
Chiral dirhodium catalysts derived from L-serine, L-threonine and L-cysteine: design, synthesis and application†
Jian
Kang
a,
Baofu
Zhu
a,
Jiewei
Liu
a,
Bo
Wang
a,
Li
Zhang
*a and
Cheng-Yong
Su
 *ab
*ab
aMOE Laboratory of Bioinorganic and Synthetic Chemistry, Lehn Institute of Functional Materials, School of Chemistry and Chemical Engineering, Sun yat-sen University, Guangzhou, 510275, P. R. China. E-mail: zhli99@mail.sysu.edu.cn; cesscy@mail.sysu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 6th May 2015
Abstract
A series of dirhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate), dirhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate) and dirhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide) complexes with different para-substituted arylsulfonyl groups (e.g. –NO2, –F, –CF3, –Me, –tBu, –OMe and –nC12H25) derived from L-serine, L-threonine and L-cysteine, respectively, were prepared with yields in the range of 40–87% through refluxing ligands in water with Na4Rh2(CO3)4. These chiral Rh(II) complexes have been fully characterized by EA, IR, UV-vis, NMR and specific rotation measurements. They are found to be effective chiral catalysts for asymmetric aziridination and cyclopropanation reactions in terms of reactivity and enantioselectivity. They are extremely stable and can be stored for a long period (at least 18 months) on the bench without adversely affecting their reactivity and selectivity. The heterocyclic rings as well as the substituents on the arylsulfonyl groups have critical effects on the degree of asymmetric induction. In general, a higher enantioselectivity was observed in the reactions catalyzed by the oxazolidine-4-carboxylate-derived catalysts than the thiazolidine-4-carboxylate 1,1-dioxide-based catalysts. Among these 21 new Rh(II) catalysts, the uses of dirhodium tetrakis((4S)-3-((4-dodecylphenyl)sulfonyl)oxazolidine-4-carboxylate) (Rh2(4S-DOSO)4) and dirhodium tetrakis((4S,5R)-5-methyl-3-((4-nitrophenyl)sulfonyl)oxazolidine-4-carboxylate) (Rh2(4S,5R-MNOSO)4) resulted in the highest levels of enantioselectivity in aziridination (94% ee) and cyclopropanation (98% ee) of styrene, respectively. The successful design and syntheses of these novel Rh(II) complexes enlarged the scope of accessible chiral dirhodium(II) catalysts.
Introduction
Chiral dirhodium(II) paddlewheel complexes are exceptional catalysts for a wide range of transformations.1–5 They can react with iminoiodane and diazo compounds efficiently to form the important Rh(II) nitrenoid and carbenoid intermediates, respectively, which then undergo a number of highly selective reactions, including C–H bond functionalization and cycloaddition such as aziridination and cyclopropanation. Dirhodium carboxylates, dirhodium carboxamidates, dirhodium phosphates and ortho-metalated dirhodium(II) complexes are among the most efficient and widely used catalysts for metal–nitrene and metal–carbene transformations. Dirhodium carboxylates are very easy to handle, being stable towards air, heat, and moisture. Among the chiral dirhodium carboxylates that had been used in nitrenoid/carbenoid reactions, four catalyst systems stood out, including dirhodium prolinates, N-phthaloylaminocarboxylates, N-naphthaloylaminocarboxylates and cyclopropanecarboxylates (Fig. 1).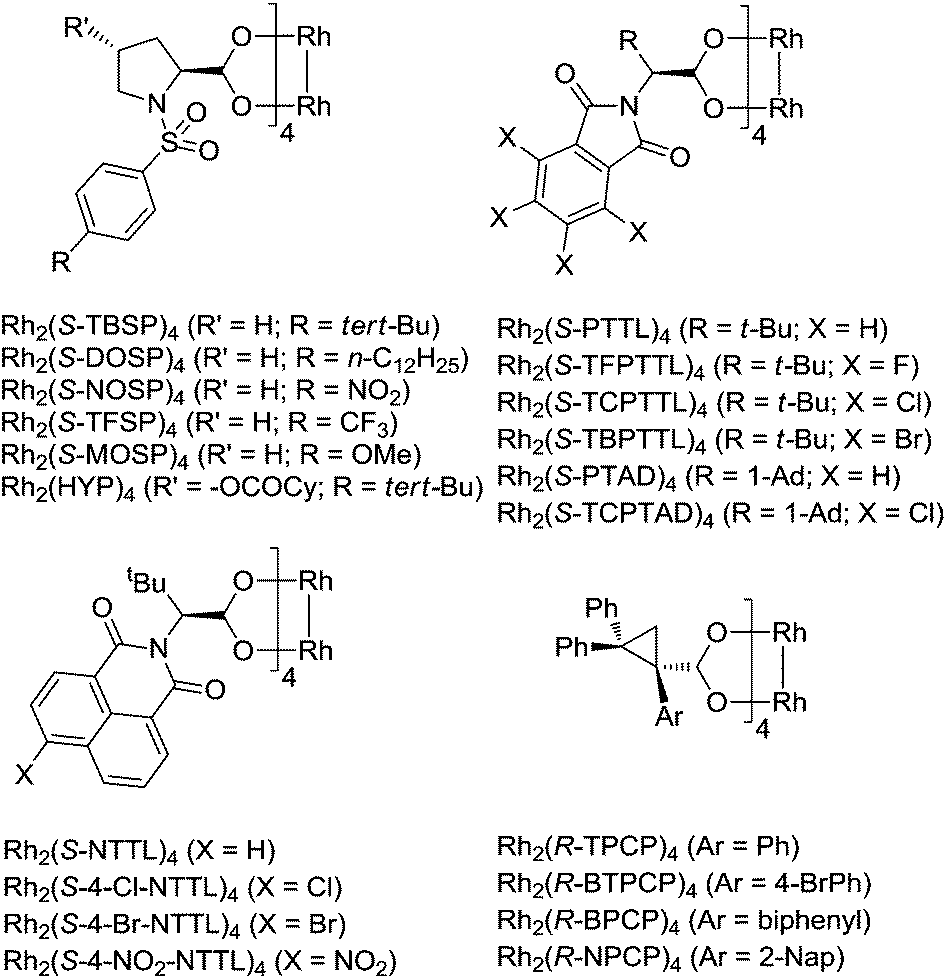 | ||
| Fig. 1 Structures of chiral dirhodium carboxylates. | ||
The synthesis and catalytic applications of dirhodium N-sulfonylprolinates were firstly studied by the McKervey group, and then developed by the Davies group. In the early 1990s McKervy developed different N-sulfonyl functionalized Rh(II) prolinates, and they found that the tert-butyl substituted complex Rh2(S-TBSP)4 showed a remarkably improved diastereoselectivity and enantioselectivity in the cyclopropanation reactions with methyl phenyldiazoacetate.6 The Davies group developed the n-dodecyl substituted complex Rh2(S-DOSP)4, which can be dissolved in pentane even at −78 °C and displayed especially high asymmetric induction in the reactions of a variety of donor/acceptor carbenoids, including cycloaddition such as cyclopropanation, cyclopropenation, [3 + 2] annulation, tandem cyclopropanation/Cope rearrangement, and three component coupling,7 and X–H insertion such as carbenoid C–H insertions, combined C–H activation/Cope rearrangement, and tandem ylide formation/[2,3]-sigmatropic rearrangement.8 The Hansen group developed chiral Rh(II) catalysts with 4-hydroxyproline-derived ligands.9 The hydroxyl groups were O-acylated with acid chlorides such as lauroyl chloride and cyclohexylcarbonyl chloride. The formed Rh(II) catalysts efficiently promoted cyclopropanation and C–H insertion with high enantioselectivities (up to 93% ee).
