
Zinc diiodide-promoted synthesis of trisubstituted allenes from propargylic amines†
Jinqiang
Kuang
a,
Xinjun
Tang
a and
Shengming
Ma
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, P. R. China
First published on 16th March 2015
Abstract
Zinc diiodide has been identified as an effective reagent for the efficient synthesis of trisubstituted allenes from propargylic amines. Compared to CdI2 this protocol offers a green approach. Due to the easy preparation of propargylic amines through the method developed by this group, this method provides a two-step synthesis of trisubstituted allenes from 1-alkynes, ketones, and pyrrolidine. Finally, an efficient synthesis of such trisubstituted allenes from 1-alkynes, ketones, and pyrrolidine via simple filtration has been developed. Compared with the CdI2-mediated protocol, the current protocol enjoys a much wider scope for ketones and affords functionalized allenes without further cyclization in some substrates.
Introduction
Allenes are playing more and more important roles in organic synthesis as many attractive transformations of allenes have been discovered,1 thus, efficient synthetic methods of preparation of allenes from readily available starting materials are highly desirable.2 With such a consideration, we have been paying close attention to the allenylation of terminal alkyne (ATA) reactions since its starting materials, i.e., terminal alkynes, aldehydes (or ketones) and amines, are readily available. Based on the pioneering contribution of Crabbé et al. with paraformaldehyde,3,4 in the last few years, we have developed a methodology for the synthesis of mono-substituted and 1,3-disubstituted allenes from terminal alkynes, aldehydes and amines.5,6 However, it should be noted that these reactions are limited to paraformaldehyde or aldehydes: no or only a trace amount of allene was formed when ketones were used. There are only limited reports on synthesis of trisubstituted allenes from terminal alkynes. Bertrand et al. employed a cationic gold(I) complex for the catalytic coupling of enamines, which are derived from aldehydes or ketones, and terminal alkynes to non-terminal allenes,7 however, only one example of ketone, i.e., phenyl isopropyl ketone, was reported. Wang et al. developed a Cu(I)-catalyzed protocol for the reaction of 1-alkynes with N-tosylhydrazones of aldehydes or ketones to afford 1,3-disubstituted or trisubstituted allenes.8 Recently, this group reported that the allenylation of terminal alkyne (ATA) reactions with methyl alkyl ketones or cyclic ketones may be realized to afford trisubstituted allenes efficiently when promoted by CdI2,9 in which propargylic amines derived from 1-alkynes, ketones and pyrrolidine were believed to be key intermediates. However, it should be noted that CdI2 is a relatively toxic chemical, which is harmful to the environment and human beings. In this report, we would like to report that ZnI2, cheaper, non-toxic and environmentally benign, is very effective for converting corresponding propargylic amines derivatized from ketones to trisubstituted allenes. Finally a practical two-step protocol from terminal alkynes and ketones to trisubstituted allenes via simple filtration has also been developed.Results and discussion
We have recently developed a CuBr-catalyzed method for the efficient synthesis of propargylic amines.10 Through this method, various propargylic amines derived from terminal alkynes, ketones and pyrrolidine could be obtained in excellent yields. Under the mediation of CdI2, these types of propargylic amines may be converted into trisubstituted allenes efficiently (eqn (1)).9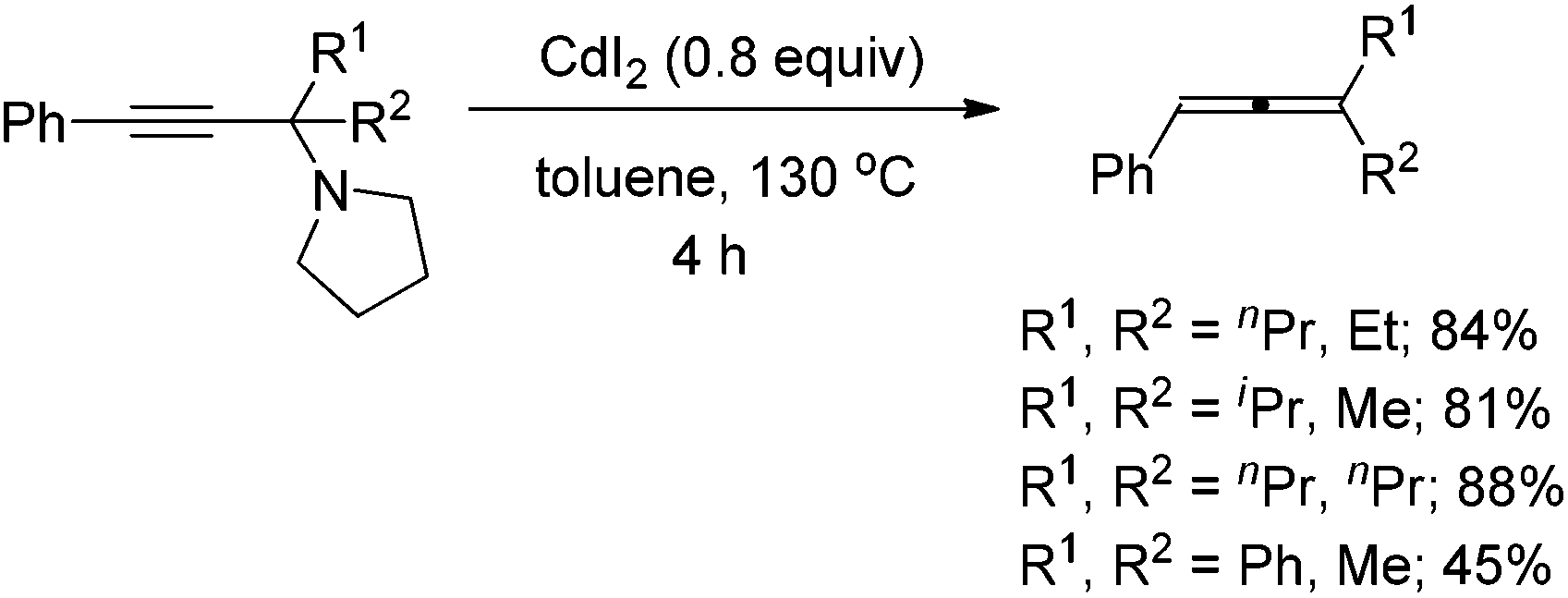 | (1) |
To our delight, ZnI2 could also promote the transformation effectively (entry 1, Table 1), which nicely provides chemists an environmentally friendly choice to produce trisubstituted allenes. In comparison, ZnBr2, Zn(OTf)2, CuBr, and CuBr2 might also be a choice, however, much less effective (entries 2–4 and 9, Table 1). CuI, KAuCl4·2H2O,11a and AgNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11b are extremely ineffective for such transformation (entries 10–12, Table 1). Further screening of reaction temperature and loading of ZnI2 led to a set of optimal conditions: 0.6 equiv. of ZnI2 in toluene at 120 °C (entry 5, Table 1). As a comparison, CdI2 provided the product in 88% NMR yield (entry 13, Table 1).
