
Concise asymmetric total synthesis of bruceolline J†
Dattatraya H.
Dethe
* and
Vijay
Kumar B
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India. E-mail: ddethe@iitk.ac.in; Fax: (+91)-512-2597436; Tel: (+91)-512-2596537
First published on 3rd March 2015
Abstract
We have developed a concise biomimetic and asymmetric approach involving Sharpless asymmetric dihydroxylation and Lewis acid catalysed cyclopenta[b]annulation as key steps to synthesize (+)-bruceolline J, and a racemic approach employing an intramolecular rhodium carbenoid C–H insertion and highly regioselective gem-dimethylation reactions as key steps to synthesize bruceolline D, E along with (±)-bruceolline J.
Bruceollines are a small group of natural products containing a highly oxygenated cyclopent[b]indole moiety.1 In 1994, Ohmoto and co-workers reported the isolation of bruceollines D–F (1–3) (Fig. 1), from the root wood of Brucea mollis Wall. var. tonkinensis Lecomte.2 Recently, in 2011 Yu and co-workers isolated bruceolline J (4) along with several other bruceollines from an ethanol extract of the stems of Brucea mollis.3 The genus B. mollis is found in southern china and traditionally used as remedy for malaria and other parasitic diseases. Despite their potential medicinal utility, bruceollines have attracted much less attention from the synthetic community.
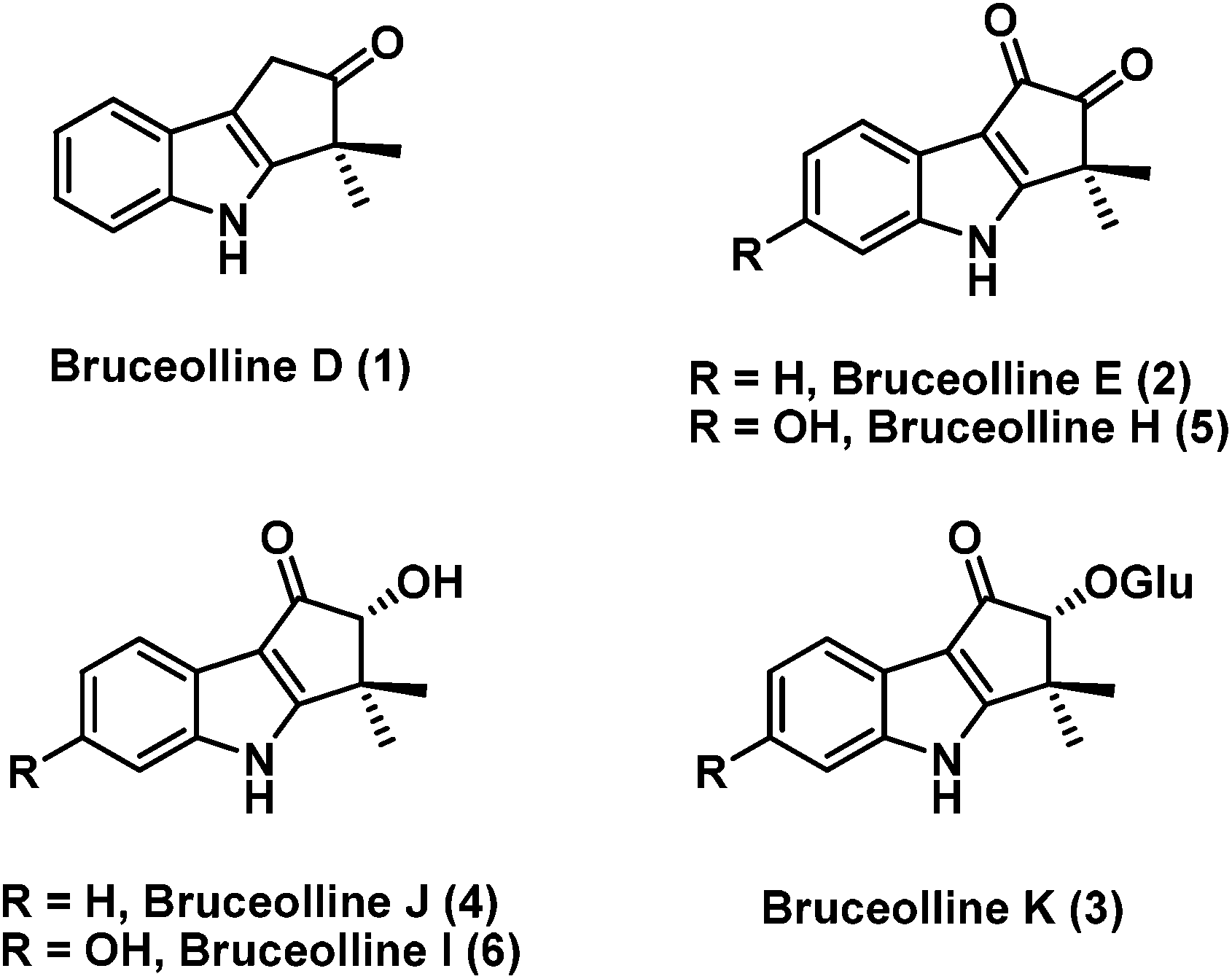 | ||
| Fig. 1 The structures of bruceolline family of natural products. | ||
So far only one synthesis of bruceolline E (2) and J (4) has been reported in the literature by Gribble and co-workers.4,5 Recently, the same group also reported a concise asymmetric synthesis of bruceolline J using a superstoichiometric amount (3 equiv.) of (+)- and (−)-DIPCl for the asymmetric induction. Herein we report racemic as well as asymmetric syntheses of bruceolline J using two different strategies and synthesis of related natural products.
