
AlCl3-catalyzed O-alkylative Passerini reaction of isocyanides, cinnamaldehydes and various aliphatic alcohols for accessing α-alkoxy-β,γ-enamides†
Longyun
Lyu
ab,
Han
Xie
a,
Huaixue
Mu
b,
Qijie
He
a,
Zhaoxiang
Bian
*b and
Jun
Wang
*a
aDepartment of Chemistry, South University of Science and Technology of China, Shenzhen, Guangdong 518055, China. E-mail: wang.j@sustc.edu.cn; Fax: (+86) 755-88018304
bSchool of Chinese Medicine, Hong Kong Baptist University, Hong Kong, China. E-mail: bzxiang@hkbu.edu.hk; Fax: (+852)-34112929
First published on 30th April 2015
Abstract
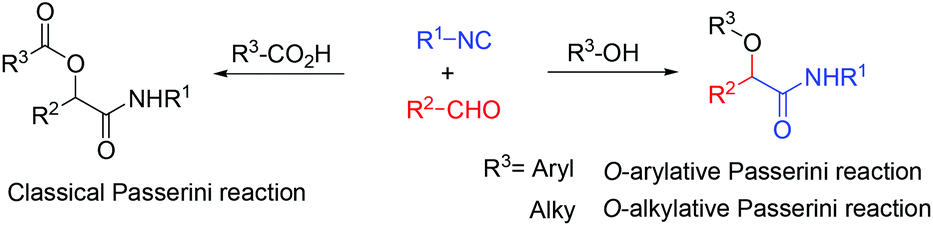
The inexpensive Lewis acid AlCl3 was found to be an efficient catalyst for the O-alkylative Passerini reaction of isocyanides, cinnamaldehydes and alcohols. Instead of carboxylic acid in the classical Passerini reaction, alcohols performed both as the solvent and substrate nicely to afford α-alkoxy-amide products in good yield (up to 91%). This method provides practical access for functional α-alkoxy-β,γ-enamide derivatives.
The Passerini three-component reaction (P-3CR), discovered in 1921, is the known isocyanide-based multicomponent reaction which gives α-acyloxy amides with isocyanides, aldehydes and carboxylic acids.1 The Passerini three-component reaction (P-3CR) plays important roles in combinatorial chemistry, for drug discovery as well as natural product synthesis.2 Besides, the application of the Passerini reaction in the preparation of polymers and peptides has also been reported.3,4 Recently, several examples of enantioselective versions for the Passerini reactions have been developed.5 Although various modifications of this reaction have already been developed, the direct Passerini reaction using a phenol or an aliphatic alcohol instead of a carboxylic acid is still less developed (Scheme 1). Only three examples using other components instead of a carboxylic acid have been reported so far. In 2006, El Kaim and Grimaud reported the O-arylative Passerini-type reaction using nitrophenol derivatives, which have a more acidic proton compared to aliphatic alcohols.6 In 2010, Soeta and Inomata used silanol instead