11b are extremely ineffective for such transformation (entries 10–12, Table 1). Further screening of reaction temperature and loading of ZnI2 led to a set of optimal conditions: 0.6 equiv. of ZnI2 in toluene at 120 °C (entry 5, Table 1). As a comparison, CdI2 provided the product in 88% NMR yield (entry 13, Table 1).
a
|
|
||||
|---|---|---|---|---|
| Entry | Metallic salt (X equiv.) | Time (h) | Yield of 2ab (%) | Recovery of 1ab (%) |
| a The reaction was carried out on a 0.5 mmol scale in 1.5 mL of toluene. b Determined by 1H NMR analysis of the crude reaction mixture with CH2Br2 as the internal standard. c The reaction was carried out at 110 °C. d The reaction was carried out at 100 °C. | ||||
| 1 | ZnI2 (0.8) | 1.1 | 78 | — |
| 2 | ZnBr2 (0.8) | 1.1 | 74 | — |
| 3 | Zn(OTf)2 (0.8) | 1.1 | 69 | — |
| 4 | CuBr (0.8) | 14.5 | 18 | 8 |
| 5 | ZnI 2 (0.6) | 0.9 | 77 | — |
| 6 | ZnI2 (0.5) | 1.6 | 65 | — |
| 7c | ZnI2 (0.6) | 1.6 | 72 | — |
| 8d | ZnI2 (0.6) | 2.5 | 64 | — |
| 9 | CuBr2 (0.6) | 10 | 17 | 6 |
| 10 | CuI (0.6) | 3 | Trace | 75 |
| 11 | AgNO3 (0.6) | 3 | 2 | 45 |
| 12 | KAuCl4·2H2O (0.1) | 3 | 4 | 60 |
| 13 | CdI2 (0.8) | 2 | 88 | — |
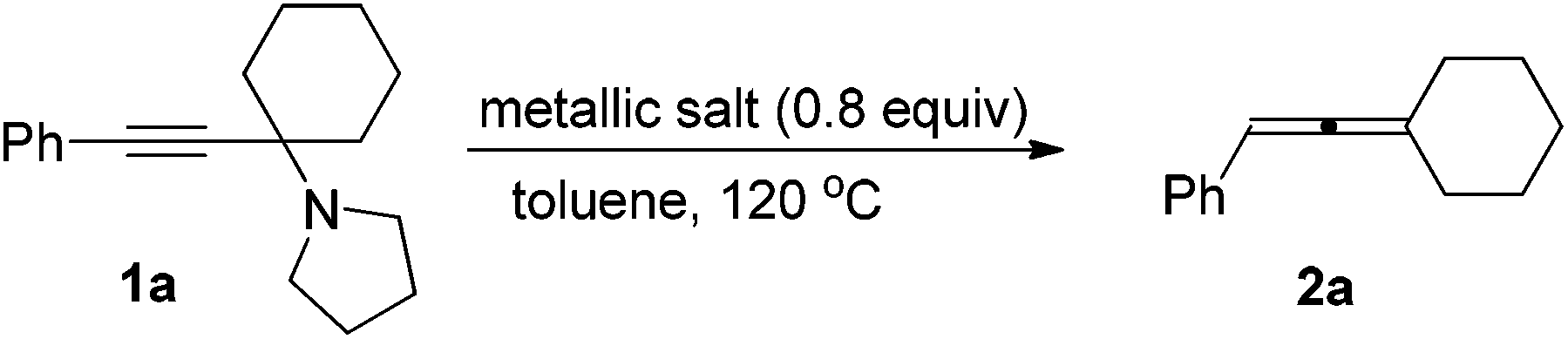
With the optimal conditions in hand, we then investigated the scope of the transformation. Under the mediation of ZnI2, propargylic amine 1a afforded trisubstituted allene 2a in 63% isolated yield (entry 1, Table 2). Propargylic amines 1b–1f derived from phenylacetylene, pyrrolidine, and different methyl ketones were transformed to allenes 2b–2f smoothly in moderate yields (entries 2–6, Table 2). Notably, the reaction of substrate 1c bearing an isopropyl group proceeded smoothly, albeit with a longer reaction time, which may be due to the steric hindrance of i-Pr (entry 3, Table 2). Surprisingly, propargylic amines 1g–1i also require longer reaction times (entries 7–9, Table 2). Since ethyl and n-propyl groups are generally considered as not so bulky, it indicates that the reaction rate is very sensitive to the steric effect of R2 and R3. It should be noted that acyclic non-methyl ketones are not suitable for our previously reported CdI2-mediated ATA reaction to afford trisubstituted allenes.9 When propargylic amines 1j–1q, which were prepared from differently substituted arylacetylenes, were applied in the reaction, allenes bearing different aryl groups were obtained in 50–65% isolated yields (entries 10–17, Table 2). Substitutes on the phenyl group have only a slight impact on the yields; a halogen substituent survived in the reaction (entries 11, 12 and 14–16, Table 2), affording products ready for further elaborations. When propargylic amines derived from alkyl-substituted acetylenes were treated under the standard conditions, the corresponding allene products were produced in higher yields (entries 18–21, Table 2), as compared to the arylacetylene-based propargylic amines.