We planned the racemic total synthesis of bruceolline J (4), D (1) and E (2) using intramolecular rhodium carbenoid C–H insertion and highly regioselective alkylation as the key steps. It was envisaged that bruceolline E (2) and J (4) could be obtained from bruceolline D (1) by functional group manipulations. Bruceolline D (1) in turn could be obtained from ketone 7 by regioselective gem-dimethylation. The ketone 7 could be accessed by intramolecular rhodium carbenoid C–H insertion reaction of diazoketone 8 which in turn could be easily synthesized from indole acetic acid. On the other hand, bruceolline E (2) was thought to be obtained from diketone 9 by gem-dimethylation. The diketone 9 could be generated by C–H insertion reaction of corresponding diazoketone made from indole glyoxyl chloride (Scheme 1).
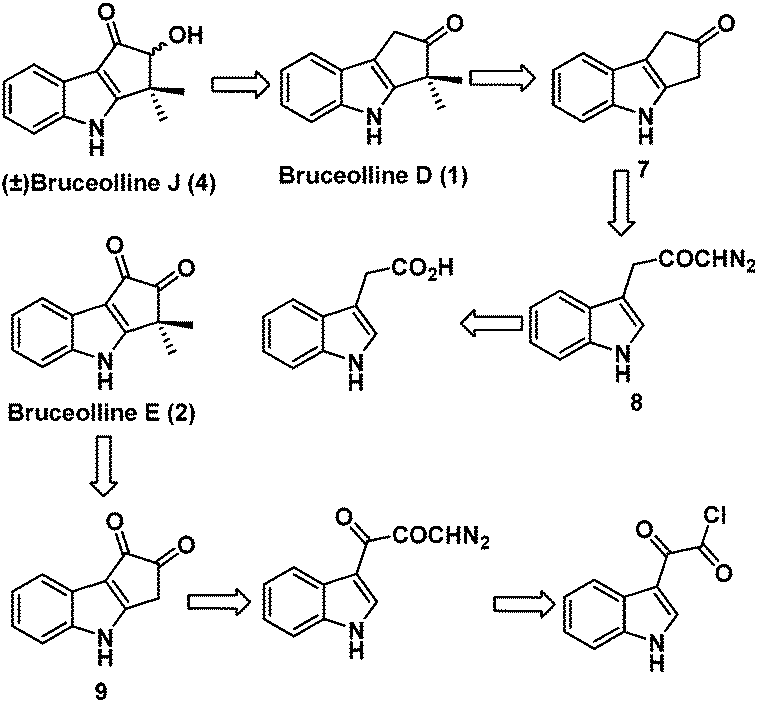 | ||
| Scheme 1 Retrosynthetic analysis of bruceolline D, E and J. | ||
First, we adopted the second strategy as it is a more direct route and would result in a short synthesis of bruceolline E (2). The synthesis commenced with the treatment of indolyl glyoxyl chloride 10 (prepared from N-benzyl indole and oxalyl chloride)6 with diazomethane to generate diazoketone 11. To our surprise, exposure of the diazoketone 11 to a catalytic amount of Rh(OAc)4 under reflux in CH2Cl2 generated the acetyl indole 12 in 70% yield instead of diketone 9 (Scheme 2). Even the usage of the more reactive catalyst Rh(OCOCF3)4 also afforded compound 12. A mechanism for the formation of the acetyl indole is proposed in Scheme 3.