of a carboxylic acid component, giving the corresponding α-siloxyamides in moderate to good yields.7 The only report using an aliphatic alcohol was developed by Taguchi.8 Catalyzed by In(OTf)3 with HC(OMe)3 as an additive, isocyanides, aldehydes and aliphatic alcohols afforded α-alkoxy amide derivatives in good yield, but there was only one example of unsubstituted cinnamaldehyde and one isocyanide reported. Herein, we developed an inexpensive Lewis acid AlCl3 catalyzed direct Passerini reaction for accessing α-alkoxy-β,γ-enamide derivatives. A large scope commonly available alcohols, isocyanides, and cinnamaldehydes are suitable substrates for this catalyst system. By this synthetic strategy, a polyfunctional molecular scaffold, α-alkoxy-β,γ-enamides, could be prepared in one step. α-Alkoxy-β,γ-enamide is the core part in many natural compounds, such as symbioramide, a type of bioactive ceramide with antileukemic activities and as inhibitors of the Dengue and West Nile virus proteases.9
 | ||
| Scheme 1 Passerini reaction. | ||
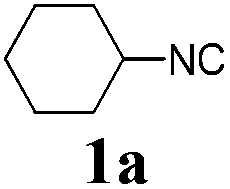
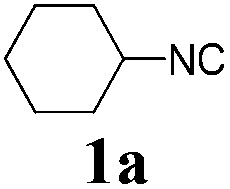
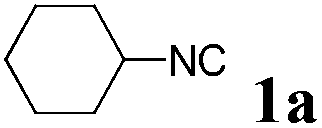
We initially started to optimize the reaction conditions using cyclohexyl isocyanide (Cy-NC) 1a and cinnamaldehyde 2a as the model substrates, MeOH 3a as the reagent as well as the solvent. Selected results of Lewis acids are summarized in Table 1. To our delight, moderate to good yields were obtained, catalyzed by several Lewis acids. Among them, AlCl3 was the most efficient catalyst which gave the expected P-3CR product in 73% yield (entry 6, Table 1). The other two Lewis acids In(OTf)3 and FeCl3 were also effective in the reaction, but resulted in lower yields. The ratios of isocyanide 1a and cinnamaldehyde 2a were also investigated (entries 10–13), leading to improvement of the yield from 73% to 85% (entry 12). 10 mol% AlCl3 can make the reaction work, but relatively lower yield (63%) was obtained (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | 1a (eq.) | 2a (eq.) | Catalyst | Yieldb (%) |
