) | 72 |
| 13 | DTBP | Benzene | K3PO4 (3 eq.) | 60 |
| 14 | DTBP | Benzene | — | 66e |
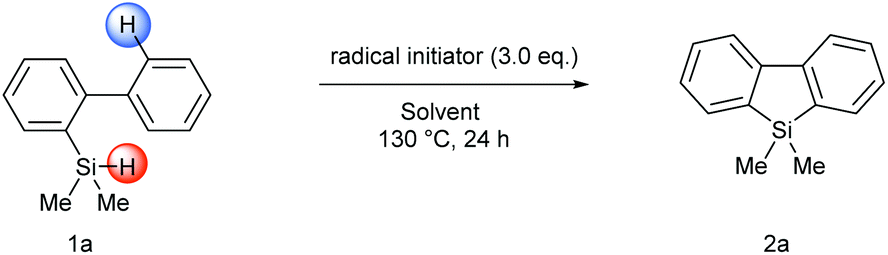
With the optimized conditions in hand, the homolytic aromatic silylation method was applied to various 2-hydrosilyl biaryls. Biphenylhydrosilanes bearing electron-withdrawing and -donating groups at the 4′-position gave the desired silafluorenes in good yields (Scheme 2, 2b–2g). A methyl group at the 2′-position was tolerated and the corresponding silafluorene derivative 2h was obtained in 63% yield. In this case, 9,10-dihydro-9-silaphenanthrene, which had been the major product of the Rh-catalyzed pathway7d due to intramolecular silylation on the methyl group, was not observed. When a biarylhydrosilane bearing a 3′-methoxy group (1i) was used as the substrate, the radical silylation occurred selectively at the more hindered position to produce 2i, presumably governed by an electronic effect. This regioselectivity was in sharp contrast with transition metal-catalyzed C–H activation wherein reactions usually take place at the least hindered position; therefore the present method is complementary in preparation of this type of silafluorenes. Furthermore, π-extended silafluorenes 2g, 2j and 2k which may impart unusual electronic properties on the molecules could be obtained by this method. Finally, the substituents on the silicon center were also variable, thus single or double phenyl substituted silanes were converted to the corresponding products under the same conditions (2l and 2m).
 | ||
| Scheme 2 Silafluorene synthesis via intramolecular homolytic aromatic silylation reaction. | ||
Radical cascade reactions are well known as powerful methods to enable quick access to complex compounds containing polycyclic frameworks.15 In this context, intermolecular annulation of an arene-containing radical species with an alkyne might form highly substituted arene-fused rings in a concise fashion.16 In fact, the selenium-centered radical has been reported to undergo this type of reaction to form a benzoselenophene as the minor product in a radical 1,2-addition process of (PhSe)2 onto an alkyne.17 More recently, benzophosphole oxides were elegantly accessed via this strategy.18 It is known that silyl radicals can add to alkynes to form a new Si–C bond and a vinyl radical intermediate in radical hydrosilylation reactions,10,19 we therefore questioned whether silaindenes (benzosiloles) might be synthesized in a similar manner. Thus arylhydrosilanes, as silyl radical precursors, would intermolecularly react with alkynes to generate the vinyl radical intermediates that may be intramolecularly intercepted by the appendant aryl groups. Because many arylhydrosilanes are readily available, this approach would represent a more practical method in preparation of silaindenes than the previous ones which necessitate ortho-halogenated or -metalated arylsilanes (Scheme 1b).
In practice, we first used commercially available triphenyl-hydrosilane (Ph3SiH, 3a) and 1,2-diphenylacetylene as the substrates and simply transplanted the reaction conditions of the above intramolecular silylation reaction (Scheme 3). Pleasingly, the corresponding silaindene 5a could be isolated in 67% yield. Based on this success, we further examined the scope and limitations of this intermolecular radical annulation reaction. Diarylacetylenes containing electron-donating and electron-withdrawing groups are all viable reaction partners, leading to multiaryl-substituted silaindenes in moderate to good yields (5a–5h). Notably, these 1,1,2,3-tetraaryl silaindenes, which had not been synthesized via any previously existing methods, could be obtained via the current reaction in just one step from simple building blocks. A dithiophen-2-yl-acetylene and a dialkylacetylene were also used under the same conditions but gave low yields of the desired products (5k, 5l). When one of the substituents on the silyl group varied from phenyl to a methyl, the corresponding silaindene 5i was obtained in 60% yield. A more hindered tert-butyl group on the silicon center could also be used, although with decreased yield (5j).