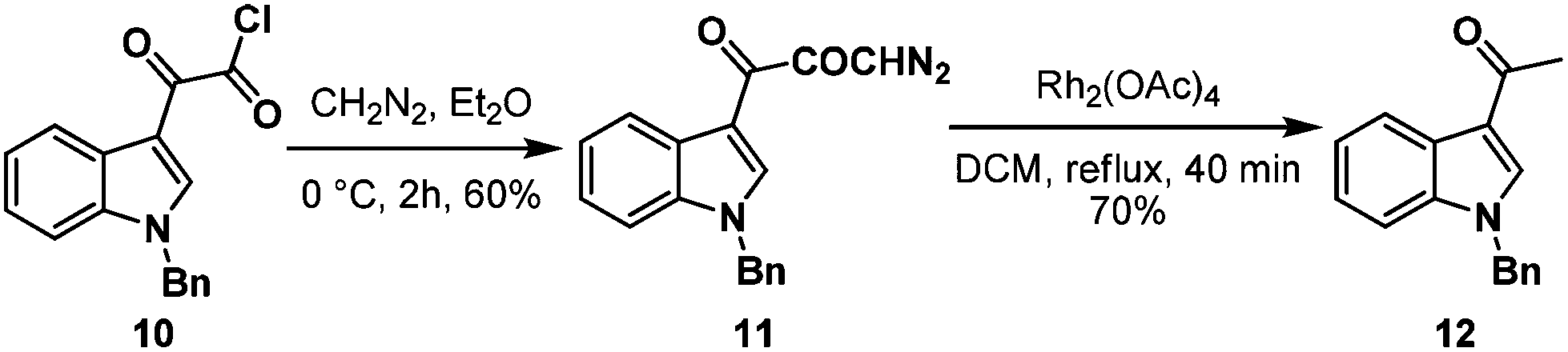 | ||
| Scheme 2 Attempts towards the synthesis of bruceolline E. | ||
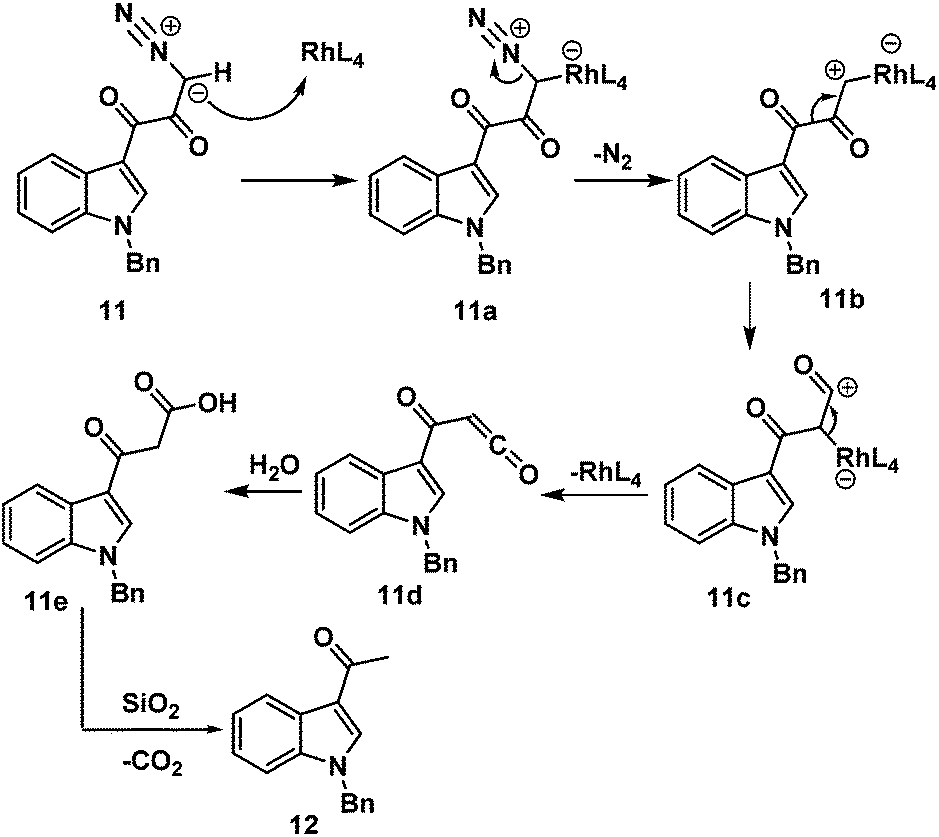 | ||
| Scheme 3 Mechanism showing the formation of 3-acetyl indole (12) from diazoketone (11). | ||
In the first step, reaction of rhodium acetate with diazoketone 11 generates rhodium carbenoid 11b. Wolff rearrangement of rhodium carbenoid 11b generates the ketene 11d. Once 11d is formed, reaction with water (during silica gel column purification) rapidly generates 3-acetyl indole 12 and carbon dioxide.
