
Conditions for palladium-catalyzed direct arylations of 4-bromo and 4-iodo N-substituted pyrazoles without C–Br or C–I bond cleavage
Mariem
Brahim
ab,
Imen
Smari
ab,
Hamed
Ben Ammar
*b,
Bechir
Ben Hassine
b,
Jean-François
Soulé
a and
Henri
Doucet
*a
aInstitut des Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1 “Organométalliques, Matériaux et Catalyse”, Campus de Beaulieu, 35042 Rennes, France. E-mail: henri.doucet@univ-rennes1.fr
bLaboratoire de Synthèse Organique Asymétrique et Catalyse Homogène (UR 11ES56), Université de Monastir Faculté des Sciences de Monastir, avenue de l'environnement, Monastir 5000, Tunisia
First published on 29th May 2015
Abstract
The Pd-catalyzed arylation at the C5 position of N-protected pyrazole derivatives bearing bromo or iodo substituents at the C4 position is described. A simple phosphine-free catalytic system was used, namely, 1 mol% Pd(OAc)2 in DMA in the presence of KOAc as the base. A wide aryl bromide scope as a coupling partner has been coupled with pyrazole derivatives. The reaction was very chemoselective as the C–halogen bonds of the pyrazole units were not involved in the C–H bond arylation process. Some examples demonstrating the synthetic potential of the bromo and iodo pyrazole substituents for chemical transformations are reported.
Pyrazole derivatives including molecules containing a 5-arylpyrazole motif are well represented in pharmaceutical drugs. For example, Deracoxib (Deramaxx® drug developed by Novartis) is employed in veterinary medicine as a non-steroidal anti-inflammatory drug of the coxib class. Temonogrel is an inverse agonist of the serotonin 2A receptor in phase II. Nelotanserin is an inverse agonist of the serotonin receptor subtype 5-HT2A developed by Arena Pharmaceuticals (Fig. 1).
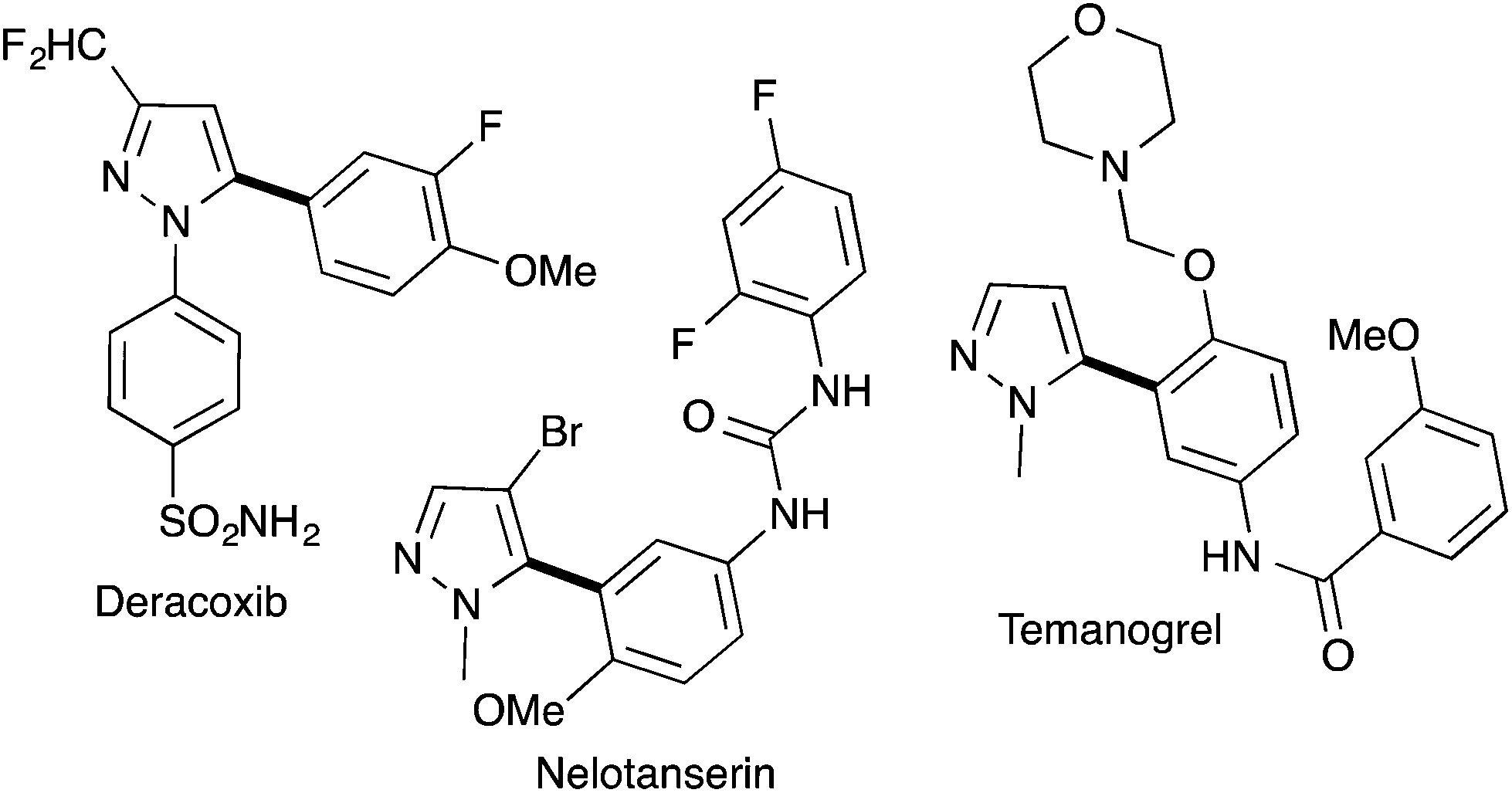 | ||
| Fig. 1 Examples of bioactive molecules containing a 5-arylpyrazole unit. | ||
Traditionally, 5-arylpyrazole derivatives have been synthesized using cross-coupling reactions between an aryl halide with an organometallic pyrazole derivative,1 or a halopyrazole with an organometallic aryl derivative using palladium catalysts.2 More recently, metal-catalyzed direct C–H bond arylation has appeared as one of the most suitable alternatives to such traditional cross-coupling reactions for the C–C bond formation with respect to the environment.3 This strategy has been employed for the functionalization of a large number of different heteroarenes, however, examples with pyrazole remain scarce. Indeed, using pyrazoles the reaction generally suffers from regioselectivity issue. As examples, in 2009, Sames reported that the palladium-catalyzed direct arylation of N-protected pyrazoles led to a mixture of C4 and C5 arylated pyrazoles with also the formation of large amounts of C4,C5-diarylated pyrazoles (Fig. 2a).4 Later, Doucet and co-workers reported Pd(OAc)2 phosphine-free conditions for the direct arylation of 1-methylpyrazole.5 Again, the reaction was not regioselective and a mixture of C5,C4 arylated and diarylated products was obtained in a 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16:6 ratio. Moreover, a large excess of 1-methylpyrazole (4 equiv.) was employed (Fig. 2a). In 2013, Bellina obtained a higher C5:C4 ratio (i.e., 86:14) without formation of a diarylated product, using Bu4NOAc as the base (Fig. 2a).6 According to Gorelsky calculations, in the concerted metallation deprotonation (CMD) process, this regioselectivity issue can be explained by similar energies of activation of C4 and C5 protons (28.5 vs. 27.3).7 In 2014, Kumpulainen and co-workers reported that Pd(OAc)2 associated with PPh3 catalyzes highly regioselective C5 arylation of N-dimethylaminosulfamoyl-protected pyrazoles; whereas, other N-protected pyrazoles such as 1-methyl or 1-benzylpyrazoles lead to mixtures of C5 and C4 arylated products and also to diarylated pyrazoles.8 In 2010, Mateos and Mendiola, after a large screening of the reaction conditions, successfully arylated 4-chloro-1-methylpyrazole at the C5 position in good yield (Fig. 2c).9 However, the chloro function is generally not appropriate for further transformations. 4-Bromo-1-methylpyrazole had also been tested under similar reaction conditions; albeit a poor yield was obtained and a diarylated pyrazole was also formed (Fig. 2c). Similar strategies, in which the C4 or C5 position was blocked by a substituent, were reported using chloro,10 formyl,11 and nitro substituents,12 or using indazoles as starting materials.13 The diarylation of pyrazole derivatives at C4 and C5 positions has also been reported using an excess of arylbromides.14 Similar approaches, namely, regioselective Pd-catalyzed direct arylations of halo-heteroarenes have also been reported.15