The phthalimide derived Rh(II) complexes were developed by Hashimoto and co-workers.10 The optimum catalyst can vary depending on the specific reaction, but usually the tert-leucine derived catalyst Rh2(S-PTTL)4,10a and its halogen-substituted complexes Rh2(S-TFPTTL)4 (X = F),10b,c Rh2(S-TCPTTL)4 (X = Cl)10d–f and Rh2(S-TBPTTL)4 (X = Br)10g,h gave the highest asymmetric induction. Later, the Davies group developed the adamantylglycine derived complexes Rh2(S-PTAD)4 and Rh2(S-TCPTAD)4, which complemented the tert-leucine derived catalysts in nitrenoid and carbenoid reactions.11 Impressively, Rh2(S-TCPTAD)4 (X = Cl) is an exceptional catalyst for enantioselective cyclopropanation of electron-deficient alkenes11e and C–H amination.11b
The Müller group designed and synthesized N-naphthaloyl-tethered chiral dirhodium tetracarboxylates, among which Rh2(S-NTTL)4 and bromo-substituted Rh2(S-4-Br-NTTL)4 are the most efficient in terms of reactivities and selectivities.12a–d In conjunction with chiral sulfonimidamide-derived iminoiodanes, Rh2(S-NTTL)4 has been successfully employed in efficient diastereoselective intermolecular C–H amination.12e,f Compared to the former two chiral templates, rhodium N-naphthaloylaminocarboxylates have been less employed in asymmetric catalytic carbene transfer reactions.13 The development of azavinyl carbenes highlighted its importance in organic synthesis. Rh2(S-NTTL)4 outperformed Rh2(S-DOSP)4 and Rh2(S-PTTL)4 in cyclopropanation and C–H bond activation when 1,2,3-triazoles were used as the azavinyl carbene sources.14,15
The Davies group for the first time prepared dirhodium triarylcyclopropanecarboxylate complexes.16 Similar to prolinate catalysts such as Rh2(DOSP)4, these catalysts showed high enantioselectivity in a series of donor/acceptor carbene transfer reactions. The advantages of Rh(II) cyclopropanecarboxylates include their ease of synthesis and compatibility in dichloromethane as a solvent, and most importantly, they can promote highly selective C–H functionalization of primary C–H bonds.16c,d
Our interest is in enlarging the scope of accessible chiral dirhodium catalysts, considering the high need of new dirhodium(II) catalysts with different electronic and steric environments for the powerful C–C and C–N bond-forming processes in synthetic organic chemistry.17–23 Herein, we report the design and synthesis of a series of dirhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate), dirhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate) and dirhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide) complexes with different para-substituted arylsulfonyl groups (e.g. −NO2, −F, −CF3, −Me, −tBu, −OMe and −nC12H25) derived from L-serine, L-threonine and L-cysteine, respectively (Fig. 2). Their catalytic activities have been tested in nitrene and carbene transfer reactions.
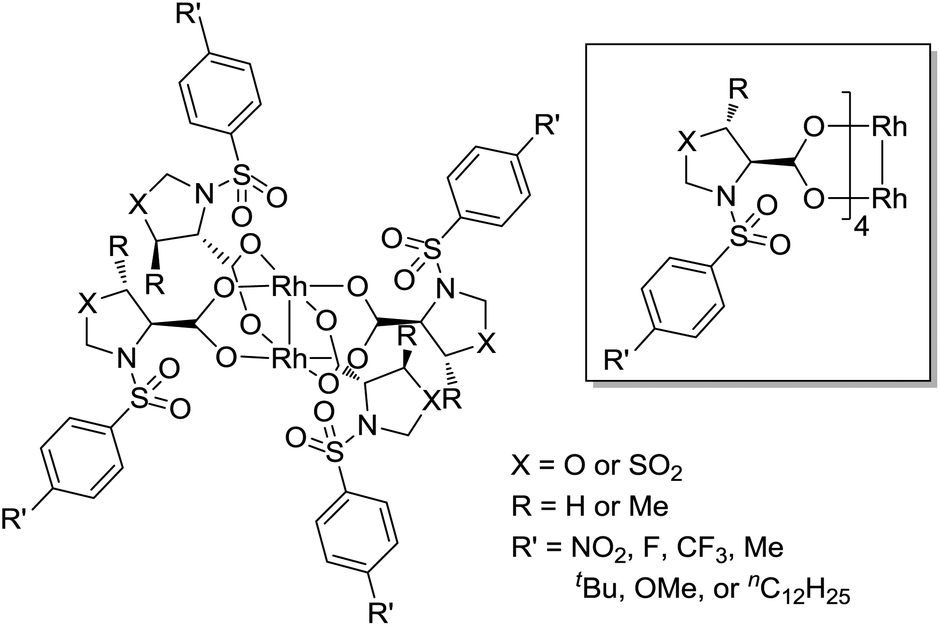 | ||
| Fig. 2 Dirhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate) (X = O, R = H), dirhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate) (X = O, R = Me) and dirhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide) (X = SO2, R = H). | ||
Results and discussion
Ligand design
Rhodium(II) prolinates have been shown to be effective in carbene transfer reactions with high stereoselectivity and excellent enantioselectivity.6–9 The catalytic results depended on the N-sulfonyl functionalities of the rhodium(II) prolinate complexes. Neither enlargement nor shrinkage in the ring size of the proline ring has necessarily affected the outcomes, and high levels of asymmetric induction were observed for both the N-4-tert-butylphenylsulfonyl azetidinecarboxylate complex and the pipecolinate complex.7a,24 Further modifications with the addition of the second heteroatom such as O, N, and S into the proline ring, that is 1,3-oxazolidine-4-carboxylic acid, 1,3-imidazolidine-4-carboxylic acid and 1,3-thiazolidine-4-carboxylic acid and their derivatives, however, have been seldom studied.25 Nevertheless, chiral dirhodium oxazolidinones and imidazolidinones have been prepared and applied in nitrene and carbene transfer reactions, which offer advantages in the diastereo/enantio-control than Rh(II) pyrrolidones.26–28Our catalysts are based on oxazolidine-4-carboxylate and thiazolidine-4-carboxylate, which are derived from naturally available amino acids. Their structures are related to prolinates. The major advantages of our catalysts over Rh(II) prolinates lie in much more modifications that can be made on them, as shown in Fig. 3. Synergistic effects of different hetero atoms of X (electronic effects), substituents of R on the methylene carbon (steric effects), and different R′ groups attached to the nitrogen (electronic effects and solubilities) could enrich the catalysis chemistry of the corresponding Rh(II) complexes. Appropriate combinations might endow the ligands with unique properties.