| a Reaction conditions: to a solution of the catalyst (0.2 eq.) in MeOH (1 mL) in a sealed vial were added cinnamaldehydes (0.2 mmol) and isocyanides (0.3 mmol) in sequence. The reaction mixtures were stirred at 60 °C for 12 h. b Isolated yield. | ||||
| 1 | 1 | 1.2 | None | Trace |
| 2 | 1 | 1.2 | Zn(OTf)2/0.2 | Trace |
| 3 | 1 | 1.2 | AgOTf/0.2 | Trace |
| 4 | 1 | 1.2 | In(OTf)3/0.2 | 65 |
| 5 | 1 | 1.2 | ZnCl2/0.2 | Trace |
| 6 | 1 | 1.2 | AlCl3/0.2 | 73 |
| 7 | 1 | 1.2 | FeCl3/0.2 | 43 |
| 8 | 1 | 1.2 | CuCl2/0.2 | Trace |
| 9 | 1 | 1.2 | CH3COOH/0.2 | Trace |
| 10 | 1 | 1 | AlCl3/0.2 | 76 |
| 11 | 1.2 | 1 | AlCl3/0.2 | 79 |
| 12 | 1.5 | 1 | AlCl3/0.2 | 85 |
| 13 | 1.8 | 1 | AlCl3/0.2 | 80 |
| 14 | 1.5 | 1 | AlCl3/0.1 | 63 |
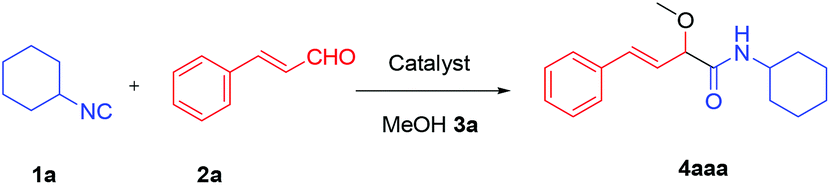
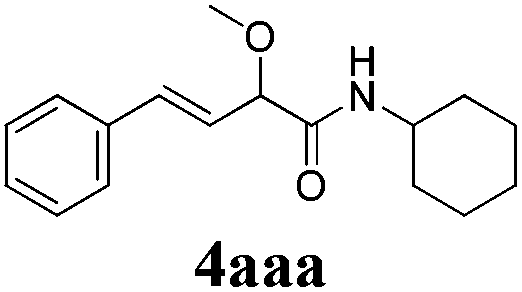
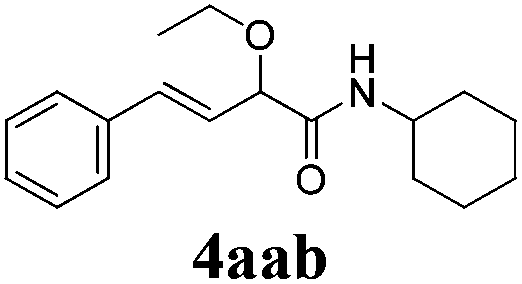
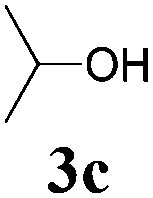
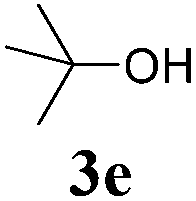
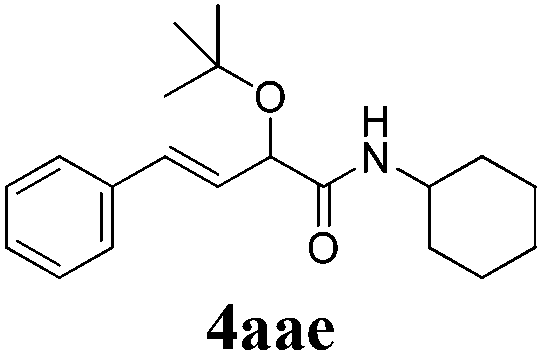
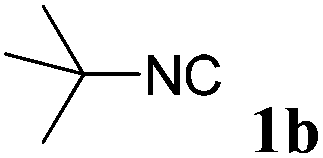
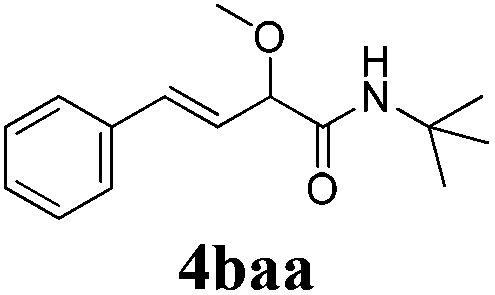
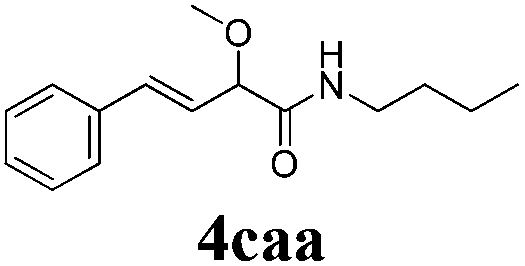
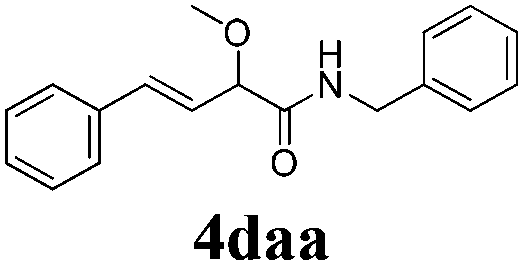
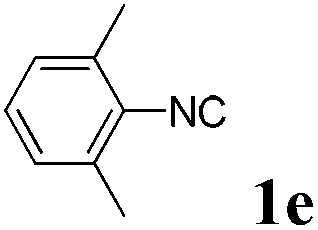
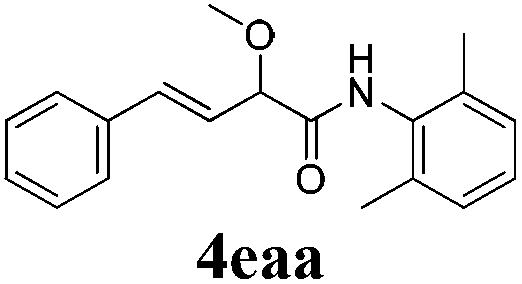
With the optimized reaction conditions in hand, various α-alkoxy-β,γ-enamides were successfully synthesized via the O-alkylative Passerini reaction of various isocyanides 1 and alcohols 3 with cinnamaldehyde 2a. As shown in Table 2, the reaction proceeded smoothly in many primary and secondary alcohols (entries 1–4). The corresponding α-alkoxy-β,γ-enamides were obtained in 61–85% yields. However, when the sterically hindered tert-butyl alcohol was used as the substrate as well as the solvent, only 53% yield was obtained even in the presence of 100 mol% AlCl3 with stirring at 80 °C for 36 h (entry 5). The steric effect of isocyanides showed no obvious influence on the yields of products. 81% and 82% yields were obtained for n-butyl isocyanide and tert-butyl isocyanide separately (entries 6 and 7). Aromatic isocyanides 1e, also suitable substrates under these reaction conditions, afforded the corresponding product 4eaa in 72% yield, but a longer reaction time (24 h) was needed (entry 9).