|
|
||||
|---|---|---|---|---|
| Entry | 1 | Time (h) | Yield of 2b (%) | |
| R1 | R2; R3 | |||
| a The reaction was carried out on a 1.0 mmol scale in 3 mL of toluene. b Isolated yield. | ||||
| 1 | Ph | –(CH2)5– (1a) | 0.8 | 63 (2a) |
| 2 | Ph | Me; Et (1b) | 1.1 | 57 (2b) |
| 3 | Ph | Me; i-Pr (1c) | 5.3 | 62 (2c) |
| 4 | Ph | Me; n-Bu (1d) | 1.5 | 53 (2d) |
| 5 | Ph | Me; n-Hex (1e) | 1.0 | 55 (2e) |
| 6 | Ph | Me; BnCH2 (1f) | 1.0 | 56 (2f) |
| 7 | Ph | Et; n-Pr (1g) | 7.8 | 52 (2g) |
| 8 | Ph | n-Pr; n-Pr (1h) | 5.0 | 56 (2h) |
| 9 | Ph | Et; Et (1i) | 4.0 | 60 (2i) |
| 10 | 4-MeOC6H4 | –(CH2)5– (1j) | 2.0 | 65 (2j) |
| 11 | 4-BrC6H4 | –(CH2)5– (1k) | 2.0 | 63 (2k) |
| 12 | 3-BrC6H4 | –(CH2)5– (1l) | 2.0 | 60 (2l) |
| 13 | 4-MeOC6H4 | Me; n-Bu (1m) | 0.9 | 50 (2m) |
| 14 | 4-BrC6H4 | Me; n-Bu (1n) | 2.7 | 52 (2n) |
| 15 | 3-BrC6H4 | Me; n-Bu (1o) | 1.0 | 55 (2o) |
| 16 | 2-ClC6H4 | Me; n-Bu (1p) | 1.0 | 56 (2p) |
| 17 | 4-MeC6H4 | Me; n-Bu (1q) | 1.7 | 57 (2q) |
| 18 | n-C6H13 | Me; n-Bu (1r) | 2.0 | 67 (2r) |
| 19 | n-C8H17 | –(CH2)5– (1s) | 1.3 | 81 (2s) |
| 20 | n-C8H17 | Me; Me (1t) | 1.3 | 61 (2t) |
| 21 | n-C8H17 | –(CH2)4– (1u) | 1.6 | 58 (2u) |
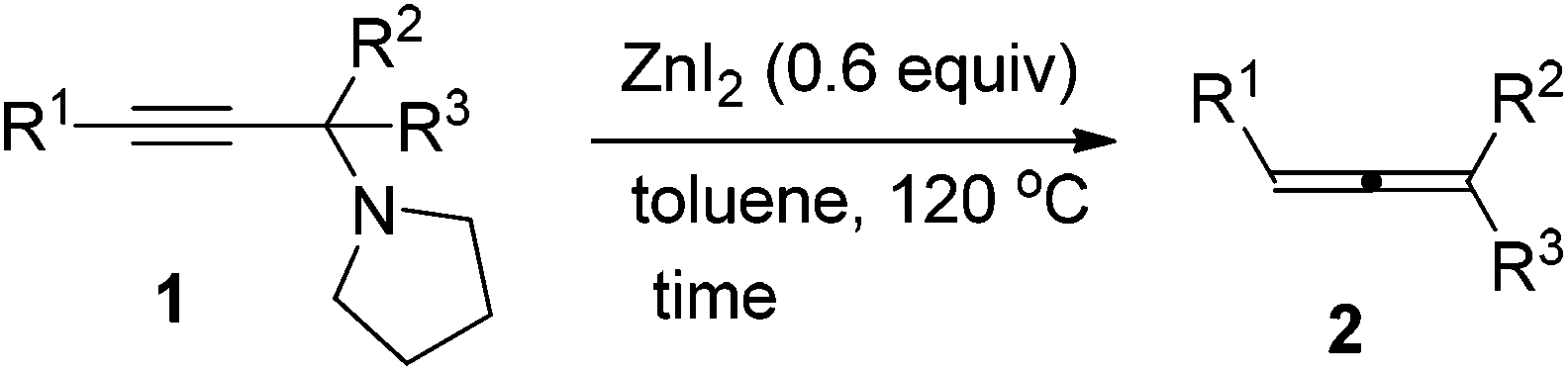
The method could also tolerate substrates bearing reactive functional groups such as hydroxyl and amide functionalities (Table 3), which could easily undergo further transformations for the preparation of other functionalized allenes.1,2c,12
|
|
|||
|---|---|---|---|
| Entry | 1 | Time (h) | Yield of 2 (%) |
| R1; R2; R3 | |||
| a The reaction was carried out on a 1.0 mmol scale in 3 mL of toluene. Ts = p-toluenesulfonyl. b Isolated yield. | |||
| 1 | Me2C(OH); Me; n-Bu (1v) | 2.0 | 58b (2v) |
| 2 | HO(CH2)2; Me; n-Bu (1w) | 1.0 | 53b(2w) |
| 3 | TsNHCH2; –(CH2)5– (1x) | 1.0 | 79b (2x) |
| 4 | TsNHCH(n-C7H15); –(CH2)5– (1y) | 1.8 | 92b (2y) |
| 5 | TsNHPhCH; –(CH2)5– (1z) | 1.3 | 85b (2z) |
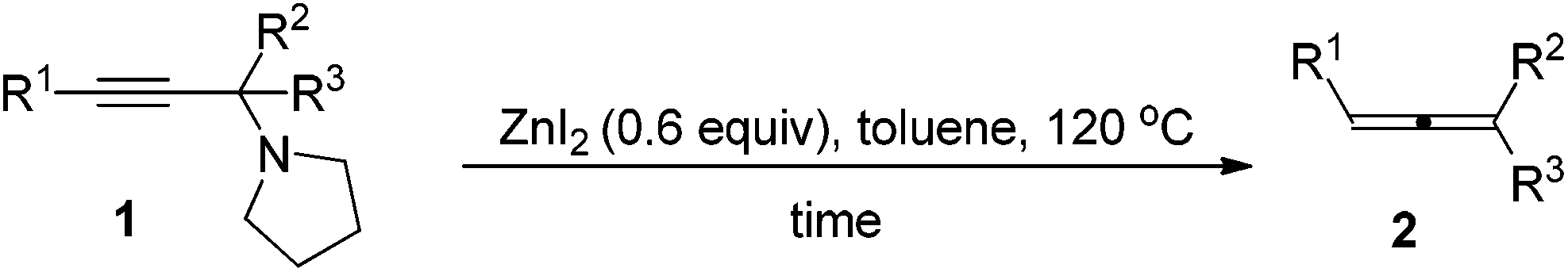
In fact, as a comparison, when 1x was treated with CdI2, in addition to the corresponding allenyl amide 2x, the related cyclization product 4x was also obtained in 60% yield as the major product (eqn (2)).
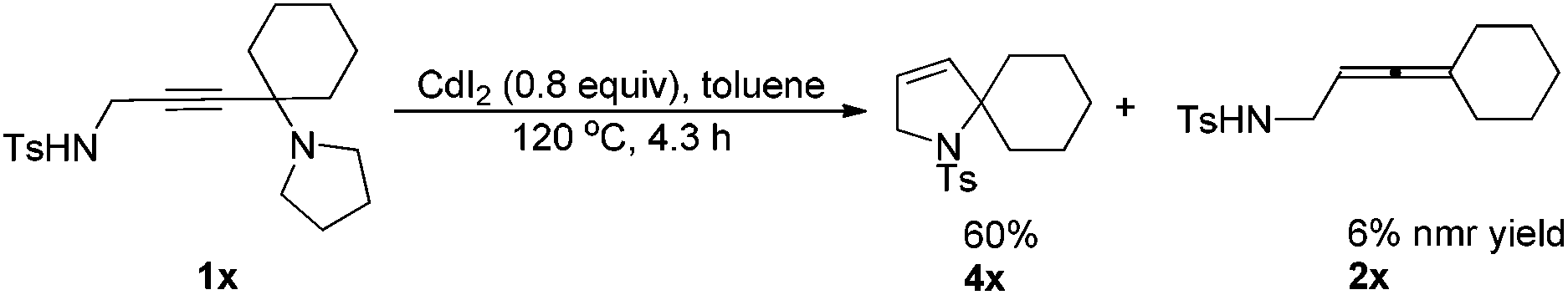 | (2) |
Furthermore, it is interesting to observe that the reaction depends highly on the structure of the amine applied: propargylic amines 1d′ and 1d′′ from phenylacetylene, 2-hexanone, and six-membered morpholine or piperidine may also afford corresponding trisubstituted allenes, however, with a very low efficiency (Scheme 1).