Next, we turned our attention towards the synthesis of ketone 13 from diazoketone 14. Thus N-benzyl indole acetic acid 15 on reaction with oxalyl chloride followed by treatment with diazomethane afforded diazoketone 14 in moderate yield. Exposure of diazoketone 14 to a catalytic amount of rhodium(II) acetate in CH2Cl2 under reflux conditions afforded the ketone 13 in 78% yield. In an effort to synthesize diketone 16, ketone 13 was treated with SeO2 in dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (6:1) for 16 hours.7 Unfortunately, it furnished the undesired regioisomer 17 in 92% yield (Scheme 4). The structure of 17 was unambiguously confirmed by single crystal X-ray analysis (Fig. 2).8
:H2O (6:1) for 16 hours.7 Unfortunately, it furnished the undesired regioisomer 17 in 92% yield (Scheme 4). The structure of 17 was unambiguously confirmed by single crystal X-ray analysis (Fig. 2).8
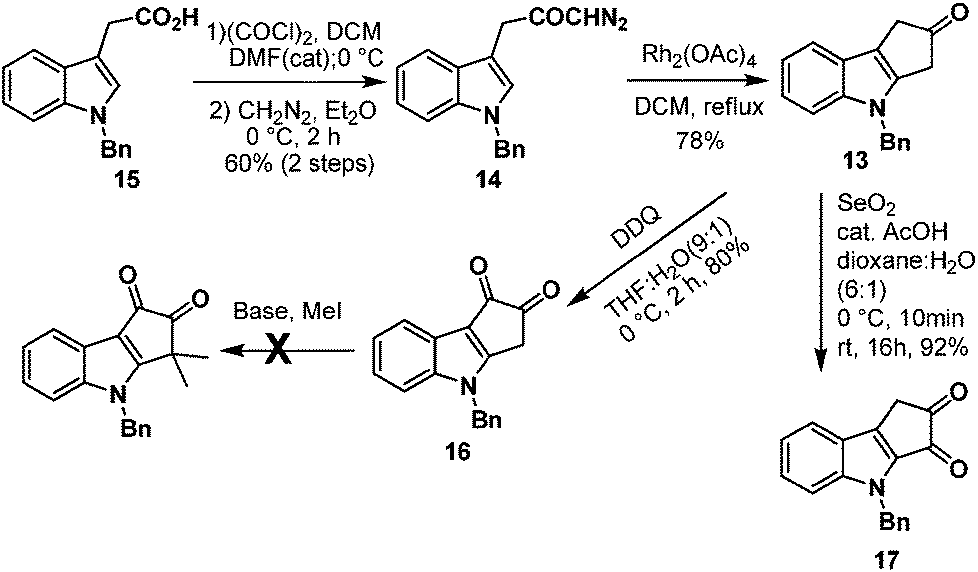 | ||
| Scheme 4 Synthesis of ketone 13. | ||
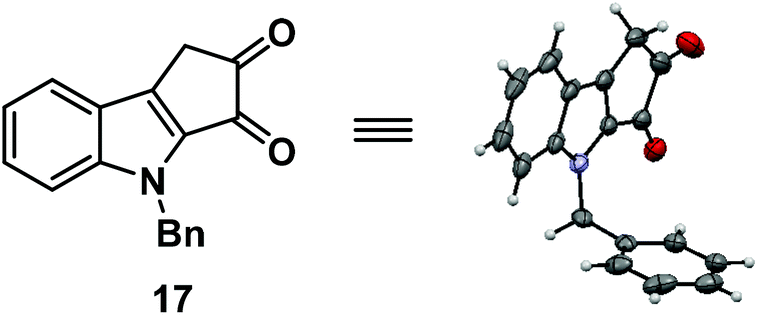 | ||
| Fig. 2 X-ray crystal structure of compound 17. | ||
Benzylic oxidation of ketone 13 on treatment with DDQ9 in THF–H2O (9:1) afforded the required diketone 16 in 80% yield. However gem-dimethylation of diketone 16 to generate bruceolline E (2) under various basic conditions, such as LDA, NaH, t-BuOK, NaOMe, encountered a deadlock as the diketone 16 decomposed under these reaction conditions. But, to our delight, treatment of ketone 13 with NaH and MeI furnished compound 18 in a highly regioselective manner with 80% yield. The nucleophilic C-3 position of indole might be making the enolate formation at the other side of ketone 13 less favourable, resulting in the excellent regioselective gem-dimethylation. Hydrogenolysis of the N-benzyl group using H2/Pd(OH)2 afforded the natural product bruceolline D (1) in 70% yield. A simple benzylic oxidation of bruceolline D (1) assisted by DDQ furnished bruceolline E (2) in 92% yield.
Next we targeted the synthesis of (±)-bruceolline J (4) from the common intermediate 18. Reduction of the ketone group of compound 18 and acetylation of the resulting hydroxyl group gave rise to (±)-19. A successive benzylic oxidation using DDQ, debenzylation10 using H2/Pd(OH)2, followed by acetate hydrolysis under basic conditions afforded the natural product (±)-bruceolline J (4) in 81% overall yield from 19 (Scheme 5).