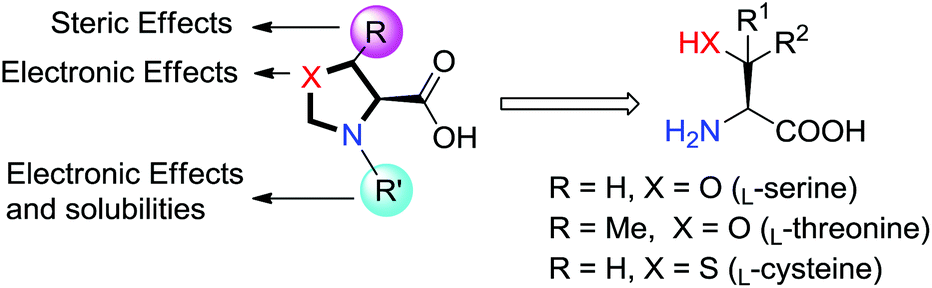 | ||
| Fig. 3 Ligand design. | ||
Syntheses and characterization of ligands
Ligands (4S)-3-(arylsulfonyl)oxazolidine-4-carboxylic acids were synthesized from the reactions of (S)-oxazolidine-4-carboxylic acid and arylsulfonyl chlorides, whereas the former was prepared from the natural amino acid L-serine (Fig. 4).29L-Serine and formaldehyde, in aqueous medium, are in equilibrium with 1,3-oxazolidine-4-carboxylate: at a pH value greater than 7 the oxazolidine adduct can be trapped under Schotten–Baumann conditions leading to the corresponding N-protected derivatives, which are then converted to 4-oxaproline compounds via the removal of the protecting group under acidic conditions. (4S,5R)-5-Methyl-3-(arylsulfonyl)oxazolidine-4-carboxylic acids and (4R)-3-(arylsulfonyl)thiazolidine-4-carboxylic acids were prepared in a similar way, starting from the natural amino acid L-threonine and L-cysteine, respectively. Ligands (R)-3-(arylsulfonyl)thiazolidine-4-carboxylic acid 1,1-dioxides were then obtained from the oxidation of (R)-3-(arylsulfonyl)thiazolidine-4-carboxylic acids with urea-hydrogen peroxide (UHP).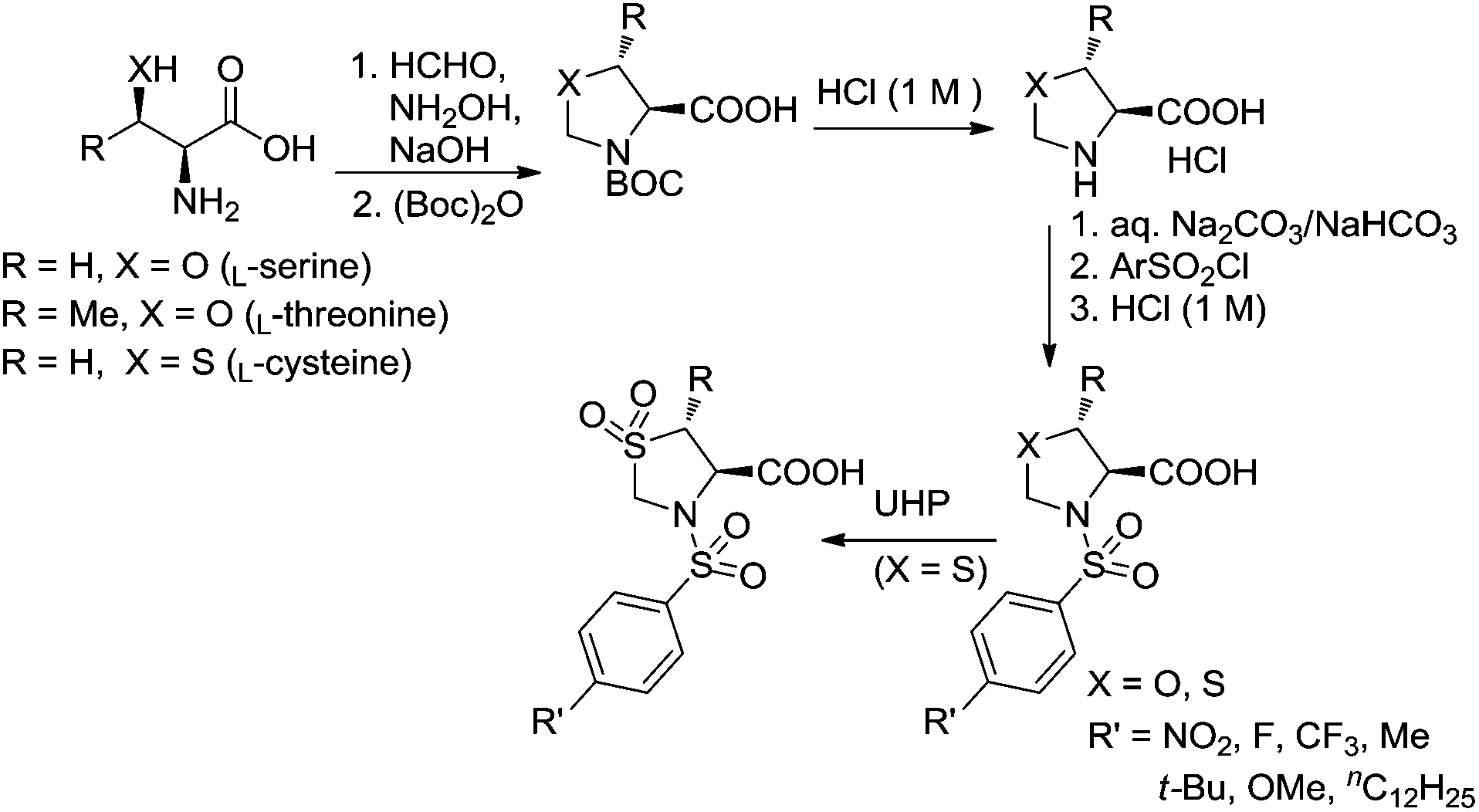 | ||
| Fig. 4 Syntheses of ligands. | ||
These ligands have been characterized by FTIR, NMR, HRMS-ESI, and optical rotation measurements. The IR spectra of these carboxylic acids displayed the characteristic asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching adsorption in the range of 1713–1770 cm−1. The 1H NMR (DMSO-d6) spectra clearly exhibited the characteristic peaks of the protons in the oxazolidine or thiazolidine 1,1-dioxide heterocycles. Due to the larger electronegativity of the atom O than S, the protons on the carbon atoms next to these heteroatoms appeared at the lower field in the oxazolidine ring than the thiazolidine 1,1-dioxide ring. For example, (4S)-3-((4-(trifluoromethyl)phenyl)sulfonyl)oxazolidine-4-carboxylic acid (4S-TFSO) exhibited two doublet peaks at 5.17 and 4.57 ppm for the two protons attached to the carbon at the 2-position of the oxazolidine ring, and two doublet–doublet peaks (one of the peaks appeared as a pseudo triplet) at 3.93 and 3.73 ppm for the two protons attached to the carbon at the 5-position. In comparison, (4R)-3-((4-(trifluoromethyl)phenyl)sulfonyl) thiazolidine-4-carboxylic acid 1,1-dioxide (4R-TFST) displayed two doublet peaks at 4.96 and 4.30 ppm for the protons attached to the carbon at the 2-position of the thiazolidine ring, and two doublet–doublet peaks at 3.68 and 3.56 ppm for the two protons attached to the carbon at the 5-position. In a similar way, as suggested by 13C NMR spectra and DEPT 13C NMR spectra, the two carbon nuclei next to the heteroatom O in the oxazolidine ring appeared with higher chemical shifts than the two carbon nuclei next to the atom S in the thiazolidine 1,1-dioxide ring. For example, as shown in 13C NMR (DMSO-d6) spectra, the carbon nuclei at the 2- and 5-position of the oxazolidine ring in 4S-TFSO appeared at 80.87 and 69.09 ppm, respectively, whereas the related carbon nuclei in 4S-TFST appeared at 62.40 and 51.33 ppm respectively.