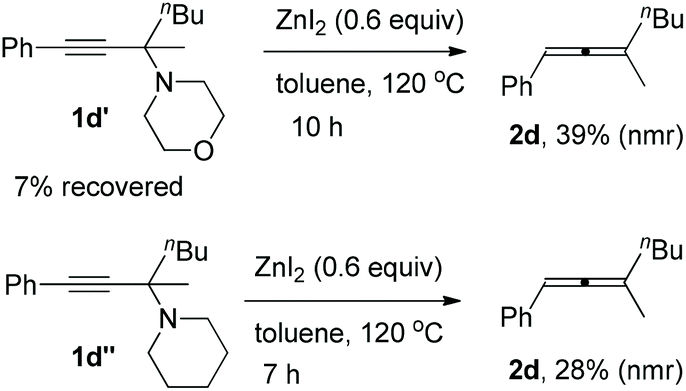 | ||
| Scheme 1 Synthesis of trisubstituted allenes from propargylic amines derived from morpholine or piperidine. | ||
Since CuBr-catalyzed synthesis of propargylic amines from 1-alkynes, ketones, and pyrrolidine10 and ZnI2-promoted the synthesis of trisubstituted allenes from propargylic amines both proceeded smoothly in the same solvent, we tried to combine them in one pot, however, a poor result was obtained (eqn (3)).
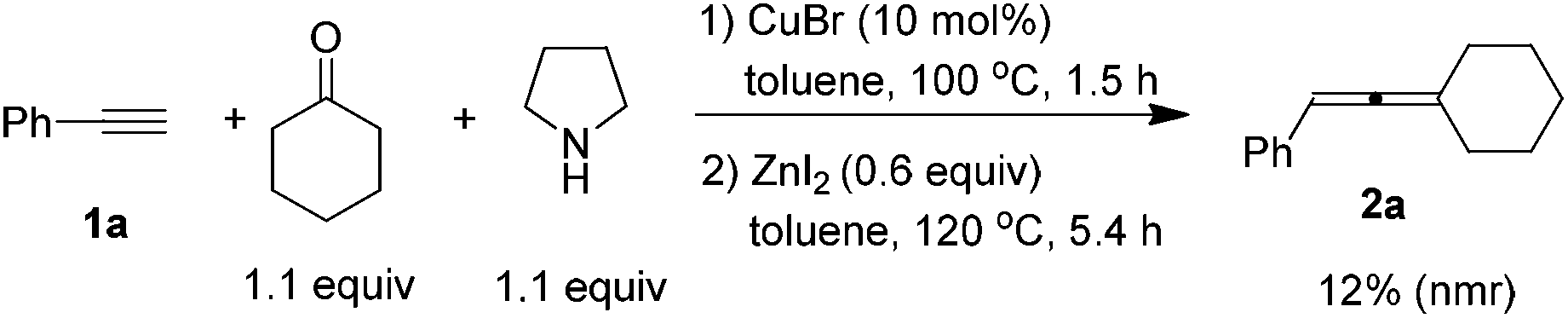 | (3) |
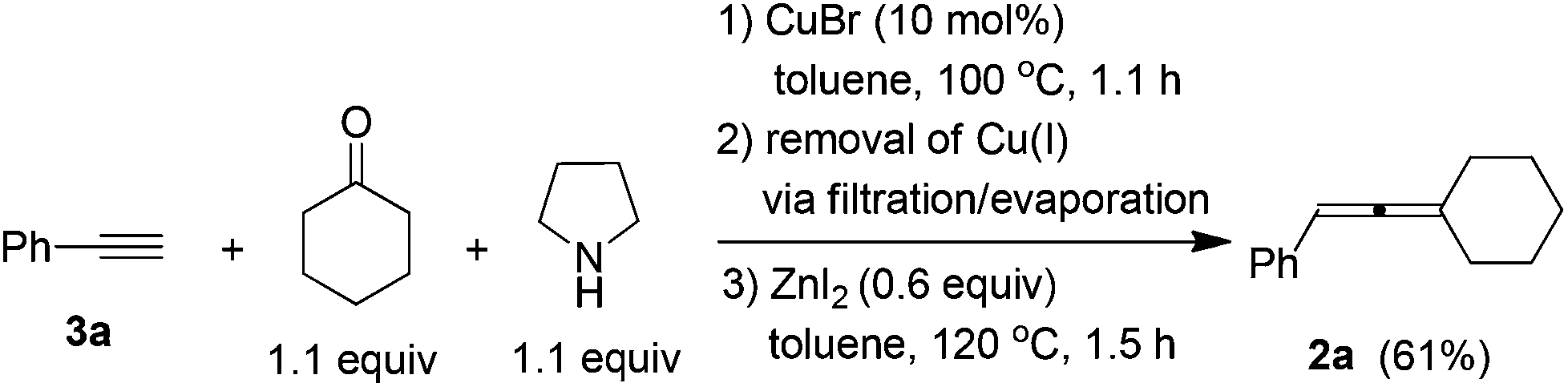
After careful study and optimizations, we found that filtration to remove Cu(I) after the first step is essential for a successful second step. Based on this, we developed a convenient two-step synthesis of trisubstituted allenes from 1-alkynes, ketones, and pyrrolidine. Propargylic amines were firstly synthesized through the CuBr-catalyzed KA2 reaction10 and then subjected to the ZnI2-mediated transformation after a simple filtration through a short pad of silica gel to remove Cu(I). The substrate scope of the reaction was evaluated for various 1-alkynes and ketones and the typical results are summarized in Table 4. Besides phenylacetylene 3a, substituted phenylacetylenes bearing 4-Cl, 4-Br, and 4-Me substituents reacted with cyclohexanone to afford the corresponding allenes in moderate yields (entries 1–4, Table 4). Alkyl-substituted 1-alkyne 3f could react with cyclohexanone to afford alkyl-substituted allene 2s in 74% isolated yield (entry 5, Table 4). Since 2,3-allenols and 2,3-allenyl amines are useful in organic synthesis,1,2c,11 we also investigated the tolerance of amino and hydroxyl groups in the reaction: encouragingly, terminal propargylic toluenesulfonylamide 3g may be applied, affording 2,3-allenyl tosylamide 2x in 30% isolated yield (entry 6, Table 4). The reaction of terminal homopropargylic alcohol 3h with cyclohexanone afforded 2,3-allenol 2w in 53% isolated yield (entry 7, Table 4). Moreover, terminal alkynes with common protecting groups such as THP and TBS were also tolerated, producing corresponding allenes 2ad and 2ae in moderate yields (entries 8 and 9, Table 4). Cyclopentanone reacted with 1-decyne 3f to afford trisubstituted allene 2u smoothly, however, with an obviously decreased yield as compared to that of cyclohexanone (entry 10 vs. 5, Table 4). When acetone was used, allene 2t bearing two methyl groups on the allene moiety could be obtained in 49% isolated yield, although two equivalents of acetone were required to ensure the yield due to the lower boiling point of acetone (entry 11, Table 4). 2-Octanone may also be used in the reaction, affording allene 2af in moderate yield (entry 12, Table 4). Meanwhile, aromatic ketones gave a disappointing result in this reaction.