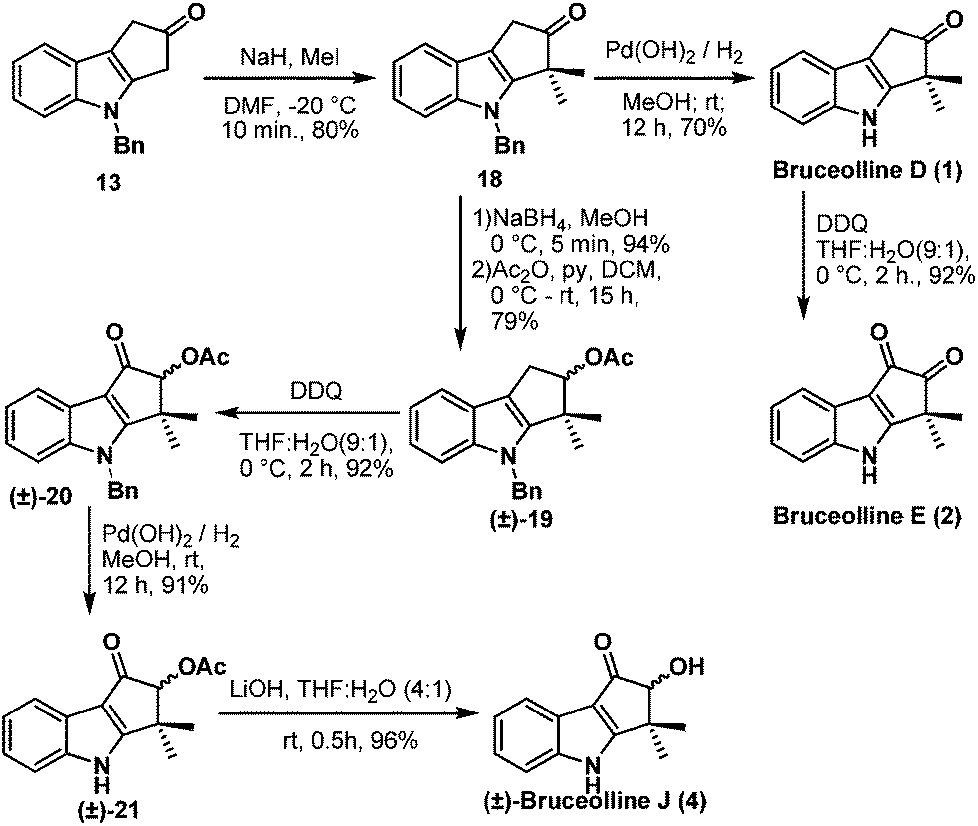 | ||
| Scheme 5 Total synthesis of bruceolline D (1), E (2) and J (4). | ||
Our task then was a concise catalytic asymmetric synthesis of (+)-bruceolline J. It was contemplated that (+)-bruceolline J (4) could be obtained from cyclopenta[b]indole 19 by benzylic oxidation (Scheme 6). Inspired by the biosynthetic pathway of bruceolline alkaloids (Scheme 7),3 it was envisaged that compound 19 could be accessed from diol 22 by a Lewis acid catalysed cyclopentannulation reaction. The chiral diol 22 could be prepared by Sharpless asymmetric dihydroxylation of compound 23, which in turn could be obtained by prenylation of indole.