O stretching adsorption in the range of 1713–1770 cm−1. The 1H NMR (DMSO-d6) spectra clearly exhibited the characteristic peaks of the protons in the oxazolidine or thiazolidine 1,1-dioxide heterocycles. Due to the larger electronegativity of the atom O than S, the protons on the carbon atoms next to these heteroatoms appeared at the lower field in the oxazolidine ring than the thiazolidine 1,1-dioxide ring. For example, (4S)-3-((4-(trifluoromethyl)phenyl)sulfonyl)oxazolidine-4-carboxylic acid (4S-TFSO) exhibited two doublet peaks at 5.17 and 4.57 ppm for the two protons attached to the carbon at the 2-position of the oxazolidine ring, and two doublet–doublet peaks (one of the peaks appeared as a pseudo triplet) at 3.93 and 3.73 ppm for the two protons attached to the carbon at the 5-position. In comparison, (4R)-3-((4-(trifluoromethyl)phenyl)sulfonyl) thiazolidine-4-carboxylic acid 1,1-dioxide (4R-TFST) displayed two doublet peaks at 4.96 and 4.30 ppm for the protons attached to the carbon at the 2-position of the thiazolidine ring, and two doublet–doublet peaks at 3.68 and 3.56 ppm for the two protons attached to the carbon at the 5-position. In a similar way, as suggested by 13C NMR spectra and DEPT 13C NMR spectra, the two carbon nuclei next to the heteroatom O in the oxazolidine ring appeared with higher chemical shifts than the two carbon nuclei next to the atom S in the thiazolidine 1,1-dioxide ring. For example, as shown in 13C NMR (DMSO-d6) spectra, the carbon nuclei at the 2- and 5-position of the oxazolidine ring in 4S-TFSO appeared at 80.87 and 69.09 ppm, respectively, whereas the related carbon nuclei in 4S-TFST appeared at 62.40 and 51.33 ppm respectively.
The structures of (4S,5R)-5-methyl-3-((4-nitrophenyl)sulfonyl)-oxazolidine-4-carboxylic acid (4S,5R-MNOSO) and (4R)-3-((4-methoxyphenyl)sulfonyl)thiazolidine-4-carboxylic acid 1,1-dioxide (4R-MOST) have been confirmed by the X-ray diffraction study, revealing that the sulfonamide moiety and the carboxylic acid group (–COOH) are pointing to different directions (Fig. 5). In contrast, an analysis of the Cambridge Crystallographic Database (CCDB) indicated that the sulfonamide moiety and the carboxylic acid group (–COOH) (or ester group (–COOR)) in (S)-(arylsulfonyl)pyrrolidine-2-carboxylic acid (or their ester relatives) are pointing to the same direction.30 This indicated the importance of the heteroatom on the rings to their solid structures.
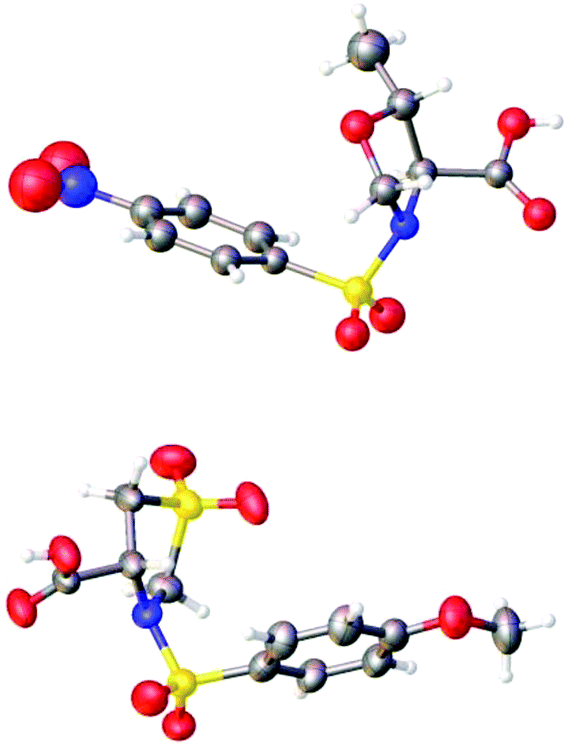 | ||
| Fig. 5 Crystal structures of 4S,5R-MNOSO (up) and 4R-MOST (down). | ||
Rhodium complex syntheses
The present chiral rhodium complexes (e.g. oxazolidine-4-carboxylate, 5-methyloxazolidine-4-carboxylate and thiazolidine-4-carboxylate 1,1-dioxide) were prepared in a similar procedure to the rhodium prolinate complexes by carbonate displacement using Na4Rh2(CO3)4 (Fig. 6).31a We ever tried high temperature ligand exchange with rhodium(II) acetate, which is another common synthesis method.31b However, the latter method has been discarded due to the much lower yields. In order to circumvent solubility problems in hydrocarbons, which seemed to be crucial for the high selectivities in the catalytic cyclopropanation reactions,7 4-n-dodecylphenylsulfonyl substituted Rh(II) complexes have been prepared. In addition, the strong electron-withdrawing (e.g. –NO2) and electron-donating (e.g. –OMe) substituted Rh(II) complexes have been prepared to explore the electronic effects of N-sulfonyl functionalities on the catalytic results. After isolation and purification, these complexes are extremely stable and can be stored for long periods (at least 18 months) of time on the bench without adversely affecting their reactivity and selectivity in catalytic reactions. An endeavor to prepare Rh(II) thiazolidine-4-carboxylate failed, due to the strong coordination ability of the S atoms in the thiazolidine ring to metal centers. As soon as the thiazolidine-4-carboxylic acid and Na4Rh2(CO3)4 were mixed in solution, the reaction solution turned purple, indicative of the coordination of S to Rh(II) atoms.32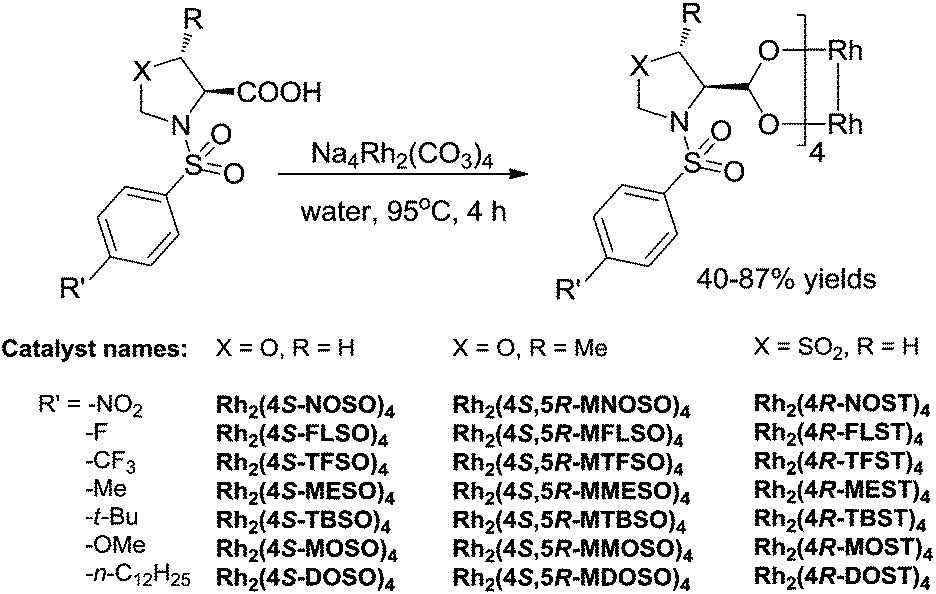 | ||
| Fig. 6 Rh(II) complex syntheses. | ||
The prepared Rh(II) complexes have been characterized by EA, FTIR, NMR and optical rotation measurements. The IR spectra of these Rh(II)-carboxylate complexes exhibited a strong asymmetric CO stretching adsorption at 1612–1625 cm−1 and a relatively less strong symmetric CO stretching adsorption at 1408–1420 cm−1. Coordination of the carboxylate groups with Rh(II) atoms weakens the CO bond, and thus resulting in absorption at a lower frequency than the free ligands (Rh2(4S,5R-MNOSO)4, 1613 cm−1vs. 4S,5R-MNOSO, 1713 cm−1).