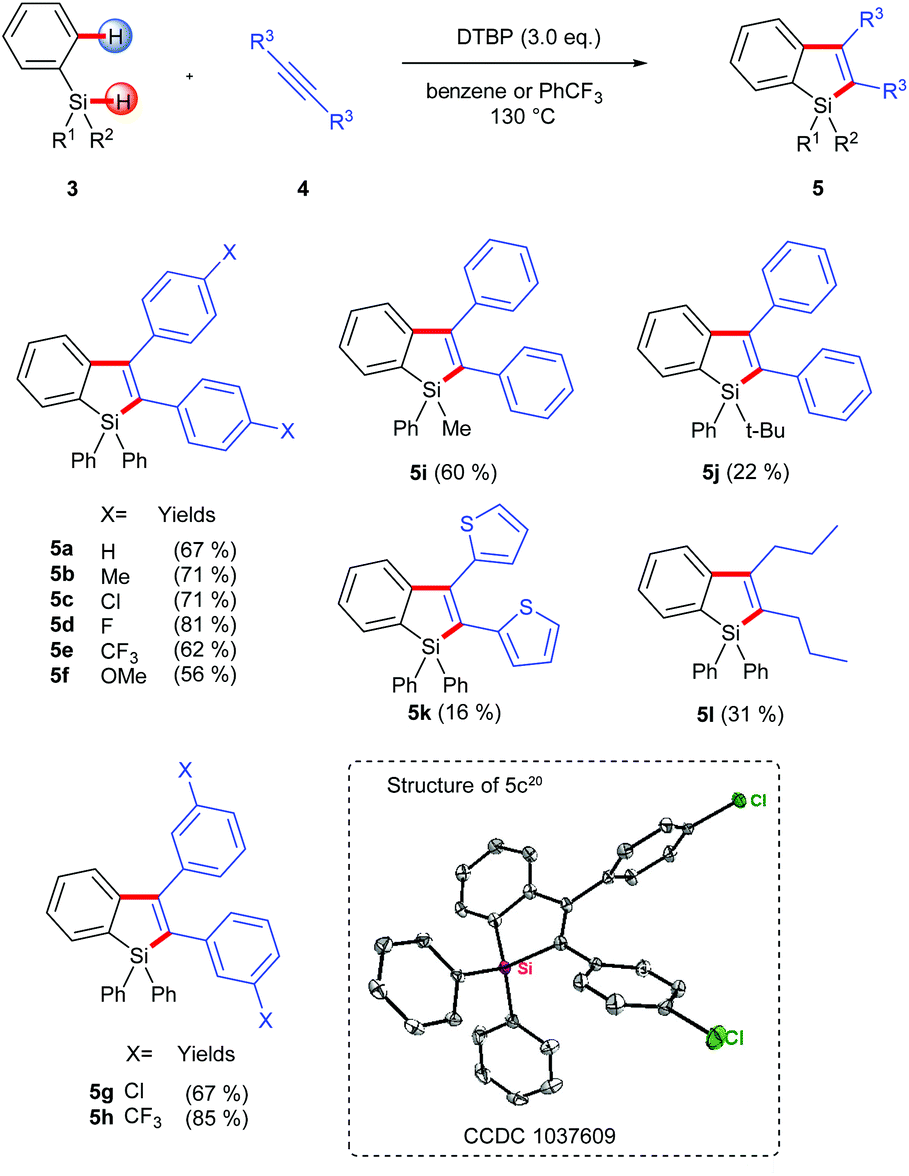 | ||
| Scheme 3 Silaindene synthesis via intermolecular radical annulation. | ||
On the basis of the established silyl radical chemistry10 and other aromatic homolytic substitution11 and radical cascade reactions,15 we propose two possible reaction pathways as shown in Scheme 4. In both pathways, the silyl radical is generated through a homolytic Si–H bond cleavage. For the formation of silafluorenes, the intramolecular addition of the silyl radical onto the proximal arene generates the cyclohexadienyl radical A2. Hydrogen elimination of A2 assisted by the tert-butoxyl radical will give the rearomatized final product 2. For silaindenes, the addition of the silyl radical across the alkyne triple bond generates the highly reactive alkenyl radical B2 which then undergoes intramolecular addition onto the phenyl moiety, forming another cyclohexadienyl radical B3. This radical eventually leads to silaindene 5, again by tert-butoxyl radical mediated hydrogen abstraction.