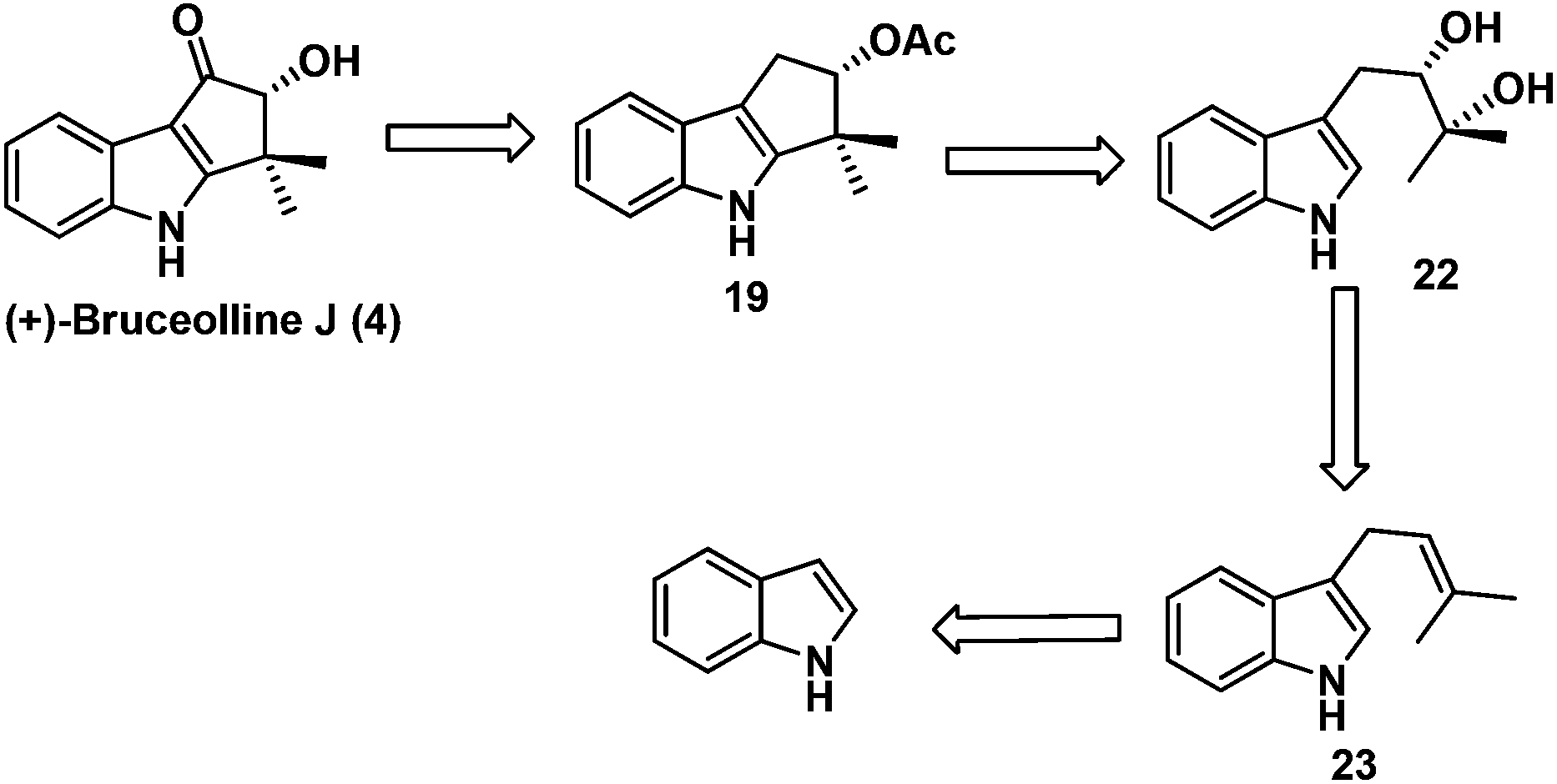 | ||
| Scheme 6 Retrosynthetic analysis of bruceolline J. | ||
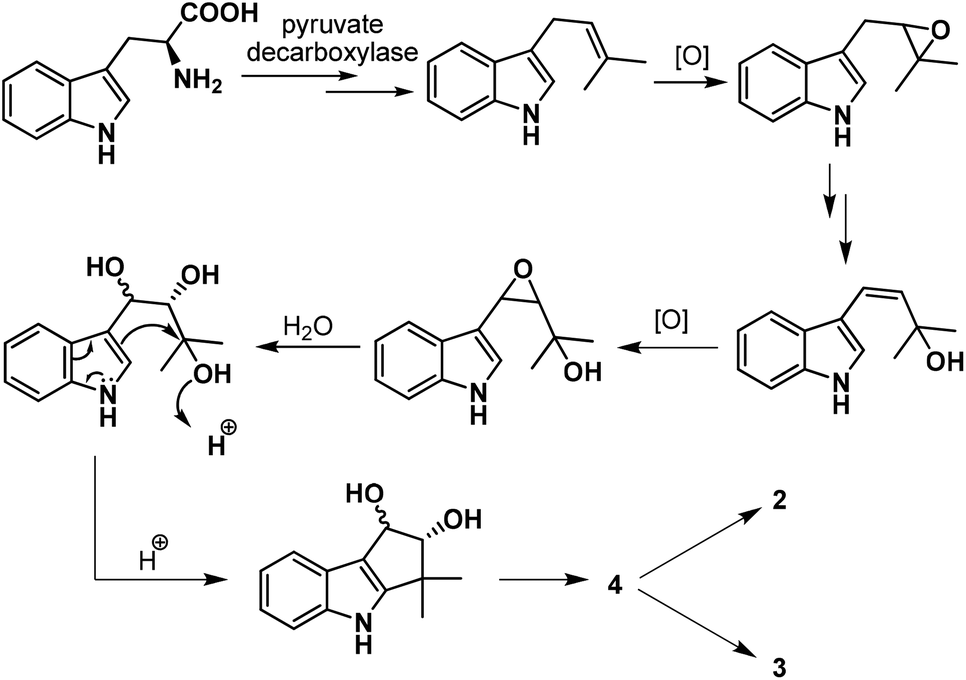 | ||
| Scheme 7 Plausible biosynthetic pathway. | ||
Thus, treatment of N-benzyl protected indole 24 with prenyl bromide in the presence of base11 furnished compound 23, which was then subjected to Sharpless asymmetric dihydroxylation protocol12 to get diol (+)-22 in 93% yield and 90% ee. Selective acetylation of secondary alcohol followed by BF3·OEt2 catalysed cyclopentannulation allowed a rapid construction of the cyclopentane ring to afford cyclopenta-[b]-indole (−)-19 in good yield which represents the key intermediate of the scheme (Scheme 8).