:16:6 ratio. Moreover, a large excess of 1-methylpyrazole (4 equiv.) was employed (Fig. 2a). In 2013, Bellina obtained a higher C5:C4 ratio (i.e., 86:14) without formation of a diarylated product, using Bu4NOAc as the base (Fig. 2a).6 According to Gorelsky calculations, in the concerted metallation deprotonation (CMD) process, this regioselectivity issue can be explained by similar energies of activation of C4 and C5 protons (28.5 vs. 27.3).7 In 2014, Kumpulainen and co-workers reported that Pd(OAc)2 associated with PPh3 catalyzes highly regioselective C5 arylation of N-dimethylaminosulfamoyl-protected pyrazoles; whereas, other N-protected pyrazoles such as 1-methyl or 1-benzylpyrazoles lead to mixtures of C5 and C4 arylated products and also to diarylated pyrazoles.8 In 2010, Mateos and Mendiola, after a large screening of the reaction conditions, successfully arylated 4-chloro-1-methylpyrazole at the C5 position in good yield (Fig. 2c).9 However, the chloro function is generally not appropriate for further transformations. 4-Bromo-1-methylpyrazole had also been tested under similar reaction conditions; albeit a poor yield was obtained and a diarylated pyrazole was also formed (Fig. 2c). Similar strategies, in which the C4 or C5 position was blocked by a substituent, were reported using chloro,10 formyl,11 and nitro substituents,12 or using indazoles as starting materials.13 The diarylation of pyrazole derivatives at C4 and C5 positions has also been reported using an excess of arylbromides.14 Similar approaches, namely, regioselective Pd-catalyzed direct arylations of halo-heteroarenes have also been reported.15
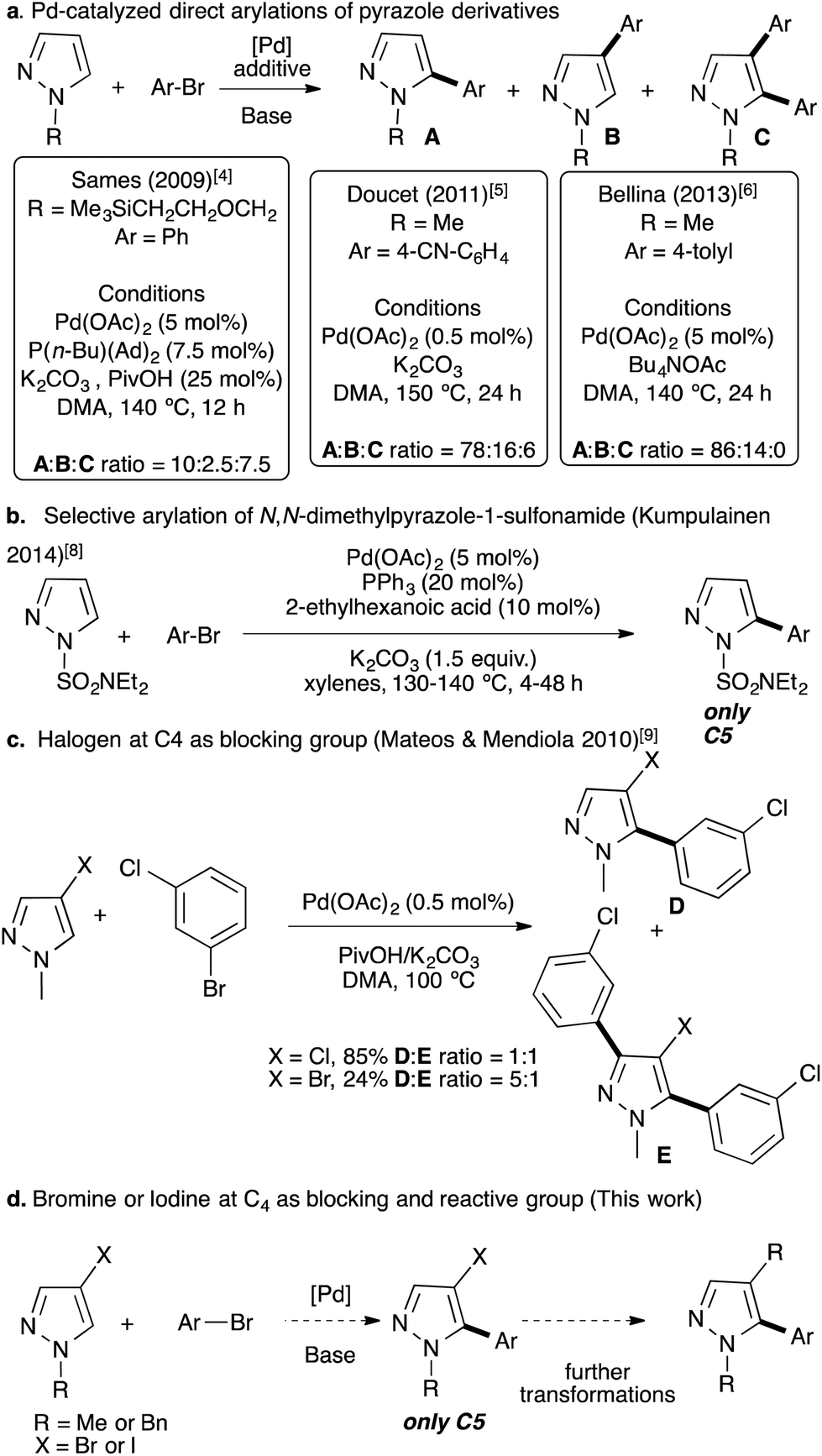 | ||
| Fig. 2 Previous examples of Pd-catalyzed direct arylations of pyrazoles using aryl bromides. (a) Pd-catalyzed direct arylations of pyrazole derivatives.4–6 (b) Selective arylation of N,N-dimethylpyrazole-1-sulfonamide (Kumpulainen, 2014).8 (c) Halogen at C4 as the blocking group (Mateos & Mendiola, 2010).9 (d) Bromine or iodine at C4 as blocking and reactive groups (this work). | ||
Here, we investigated the direct arylation of 4-bromo-1-(protected)pyrazole derivatives using a simple catalytic system based on palladium and also extended this reaction to more challenging 4-iodo-1-(protected)pyrazole derivatives (Fig. 2d).
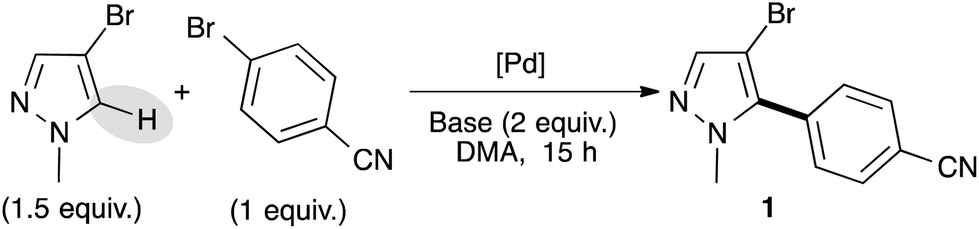
We selected 4-bromobenzonitrile and 3-bromo-1-methylpyrazole as model substrates for this reaction and used a small excess of pyrazole derivatives in order to prevent the side diarylation reaction. Based on our previous results,16 we firstly started our optimization using a palladium–diphosphine complex catalyst, namely PdCl(C3H5)(dppb), in the presence of KOAc as the base in DMA at 150 °C. Under these reaction conditions, the desired C5-arylated pyrazole 1 was obtained in an excellent yield of 85% (Table 1, entry 1). Using lower reaction temperature (130 °C), the reaction was not complete and the pyrazole 1 was obtained in only 65% yield (Table 1, entry 2). Interestingly, using 1 mol% of Pd(OAc)2 without phosphines instead of the PdCl(C3H5)(dppb) catalyst allowed a full conversion and 1 was isolated in 89% yield (Table 1, entry 3). Potassium pivalate (PivOK) instead of KOAc did not significantly affect the reaction, whereas, when the reaction was performed in the presence of K2CO3 as the base, lower conversion and yield of 1 were observed (Table 1, entries 4 and 5). Using a lower catalyst loading (i.e., 0.5 mol% of Pd(OAc)2), the reaction was not complete and 1 was obtained in only 56% yield (Table 1, entry 6). The same result was observed when the reaction was performed at only 100 °C, whatever the catalyst (Table 1, entries 7 and 8). Finally, the reaction could also be performed using only 1.1 equivalent of the pyrazole derivative without influence on the reaction yield (Table 1, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (x mol%) | Base | Temp. (°C) | Conv.a (%) | Yield of 1b (%) |
| a Based on the consumption of 4-bromobenzonitrile. b Determined using crude 1H-NMR, the number in parentheses shows the isolated yield. c The reaction was performed using 1.1 equiv. of 4-bromo-1-methylpyrazole. | |||||
| 1 | PdCl(C3H5)(dppb) (1) | KOAc | 150 | 100 | 85 |
| 2 | PdCl(C3H5)(dppb) (1) | KOAc | 130 | 92 | 65 |
| 3 | Pd(OAc)2 (1) | KOAc | 130 | 100 | 95 (89) |
| 4 | Pd(OAc)2 (1) | KOPiv | 130 | 100 | 95 |
| 5 | Pd(OAc)2 (1) | K2CO3 | 130 | 73 | 68 |
| 6 | Pd(OAc)2 (0.5) | KOAc | 130 | 58 | 56 |
| 7 | Pd(OAc)2 (1) | KOAc | 100 | 67 | 66 |
| 8 | PdCl(C3H5)(dppb) (1) | KOAc | 100 | 85 | 83 |
| 9c | Pd(OAc)2 (1) | KOAc | 130 | 100 | 95 |
With the best reaction conditions in hand, we decided to turn our attention to the scope and limitation of the direct arylation of 4-bromo-1-methylpyrazole using a range of aryl bromides (Scheme 1). We started by a set of para-substituted aryl bromides. Electron-withdrawing substituents such as nitro, formyl, and propionyl on the aryl bromide partner allowed the formation of the C5 arylated pyrazoles 2–4 in 90%, 84% and 72% yields, respectively. Using an electron-donating group such as 4-methoxy, Pd(OAc)2 was ineffective, while the use of a 1 mol% PdCl(C3H5)(dppb) catalyst afforded the desired arylated product 5 in 42% yield. The reaction also tolerated heteroaryl bromides as coupling partners. For example, from 3-bromopyridine, 5-bromopyrimidine, or 3-bromoquinoline the C5 arylpyrazoles 6–8 were isolated in 52%, 64%, and 77% yields, respectively. Finally, the reaction was found to be slightly sensitive to the steric hindrance of the aryl bromide partners, as the use of 2-bromobenzonitrile or 2-bromobenzaldehyde gave the arylated pyrazoles 9 and 10 in lower yields than their para-substituted homologues.