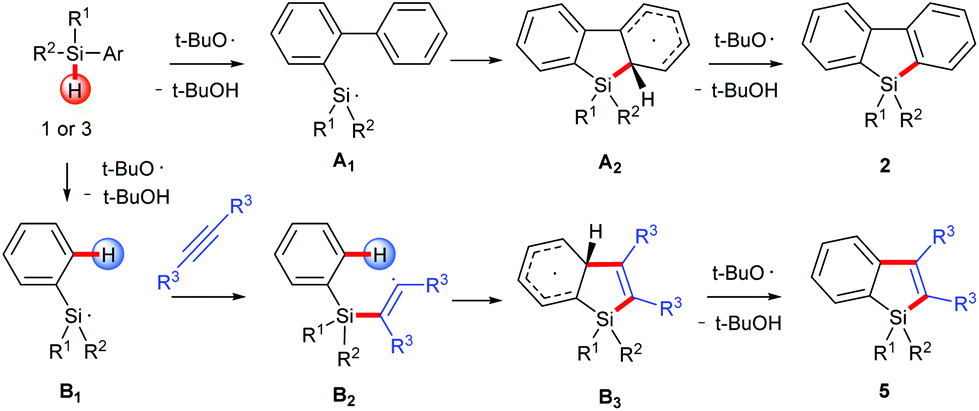 | ||
| Scheme 4 Reaction mechanism. | ||
In conclusion, we have developed a direct and efficient strategy employing aryl silyl radical intermediates towards the synthesis of silafluorenes and silaindenes. These reactions start from readily available arylhydrosilanes, proceed in a transition metal, base or acid-free system and furnish the products via direct Si–H and C–H cleavage pathways in one step. These synthetic methods are complementary to the existing ones towards benzosilole derivatives. Further extension and application of these reactions are underway in our laboratory.
Acknowledgements
We are grateful for financial support by the National Science Foundation of China (no. 21202129 and 21472146) and the Ministry of Science and Technology of PRC (973 program for young scientists, no. 2014CB548200).Notes and references
- For reviews about the synthesis, properties and application of silole derivatives, see: (a) S. Yamaguchi and K. Tamao, J. Chem. Soc., Dalton Trans., 1998, 3693 RSC; (b) S. Yamaguchi and K. Tamao, J. Organomet. Chem., 2002, 653, 223 CrossRef CAS; (c) M. Hissler, P. W. Dyer and R. Réau, Coord. Chem. Rev., 2003, 244, 1 CrossRef CAS; (d) S. Yamaguchi and K. Tamao, Chem. Lett., 2005, 34, 2 CrossRef CAS; (e) J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714 CrossRef CAS; (f) J. Y. Corey, Adv. Organomet. Chem., 2011, 59, 1 CrossRef CAS; (g) J. Y. Corey, Adv. Organomet. Chem., 2011, 59, 181 CrossRef CAS.
- For recent application of silole derivatives, see: (a) J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144 CrossRef CAS PubMed; (b) G. Lu, H. Usta, C. Risko, L. Wang, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 7670 CrossRef CAS PubMed; (c) P. M. Beaujuge, W. Pisula, H. N. Tsao, S. Ellinger, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 7514 CrossRef CAS PubMed; (d) T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250 CrossRef CAS PubMed; (e) X. Shen, G. Zhang and D. Zhang, Org. Lett., 2012, 14, 1744 CrossRef CAS PubMed; (f) H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2013, 15, 2104 CrossRef CAS PubMed.