The chemical shifts of the proton and carbon nuclei on the oxazoline rings of dirhodium complexes haven't displayed noticeable deviations from those of free ligands, whereas the carboxylate carbons of the Rh(II) complex shifted to the lower field with a large extent compared to those of the free ligands in the NMR spectra, indicative of the coordination of Rh(II)-carboxylate. For example, the carboxyl carbon in 4S,5R-MNOSO appeared at 170.40 ppm, whereas the carboxylate carbon in Rh2(4S,5R-MNOSO)4 appeared at 189.51 ppm in the 13C NMR (acetone-d6) spectra.
In order to explore the role of ligands on the electronic properties of these new dirhodium(II) complexes, the UV-vis spectra were examined (Fig. 7). The λmax for the two peaks in the visible region were ∼590 and ∼450 nm in acetone, which did not change greatly with different N-substituted oxazolidine-4-carboxylate or N-substituted thiazolidine-4-carboxylate 1,1-dioxide ligands, suggesting that these ligands did not significantly affect the electronic properties of the complex.33
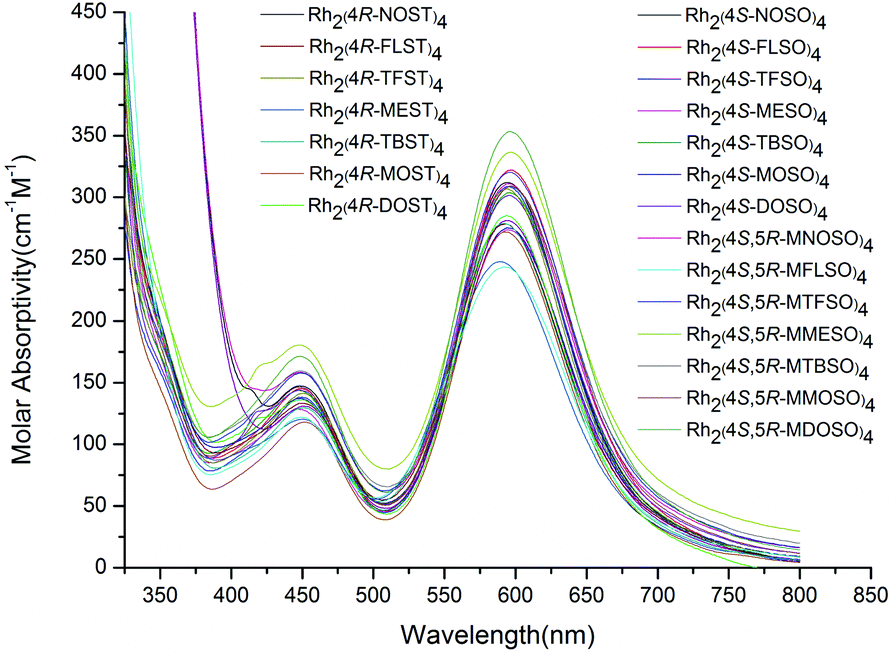 | ||
| Fig. 7 UV-vis spectra of Rh(II) complexes in acetone (R.T.). | ||
Catalytic nitrene transfer reactions
A limited number of chiral Rh(II) catalysts have been examined in aziridination reactions. A survey on the Rh(II)-catalyzed enantioselective aziridination of styrene disclosed that the rhodium(II) phosphate catalyst Rh2(R-bnp)4 gave the highest enantioselectivity (55% ee).34 These results were unsatisfactory, considering that copper complexes with chiral bis(oxazoline) or salen ligands promoted the aziridination of olefins with exceptional enantioselectivities (up to 98% ee).35 With our new catalysts in hand, we want to investigate their asymmetric induction in the aziridination reactions of olefins.The Rh(II)-catalyzed aziridination of olefins proceed via intermediate metal nitrenes (M = NR), which are generated upon decomposition of iminoiodanes (PhI = NR) directly or formed in situ from the amine and oxidant (e.g. PhI(OAc)2, PhIO) pair. In this work, the efficiency and enantioselectivity of the aziridination were tested with styrene, using NsNH2/PhI(OAc)2 as the nitrogen source (Table 1). The yields of the 1-((4-nitrophenyl)sulfonyl)-2-phenylaziridine (1) in the presence of all of the Rh(II) catalysts were rather high (up to 96%). The dodecyl substituted Rh(II) complexes (e.g. Rh2(4S-DOSO)4, Rh2(4S,5R-MDOSO)4 and Rh2(4R-DOST)4) gave high asymmetric inductions of 94%, 89% and 79% ee, respectively (entries 7, 14 and 21), which might be due to their good solubilities in organic solvents. In addition to these catalysts, another three 5-methyloxazolidine-4-carboxylate complexes (e.g. Rh2(4S,5R-MNOSO)4, Rh2(4S,5R-MFLSO)4 and Rh2(4S,5R-MTFSO)4) promote the aziridination reaction with high enantioselectivities (≥80% ee, entries 8–10). In general, Rh(II) oxazolidine-4-carboxylate catalysts displayed higher enantioselectivities than Rh(II) thiazolidine-4-carboxylate 1,1-dioxide catalysts. An additional methyl group at the 5-position endowed the 5-methyloxazolidine-4-carboxylate complexes with higher asymmetric inductions than the corresponding Rh(II) oxazolidine-4-carboxylate catalysts.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield, % | ee, % |
| a Reaction conditions: to 2 mL of DCM were added sequentially MgO (29 mg, 0.72 mmol, 2.4 eq.), PhI(OAc)2 (152 mg, 0.45 mmol, 1.5 eq.), styrene (47 mg, 0.45 mmol, 1.5 eq.), NsNH2 (61 mg, 0.30 mmol, 1.0 eq.) and 2 mol% catalyst. The suspension was stirred vigorously overnight at room temperature until complete consumption of most starting material was indicated by TLC. b PhCF3. c DCE. d CH2Br2. e Toluene. | |||
| 1 | Rh2(4S-NOSO)4 | 85 | 47 |
| 2 | Rh2(4S-FLSO)4 | 79 | 42 |
| 3 | Rh2(4S-TFSO)4 | 96 | 46 |
| 4 | Rh2(4S-MESO)4 | 68 | 35 |
| 5 | Rh2(4S-TBSO)4 | 90 | 41 |
| 6 | Rh2(4S-MOSO)4 | 66 | 50 |
| 7 | Rh2(4S-DOSO)4 | 86 | 94 |
| 8 | Rh2(4S,5R-MNOSO)4 | 92 | 88 |
| 9 | Rh2(4S,5R-MFLSO)4 | 81 | 80 |
| 10 | Rh2(4S,5R-MFSO)4 | 48 | 80 |
| 11 | Rh2(4S,5R-MMESO)4 | 93 | 49 |
| 12 | Rh2(4S,5R-MTBSO)4 | 55 | 55 |
| 13 | Rh2(4S,5R-MMOSO)4 | 55 | 56 |
| 14 | Rh2(4S,5R-MDOSO)4 | 92 | 89 |
| 15 | Rh2(4R-NOST)4 | 89 | 13 |
| 16 | Rh2(4R-FLST)4 | 93 | 16 |
| 17 | Rh2(4R-TFST)4 | 90 | 9 |
| 18 | Rh2(4R-MEST)4 | 90 | 14 |
| 19 | Rh2(4R-TBST)4 | 88 | 55 |
| 20 | Rh2(4R-MOST)4 | 91 | 13 |
| 21 | Rh2(4R-DOST)4 | 90 | 79 |
| 22 | Rh2(S-NOSP)4 | 69 | −46 |