|
|
||||
|---|---|---|---|---|
| Entry | 3 | R2; R3 | t 1/t2 (h) | Yield of 2b (%) |
| R1 | ||||
| a Ts = p-toluenesulfonyl; THP = 2-tetrahydropyranyl; TBS = t-butyldimethylsilyl. b Isolated yield. c 2.0 equiv. of acetone were used. d 1.3 equiv. of 2-octanone were used. | ||||
| 1 | Ph (3a) | –(CH2)5– | 1.1/1.5 | 61 (2a) |
| 2 | 4-ClC6H4 (3c) | –(CH2)5– | 1.5/1.5 | 57 (2aa) |
| 3 | 4-BrC6H4 (3d) | –(CH2)5– | 1.5/0.5 | 51 (2k) |
| 4 | 4-MeC6H4 (3e) | –(CH2)5– | 1.1/0.8 | 51 (2ab) |
| 5 | n-C8H17 (3f) | –(CH2)5– | 1.0/0.7 | 74 (2s) |
| 6 | TsNHCH2 (3g) | –(CH2)5– | 1.9/3.5 | 30 (2x) |
| 7 | HO(CH2)2 (3h) | –(CH2)5– | 0.9/1.0 | 53 (2ac) |
| 8 | TBSOCH2 (3i) | –(CH2)5– | 1.2/0.8 | 52 (2ad) |
| 9 | THPOCH2 (3j) | –(CH2)5– | 1.1/0.9 | 43 (2ae) |
| 10 | n-C8H17 (3f) | –(CH2)4– | 1.3/1.3 | 50 (2u) |
| 11c | n-C8H17 (3f) | Me; Me | 3.1/0.9 | 49 (2t) |
| 12d | n-C8H17 (3f) | Me; n-C6H13 | 1.9/0.6 | 45 (2af) |
When the reaction was carried out on a 50 mmol-scale, only 1.5 mol% of CuBr was needed10 and allene 2a was afforded in 67% isolated yield (eqn (4)).
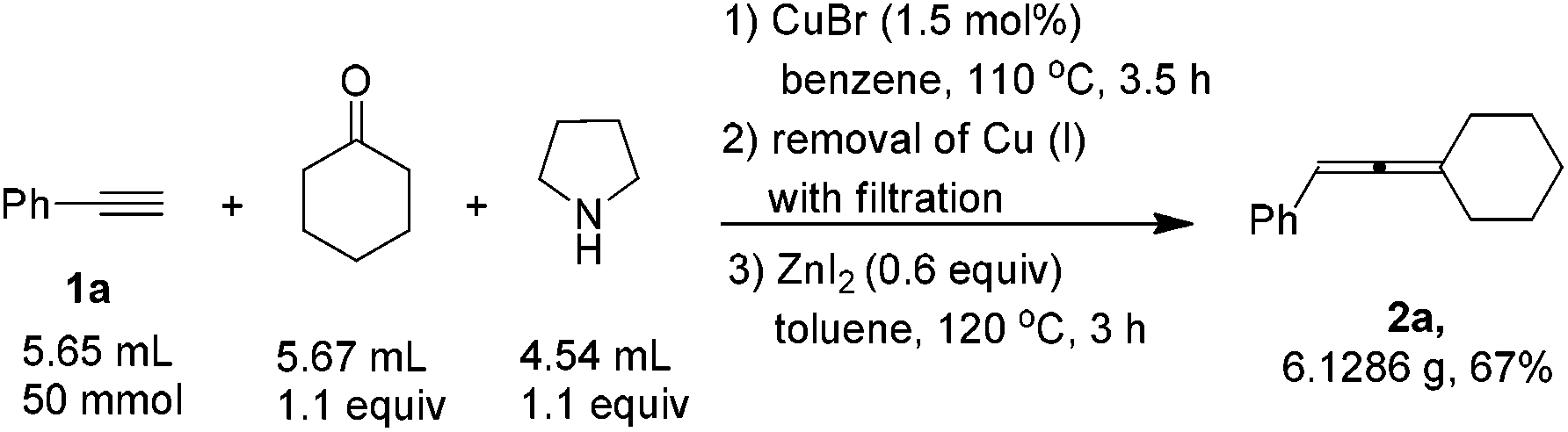 | (4) |
Based on recent reports of the ATA reactions,9 we proposed a plausible mechanism for this reaction. In the first place, ketoniminium 7, formed in situ from ketone and pyrrolidine, would react with alkynyl copper species 6 to give propargylic amine 1. Then the triple in propargylic amine 1 coordinates with ZnI2 which was followed by 1,5-hydride transfer and β-elimination to afford the corresponding trisubstituted allene 2 (Scheme 2). We reasoned that the coordination of Zn2+ with the in situ generated imine makes the Zn2+ much less active after the reaction.