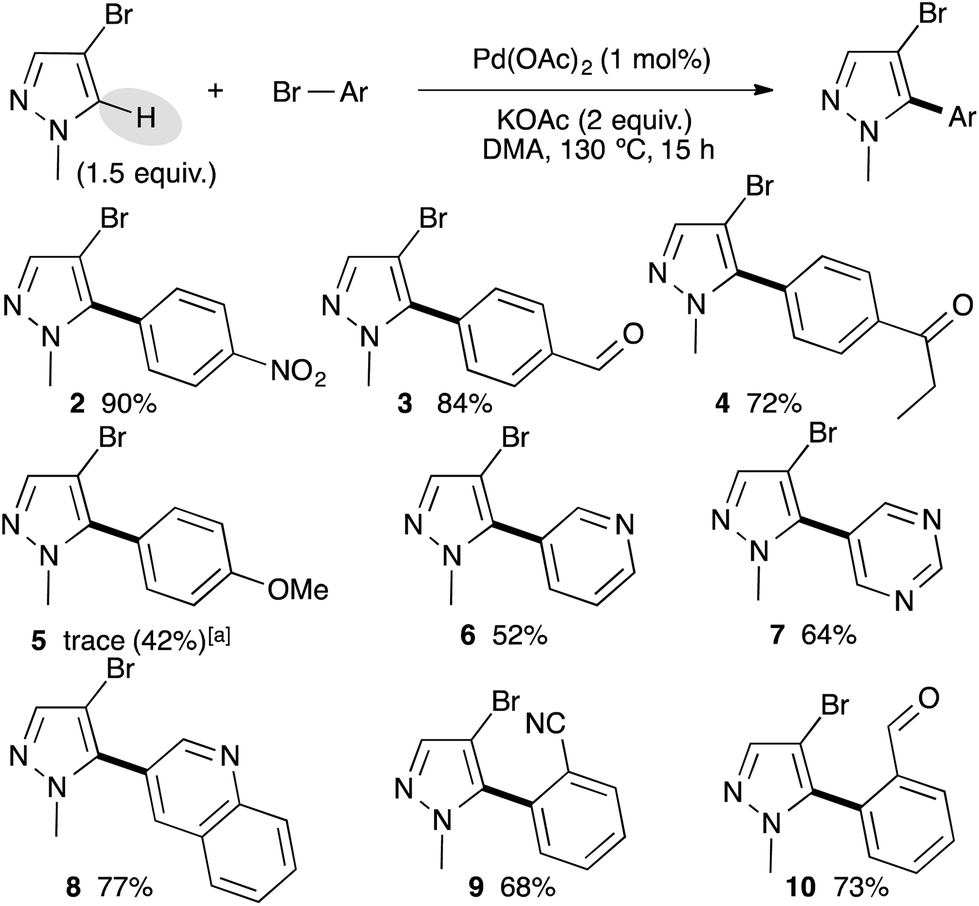 | ||
| Scheme 1 Pd-catalyzed direct arylation of 4-bromo-1-methylpyrazole. aReaction performed using 1 mol% PdCI(C3H5)(dppb) instead of Pd(OAc)2. | ||
After successfully arylating 4-bromo-1-methylpyrazole at the C5 position, we investigated the reactivity of 4-bromopyrazole without the N-H substituent. Unfortunately the reaction was completely inhibited. This lack of reactivity might be explained by the coordination of N-H to palladium resulting into a catalyst poisoning. Next, we examined the reactivity of 4-bromo-1-benzylpyrazole with a set of bromobenzene derivatives using our optimized reaction conditions (Scheme 2). Again, using bromobenzenes bearing electron-withdrawing groups at the para position, the desired C5 arylated 4-bromo-1-benzylpyrazoles were isolated as a single regioisomer in high yields, albeit using 4-bromonitrobenzene the arylated product 12 was obtained in only 54% yield. Under these reaction conditions, we never observed a debenzylation side-reaction. ortho-Substituted bromobenzenes, such as 2-bromobenzonitrile or 2-bromobenzaldehyde, also allowed the formation of C5 arylated products 16 and 17 in 69% and 62% yields, respectively. However, a more bulky substituent such as bromo at the ortho-position inhibited the reaction and only a trace amount of coupling product 18 was detected. Even a reverse stoichiometry did not afford the desired coupling product 18 or dipyrazoles. Bromoheteroarenes, such as 3-bromopyridine, 5-bromopyrimidine and 3-bromoquinoline were coupled with 4-bromo-1-benzylpyrazole to afford the C5 arylated products 19–21 in moderate yields.
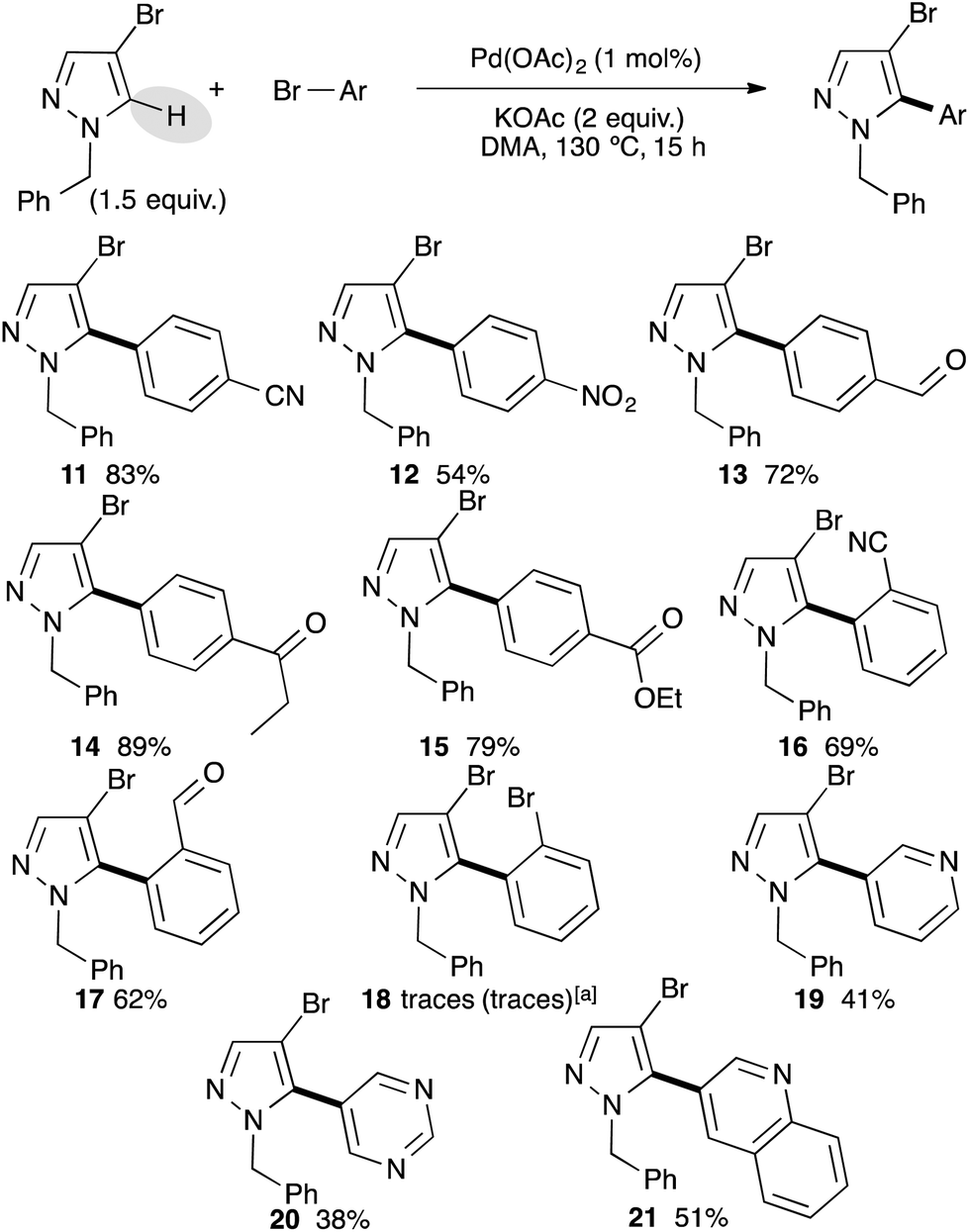 | ||
| Scheme 2 Pd-catalyzed direct arylation of 4-bromo-1-benzylpyrazole. aReaction performed using 1 equiv. of pyrazole and 2 equiv. of 1,2-dibromobenzene. | ||
Then, 4-bromo-1-phenylpyrazole was used as the starting material. It displayed a lower reactivity than its 1-methyl or 1-benzyl substituted analogues (Scheme 3). Indeed, using the same bromobenzene derivatives, only moderate yields of the desired cross-coupling products 22–25 were obtained. This lower reactivity might be explained by the steric hindrance of the phenyl group, which might partially block the C5 position of the pyrazole. An electronic influence, which modifies the nucleophilicity of such N-arylated pyrazoles due to delocalization of a lone pair of nitrogen to the aryl group, might also explain this lower reactivity. The poor reactivity of 2-bromobenzonitrile seems to confirm this trend.
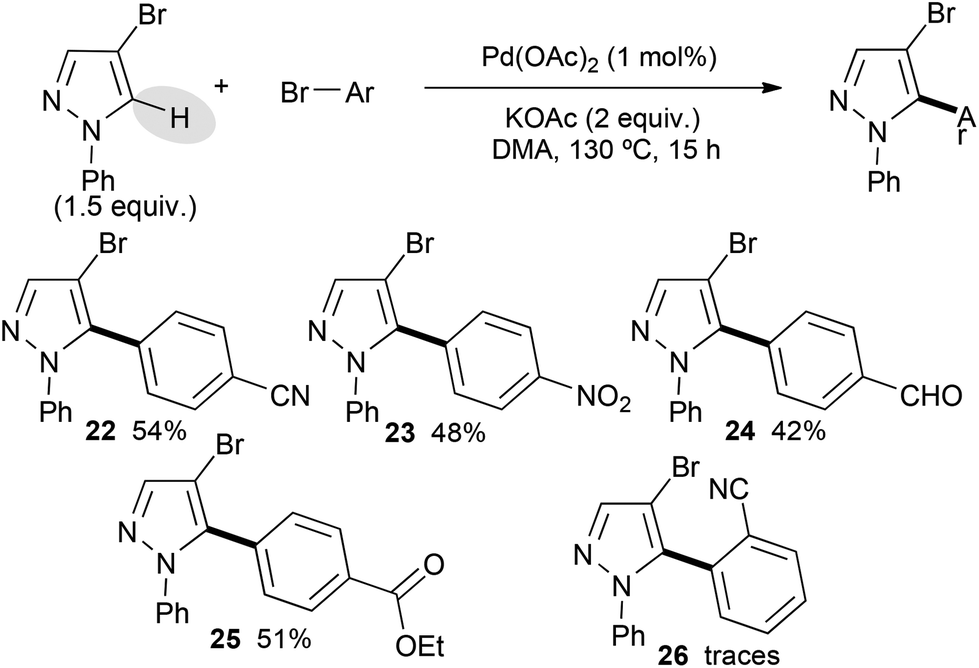 | ||
| Scheme 3 Pd-catalyzed direct arylation of 4-bromo-1-phenylpyrazole. | ||
After successfully arylating pyrazoles bearing a bromo substituent at the C4 position, we investigated the reactivity of more challenging pyrazoles bearing an iodo substituent at the C4 position (Scheme 4). We used 1-benzyl-4-iodopyrazole as a model substrate, which was easily prepared from pyrazoles via iodation using a I2/CAN system followed by benzylation.17 Using electron-deficient para-substituted bromoarenes as cyano, ethyl ester, of formyl, the C5 arylated pyrazole derivatives 27–29 were isolated in good yields. The reaction is highly chemoselective and the C–I bond was not involved, allowing further transformations. 3-Bromoquinoline has also been coupled with 1-benzyl-4-iodopyrazole to give the desired product 30 in 47% yield. The reaction is slightly sensitive to the steric hindrance, as from 2-bromobenzonitrile the 5-arylated pyrazole 31 was isolated in only 63% yield.