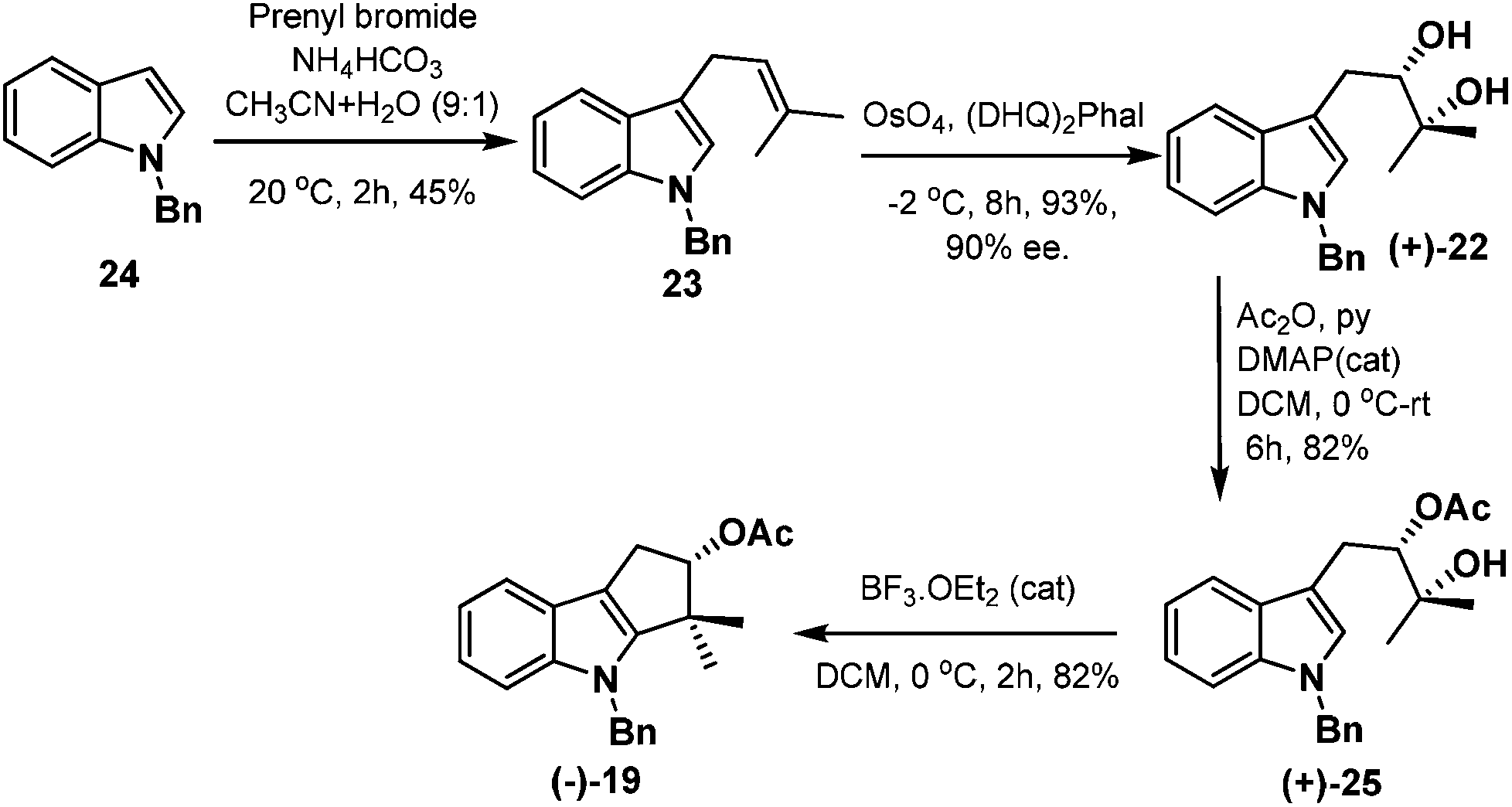 | ||
| Scheme 8 Synthesis of key intermediate 19. | ||
Benzylic oxidation of the compound (−)-19 using DDQ in THF–H2O (9:1) afforded ketone (+)-20 in good yield which was then employed in debenzylation13 using palladium hydroxide to give (+)-21 in 91% yield. Base mediated hydrolysis of acetyl group of (+)-21 furnished the natural product, (+)-bruceolline J (4), in excellent yield with 84% ee (Scheme 9). The 1H, 13C spectroscopic data, optical rotations of (+)-bruceolline J (4) are identical with that of the natural (+)-bruceolline.
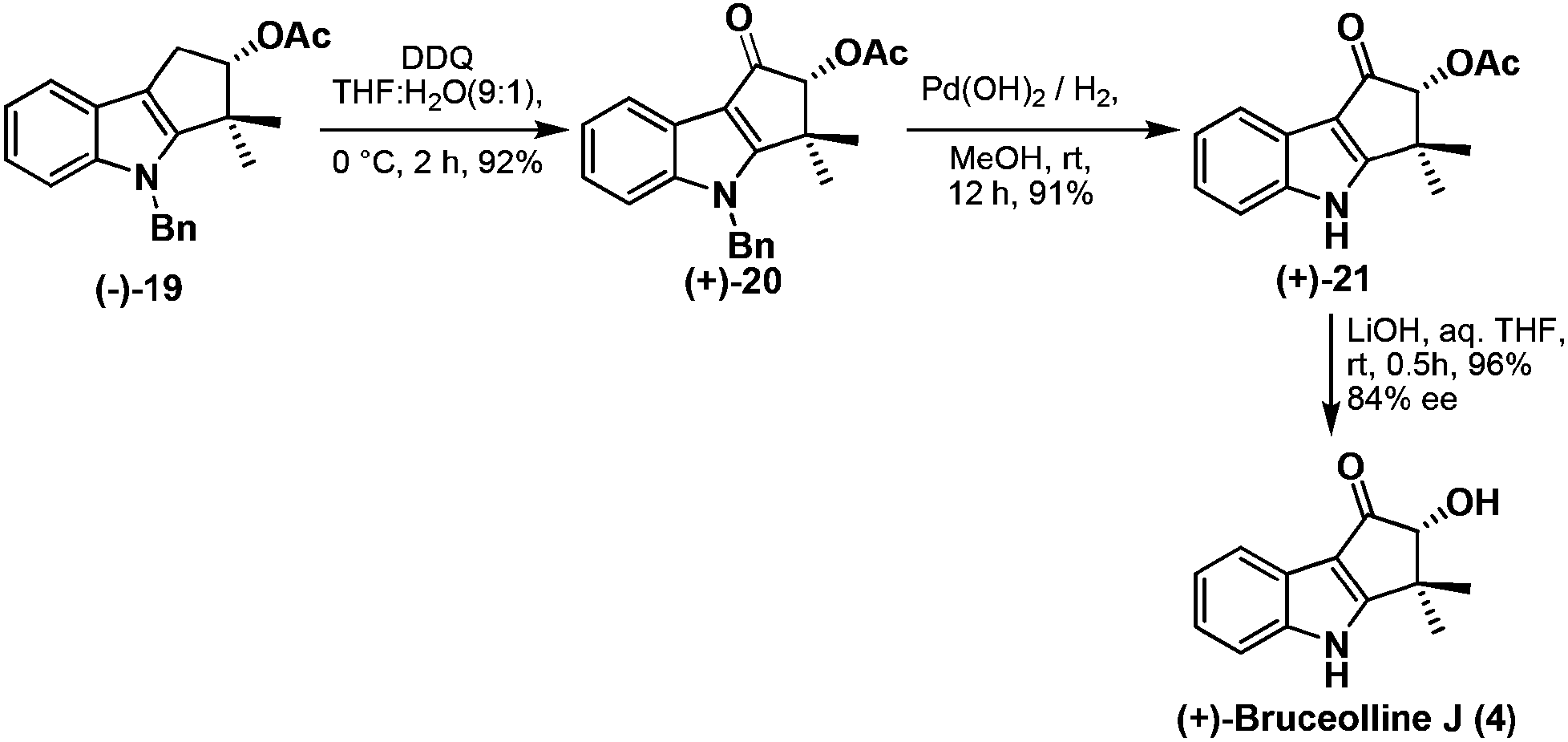 | ||
| Scheme 9 Synthesis of (+)-bruceollines J from (−)-19. | ||
In conclusion, we have reported a successful integration of two divergent synthetic strategies that allow the efficient total syntheses of racemic as well as chiral bruceolline J and bruceollines D, E. Asymmetric synthesis of bruceolline J was accomplished using a Sharpless asymmetric dihydroxylation and a Lewis acid catalysed cyclopentannulation as the key steps. A racemic approach constitutes a common route for the synthesis of bruceolline D, E and J in 4, 5 and 8 steps, respectively, in very good overall yield using an intramolecular rhodium carbenoid C–H insertion and a highly regioselective methylation as the key steps. The racemic and asymmetric approaches also give ready access to other natural products such as bruceollines H (5), I (6) and K (3) for further biological studies.
Acknowledgements
We thank the reviewers for their valuable suggestions and corrections which helped a lot in improving the manuscript. V.K.B. thanks CSIR, New Delhi, India, for the award of a research fellowship. We thank Dr Sakiat Hossain and Dr Atanu Dey from IIT Kanpur for their assistance in solving the crystal structure. Financial support from IIT Kanpur, India, is gratefully acknowledged.Notes and references