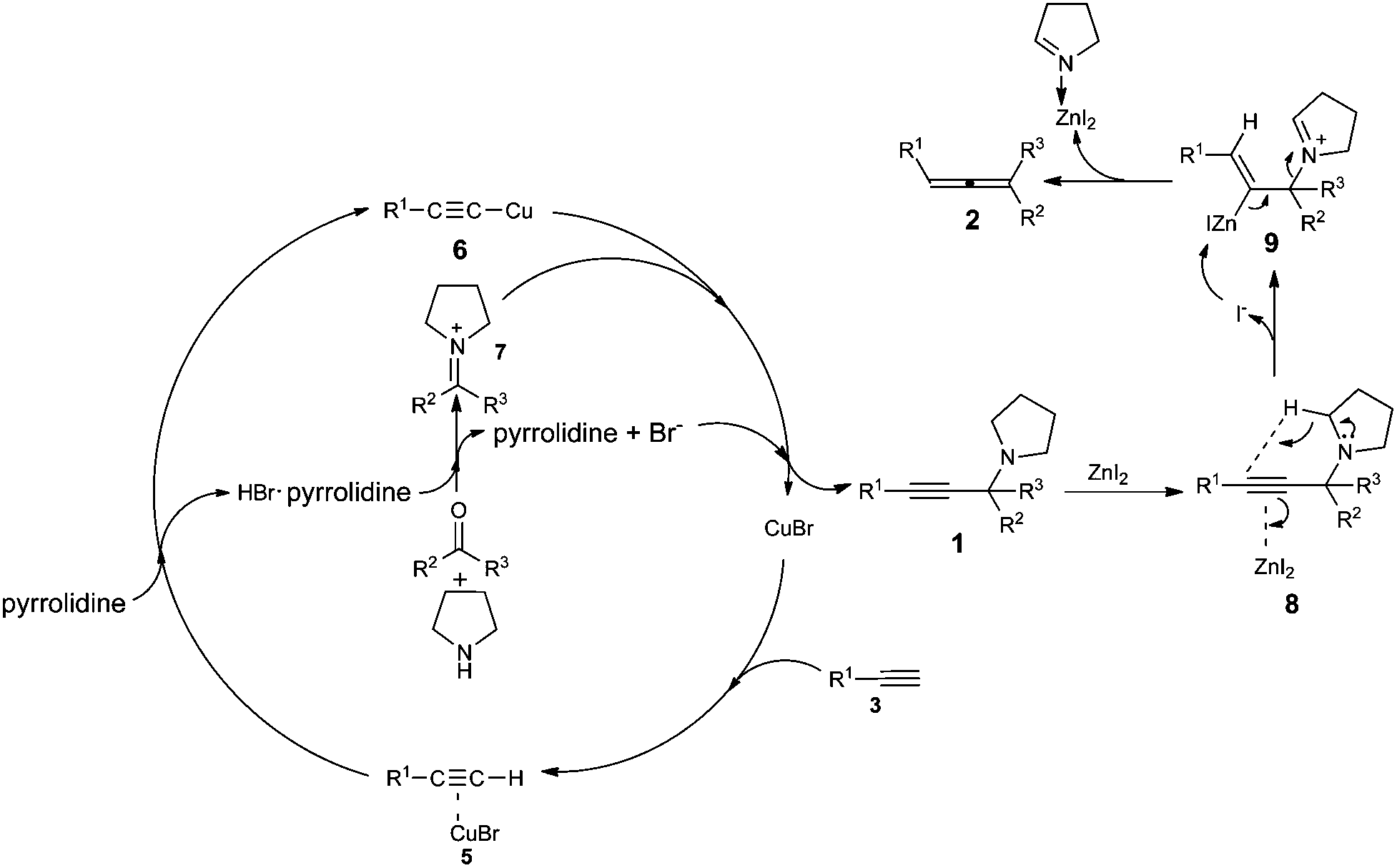 | ||
| Scheme 2 Proposed mechanism for the reaction. | ||
Conclusions
In conclusion, we have developed an efficient ZnI2-mediated method for the synthesis of trisubstituted allenes from propargylic amines bearing a quaternary carbon center, which are readily available from 1-alkynes, ketones and amines through our recently established method.10 This ZnI2-mediated method meets our long-term goal of efficient synthesis of allenes from readily available chemicals in common chemistry laboratories. Based on this, a two-step synthesis of trisubstituted allenes from terminal alkynes and ketones has been developed. Due to the fact that ZnI2 is cheaper, non-toxic and environmentally benign, and enjoys a much wider scope as compared to CdI2, the method constitutes an urgent supplement to the previously reported CdI2-promoted synthesis of trisubstituted allenes and will be of great interest to chemists. Further studies including the asymmetric version of this reaction are being pursued in our laboratory.Experimental section
General information
CuBr (98.5%) and pyrrolidine were purchased from Sinopharm Chemical Reagent Co., Ltd and used without further treatment. ZnI2 (98%) was purchased from Acros and kept in a glove box. Toluene was dried over a sodium wire with benzophenone as the indicator and distilled freshly before use. Anhydrous benzene was purchased from Aladdin and used without further treatment. Other reagents were used without further treatment. All the temperatures are referred to the oil baths used.To a flame-dried Schlenk tube was added anhydrous ZnI2 (191.3 mg, 0.6 mmol). The Schlenk tube was then taken out and dried under vacuum with a heating gun. 1a (252.9 mg. 1.0 mmol) and 3 mL of toluene were added sequentially under an Ar atmosphere. The Schlenk tube was then equipped with a condenser and placed in a pre-heated oil bath of 120 °C with stirring for 0.8 h and monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtered through a short pad of silica gel with a sand-core funnel eluted with ethyl acetate (15 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: 30–60 °C petroleum ether) to afford 2a13 (116.7 mg, 63%) as a liquid. 1H NMR (300 MHz, CDCl3) δ 7.32–7.23 (m, 4 H, ArH), 7.18–7.10 (m, 1 H, ArH), 6.02–5.96 (m, 1 H,
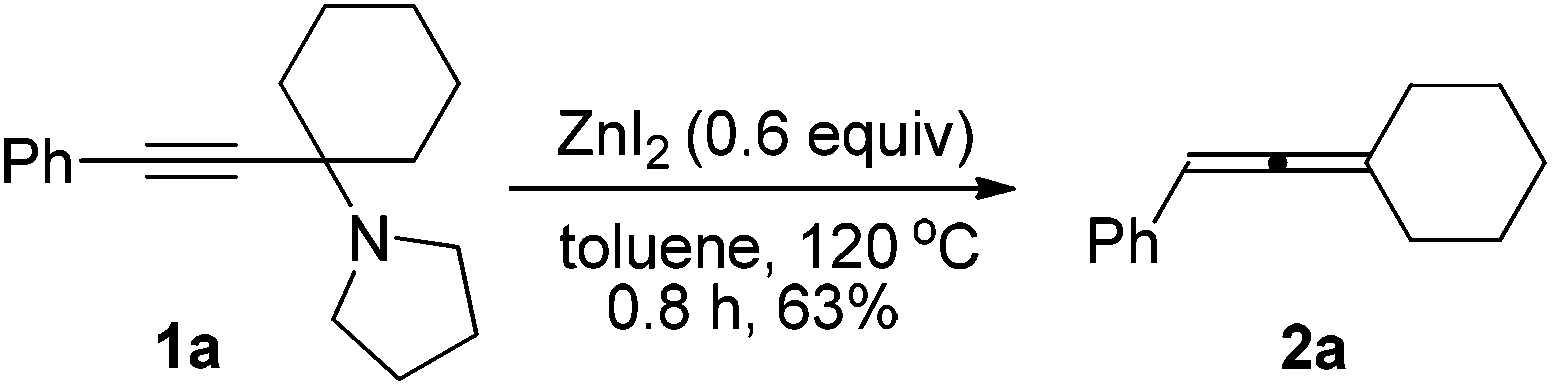
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2.32–2.12 (m, 4 H, 2 × CH2), 1.77–1.48 (m, 6 H, 3 × CH2); 13C NMR (75.4 MHz, CDCl3) δ 199.6, 136.1, 128.5, 126.5, 126.2, 106.4, 92.3, 31.3, 27.7, 26.1; MS (EI) m/z 184 (M+, 70.53), 141 (100); IR (neat) 3030, 2929, 2887, 2853, 1951, 1598, 1496, 1459, 1446, 1256, 1237, 1198, 1069, 1027 cm−1.