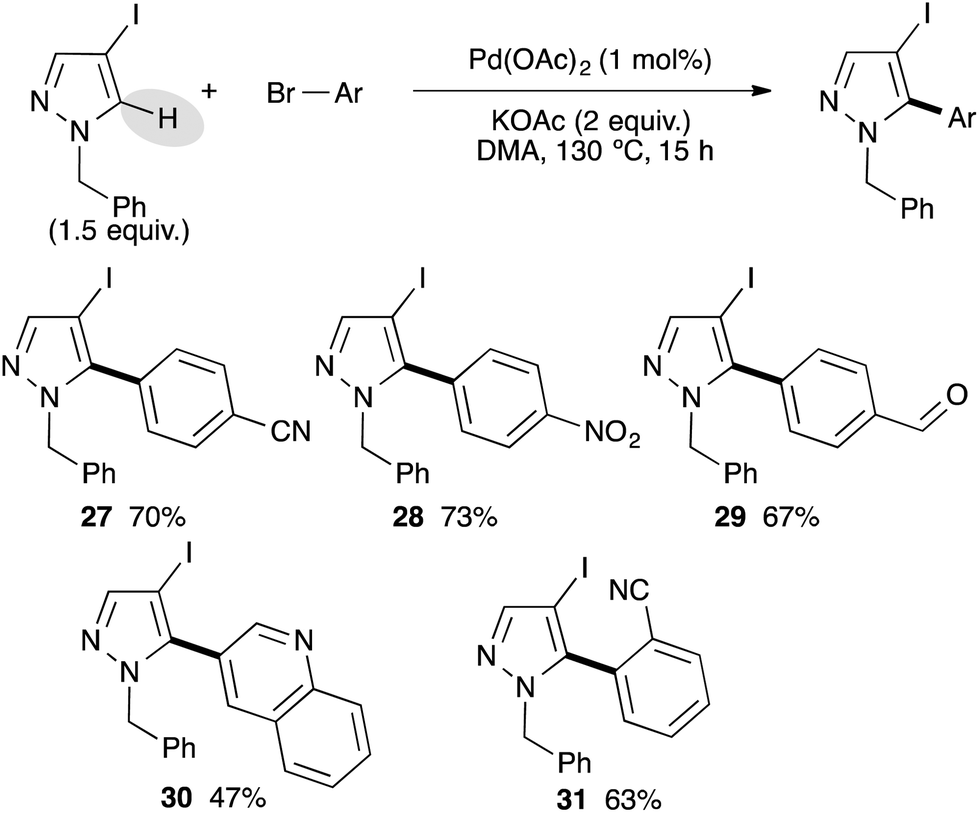 | ||
| Scheme 4 Pd-catalyzed direct arylation of 4-iodo-1-benzylpyrazole. | ||
The debromination of a C5-arylated pyrazole was then studied (Scheme 5). In the presence of Pd/C (10 mass% of the starting materials) in ethanol and triethylamine under a hydrogen atmosphere (3–5 bar) at 70 °C for 5 h, the 4-bromo-pyrazole 4 was debrominated to afford the 5-mono-arylated pyrazole 32 in excellent 94% yield.18
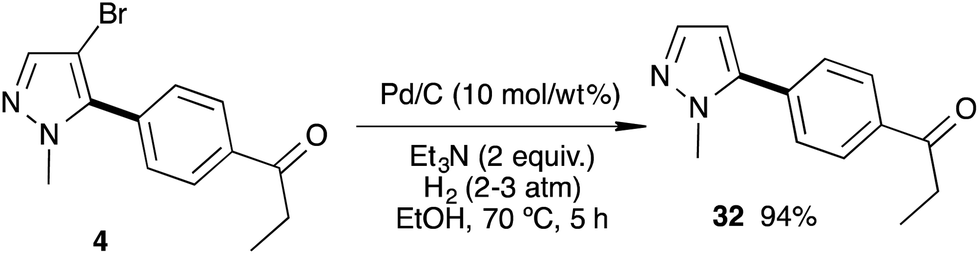 | ||
| Scheme 5 Cleavage of the C–Br bond. | ||
Then, we investigated the reactivity of the C–I bond of the previously synthesized C5 arylated pyrazole derivative 27 (Scheme 6). Firstly, the 4-iodopyrazole 27 was arylated via a Suzuki–Miyaura reaction.19 Using phenylboronic acid in the presence of 2 mol% Pd(OAc)2 without phosphines and 3 equiv. of K2CO3 in DMA, the unsymmetrical C5,C4-diarylpyrazole 33 was isolated in 74% yield. Using the conditions described by Janin,20 namely, HCl under air atmospheric conditions, selective deiodination of 27 could be performed without the cleavage of the N-benzyl group to afford the 5-arylated pyrazole 34 in 86% yield. Finally, we also performed a C–H bond heteroarylation of this iodopyrrole with 2-ethyl-4-methylthiazole. Using 1 mol% PdCl(C3H5)(dppb) in the presence of KOAc as the base in DMA at 150 °C, 4-(1-benzyl-4-(2-ethyl-4-methylthiazol-5-yl)pyrazol-5-yl)benzonitrile (35) was isolated in 54% yield.
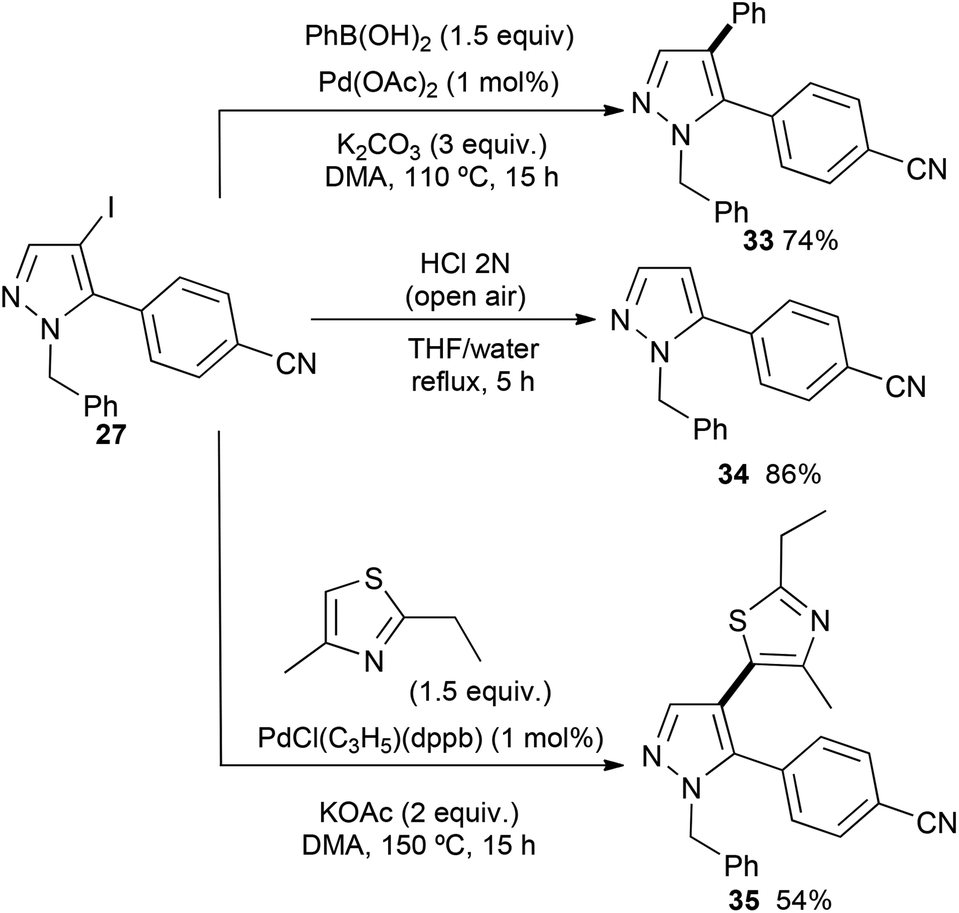 | ||
| Scheme 6 Transformation of C–I bond of the pyrazole unit. | ||
Conclusions
In summary, we have demonstrated that using appropriate reaction conditions, C4 halosubstituted N-protected pyrazole derivatives were regioselectively arylated at the C5 position using aryl bromides as coupling partners. The reaction is very chemoselective as the C–X bonds (X = Br and I) on the pyrazole unit were never involved during the C–H bond arylation process. The reaction proceeds in moderate to very high yields in the presence of electron-deficient aryl bromides or heteroaryl bromides using 1 mol% of Pd(OAc)2 as the catalyst precursor. Electron-rich aryl bromides could also be employed with a high chemoselectivity using 1 mol% of PdCl(C3H5)(dppb) as the catalyst. We also showed that bromo or iodo substituents could be used as traceless protecting groups for the formation of regioselective C5-arylated pyrazoles. Moreover, using 4-iodopyrazole derivatives, orthogonal arylations were performed to allow the formation of unsymmetrical C4,C5-diarylated pyrazoles in high yields.Experimental section
All reactions were carried out under an argon atmosphere with standard Schlenk techniques. DMA (N,N-dimethylacetamide) (99%) was purchased from Acros. KOAc (99%), and Pd(OAc)2 (98%) were purchased from Alfa Aesar. These compounds were not purified before use. 1H NMR spectra were recorded on a Bruker GPX (400 MHz) spectrometer. Chemical shifts (δ) were reported in parts per million relative to residual chloroform (7.26 ppm for 1H; 77.0 ppm for 13C), constants were reported in hertz. 1H NMR assignment abbreviations were the following: singlet (s), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), doublet of triplets (dt), and multiplet (m). 13C NMR spectra were recorded at 100 MHz on the same spectrometer and reported in ppm. All reagents were weighed and handled in air.General procedure
As a typical experiment, the reaction of the aryl bromide (1 mmol), 4-bromo-1-methylpyrazole, 1-benzyl-4-bromopyrazole, 4-bromo-1-phenylpyrazole, or 1-benzyl-4-iodopyrazole (1.5 mmol) and KOAc (0.196 g, 2 mmol) at 130 °C for 16 h in DMA (2 mL) in the presence of Pd(OAc)2 (0.56 mg, 0.0025 mmol) (see tables or schemes) under argon affords the arylation product after evaporation of the solvent and filtration on silica gel.4-(4-Bromo-1-methylpyrazol-5-yl)benzonitrile 1
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 4-bromobenzonitrile (0.182 g, 1 mmol), 1 was obtained in 89% (0.233 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.80 (d, J = 8.3 Hz, 2H), 7.56–7.53 (m, 3H), 3.83 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 139.5, 139.2, 132.9, 132.4, 130.4, 118.1, 113.0, 94.1, 38.5.