| 23 | Rh2(S-TFSP)4 | 96 | 60 |
| 24 | Rh2(S-MOSP)4 | 95 | 81 |
| 25 | Rh2(S-DOSP)4 | 95 | 73 |
| 26 | Rh2(S-PTAD)4 | 90 | 25 |
| 27 | Rh2(S-NTTL)4 | 92 | 60 |
| 28b | Rh2(4S,5R-MNOSO)4 | 84 | 78 |
| 29c | Rh2(4S,5R-MNOSO)4 | 75 | 65 |
| 30d | Rh2(4S,5R-MNOSO)4 | 70 | 55 |
| 31e | Rh2(4S,5R-MNOSO)4 | 78 | 15 |
In both systems of rhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate) and rhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide), the electronic effects of the N-sulfonyl functionalities have negligible influence on the enantioselectivity results, as the nitro- and methoxy-substituted oxazolidine carboxylate complexes gave similar asymmetric induction (entries 1 vs. 6, and 15 vs. 20).
In contrast, the arylsulfonyl groups with different electronic effects have critical effects on the degree of asymmetric induction catalyzed by rhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate). The asymmetric induction was good in the reactions catalyzed by the –NO2, –F or –CF3 functionalized catalysts (80–88% ee, entries 8–10), but only modest in the presence of –Me, –tert-Bu or –OMe substituted Rh(II) complexes (49–56% ee, entries 11–13).
Sulfonated rhodium prolinates Rh2(S-NOSP)4, Rh2(S-TFSP)4, Rh2(S-MOSP)4 and Rh2(S-DOSP)4 have been prepared according to the reported procedures (Fig. 1),31 and their activities have been tested in the aziridination of styrene. Under our standard reaction conditions, they gave rise to −46, 60, 81 and 73% ee, respectively (entries 21–25). Next, we have also examined the catalytic activities of Rh2(S-PTAD)4 and Rh2(S-NTTL)4, which are popular catalysts in nitrene transfer reactions,11,12 and they led to the formation of the aziridine product in 25% and 60% ee, respectively (entries 26 and 27).
The effects of other solvents (e.g. PhCF3, DCE, CH2Br2, toluene) on the enantioselectivity were also tested, and the enantiomeric excess was in the range of 15–78% ee using Rh2(4S,5R-MNOSO)4 as the catalyst (entries 28–31). Among the solvents that have been tested for catalytic aziridination reactions, it seemed that toluene was the worst choice and the asymmetric induction was only 15% ee.
Comparison of the catalytic data in the presence of different chiral Rh(II) catalysts show that, our catalysts Rh2(4S-DOSO)4 and Rh2(4S,5R-MDOSO)4 are so far the most effective Rh(II) catalysts in the aziridination reaction of styrene in terms of yields and enantioselectivities. We have further tested the catalytic capabilities of our catalysts in intramolecular C–H bond amination of sulfamate esters such as 2,3-dihydro-1H-inden-2-yl sulfamate developed by Du Bois.19 The asymmetric inductions, however, were not satisfactory. The best result was only in modest asymmetric induction (40% ee) and achieved in Rh2(4S,5R-MNOSO)4-catalyzed amination reaction.
Catalytic cyclopropanation reactions
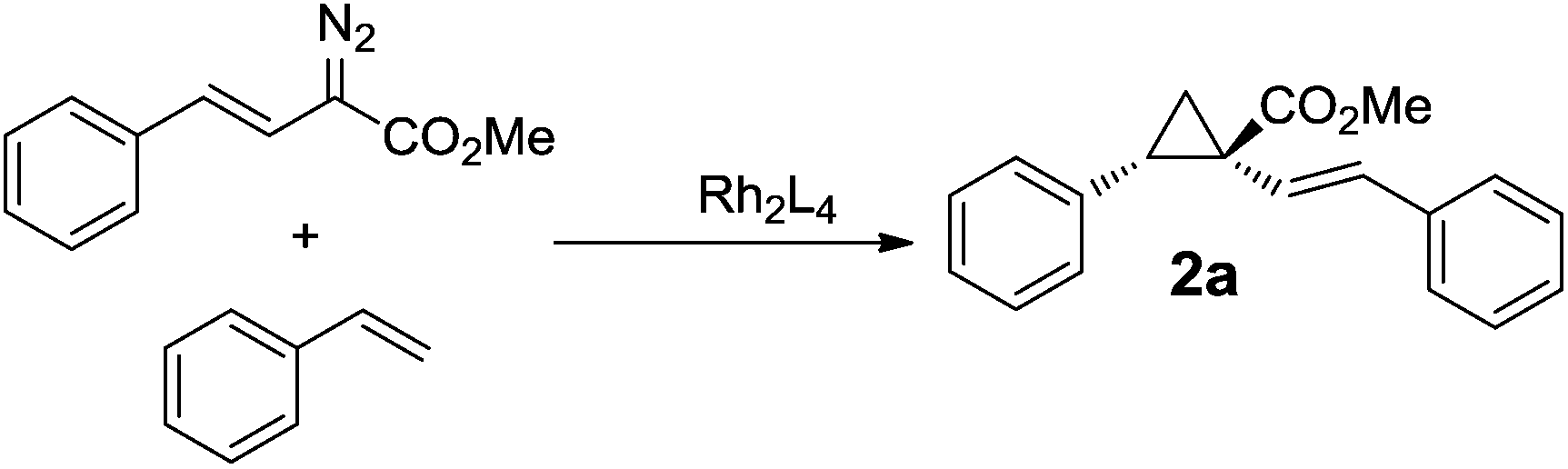
Cyclopropanation of alkenes by transition metal catalysts with diazo compounds represents a straightforward approach to cyclopropanes, which are important in diverse areas of organic chemistry and biology. In particular, the optically pure cyclopropanes are critical to many applications, for example, asymmetric syntheses of cyclopropane amino acids and sertraline.36 In the last two decades, the catalytic asymmetric cyclopropanation of alkenes with diazo carbonyl compounds in the presence of chiral rhodium catalysts offers significant potential as a general method for the synthesis of optically pure cyclopropanes.We have shown that our Rh(II) catalysts can efficiently promote aziridination reactions with moderate to high asymmetric inductions (up to 94% ee). Next, we explored the catalytic capabilities of our catalysts in cyclopropanation reactions. As the test reaction cyclopropanation with vinyldiazoacetates such as (E)-methyl 2-diazo-4-phenylbut-3-enoate was chosen. This protocol employing Rh2(DOSP)4 as the catalyst and pentane as the solvent at −78 °C has been successfully elaborated by Davies et al. and has transformed donor/acceptor substituted diazo compounds into cyclopropanes with different alkenes in high to excellent enantioselectivities (up to 98% ee).7a No attempt was made on our part to improve the Davies protocol; rather, it served as a well-established test reaction to investigate the effects of heterocycles and N-sulfonyl functionalities on the catalytic results of carbene transfer reactions.