CH), 2.32–2.12 (m, 4 H, 2 × CH2), 1.77–1.48 (m, 6 H, 3 × CH2); 13C NMR (75.4 MHz, CDCl3) δ 199.6, 136.1, 128.5, 126.5, 126.2, 106.4, 92.3, 31.3, 27.7, 26.1; MS (EI) m/z 184 (M+, 70.53), 141 (100); IR (neat) 3030, 2929, 2887, 2853, 1951, 1598, 1496, 1459, 1446, 1256, 1237, 1198, 1069, 1027 cm−1.
To a flame-dried Schlenk tube were added CuBr (14.6 mg, 0.1 mmol), phenyl acetylene 3a (102.4 mg, 1.0 mmol)/toluene (0.5 mL), cyclohexanone (107.8 mg, 1.1 mmol)/toluene (0.5 mL), and pyrrolidine (93.0 μL, d = 0.8618 g mL−1, 80.1 mg, 1.1 mmol) sequentially. The Schlenk tube was then stirred at 100 °C until the completion of the reaction as monitored by TLC (1.0 h). After cooling to room temperature, the crude reaction mixture was filtered through a short pad of silica gel with a sand-core funnel eluted with acetone (20 mL). After evaporation, the crude product was used in the next step without further treatment.
To another Schlenk tube was added anhydrous ZnI2 (191.4 mg, 0.6 mmol). The Schlenk tube was then dried under vacuum with a heating gun. The above crude product was then dissolved in toluene (3 mL) and transferred to the Schlenk tube via a syringe under an Ar atmosphere. The Schlenk tube was then equipped with a condenser and placed in a pre-heated oil bath of 120 °C with stirring. After 1.5 h, the reaction was complete as monitored by TLC, the crude reaction mixture was cooled to room temperature and filtrered through a short pad of silica gel with a sand-core funnel eluted with ethyl ether (20 mL). After evaporation, the residue was purified by chromatography on silica gel to afford 2a13 (112.1 mg, 61%) as a liquid (eluent: petroleum ether). 1H NMR (300 MHz, CDCl3) δ 7.33–7.22 (m, 4 H, ArH), 7.20–7.10 (m, 1 H, ArH), 6.02–5.96 (m, 1 H, CH), 2.34–2.12 (m, 4 H, 2 × CH2), 1.80–1.46 (m, 6 H, 3 × CH2).
To a three-necked flask equipped with a Dean-Stark trap and a condenser dried under vacuum with a heating gun, were added CuBr (11 mg, 0.75 mmol), 3a (5.65 mL, d = 0.93 g mL−1, 5.25 g, 97%, 50 mmol), cyclohexanone (5.67 mL, d = 0.95 g mL−1, 5.39 g, 55 mmol), pyrrolidine (4.54 mL, d = 0.86 g mL−1, 3.90 g, 55 mmol), and benzene (50 mL) sequentially under an Ar atmosphere. The flask was then placed in a pre-heated oil bath at 110 °C with stirring for 3.5 h and monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtered through a short pad of silica gel with a sand-core funnel eluted with ethyl acetate (120 mL). After evaporation, the crude product was used in the next step without further treatment.
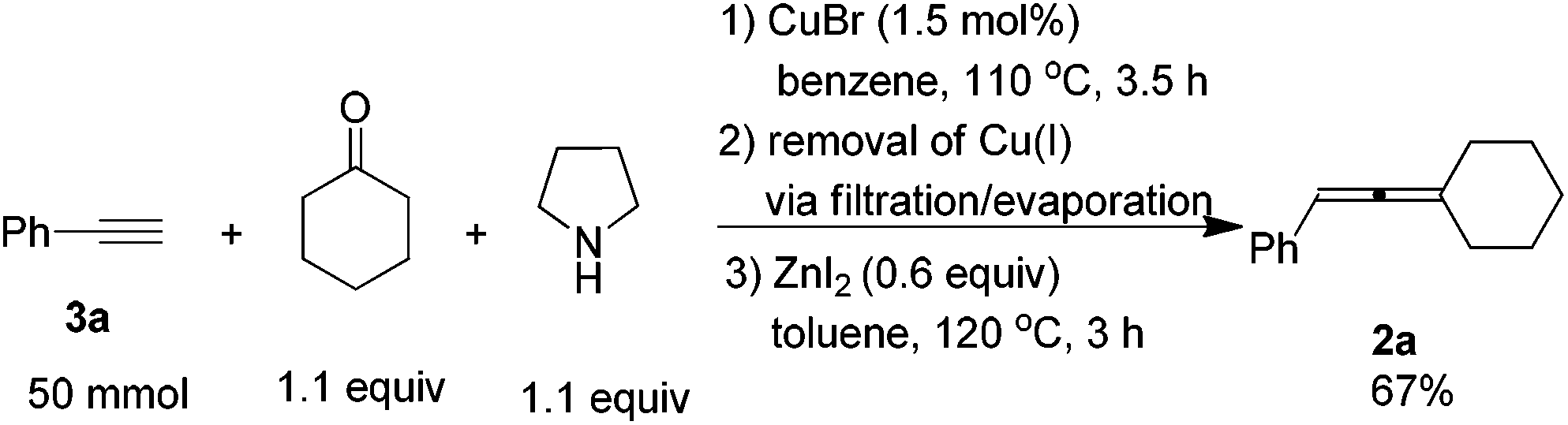
To another three-necked flask equipped with a condenser was added anhydrous ZnI2 (9.78 g, 30 mmol). The flask was then dried under vacuum with a heating gun. The above crude product was then dissolved in toluene (150 mL) and transferred to the flask via a syringe under an Ar atmosphere. The flask was then placed in a pre-heated oil bath of 120 °C with stirring. After 3.0 h, the reaction was complete as monitored by TLC, the crude reaction mixture was cooled to room temperature and filtered through a short pad of silica gel with a sand-core funnel eluted with ethyl ether (100 mL). After evaporation, the residue was purified by chromatography on silica gel to afford 2a13 (6.13 g, 67%) as a liquid (eluent: petroleum ether). 1H NMR (400 MHz, CDCl3) δ 7.32–7.25 (m, 4 H, ArH), 7.20–7.12 (m, 1 H, ArH), 6.02–5.97 (m, 1 H, CH), 2.32–2.12 (m, 4 H, 2 × CH2), 1.77–1.47 (m, 6 H, 3 × CH2).