- For representative contributions, see: (a) H. Gilman and R. D. Gorsich, J. Am. Chem. Soc., 1955, 77, 6380 CrossRef CAS; (b) K. L. Chan, M. J. McKiernan, C. R. Towns and A. B. Holmes, J. Am. Chem. Soc., 2005, 127, 7662 CrossRef CAS PubMed; (c) S. H. Lee, B.-B. Jang and Z. H. Kafafi, J. Am. Chem. Soc., 2005, 127, 9071 CrossRef CAS PubMed; (d) R.-F. Chen, Q.-L. Fan, C. Zheng and W. Huang, Org. Lett., 2006, 8, 203 CrossRef CAS PubMed; (e) L. Li, J. Xiang and C. Xu, Org. Lett., 2007, 9, 4877 CrossRef CAS PubMed; (f) C. Wang, Q. Luo, H. Sun, X. Guo and Z. Xi, J. Am. Chem. Soc., 2007, 129, 3094 CrossRef CAS PubMed; (g) N. Yu, C. Wang, F. Zhao, L. Liu, W.-X. Zhang and Z. Xi, Chem. – Eur. J., 2008, 14, 5670 CrossRef CAS PubMed.
- For representative contributions, see: (a) K. Tamao, S. Yamaguchi and M. Shiro, J. Am. Chem. Soc., 1994, 116, 11715 CrossRef CAS; (b) K. Tamao, M. Uchida, T. Izumizawa, K. Furukawa and S. Yamaguchi, J. Am. Chem. Soc., 1996, 118, 11974 CrossRef CAS; (c) S. Yamaguchi, T. Goto and K. Tamao, Angew. Chem., Int. Ed., 2000, 39, 1695 CrossRef CAS; (d) S. Yamaguchi, C. Xu and K. Tamao, J. Am. Chem. Soc., 2003, 125, 13662 CrossRef CAS PubMed; (e) C. Xu, A. Wakamiya and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 1638 CrossRef CAS PubMed; (f) L. Ilies, H. Tsuji, Y. Sato and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 4240 CrossRef CAS PubMed.
- (a) K. Tamao, S. Yamaguchi, M. Shiozaki, Y. Nakagawa and Y. Ito, J. Am. Chem. Soc., 1992, 114, 5867 CrossRef CAS; (b) K. Ikenaga, K. Hiramatsu, N. Nasaka and S. Matsumoto, J. Org. Chem., 1993, 58, 5045 CrossRef CAS; (c) W. S. Palmer and K. A. Woerpel, Organometallics, 1997, 16, 1097 CrossRef CAS; (d) W. S. Palmer and K. A. Woerpel, Organometallics, 2001, 20, 3691 CrossRef CAS; (e) T. Matsuda, S. Kadowaki and M. Murakami, Chem. Commun., 2007, 2627 RSC; (f) T. Ohmura, K. Masuda and M. Suginome, J. Am. Chem. Soc., 2008, 130, 1526 CrossRef CAS PubMed; (g) T. Ohmura, K. Masuda, I. Takase and M. Suginome, J. Am. Chem. Soc., 2009, 131, 16624 CrossRef CAS PubMed.
- (a) T. Matsuda, S. Kadowaki, Y. Yamaguchi and M. Murakami, Chem. Commun., 2008, 2744 RSC; (b) M. Tobisu, M. Onoe, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 7506 CrossRef CAS PubMed; (c) T. Matsuda, Y. Yamaguchi, M. Shigeno, S. Sato and M. Murakami, Chem. Commun., 2011, 47, 8697 RSC; (d) E. Shirakawa, S. Masui, R. Narui, R. Watabe, D. Ikeda and T. Hayashi, Chem. Commun., 2011, 47, 9714 RSC; (e) Y. Liang, W. Geng, J. Wei and Z. Xi, Angew. Chem., Int. Ed., 2012, 51, 1934 CrossRef CAS PubMed; (f) M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477 CrossRef CAS PubMed; (g) K. Ouyang, Y. Liang and Z. Xi, Org. Lett., 2012, 14, 4572 CrossRef CAS PubMed; (h) T. Meng, K. Ouyang and Z. Xi, RSC Adv., 2013, 3, 14273 RSC.