- G. W. Gribble, Pure Appl. Chem., 2003, 75, 1417 CrossRef CAS.
- Y. Ouyang, K. Koike and T. Ohmoto, Phytochemistry, 1994, 36, 1543 CrossRef CAS.
- H. Chen, J. Bai, Z. F. Fang, S. S. Yu, S. G. Ma, S. Xu, Y. Li, J. Qu, J. H. Ren, L. Li, Y. K. Si and X. G. Chen, J. Nat. Prod., 2011, 74, 2438 CrossRef CAS PubMed.
- J. A. Jordan, G. W. Gribble and J. C. Badenock, Tetrahedron Lett., 2011, 52, 6772 CrossRef CAS PubMed.
- J. M. Lopchuk, I. L. Green, J. C. Badenock and G. W. Gribble, Org. Lett., 2013, 15, 4485 CrossRef CAS PubMed.
- X. Guinchard, Y. Vallee and J. N. Denis, Org. Lett., 2007, 9, 3761 CrossRef CAS PubMed.
- G. Mehta and H. M. Shinde, Tetrahedron Lett., 2003, 44, 7049 CrossRef CAS.
- CCDC 1004473 (17) contains the supplementary crystallographic data for this paper.
- K. C. Nicolaou, D. Y. K. Chen, X. Huang, T. Ling, M. Bella and S. A. Snyder, J. Am. Chem. Soc., 2004, 126, 12888 CrossRef CAS PubMed.
- T. Fekner, J. Gallucci and M. K. Chan, Org. Lett., 2003, 5, 4795 CrossRef CAS PubMed.
- M. Westermaier and H. Mayr, Org. Lett., 2006, 8, 4791 CrossRef CAS PubMed.
- K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H. L. Kwong, K. Morikawa, Z.-M. Wang, D. Xu and X. L. Zhang, J. Org. Chem., 1992, 57, 2768 CrossRef CAS.
- T. Fujiwara, M. Sasaki, K. Omata, C. Kebuto, K. Kabuto and Y. Takeuchi, Tetrahedron: Asymmetry, 2004, 15, 555 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures giving 1H and 13C NMR spectra for all compounds and a CIF file giving crystallographic data for compound 17. CCDC 1004473. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00030k |
| This journal is © the Partner Organisations 2015 |