The evaluation of our catalysts was initially carried out using the cyclopropanation between the (E)-methyl 2-diazo-4-phenylbut-3-enoate (1.0 eq.) and styrene (5.0 eq.) with 0.02 equiv. of the catalyst and 1.0 eq. of methyl benzoate as the additive and toluene as the solvent at −40 °C as the standard reaction.8m The catalytic results are shown in Table 2. GC-MS analyses of the reaction mixtures suggest that excellent E-/Z-diastereoselectivities (ranging from 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 to 96:4, entries 1–21) were obtained. All the reactions proceeded in high to excellent yields ranging from 80 to 96%, and in moderate to excellent asymmetric induction (up to 98% ee). The absolute stereochemistry of the major isomer in all cases was 1S,2S, which was determined by comparison of the HPLC spectra with that of an authentic sample.7a
:8 to 96:4, entries 1–21) were obtained. All the reactions proceeded in high to excellent yields ranging from 80 to 96%, and in moderate to excellent asymmetric induction (up to 98% ee). The absolute stereochemistry of the major isomer in all cases was 1S,2S, which was determined by comparison of the HPLC spectra with that of an authentic sample.7a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yield, % | E-/Z- | ee, % |
| a Reaction conditions: a solution of diazo compound (40 mg, 0.2 mmol, 1.0 eq.) in 1 mL toluene was added slowly to the solution of styrene (104 mg, 1.0 mmol, 5.0 eq.), methyl benzoate (27 mg, 0.2 mmol, 1.0 eq.) and 2 mol% catalyst in 1 mL toluene at −40 °C. The resulting solution was vigorously stirred at −40 °C for two days until most diazo compound was completely consumed. b −40 °C, no PhCO2Me. c −30 °C, no PhCO2Me. d −20 °C, no PhCO2Me. e 0 °C, no PhCO2Me. f Room temperature, no PhCO2Me. g With 0.1 mol% catalyst. h With 0.01 mol% catalyst. | ||||
| 1 | Rh2(4S-NOSO)4 | 80 | 96:4 |
63 |
| 2 | Rh2(4S-FLSO)4 | 92 | 95:5 |
81 |
| 3 | Rh2(4S-TFSO)4 | 90 | 95:5 |
49 |
| 4 | Rh2(4S-MESO)4 | 82 | 95:5 |
61 |
| 5 | Rh2(4S-TBSO)4 | 90 | 96:4 |
71 |
| 6 | Rh2(4S-MOSO)4 | 88 | 94:6 |
60 |
| 7 | Rh2(4S-DOSO)4 | 84 | 95:5 |
65 |
| 8 | Rh2(4S,5R-MNOSO)4 | 96 | 93:7 |
98 |
| 9 | Rh2(4S,5R-MFLSO)4 | 90 | 93:7 |
83 |
| 10 | Rh2(4S,5R-MFSO)4 | 85 | 92:8 |
94 |
| 11 | Rh2(4S,5R-MMESO)4 | 93 | 93:7 |
88 |
| 12 | Rh2(4S,5R-MTBSO)4 | 95 | 94:6 |
88 |
| 13 | Rh2(4S,5R-MMOSO)4 | 86 | 93:7 |
92 |
| 14 | Rh2(4S,5R-MDOSO)4 | 92 | 95:5 |
90 |
| 15 | Rh2(4R-NOST)4 | 91 | 93:7 |
40 |
| 16 | Rh2(4R-FLST)4 | 94 | 95:5 |
42 |
| 17 | Rh2(4R-TFST)4 | 90 | 94:6 |
55 |
| 18 | Rh2(4R-MEST)4 | 90 | 95:5 |
47 |
| 19 | Rh2(4R-TBST)4 | 93 | 93:7 |
67 |
| 20 | Rh2(4R-MOST)4 | 90 | 94:6 |
53 |
| 21 | Rh2(4R-DOST)4 | 93 | 96:4 |
64 |
| 22 | Rh2(S-NOSP)4 | 90 | 93:7 |
90 |
| 23b | Rh2(4S,5R-MNOSO)4 | 90 | 95:5 |
95 |
| 24c | Rh2(4S,5R-MNOSO)4 | 85 | 96:4 |
92 |
| 25d | Rh2(4S,5R-MNOSO)4 | 87 | 93:7 |
86 |
| 26e | Rh2(4S,5R-MNOSO)4 | 75 | 94:6 |
81 |
| 27f | Rh2(4S,5R-MNOSO)4 | 80 | 93:7 |
80 |
| 28g | Rh2(4S,5R-MNOSO)4 | 75 | 96:4 |
84 |
| 29h | Rh2(4S,5R-MNOSO)4 | 42 | 95:5 |
82 |
In general, a higher enantioselectivity was observed in the reactions catalyzed by the oxazolidine-4-carboxylate-derived catalysts (49–81% ee, entries 1–7) than the thiazolidine-4-carboxylate 1,1-dioxide-based catalysts (40–67% ee, entries 15–21). An additional methyl group in the oxazolidine ring has largely enhanced the asymmetric inductions of the corresponding catalysts (83–98% ee, entries 8–14). Among the 21 Rh(II) complexes, dirhodium tetrakis((4S,5R)-5-methyl-3-((4-nitrophenyl)sulfonyl)oxazolidine-4-carboxylate) (Rh2(4S,5R-MNOSO)4) displayed the highest efficiency in terms of yields and enantioselectivities (96% yield and 98% ee, entry 8). Electronic changes on the aryl ring had a minimal effect, considering that the electron-rich 4-methoxyphenyl derivative Rh2(4S,5R-MMOSO)4 also resulted in excellent asymmetric induction (92% ee, entry 13).