Elemental analysis: calcd (%) C11H8BrN3 for (262.11): C 50.41, H 3.08; found: C 50.56, H 3.21.
4-Bromo-1-methyl-5-(4-nitrophenyl)pyrazole 2
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 4-bromonitrobenzene (0.202 g, 1 mmol), 2 was obtained in 80% (0.226 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.36 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.7 Hz, 2H), 7.57 (s, 1H), 3.86 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 148.0, 139.6, 138.9, 134.7, 130.7, 123.9, 94.3, 38.6.
Elemental analysis: calcd (%) C10H8BrN3O2 for (282.10): C 42.58, H 2.86; found: C 42.74, H 3.12.
4-(4-Bromo-1-methylpyrazol-5-yl)benzaldehyde 3
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 4-bromobenzaldehyde (0.185 g, 1 mmol), 3 was obtained in 84% (0.223 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 10.10 (s, 1H), 8.03 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.57 (s, 1H), 3.86 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 191.4, 139.9, 139.6, 136.4, 134.3, 130.4, 129.9, 94.1, 38.6.
Elemental analysis: calcd (%) C11H9BrN2O for (265.11): C 49.84, H 3.42; found: C 50.11, H 3.75.
1-(4-(4-Bromo-1-methylpyrazol-5-yl)phenyl)propan-1-one 4
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 4-bromopropiophenone (0.213 g, 1 mmol), 4 was obtained in 72% (0.211 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.09 (d, J = 8.3 Hz, 2H), 7.56–7.51 (m, 3H), 3.84 (s, 3H), 3.05 (q, J = 7.2 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 200.0, 140.2, 139.5, 137.2, 132.7, 130.0, 128.3, 93.8, 38.5, 31.9, 8.2.
Elemental analysis: calcd (%) C13H13BrN2O for (293.16): C 53.26, H 4.47; found: C 53.58, H 4.71.
4-Bromo-5-(4-methoxyphenyl)-1-methylpyrazole 5
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 4-bromoanisole (0.187 g, 1 mmol), 5 was obtained in 42% (0.112 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.51 (s, 1H), 7.33 (d, J = 8.9 Hz, 2H), 7.02 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 3.80 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 160.2, 141.1, 139.1, 131.1, 120.5, 114.2, 93.3, 55.3, 38.2.
Elemental analysis: calcd (%) C11H11BrN2O for (267.13): C 49.46, H 4.15; found: C 49.85, H 4.01.
3-(4-Bromo-1-methylpyrazol-5-yl)pyridine 6
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 3-bromopyridine (0.158 g, 1 mmol), 6 was obtained in 52% (0.124 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) 8.71 (brs, 1H), 8.68 (brs, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.56 (s, 1H), 7.45 (dd, J = 4.8 and 7.9 Hz, 1H), 3.84 (s, 3H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 150.2, 139.5, 138.0, 137.3, 137.1, 124.8, 123.5, 94.4, 38.4.
Elemental analysis: calcd (%) C9H8BrN3 for (238.09): C 45.40, H 3.39; found: C 45.67, H 3.31.
5-(4-Bromo-1-methyl-1H-pyrazol-5-yl)pyrimidine 7
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 5-bromopyrimidine (0.159 g, 1 mmol), 7 was obtained in 64% (0.153 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 9.31 (s, 1H), 8.85 (s, 2H), 7.60 (s, 1H), 3.88 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 158.8, 157.2, 139.8, 134.8, 123.5, 95.2, 38.6.
Elemental analysis: calcd (%) C8H7BrN4 for (239.08): C 40.19, H 2.95; found: C 40.33, H 3.18.
3-(4-Bromo-1-methylpyrazol-5-yl)quinoline 8
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 3-bromoquinoline (0.208 g, 1 mmol), 8 was obtained in 77% (0.222 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.94 (d, J = 2.3 Hz, 1H), 8.23 (d, J = 2.3 Hz, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.81 (ddd, J = 1.8, 6.9 and 8.4 Hz, 1H), 7.64 (d, J = 7.2 Hz, 1H), 7.60 (s, 1H), 3.89 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 150.2, 147.8, 139.5, 138.1, 137.2, 130.7, 129.4, 128.1, 127.5, 127.2, 121.7, 94.6, 38.5.
Elemental analysis: calcd (%) C13H10BrN3 for (288.15): C 54.19, H 3.50; found: C 54.36, H 3.32.
2-(4-Bromo-1-methylpyrazol-5-yl)benzonitrile 9
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 2-bromobenzonitrile (0.182 g, 1 mmol), 9 was obtained in 68% (0.178 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.85 (d, J = 7.8 Hz, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.58 (s, 1H), 7.48 (d, J = 7.8 Hz, 1H), 3.80 (s, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 139.4, 137.8, 133.5, 133.0, 132.1, 131.8, 130.0, 116.9, 113.9, 95.3, 38.3.
Elemental analysis: calcd (%) C11H8BrN3 for (262.11): C 50.41, H 3.08; found: C 50.29, H 3.33.
2-(4-Bromo-1-methylpyrazol-5-yl)benzaldehyde 10
From 4-bromo-1-methylpyrazole (0.241 g, 1.5 mmol) and 2-bromobenzaldehyde (0.185 g, 1 mmol), 10 was obtained in 73% (0.193 g) yield.1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 9.81 (s, 1H), 8.11 (dd, J = 1.6 and 7.7 Hz, 1H), 7.75 (dt, J = 1.8 and 7.5 Hz, 1H), 7.67 (t, J = 7.7 Hz, 1H), 7.60 (s, 1H), 7.39 (dd, J = 1.4 and 7.7 Hz, 1H), 3.72 (s, 3H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 190.4, 139.2, 137.9, 134.8, 134.1, 131.6, 131.1, 130.3, 128.8, 95.8, 38.2.
Elemental analysis: calcd (%) C11H9BrN2O for (265.11): C 49.84, H 3.42; found: C 59.99, H 3.27.
4-(1-Benzyl-4-bromopyrazol-5-yl)benzonitrile 11
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 4-bromobenzonitrile (0.182 g, 1 mmol), 11 was obtained in 83% (0.281 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.72 (d, J = 8.1 Hz, 2H), 7.65 (s, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.29–7.24 (m, 3H), 6.99–6.95 (m, 2H), 5.27 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 140.1, 139.7, 136.2, 133.1, 132.4, 130.6, 128.8, 128.1, 126.8, 118.1, 113.2, 94.8, 54.8.
Elemental analysis: calcd (%) C17H12BrN3 for (338.21): C 60.37, H 3.58; found: C 60.84, H 3.71.
1-Benzyl-4-bromo-5-(4-nitrophenyl)pyrazole 12
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 4-bromonitrobenzene (0.202 g, 1 mmol), 12 was obtained in 54% (0.193 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.29 (d, J = 8.6 Hz, 2H), 7.66 (s, 1H), 7.47 (d, J = 8.6 Hz, 2H), 7.29–7.25 (m, 3H), 7.00–6.96 (m, 2H), 5.29 (s, 2H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 148.1, 140.2, 139.4, 136.2, 134.8, 130.9, 128.8, 128.1, 126.8, 123.9, 95.0, 54.8.
Elemental analysis: calcd (%) C16H12BrN3O2 for (358.20): C 53.65, H 3.38; found: C 53.89, H 3.11.
4-(1-Benzyl-4-bromopyrazol-5-yl)benzaldehyde 13
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 4-bromobenzaldehyde (0.185 g, 1 mmol), 13 was obtained in 72% (0.246 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 10.01 (s, 1H), 7.95 (d, J = 7.8 Hz, 2H), 7.65 (s, 1H), 7.48 (d, J = 7.8 Hz, 2H), 7.28–7.23 (m, 3H), 7.01–6.97 (m, 2H), 5.28 (s, 2H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 191.3, 140.4, 140.1, 136.6, 136.4, 134.4, 130.6, 129.8, 128.8, 128.0, 127.0, 94.7, 54.7.
Elemental analysis: calcd (%) C17H13BrN2O for (341.21): C 59.84, H 3.84; found: C 60.17, H 4.02.