|
|
||||
|---|---|---|---|---|
| Entry | Isocyanides | Alcohols | Products | Yieldd (%) |
| a To a solution of AlCl3 (0.2 mmol, 0.20 eq.) in indicated alcohols (1 mL) in a sealed vial were added cinnamaldehyde 2a (1 mmol) and isocyanides 1 (1.5 mmol, 1.5 eq.) in sequence. The reaction mixtures were stirred at 60 °C for 12 h. b The reaction was carried out in t-BuOH in the presence of 100 mol% AlCl3, stirred at 80 °C for 36 h. c The reaction was carried out for 24 h. d Isolated yield. | ||||
| 1 |
|
MeOH 3a |
|
85 |
| 2 |
|
EtOH 3b |
|
63 |
| 3 |
|
|
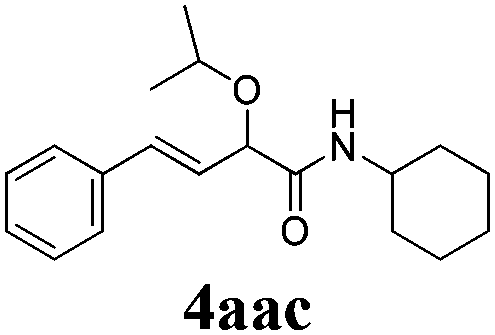
|
64 |
| 4 |
|
|
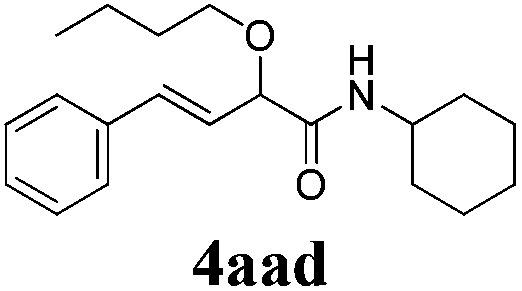
|
61 |
| 5b |
|
|
|
53 |
| 6 |
|
MeOH 3a |
|
81 |
| 7 |
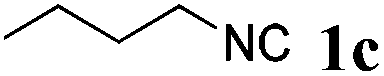
|
MeOH 3a |
|
82 |
| 8 |
|
MeOH 3a |
|
75 |
| 9c |
|
MeOH 3a |
|
72 |
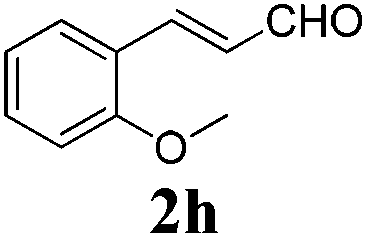
To further investigate the substrate scope, a series of substituted cinnamaldehydes 2b–2l were investigated as shown in Table 3. The results indicated that the electronic effect of cinnamaldehyde 2 has a serious influence on this reaction. Cinnamaldehydes with electron donating groups provided the formation of α-alkoxy-β,γ-enamides in good yields (entries 1, 5–8). Under the same optimized conditions, substrates with weak electron withdrawing groups (F, Cl, Br) also performed nicely to give the corresponding products in moderate yields (entries 2–4). The reactions of cinnamaldehydes with strong electron withdrawing groups (NO2, CN) were sluggish in this catalyst system. After further optimization, the desired products 4aja and 4aka were obtained in moderate yields when 50 mol% AlCl3 was added in two portions (entries 9 and 10). In addition, 2-hydroxy-cinnamaldehyde with no protection at the hydroxy group was also suitable for the reaction, indicating good substrate tolerance for this synthetic methodology (entry 11). The influence of the steric effect was also investigated. We found that ortho-substitutes in the phenyl ring showed no significant effects on the yields of products due to the distance from the functional group of aldehyde. Therefore, it was not surprising that 2-methylcinnamaldehyde 2g and 2-methoxycinnamaldehyde 2h with electron-donating groups but with steric hindrance could still give the desired products 4aga and 4aha in 80% and 83% yield separately (entries 6 and 7).
|
|
||||
|---|---|---|---|---|
| Entry | Cinnamaldehyde | Time (h) | Products | Yieldc (%) |
| a To a solution of AlCl3 (0.2 mmol, 0.20 eq.) in indicated alcohols (1 mL) in a sealed vial were added cinnamaldehyde 2a (1 mmol) and isocyanide 1 (1.5 mmol, 1.5 eq.) in sequence. The reaction mixtures were stirred at 60 °C for the indicated time. b 50 mol% AlCl3 was added in two portions. c Isolated yield. | ||||
| 1 |
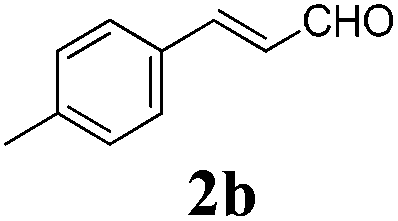
|
12 |
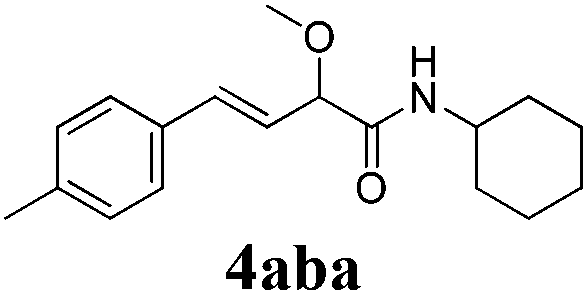
|
85 |
| 2 |
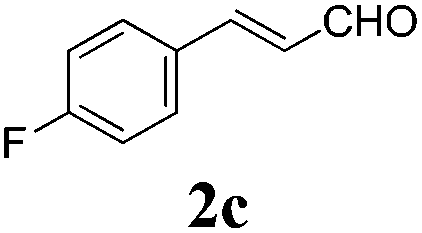
|
24 |
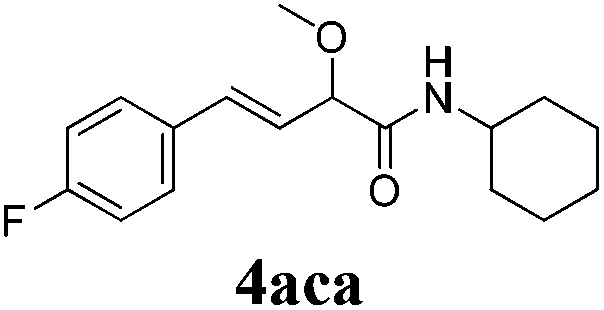
|
64 |
| 3 |