Acknowledgements
Financial support from the National Basic Research Program of China (2011CB808700) and National Natural Science Foundation of China (21232006) is greatly appreciated. We thank Mr Dengke Ma in this group for reproducing the results of entry 19 in Table 2, entry 5 in Table 3, and entry 5 in Table 4 presented in the text.Notes and references
- For reviews on the chemistry of allenes, see: (a) K. K. Wang, Chem. Rev., 1996, 96, 207 CrossRef CAS PubMed; (b) J. A. Marshall, Chem. Rev., 2000, 100, 3163 CrossRef CAS PubMed; (c) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS; (d) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef CAS PubMed; (e) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (f) R. W. Bates and V. Satcharoen, Chem. Soc. Rev., 2002, 31, 12 RSC; (g) S. Ma, Acc. Chem. Res., 2003, 36, 701 CrossRef CAS PubMed; (h) L. Brandsma and N. A. Nedolya, Synthesis, 2004, 735 CrossRef CAS; (i) M. A. Tius, Acc. Chem. Res., 2003, 36, 284 CrossRef CAS PubMed; (j) L. L. Wei, H. Xiong and R. P. Hsung, Acc. Chem. Res., 2003, 36, 773 CrossRef CAS PubMed; (k) S. Ma, in Palladium-Catalyzed Two- or Three-Component Cyclization of Functionalized Allenes in Palladium in Organic Synthesis, ed. J. Tsuji, Springer, Berlin, Heidelberg, 2005, pp. 183–210 Search PubMed; (l) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed; (m) S. Ma, Aldrichimica Acta, 2007, 40, 91 CAS; (n) H. Kim and L. J. Williams, Curr. Opin. Drug Discovery Dev., 2008, 11, 870 CAS; (o) M. Brasholz, H.-U. Reissig and R. Zimmer, Acc. Chem. Res., 2009, 42, 45 CrossRef CAS PubMed; (p) S. Ma, Acc. Chem. Res., 2009, 42, 1679 CrossRef CAS PubMed; (q) B. Alcaide, P. Almendros and T. M. Campo, Chem. – Eur. J., 2010, 16, 5836 CrossRef CAS PubMed; (r) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed; (s) F. Inagaki, S. Kitagaki and C. Mukai, Synlett, 2011, 594 CAS; (t) F. López and J. L. Mascareñas, Chem. – Eur. J., 2011, 17, 418 CrossRef PubMed; (u) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed.
- For reviews or highlights on the synthesis of allenes, see: (a) L. K. Sydnes, Chem. Rev., 2003, 103, 1133 CrossRef CAS PubMed; (b) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, vol. 1, p. 2 Search PubMed; (c) N. Krause and A. Hoffmann-Röder, Tetrahedron, 2004, 60, 11671 CrossRef CAS; (d) G. B. Hammond, ACS Symp. Ser., 2005, 911, 204 CrossRef CAS; (e) R. Saeeng and M. Isobe, Chem. Lett., 2006, 35, 552 CrossRef CAS; (f) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795 CrossRef CAS; (g) M. Ogasawara, Tetrahedron: Asymmetry, 2009, 20, 259 CrossRef CAS; (h) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC.
- (a) P. Crabbé, H. Fillion, D. André and J.-L. Luche, J. Chem. Soc., Chem. Commun., 1979, 859 RSC; (b) S. Searles, Y. Li, B. Nassim, M.-T. R. Lopes, P. T. Tran and P. Crabbé, J. Chem. Soc., Perkin Trans. 1, 1984, 747 RSC; (c) S. Ma, H. Hou, S. Zhao and G. Wang, Synthesis, 2002, 1643 CrossRef CAS; (d) U. Kazmaier, S. Lucas and M. Klein, J. Org. Chem., 2006, 71, 2429 CrossRef CAS PubMed.
- For microwave-promoted synthesis of allenes from terminal alkynes, see: (a) H. Nakamura, T. Sugiishi and Y. Tanaka, Tetrahedron Lett., 2008, 49, 7230 CrossRef CAS; (b) S. Kitagaki, M. Komizu and C. Mukai, Synlett, 2011, 1129 CrossRef CAS.
- (a) J. Kuang and S. Ma, J. Org. Chem., 2009, 74, 1763 CrossRef CAS PubMed; (b) J. Kuang and S. Ma, J. Am. Chem. Soc., 2010, 132, 1786 CrossRef CAS PubMed; (c) J. Kuang, H. Luo and S. Ma, Adv. Synth. Catal., 2012, 354, 933 CrossRef CAS; (d) J. Kuang, X. Xie and S. Ma, Synthesis, 2013, 592 CAS; (e) H. Luo and S. Ma, Eur. J. Org. Chem., 2013, 3041 CrossRef CAS.
- For asymmetric synthesis of optically active allenes from terminal alkynes, see: (a) J. Ye, S. Li, B. Chen, W. Fan, J. Kuang, J. Liu, Y. Liu, B. Miao, B. Wan, Y. Wang, X. Xie, Q. Yu, W. Yuan and S. Ma, Org. Lett., 2012, 14, 1346 CrossRef CAS PubMed; (b) J. Ye, W. Fan and S. Ma, Chem. – Eur. J., 2013, 19, 716 CrossRef CAS PubMed; (c) J. Ye, R. Lü, W. Fan and S. Ma, Tetrahedron, 2013, 69, 8959 CrossRef CAS; (d) R. Lü, J. Ye, T. Cao, B. Chen, W. Fan, W. Lin, J. Liu, H. Luo, B. Miao, S. Ni, X. Tang, N. Wang, Y. Wang, X. Xie, Q. Yu, W. Yuan, W. Zhang, C. Zhu and S. Ma, Org. Lett., 2013, 15, 2254 CrossRef PubMed.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569 CrossRef CAS PubMed.
- Q. Xiao, Y. Xia, H. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2011, 50, 1114 CrossRef CAS PubMed.
- X. Tang, C. Zhu, T. Cao, J. Kuang, W. Lin, S. Ni, J. Zhang and S. Ma, Nat. Commun., 2013, 4, 2450 Search PubMed.
- X. Tang, J. Kuang and S. Ma, Chem. Commun., 2013, 49, 8976 RSC.
- For reports on the Ag+, Au+ or Pd2+-mediated synthesis of 1,3-disubstituted allenes from propargylic amines derived from aldehydes, see: (a) V. K. Y. Lo, M.-K. Wong and C.-M. Che, Org. Lett., 2008, 10, 517 CrossRef CAS PubMed; (b) V. K.-Y. Lo, C.-Y. Zhou, M.-K. Wong and C.-M. Che, Chem. Commun., 2010, 46, 213 RSC; (c) H. Nakamura, M. Ishikura, T. Sugiishi, T. Kamakura and J. F. Biellmann, Org. Biomol. Chem., 2008, 6, 1471 RSC.
- (a) J. A. Marshall and J. Perkins, J. Org. Chem., 1994, 59, 3509 CrossRef CAS; (b) J. A. Marshall and N. D. Adams, J. Org. Chem., 1997, 62, 8976 CrossRef CAS; (c) T. Miura, M. Shimada, S.-Y. Ku, T. Tamai and M. Murakami, Angew. Chem., Int. Ed., 2007, 46, 7101 CrossRef CAS PubMed; (d) A. Fürstner and M. Méndez, Angew. Chem., Int. Ed., 2003, 42, 5355 CrossRef PubMed; (e) S. Redon, A.-L. B. Berkaoui, X. Pannecoucke and F. Outurquin, Tetrahedron, 2007, 63, 3707 CrossRef CAS.
- C.-M. Ting, Y.-L. Hsu and R.-S. Liu, Chem. Commun., 2012, 48, 6577 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00047e |
| This journal is © the Partner Organisations 2015 |