1-(4-(1-Benzyl-4-bromopyrazol-5-yl)phenyl)propan-1-one 14
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 4-bromopropiophenone (0.213 g, 1 mmol), 14 was obtained in 89% (0.329 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.02 (d, J = 8.2 Hz, 2H), 7.64 (s, 1H), 7.40 (d, J = 8.2 Hz, 2H), 7.27–7.24 (m, 3H), 7.02–6.98 (m, 2H), 5.27 (s, 2H), 3.03 (q, J = 7.2 Hz, 2H), 1.25 (t, J = 7.2 Hz, 2H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 200.0, 140.6, 140.0, 137.2, 136.4, 132.7, 130.1, 128.7, 128.2, 127.9, 126.9, 94.4, 54.5, 31.9, 8.1.
Elemental analysis: calcd (%) C19H17BrN2O for (369.26): C 61.80, H 4.64; found: C 62.17, H 4.46.
Ethyl 4-(1-benzyl-4-bromopyrazol-5-yl)benzoate 15
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and ethyl 4-bromobenzoate (0.229 g, 1 mmol), 15 was obtained in 79% (0.304 g) yield.1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 8.13 (d, J = 8.2 Hz, 2H), 7.65 (s, 1H), 7.40 (d, J = 8.2 Hz, 2H), 7.29–7.25 (m, 3H), 7.03–6.98 (m, 2H), 5.29 (s, 2H), 4.43 (q, J = 7.2 Hz, 2H), 1.43 (t, J = 7.2 Hz, 3H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 165.9, 140.7, 140.0, 136.5, 132.8, 131.3, 129.9, 129.8, 128.7, 127.9, 127.0, 94.4, 61.2, 54.6, 14.3.
Elemental analysis: calcd (%) C19H17BrN2O2 for (385.26): C 59.23, H 4.45; found: C 59.48, H 4.11.
2-(1-Benzyl-4-bromopyrazol-5-yl)benzonitrile 16
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 2-bromobenzonitrile (0.182 g, 1 mmol), 16 was obtained in 69% (0.234 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.77 (d, J = 7.6 Hz, 1H), 7.66 (s, 1H), 7.63 (t, J = 7.9 Hz, 1H), 7.56 (t, J = 7.5 Hz, 1H), 7.28–7.26 (m, 1H), 7.23–7.19 (m, 3H), 6.92–6.88 (m, 2H), 5.32 (d, J = 15.4 Hz, 1H), 5.18 (d, J = 15.4 Hz, 1H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 139.9, 138.0, 135.9, 133.4, 132.8, 132.2, 131.7, 130.0, 128.7, 128.1, 127.2, 116.7, 114.3, 96.3, 55.2.
Elemental analysis: calcd (%) C17H12BrN3 for (338.21): C 60.37, H 3.58; found: C 60.12, H 3.18.
2-(1-Benzyl-4-bromopyrazol-5-yl)benzaldehyde 17
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 2-bromobenzaldehyde (0.185 g, 1 mmol), 19 was obtained in 62% (0.212 g) yield.1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 9.52 (s, 1H), 8.02 (dd, J = 1.9 and 7.5 Hz, 1H), 7.72–7.60 (m, 3H), 7.28–7.18 (m, 4H), 6.91–6.86 (m, 2H), 5.20 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 189.9, 139.4, 137.8, 135.8, 134.7, 133.9, 131.4, 131.2, 130.3, 128.7, 128.3, 128.1, 127.3, 96.8, 55.1.
Elemental analysis: calcd (%) C17H13BrN2O for (341.21): C 59.84, H 3.84; found: C 60.08, H 3.49.
3-(1-Benzyl-4-bromopyrazol-5-yl)pyridine 19
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 3-bromopyridine (0.158 g, 1 mmol), 19 was obtained in 41% (0.129 g) yield.1H NMR (400 MHz, DMSO-d6, 25 °C): δ (ppm) = 8.66 (d, J = 4.8 Hz, 1H), 8.53 (s, 1H), 7.82 (s, 1H), 7.80 (t, J = 2.1 Hz, 1H), 7.53 (dd, J = 4.8 and 7.9 Hz, 1H), 7.28–7.21 (m, 3H), 6.91 (dd, J = 1.8 and 7.5 Hz, 2H), 5.33 (s, 2H).
13C NMR (75 MHz, DMSO-d6, 25 °C): δ (ppm) = 150.3, 149.8, 139.4, 138.2, 137.3, 136.7, 128.5, 127.6, 126.8, 124.2, 123.8, 94.2, 54.0.
Elemental analysis: calcd (%) C15H12BrN3 for (314.19): C 57.34, H 3.85; found: C 57.69, H 4.10.
5-(1-Benzyl-4-bromopyrazol-5-yl)pyrimidine 20
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 5-bromopyrimidine (0.159 g, 1 mmol), 20 was obtained in 38% (0.120 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 9.25 (s, 1H), 8.62 (s, 2H), 7.69 (s, 1H), 7.29–7.25 (m, 3H), 7.00–6.96 (m, 2H), 5.30 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 158.9, 157.3, 140.2, 135.9, 135.1, 129.0, 128.4, 126.8, 123.6, 96.0, 55.1.
Elemental analysis: calcd (%) C14H11BrN4 for (315.17): C 53.35, H 3.52; found: C 53.56, H 3.17.
3-(1-Benzyl-4-bromopyrazol-5-yl)quinoline 21
From 1-benzyl-4-bromopyrazole (0.356 g, 1.5 mmol) and 3-bromoquinoline (0.208 g, 1 mmol), 21 was obtained in 52% (0.189 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.82 (br, 1H), 8.20 (d, J = 8.5 Hz, 1H), 8.05 (d, J = 2.3 Hz, 1H), 7.81 (ddd, J = 1.5, 8.4 and 16.6 Hz, 2H), 7.71 (s, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.29–7.24 (m, 3H), 7.04–7.00 (m, 2H), 5.32 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 149.6, 146.9, 140.1, 138.3, 138.1, 136.3, 131.2, 128.9, 128.8, 128.1, 127.8, 127.2, 126.9, 121.8, 95.5, 54.9.
Elemental analysis: calcd (%) C19H14BrN3 for (364.25): C 62.65, H 3.87; found: C 62.96, H 3.61.
4-(4-Bromo-1-phenylpyrazol-5-yl)benzonitrile 22
From 4-bromo-1-phenylpyrazole (0.334 g, 1.5 mmol) and 4-bromobenzonitrile (0.182 g, 1 mmol), 22 was obtained in 54% (0.175 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.78 (s, 1H), 7.65 (d, J = 8.6 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 7.35–7.33 (m, 3H), 7.20–7.17 (m, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 141.4, 139.4, 138.4, 133.1, 132.2, 130.6, 129.2, 128.3, 124.9, 118.1, 112.7, 96.6.
Elemental analysis: calcd (%) C16H10BrN3 for (324.18): C 59.28, H 3.11; found: C 59.10, H 3.32.
4-Bromo-5-(4-nitrophenyl)-1-phenylpyrazole 23
From 4-bromo-1-phenylpyrazole (0.334 g, 1.5 mmol) and 4-bromonitrobenzene (0.202 g, 1 mmol), 23 was obtained in 48% (0.165 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.22 (d, J = 8.7 Hz, 2H), 7.80 (s, 1H), 7.47 (d, J = 8.7 Hz, 2H), 7.37–7.33 (m, 3H), 7.22–7.18 (m, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 147.7, 141.4, 139.2, 138.0, 134.9, 130.8, 129.3, 128.3, 124.9, 123.7, 96.7.
Elemental analysis: calcd (%) C15H10BrN3O2 for (344.17): C 52.35, H 2.93; found: C 52.71, H 2.98.
4-(4-Bromo-1-phenylpyrazol-5-yl)benzaldehyde 24
From 4-bromo-1-phenylpyrazole (0.334 g, 1.5 mmol) and 4-bromobenzaldehyde (0.185 g, 1 mmol), 24 was obtained in 42% (0.145 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 10.03 (s, 1H), 7.87 (d, J = 8.2 Hz, 2H), 7.79 (s, 1H), 7.46 (d, J = 8.2 Hz, 2H), 7.34–7.30 (m, 3H), 7.23–7.18 (m, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 191.5, 141.4, 139.5, 139.0, 136.1, 134.4, 130.6, 129.6, 129.1, 128.1, 124.9, 96.5.
Elemental analysis: calcd (%) C16H11BrN2O for (327.18): C 58.74, H 3.39; found: C 58.59, H 3.47.
Ethyl 4-(4-bromo-1-phenylpyrazol-5-yl)benzoate 25
From 4-bromo-1-phenylpyrazole (0.334 g, 1.5 mmol) and ethyl 4-bromobenzoate (0.229 g, 1 mmol), 25 was obtained in 51% (0.189 g) yield.1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 8.03 (d, J = 8.4 Hz, 2H), 7.77 (s, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.32–7.28 (m, 3H), 7.23–7.17 (m, 2H), 4.38 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 166.2, 141.5, 139.9, 139.7, 133.1, 131.0, 130.2, 129.8, 129.3, 128.2, 125.1, 96.5, 61.4, 14.5.
Elemental analysis: calcd (%) C18H15BrN2O2 for (371.23): C 58.24, H 4.07; found: C 58.46, H 4.25.
4-(1-Benzyl-4-iodopyrazol-5-yl)benzonitrile 27
From 1-benzyl-4-iodopyrazole (0.426 g, 1.5 mmol) and 4-bromobenzonitrile (0.182 g, 1 mmol), 27 was obtained in 70% (0.270 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.74 (d, J = 8.3 Hz, 2H), 7.70 (s, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.30–7.26 (m, 3H), 6.99–6.96 (m, 2H), 5.30 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 144.6, 142.9, 136.3, 134.1, 132.4, 130.9, 128.8, 128.1, 126.9, 118.2, 113.3, 54.8.
Elemental analysis: calcd (%) C17H12IN3 for (385.21): C 53.01, H 3.14; found: C 53.28, H 3.01.