- (a) T. Matsuda, S. Kadowaki, T. Goya and M. Murakami, Org. Lett., 2006, 9, 133 CrossRef PubMed; (b) M. Shimizu, K. Mochida and T. Hiyama, Angew. Chem., Int. Ed., 2008, 47, 9760 CrossRef CAS PubMed; (c) K. Mochida, M. Shimizu and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 8350 CrossRef CAS PubMed; (d) T. Ureshino, T. Yoshida, Y. Kuninobu and K. Takai, J. Am. Chem. Soc., 2010, 132, 14324 CrossRef CAS PubMed; (e) Y. Yabusaki, N. Ohshima, H. Kondo, T. Kusamoto, Y. Yamanoi and H. Nishihara, Chem. – Eur. J., 2010, 16, 5581 CrossRef CAS PubMed; (f) Y. Liang, S. Zhang and Z. Xi, J. Am. Chem. Soc., 2011, 133, 9204 CrossRef CAS PubMed; (g) T. Shibata, T. Uchiyama, Y. Yoshinami, S. Takayasu, K. Tsuchikama and K. Endo, Chem. Commun., 2012, 48, 1311 RSC; (h) Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520 CrossRef CAS PubMed; (i) Q.-W. Zhang, K. An and W. He, Angew. Chem., Int. Ed., 2014, 53, 5667 CrossRef CAS PubMed.
- (a) S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192 CrossRef CAS PubMed; (b) S. Furukawa, J. Kobayashi and T. Kawashima, Dalton Trans., 2010, 39, 9329 RSC.
- (a) D. Lv, C. Wu and P. Li, Org. Lett., 2014, 16, 1486 CrossRef PubMed; (b) D. Lv, C. Wu and P. Li, Chem. – Eur. J., 2014, 20, 163 CrossRef PubMed; (c) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962 CrossRef CAS PubMed.
- (a) D. Leifert and A. Studer, Org. Lett., 2015, 17, 386 CrossRef CAS PubMed. For selected general reviews on silyl radical chemistry, see: (b) C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229 CrossRef CAS; (c) C. Chatgilialoglu and V. I. Timokhin, Adv. Organomet. Chem., 2008, 57, 117 CrossRef CAS.
- W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803 RSC.
- S. Furukawa, S. Haga, J. Kobayashi and T. Kawashima, Org. Lett., 2014, 16, 3228 CrossRef CAS PubMed.
- (a) Z. Shi and F. Glorius, Chem. Sci., 2013, 4, 829 RSC; (b) S. Wertz, D. Leifert and A. Studer, Org. Lett., 2013, 15, 928 CrossRef CAS PubMed.
- For representative contributions about BHAS reactions, see: (a) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS PubMed; (b) E. Shirakawa, K.-i. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537 CrossRef CAS PubMed; (c) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS PubMed; (d) D. S. Roman, Y. Takahashi and A. B. Charette, Org. Lett., 2011, 13, 3242 CrossRef CAS PubMed; (e) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018 CrossRef CAS PubMed; (f) S. Yanagisawa and K. Itami, ChemCatChem, 2011, 3, 827 CrossRef CAS.
- U. Wille, Chem. Rev., 2012, 113, 813 CrossRef PubMed.
- A. L. J. Beckwith and D. M. O'Shea, Tetrahedron Lett., 1986, 27, 4525 CrossRef CAS.
- T. G. Back and M. V. Krishna, J. Org. Chem., 1988, 53, 2533 CrossRef CAS.
- (a) Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975 CrossRef CAS PubMed; (b) Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754 CrossRef CAS PubMed.
- For recent reports about the radical hydrosilylation reactions, see: (a) S. E. Gottschling, T. N. Grant, K. K. Milnes, M. C. Jennings and K. M. Baines, J. Org. Chem., 2005, 70, 2686 CrossRef CAS PubMed; (b) Y. Liu, S. Yamazaki and S. Yamabe, J. Org. Chem., 2004, 70, 556 CrossRef PubMed; (c) A. Postigo, S. Kopsov, S. S. Zlotsky, C. Ferreri and C. Chatgilialoglu, Organometallics, 2009, 28, 3282 CrossRef CAS; (d) A. Postigo, S. Kopsov, C. Ferreri and C. Chatgilialoglu, Org. Lett., 2007, 9, 5159 CrossRef CAS PubMed; (e) S. Amrein and A. Studer, Chem. Commun., 2002, 1592 RSC.
- CCDC 1037609 (5c) contains the supplementary crystallographic data for this paper.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials including experimental procedures, copies of 1H and 13C NMR spectra of all new products and crystallographic data for 5c. CCDC 1037609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00012b |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2015 |