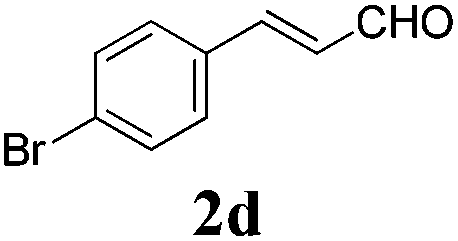
|
24 |
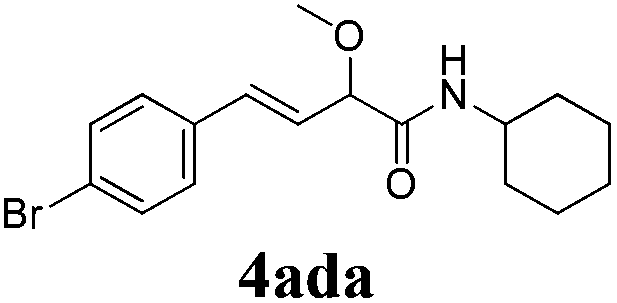
|
76 |
| 4 |
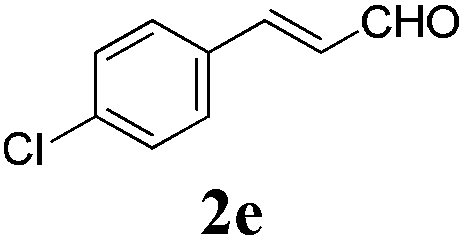
|
24 |
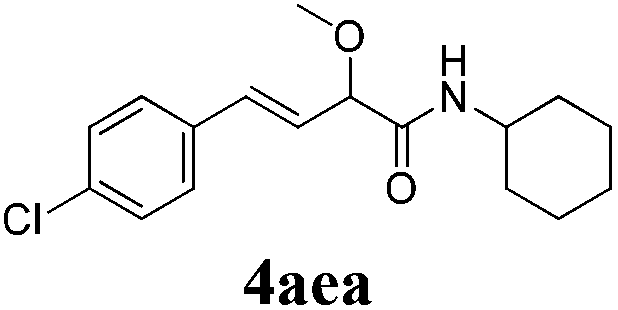
|
73 |
| 5 |
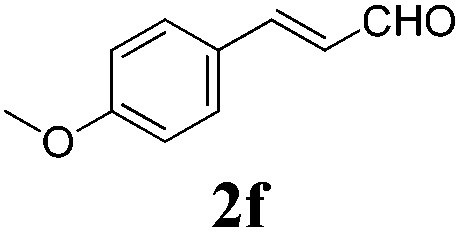
|
12 |
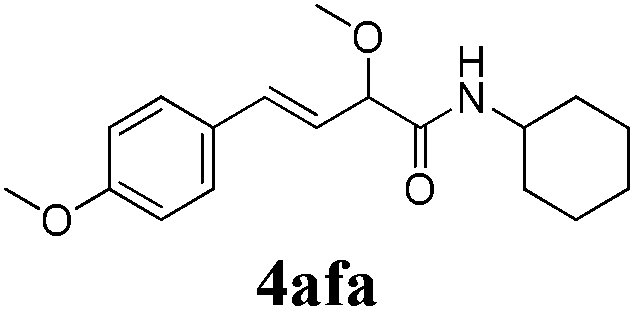
|
91 |
| 6 |
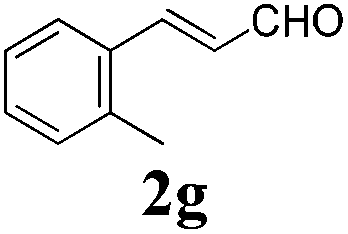
|
18 |
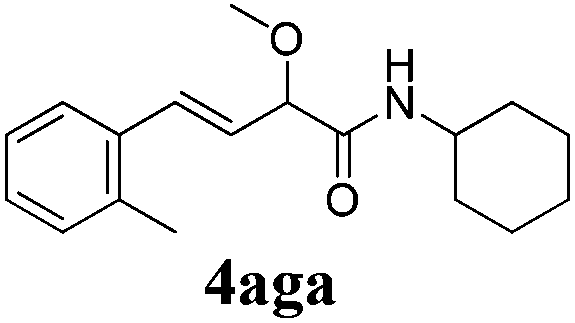
|
80 |
| 7 |
|
24 |
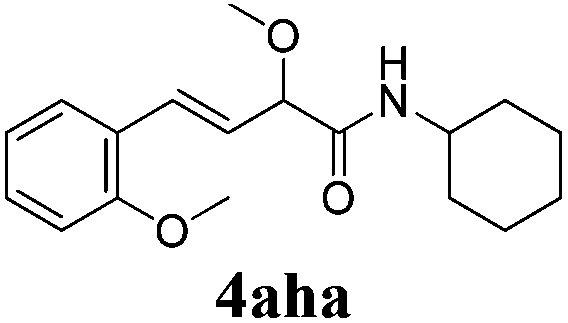
|
83 |
| 8 |
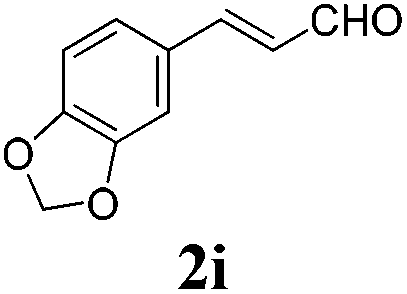
|
24 |
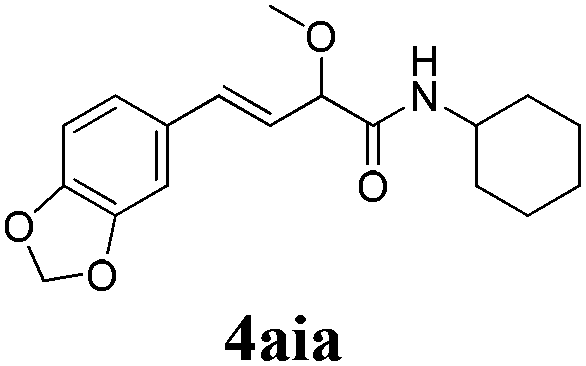
|
75 |
| 9b |
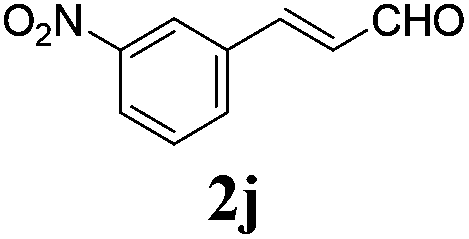
|
48 |
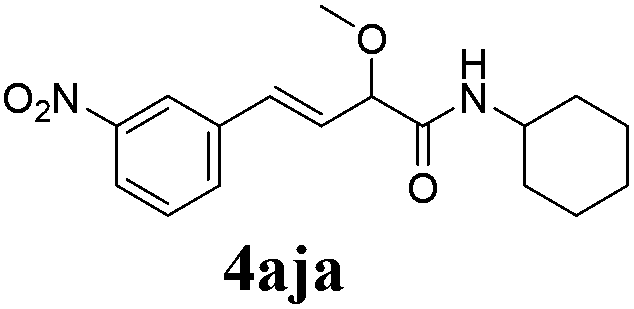
|
58 |
| 10b |
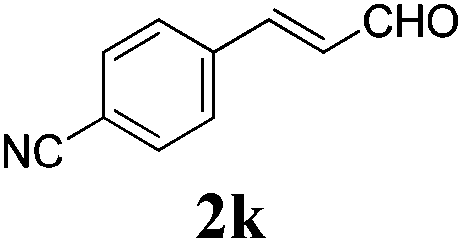
|
48 |
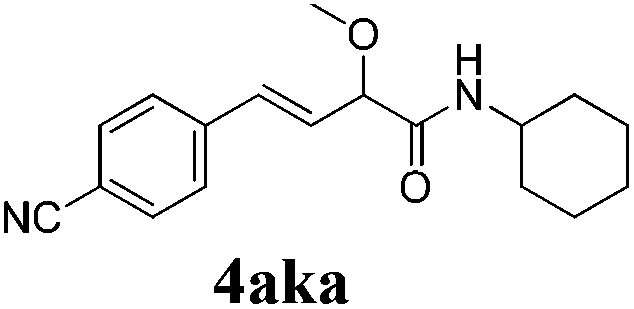
|
56 |
| 11 |
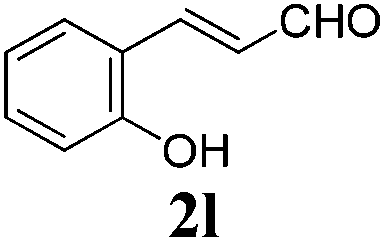
|
24 |
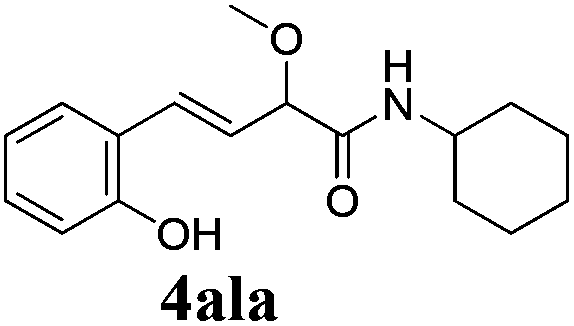
|
70 |
On the basis of the result, a mechanism for the Lewis acid-catalyzed formation of α-alkoxy-β,γ-enamide derivatives via the direct O-alkylative Passerini reaction is shown in Scheme 2. In the presence of the Lewis acid AlCl3, cinnamaldehydes react with alcohols to generate oxocarbenium species A, and lost a H2O molecule. Subsequently, A is attacked by an isocyanide to give nitrilium intermediate B. Then, the hydrolysis of the intermediate B results in the formation of C which easily isomerizes to give α-alkoxy-β,γ-enamide 2.
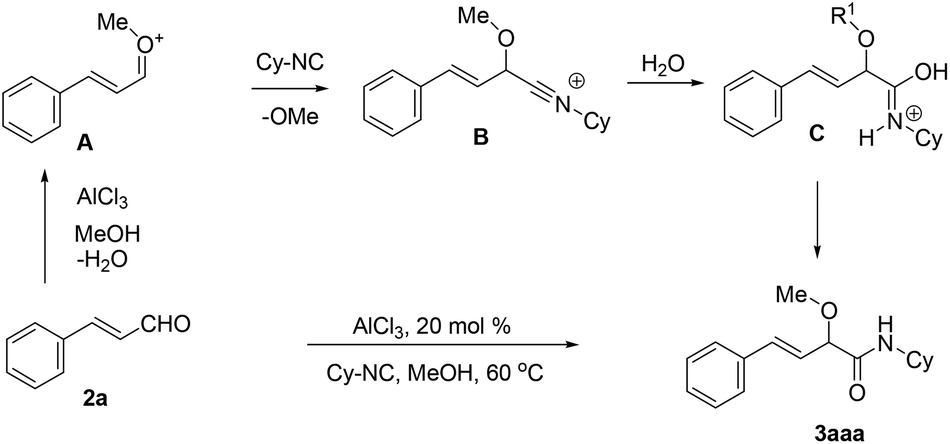 | ||