1-Benzyl-4-iodo-5-(4-nitrophenyl)pyrazole 28
From 1-benzyl-4-iodopyrazole (0.426 g, 1.5 mmol) and 4-bromonitrobenzene (0.202 g, 1 mmol), 28 was obtained in 73% (0.296 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.31 (d, J = 8.6 Hz, 2H), 7.72 (s, 1H), 7.46 (d, J = 8.6 Hz, 2H), 7.31–7.26 (m, 3H), 7.01–6.98 (m, 2H), 5.32 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 148.2, 144.7, 142.6, 136.3, 135.9, 131.2, 128.9, 128.1, 126.9, 123.9, 123.8, 54.9.
Elemental analysis: calcd (%) C16H12IN3O2 for (405.20): C 47.43, H 2.99; found: C 47.65, H 3.28.
4-(1-Benzyl-4-iodopyrazol-5-yl)benzaldehyde 29
From 1-benzyl-4-iodopyrazole (0.426 g, 1.5 mmol) and 4-bromobenzaldehyde (0.185 g, 1 mmol), 29 was obtained in 67% (0.260 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 10.09 (s, 1H), 7.96 (d, J = 8.2 Hz, 2H), 7.70 (s, 1H), 7.46 (d, J = 8.2 Hz, 2H), 7.28–7.25 (m, 3H), 7.02–6.98 (m, 2H), 5.31 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 191.3, 144.5, 143.5, 136.6, 136.4, 135.3, 130.8, 130.3, 129.7, 128.7, 127.9, 126.9, 54.7.
Elemental analysis: calcd (%) C17H13IN2O for (388.21): C 52.60, H 3.38; found: C 52.95, H 3.61.
3-(1-Benzyl-4-iodopyrazol-5-yl)quinoline 30
From 1-benzyl-4-iodopyrazole (0.426 g, 1.5 mmol) and 3-bromoquinoline (0.208 g, 1 mmol), 30 was obtained in 47% (0.193 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.82 (s, 1H), 8.26 (d, J = 8.7 Hz, 1H), 8.05 (s, 1H), 7.89–7.80 (m, 2H), 7.76 (m, 1H), 7.66 (dd, J = 7.2 and 8.3 Hz, 1H), 7.30–7.28 (m, 3H), 7.05–7.00 (m, 2H), 5.36 (s, 2H).
13C NMR (75 MHz, CDCl3, 25 °C): δ (ppm) = 150.6, 148.0, 144.5, 143.2, 141.8, 137.7, 136.5, 130.7, 129.5, 128.8, 128.1, 127.5, 127.1, 127.0, 126.6, 122.8, 54.9.
Elemental analysis: calcd (%) C19H14IN3 for (411.25): C 55.49, H 3.43; found: C 55.78, H 3.11.
2-(1-Benzyl-4-iodopyrazol-5-yl)benzonitrile 31
From 1-benzyl-4-iodopyrazole (0.426 g, 1.5 mmol) and 2-bromobenzonitrile (0.182 g, 1 mmol), 31 was obtained in 63% (0.243 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 7.79 (d, J = 7.7 Hz, 1H), 7.72 (s, 1H), 7.66 (t, J = 7.7 Hz, 1H), 7.58 (t, J = 7.5 Hz, 1H), 7.29–7.22 (m, 4H), 6.95–6.90 (m, 2H), 5.37 (d, J = 15.5 Hz, 1H), 5.22 (d, J = 15.2 Hz, 1H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 144.3, 141.3, 135.9, 133.4, 133.3, 132.8, 131.8, 130.0, 128.7, 128.0, 127.2, 116.7, 114.4, 55.2.
Elemental analysis: calcd (%) C17H12IN3 for (385.21): C 53.01, H 3.14; found: C 53.32, H 2.86.
1-(4-(1-Methylpyrazol-5-yl)phenyl)propan-1-one 32
An autoclave was charged with 1-(4-(4-bromo-1-methylpyrazol-5-yl)phenyl)propan-1-one (4) (0.293 g, 1 mmol), Et3N (270 μL; 2 mmol), Pd/C (29 mg, 10% of the weight of the pyrazole derivative) and EtOH (5 mL) and pressurized with hydrogen (3–5 bar). The crude mixture was heated at 70 °C for 5 h, and then the reaction was cooled down and filtered through a pad of Celite. After evaporation of the solvent and purification on silica gel 32 was isolated in 94% (0.201 g) yield.1H NMR (400 MHz, CDCl3, 25 °C): δ (ppm) = 8.06 (d, J = 8.2 Hz, 2H), 7.56–7.52 (m, 3H), 6.39 (d, J = 1.9 Hz, 1H), 3.94 (s, 2H), 3.05 (q, J = 7.2 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 200.1, 142.6, 138.6, 136.6, 134.9, 128.8, 128.4, 106.7, 37.8, 31.9, 8.2.
This is a known compound and the spectral data are identical to those reported in the literature.5
4-(1-Benzyl-4-phenylpyrazol-5-yl)benzonitrile 33
A Schlenk tube was charged with 4-(1-benzyl-4-iodopyrazol-5-yl)benzonitrile (27) (0.385 g, 1 mmol, 1 equiv.), phenylboronic acid (122 mg, 0.19 mmol, 1 equiv.), K2CO3 (0.415 g, 3 mmol, 3 equiv.), Pd(OAc)2 (4.5 mg, 0.002 mmol, 2 mol%) and 2–3 mL of DMA. The resulting solution was stirred under an argon atmosphere at 110 °C for 15 h. Then, the solution was poured in water/Et2O (1:1) solution. The organic phase was washed 2 times with water, dried over MgSO4 and concentrated. Then, the residue was purified using flash chromatography to afford 33 in 74% (0.248 g) yield.
1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 7.84 (s, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 7.30–7.27 (m, 3H), 7.26–7.19 (m, 3H), 7.15–7.10 (m, 2H), 7.04–7.00 (m, 2H), 5.27 (s, 2H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 138.5, 138.1, 136.9, 135.3, 132.5, 132.2, 131.1, 128.8, 128.6, 127.9, 127.6, 126.9, 126.7, 122.5, 118.2, 112.8, 53.8.
Elemental analysis: calcd (%) C23H17N3 for (335.41): C 82.36, H 5.11; found: C 82.71, H 5.32.
4-(1-Benzylpyrazol-5-yl)benzonitrile 34
In a flask equipped with a condenser, compound (27) (0.385 g, 1 mmol, 1 equiv.) was boiled in 2 N hydrochloric acid (5 mL) for 15 h. The resulting mixture was cooled, then the solution was poured in saturated solution of K2CO3 and extracted with ethyl acetate. The organic layer was washed with brine, dried over magnesium sulfate and concentrated to dryness to yield an oil. Then, the residue was purified using flash chromatography to afford 34 in 86% (0.223 g) yield.1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 7.69 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 2.0 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.35–7.28 (m, 3H), 7.05–7.03 (m, 2H), 6.45 (d, J = 2.0 Hz, 1H), 5.39 (s, 2H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 142.1, 139.6, 137.0, 135.2, 132.4, 129.4, 128.8, 127.8, 126.6, 118.3, 112.4, 107.5, 53.6.
Elemental analysis: calcd (%) C17H13N3 for (259.31): C 78.74, H 5.05; found: C 78.87, H 4.86.
4-(1-Benzyl-4-(2-ethyl-4-methylthiazol-5-yl)pyrazol-5-yl)benzonitrile 35
A Schlenk tube was charged with 4-(1-benzyl-4-iodopyrazol-5-yl)benzonitrile (27) (0.385 g, 1 mmol, 1 equiv.), 2-ethyl-4-methylthiazole (196 μL, 1.5 mmol, 1.5 equiv.), KOAc (0.196 g, 2 mmol, 2 equiv.), PdCl(C3H5)(dppb) (5 mg, 0.01 mmol, 1 mol%) and 2–3 mL of DMA. The resulting solution was stirred under an argon atmosphere at 110 °C for 15 h. Then, the solution was poured in water/Et2O (1:1) solution. The organic phase was washed 2 times with water, dried over MgSO4 and concentrated. Then, the residue was purified using flash chromatography to afford 35 in 54% (0.208 g) yield.
1H NMR (300 MHz, CDCl3, 25 °C): δ (ppm) = 7.72 (s, 1H), 7.66 (d, J = 8.2 Hz, 2H), 7.33–7.27 (m, 5H), 7.06–7.02 (m, 2H), 5.29 (s, 2H), 2.91 (q, J = 7.0 Hz, 2H), 2.17 (s, 3H), 1.32 (t, J = 7.0 Hz, 3H).
13C NMR (100 MHz, CDCl3, 25 °C): δ (ppm) = 170.9, 148.7, 139.8, 139.2, 136.5, 134.2, 132.5, 130.7, 128.9, 128.7, 128.0, 127.0, 126.9, 120.3, 108.7, 54.1, 29.7, 26.8, 14.1.
Elemental analysis: calcd (%) C23H20N4S for (384.50): C 71.85, H 5.24; found: C 72.03, H 5.32.
Acknowledgements
We thank the Centre National de la Recherche Scientifique, “Rennes Metropole”, “UTIQUE” and Scientific Ministry of Higher Education Research of Tunisia for providing financial support.Notes and references
- (a) M. J. Fray, P. Allen, P. R. Bradley, C. E. Challenger, M. Closier, T. J. Evans, M. L. Lewis, J. P. Mathias, C. L. Nichols, Y. M. Po-Ba, H. Snow, M. H. Stefaniak and H. V. Vuong, Heterocycles, 2006, 67, 489–494 CrossRef CAS; (b) F. Beaulieu, C. Ouellet, E. H. Ruediger, M. Belema, Y. Qiu, X. Yang, J. Banville, J. R. Burke, K. R. Gregor, J. F. MacMaster, A. Martel, K. W. McIntyre, M. A. Pattoli, F. C. Zusi and D. Vyas, Bioorg. Med. Chem. Lett., 2007, 17, 1233–1237 CrossRef CAS PubMed; (c) G. A. Whitlock, K. Conlon, G. McMurray, L. R. Roberts, A. Stobie and R. J. Thurlow, Bioorg. Med. Chem. Lett., 2008, 18, 2930–2934 CrossRef CAS PubMed; (d) C. I. Stathakis, S. Bernhardt, V. Quint and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9428–9432 CrossRef CAS PubMed.
- (a) X. j. Wang, J. Tan and K. Grozinger, Tetrahedron Lett., 2000, 41, 4713–4716 CrossRef CAS; (b) A. S. Paulson, J. Eskildsen, P. Vedso and M. Begtrup, J. Org. Chem., 2002, 67, 3904–3907 CrossRef CAS PubMed; (c) S. Beltran-Rodil, M. G. Edwards, D. S. Pugh, M. Reid and R. J. K. Taylor, Synlett, 2010, 602–606 CAS; (d) R. A. Khera, A. Ali, M. Hussain, J. Tatar, A. Villinger and P. Langer, Synlett, 2010, 1923–1926 CAS; (e) R. A. Khera, A. Ali, H. Rafique, M. Hussain, J. Tatar, A. Saeed, A. Villinger and P. Langer, Tetrahedron, 2011, 67, 5244–5253 CrossRef CAS PubMed; (f) Y. L. Janin, Chem. Rev., 2012, 112, 3924–3958 CrossRef CAS PubMed; (g) L. Zhang, A. Villalobos, E. M. Beck, T. Bocan, T. A. Chappie, L. Chen, S. Grimwood, S. D. Heck, C. J. Helal, X. Hou, J. M. Humphrey, J. Lu, M. B. Skaddan, T. J. McCarthy, P. R. Verhoest, T. T. Wager and K. Zasadny, J. Med. Chem., 2013, 56, 4568–4579 CrossRef CAS PubMed; (h) M. P. Winters, N. Subasinghe, M. Wall, E. Beck, M. R. Brandt, M. F. A. Finley, Y. Liu, M. L. Lubin, M. P. Neeper, N. Qin, C. M. Flores and Z. Sui, Bioorg. Med. Chem. Lett., 2014, 24, 2053–2056 CrossRef CAS PubMed.
- For reviews on C–H bond functionalizations see: (a) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed; (b) S. Pascual, P. de Mendoza and A. M. Echavarren, Org. Biomol. Chem., 2007, 5, 2727–2734 RSC; (c) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (d) F. Bellina, S. Cauteruccio and R. Rossi, Curr. Org. Chem., 2008, 12, 774–790 CrossRef CAS; (e) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013–3039 CrossRef CAS PubMed; (f) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949–957 CAS; (g) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (h) F. Bellina and R. Rossi, Chem. Rev., 2009, 110, 1082–1146 CrossRef PubMed; (i) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269–10310 CrossRef CAS PubMed; (j) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (k) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (l) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85–121 CAS; (m) J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20–40 CrossRef CAS PubMed; (n) T. Satoh and M. Miura, Synthesis, 2010, 3395–3409 CrossRef CAS PubMed; (o) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC; (p) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (q) D. Zhao, J. You and C. Hu, Chem. – Eur. J., 2011, 17, 5466–5492 CrossRef CAS PubMed; (r) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (s) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588–5598 RSC; (t) M. C. White, Synlett, 2012, 2746–2748 CAS; (u) D.-G. Yu, B.-J. Li and Z.-J. Shi, Tetrahedron, 2012, 68, 5130–5136 CrossRef CAS PubMed; (v) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886–896 RSC; (w) M. He, J.-F. Soulé and H. Doucet, ChemCatChem, 2014, 6, 1824–1859 CrossRef CAS PubMed; (x) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17–117 CrossRef CAS PubMed; (y) K. Yuan, J.-F. Soulé and H. Doucet, ACS Catal., 2015, 5, 978–991 CrossRef CAS; (z) P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS.
- R. Goikhman, T. L. Jacques and D. Sames, J. Am. Chem. Soc., 2009, 131, 3042–3048 CrossRef CAS PubMed.
- A. Beladhria, K. Beydoun, H. Ben Ammar, R. Ben Salem and H. Doucet, Synthesis, 2011, 2553–2560 CAS.
- F. Bellina, M. Lessi and C. Manzini, Eur. J. Org. Chem., 2013, 5621–5630 CrossRef CAS PubMed.
- S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS PubMed.
- E. T. T. Kumpulainen and A. Pohjakallio, Adv. Synth. Catal., 2014, 356, 1555–1561 CrossRef CAS PubMed.
- C. Mateos, J. Mendiola, M. Carpintero and J. M. Mínguez, Org. Lett., 2010, 12, 4924–4927 CrossRef CAS PubMed.
- T. Yan, L. Chen, C. Bruneau, P. H. Dixneuf and H. Doucet, J. Org. Chem., 2012, 77, 7659–7664 CrossRef CAS PubMed.
- I. Smari, C. Youssef, K. Yuan, A. Beladhria, H. Ben Ammar, B. Ben Hassine and H. Doucet, Eur. J. Org. Chem., 2014, 1778–1786 CrossRef CAS PubMed.
- (a) H. Jung, S. Bae, H.-L. Jang and J. M. Joo, Bull. Korean Chem. Soc., 2014, 35, 3009–3014 CrossRef CAS; (b) V. O. Iaroshenko, A. Gevorgyan, O. Davydova, A. Villinger and P. Langer, J. Org. Chem., 2014, 79, 2906–2915 CrossRef CAS PubMed.
- S. A. Ohnmacht, A. J. Culshaw and M. F. Greaney, Org. Lett., 2010, 12, 224–226 CrossRef CAS PubMed.
- A. Takfaoui, L. Zhao, R. Touzani, P. H. Dixneuf and H. Doucet, Tetrahedron Lett., 2014, 55, 1697–1701 CrossRef CAS PubMed.
- (a) B. Liegault, I. Petrov, S. I. Gorelsky and K. Fagnou, J. Org. Chem., 2010, 75, 1047–1060 CrossRef CAS PubMed; (b) F. Shibahara, E. Yamaguchi and T. Murai, J. Org. Chem., 2011, 76, 2680–2693 CrossRef CAS PubMed; (c) T. Yamauchi, F. Shibahara and T. Murai, J. Org. Chem., 2014, 79, 7185–7192 CrossRef CAS PubMed.
- (a) D. Roy, S. Mom, S. Royer, D. Lucas, J.-C. Hierso and H. Doucet, ACS Catal., 2012, 2, 1033–1041 CrossRef CAS; (b) N. Laidaoui, J. Roger, A. Miloudi, D. El Abed and H. Doucet, Eur. J. Org. Chem., 2011, 4373–4385 CrossRef CAS PubMed; (c) A. Takfaoui, L. Zhao, R. Touzani, P. H. Dixneuf and H. Doucet, Tetrahedron Lett., 2014, 55, 1697–1701 CrossRef CAS PubMed.
- M. I. Rodriguez-Franco, I. Dorronsoro, A. I. Hernandez-Higueras and G. Antequera, Tetrahedron Lett., 2001, 42, 863–865 CrossRef CAS.
- For other examples of pyrazole debromination see: (a) J. F. Hansen, Y. I. Kim, L. J. Griswold, G. W. Hoelle, D. L. Taylor and D. E. Vietti, J. Org. Chem., 1980, 45, 76–80 CrossRef CAS; (b) D. R. Sliskovic, B. D. Roth, M. W. Wilson, M. L. Hoefle and R. S. Newton, J. Med. Chem., 1990, 33, 31–38 CrossRef CAS; (c) X. Zhang, C. Hou, H. Hufnagel, M. Singer, E. Opas, S. McKenney, D. Johnson and Z. Sui, ACS Med. Chem. Lett., 2012, 3, 1039–1044 CrossRef CAS PubMed; (d) T. W. Johnson, P. F. Richardson, S. Bailey, A. Brooun, B. J. Burke, M. R. Collins, J. J. Cui, J. G. Deal, Y.-L. Deng, D. Dinh, L. D. Engstrom, M. He, J. Hoffman, R. L. Hoffman, Q. Huang, R. S. Kania, J. C. Kath, H. Lam, J. L. Lam, P. T. Le, L. Lingardo, W. Liu, M. McTigue, C. L. Palmer, N. W. Sach, T. Smeal, G. L. Smith, A. E. Stewart, S. Timofeevski, H. Zhu, J. Zhu, H. Y. Zou and M. P. Edwards, J. Med. Chem., 2014, 57, 4720–4744 CrossRef CAS PubMed.
- For other examples of Suzuki–Miyaura couplings of halopyrazoles see: (a) S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871–1876 CrossRef CAS PubMed; (b) T. M. Razler, Y. Hsiao, F. Qian, R. Fu, R. K. Khan and W. Doubleday, J. Org. Chem., 2009, 74, 1381–1384 CrossRef CAS PubMed; (c) A. Chartoire, M. Lesieur, L. Falivene, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem. – Eur. J., 2012, 18, 4517–4521 CrossRef CAS PubMed; (d) E. Salanouve, P. Retailleau and Y. L. Janin, Tetrahedron, 2012, 68, 2135–2140 CrossRef CAS PubMed; (e) J. Galeta, L. Tenora, S. Man and M. Potacek, Tetrahedron, 2013, 69, 7139–7146 CrossRef CAS PubMed; (f) N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. Lopez, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef CAS PubMed; (g) V. Pandarus, D. Desplantier-Giscard, G. Gingras, R. Ciriminna, P. Demma Cara, F. Beland and M. Pagliaro, Tetrahedron Lett., 2013, 54, 4712–4716 CrossRef CAS PubMed; (h) S. D. Ramgren, L. Hie, Y. Ye and N. K. Garg, Org. Lett., 2013, 15, 3950–3953 CrossRef CAS PubMed; (i) E. Bratt, O. Verho, M. J. Johansson and J.-E. Baeckvall, J. Org. Chem., 2014, 79, 3946–3954 CrossRef CAS PubMed; (j) J. Tan, Y. Chen, H. Li and N. Yasuda, J. Org. Chem., 2014, 79, 8871–8876 CrossRef CAS PubMed.
- S. Guillou, F. J. Bonhomme, M. S. Ermolenko and Y. L. Janin, Tetrahedron, 2011, 67, 8451–8457 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2015 |