| Scheme 2 Proposed mechanism of the AlCl3-catalyzed O-alkylative Passerini reaction for accessing α-alkoxy-β,γ-enamides. | ||
Conclusions
In summary, we developed an efficient and practical O-alkylative Passerini reaction of isocyanides, cinnamaldehydes and alcohols catalyzed by the inexpensive Lewis acid AlCl3. This method offers a direct synthesis of α-alkoxy-β,γ-enamide derivatives in one step. A large variety of isocyanides, cinnamaldehydes and alcohols were suitable substrates in this catalyst system to afford the α-alkoxy-β,γ-enamide derivatives up to 91% yields. Further investigations on the asymmetric synthesis methodology of α-alkoxy-β,γ-enamide derivatives and evaluation of their biological activities are ongoing in our laboratory.Acknowledgements
We gratefully thank the start-up fund from the South University of Science and Technology of China (SUSTC) for financial support.Notes and references
- (a) M. Passerini and L. Simone, Gazz. Chim. Ital., 1921, 51, 126 CAS; (b) M. Passerini and G. Ragni, Gazz. Chim. Ital., 1931, 61, 964 CAS.
- (a) A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef; (b) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (c) L. Banfi and R. Riva, Org. React., 2005, 65, 1 CAS; (d) C. de Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969 RSC; (e) S. S. van Berkel, B. G. M. Boegels, M. A. Wijdeven, B. Westermann and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2012, 3543 CrossRef CAS PubMed; (f) A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef CAS PubMed.
- Selected examples of recent Passerini-type reactions in the preparation of polymers, see: (a) O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790 CrossRef CAS PubMed; (b) X. X. Deng, Y. Cui, F. S. Du and Z. C. Li, Polym. Chem., 2014, 5, 3316 RSC; (c) O. Kreye, D. Kugele, L. Faust and M. A. R. Meier, Macromol. Rapid Commun., 2014, 35, 317 CrossRef CAS PubMed; (d) W. Lin, T. Sun, M. Zheng, Z. Xie, Y. Huang and X. Jing, RSC Adv., 2014, 4, 25114 RSC; (e) S. C. Solleder and M. A. R. Meier, Angew. Chem., Int. Ed., 2014, 53, 711 CrossRef CAS PubMed; (f) L. Li, A. Lv, X.-X. Deng, F.-S. Du and Z.-C. Li, Chem. Commun., 2013, 49, 8549 RSC.
- Selected examples of recent Passerini-type reactions in the preparation of peptides, see: (a) M. Paravidino, R. Scheffelaar, R. F. Schmitz, F. J. J. de Kanter, M. B. Groen, E. Ruijter and R. V. A. Orru, J. Org. Chem., 2007, 72, 10239 CrossRef CAS PubMed; (b) W. Szymanski, M. Zwolinska, S. Klossowski, I. Mlynarczuk-Bialy, L. Bialy, T. Issat, J. Malejczyk and R. Ostaszewski, Bioorg. Med. Chem., 2014, 22, 1773 CrossRef CAS PubMed; (c) S. Shaaban, R. Diestel, B. Hinkelmann, Y. Muthukumar, R. P. Verma, F. Sasse and C. Jacob, Eur. J. Med. Chem., 2012, 58, 192 CrossRef CAS PubMed; (d) S. Faure, T. Hjelmgaard, S. P. Roche and D. J. Aitken, Org. Lett., 2009, 11, 1167 CrossRef CAS PubMed.
- Selected examples of recent enantioselective Passerini-type reactions, see: (a) H. Mihara, Y. Xu, N. E. Shepherd, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 8384 CrossRef CAS PubMed; (b) T. Yue, M.-X. Wang, D.-X. Wang and J.-P. Zhu, Angew. Chem., Int. Ed., 2008, 47, 9454 CrossRef CAS PubMed; (c) S.-X. Wang, M.-X. Wang, D.-X. Wang and J.-P. Zhu, Angew. Chem., Int. Ed., 2008, 47, 388 CrossRef CAS PubMed; (d) S. E. Denmark and Y. Fan, J. Org. Chem., 2005, 70, 9667 CrossRef CAS PubMed; (e) P. R. Andreana, C. C. Liu and S. L. Schreiber, Org. Lett., 2004, 6, 4231 CrossRef CAS PubMed; (f) U. Kusebauch, B. Beck, K. Messer, E. Herdtweck and A. Dömling, Org. Lett., 2003, 5, 4021 CrossRef CAS PubMed; (g) S. E. Denmark and Y. Fan, J. Am. Chem. Soc., 2003, 125, 7825 CrossRef CAS PubMed; (h) A. M. Deobald, A. G. Corrêa, D. G. Rivera and M. W. Paixão, Org. Biomol. Chem., 2012, 10, 7681 RSC; (i) T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J.-P. Zhu, J. Org. Chem., 2009, 74, 8396 CrossRef CAS PubMed.
- (a) L. E. Kaim, M. Gizolme and L. Grimaud, Org. Lett., 2006, 8, 5021 CrossRef PubMed; (b) L. E. Kaim, M. Gizolme, L. Grimaud and J. Oble, J. Org. Chem., 2007, 72, 4169 CrossRef PubMed.
- T. Soeta, Y. Kojima, Y. Ukaji and K. Inomata, Org. Lett., 2010, 12, 434 CrossRef PubMed.
- H. Yanai, T. Oguchi and T. Taguchi, J. Org. Chem., 2009, 74, 3927 CrossRef CAS PubMed.
- (a) H. Azuma, R. Takao, H. Niiro, K. Shikata, S. Tamagaki, T. Tachibana and K. Ogino, J. Org. Chem., 2003, 68, 2790 CrossRef CAS PubMed; (b) C. Steuer, C. Gege, W. Fischl, K. H. Heinonen, V. Bartenschlager and C. D. Klein, Bioorg. Med. Chem., 2011, 19, 4067 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00106d |
| This journal is © the Partner Organisations 2015 |