The 4-dodecylphenyl catalysts Rh2(4S-DOSO)4, Rh2(4S,5R-MDOSO)4 and Rh2(4R-DOST)4 were prepared due to the possibly good solubility induced by these functionalities. We have tested their solubilities, finding that they can be well dissolved in a hydrocarbon solvent (e.g. toluene and pentane). It is noted that the success of the 4-dodecylphenyl prolinate catalyst Rh2(DOSP)4 was ascribed to its super solubility in the hydrocarbon solvent.7 Our results suggested that under our reaction conditions (in toluene at −40 °C), the 4-dodecylphenyl catalyst Rh2(4S,5R-MDOSO)4 cannot compete with the nitro-functionalized catalyst Rh2(4S,5R-MNOSO)4. Their ligands have the same heterocyclic ring, 5-methyloxazolidine, but they displayed 90 and 98% ee, respectively (entries 14 and 8). The other two 4-dodecylphenyl catalysts Rh2(4S-DOSO)4 and Rh2(4R-DOST) cannot rival Rh2(4S,5R-MNOSO)4, either, and the asymmetric inductions were modest (entries 7 and 21). On the other hand, the enantioselectivities of other nitro-functionalized Rh(II) catalysts Rh2(4S-NOSO)4 and Rh2(4R-NOST)4, whose ligands were composed of oxazolidine and thiazolidine 1,1-dioxide, respectively, were lower than Rh2(4S,5R-MNOSO)4 (entries 1, 8 and 15). For comparison, we have also run the same cycloaddition reaction using the nitro-functionalized Rh(II) prolinate (Rh2(S-NOSP)4, Fig. 1) as the catalyst, resulting in 90% yield and 90% ee (entry 22). We ascribe the success of Rh2(4S,5R-MNOSO)4 to the synergistic effects of the electron-withdrawing O in the oxazolidine ring (electronic effect), the methyl substituent at the 5-position of the oxazolidine ring, which is next to the carboxylate group at the 4-position of the ring (steric effect), and the highly electron-withdrawing nitro substituent attached to the sulfonylphenyl group (electronic effect). Any one of these effects cannot alone elucidate the excellent enantioselectivity achieved in the Rh2(4S,5R-MNOSO)4-catalyzed cyclopropanation reaction.
After it was determined that Rh2(4S,5R-MNOSO)4 was an exceptional catalyst, we studied the effects of the additive methyl benzoate and the temperatures on the catalytic results, finding that the presence of methyl benzoate and low temperature have positive effects on the enantioselectivity of the cyclopropane product (entries 8 and 23–27).8m Decreasing the catalyst loading from 2 to 0.1 mol% lowered the yield and enantioselectivity (75% yield and 84% ee, entry 28). Further decreasing the catalyst loading from 0.1 to 0.01 mol% did not alter the enantioselectivity (82% ee), but the yield was much poorer (42%, entry 29).
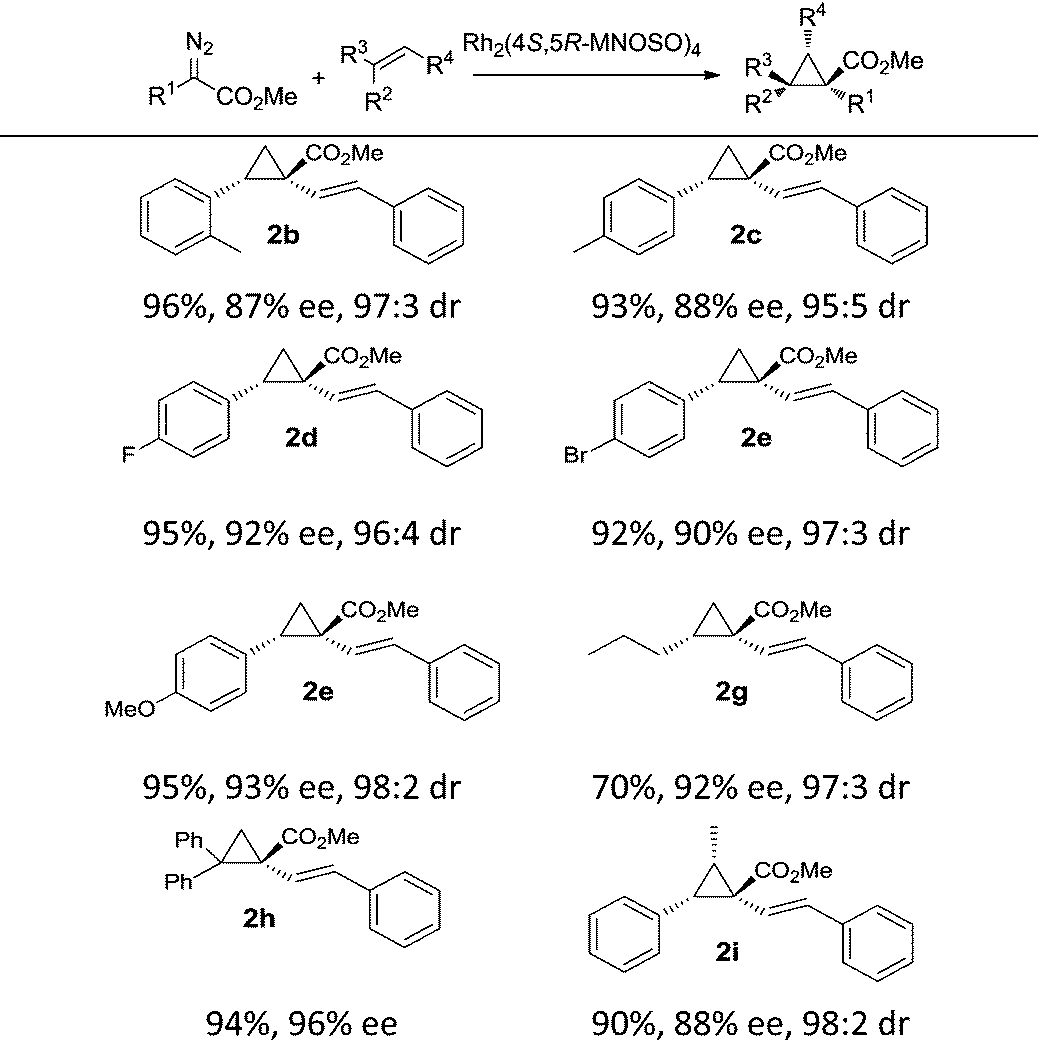
The catalytic capabilities of Rh2(4S,5R-MNOSO)4 in intermolecular cyclopropanation reactions were further evaluated with the use of alkenes other than styrene (Table 3). Exceptional stereocontrol (>95:5 E/Z) and enantiocontrol (88–93% ee) were achieved with substituted styrenes bearing either an electron-donating group (e.g. –Me and –OMe) or an electron-withdrawing group (e.g. –F and –Br) at the 4-position. The introduction of a 2-methyl group in 2-methyl styrene, and thus making the olefin substrate relatively crowded than styrene, does not largely reduce the enantioselectivity of the cyclopropane product 2b (87% ee). High asymmetric induction was also found in the cyclopropanation of an aliphatic alkene (n-hexene), and the corresponding cyclopropane product 2g was in 92% ee, although the yield is modest (70%). 1,1-Diphenylethene was also effective in Rh2(4S,5R-MNOSO)4-catalyzed cyclopropanation, leading to the formation of the cyclopropane 2h in 94% yield and 96% ee. (Z)-Methyl styrene gave the corresponding cyclopropane 2i in high enantioselectivity (88% ee). However, the reaction with (E)-methyl styrene failed.
| a Reaction conditions: a solution of diazo compound (40 mg, 0.2 mmol, 1.0 eq.) in 1 mL toluene was added slowly to the solution of olefin (1.0 mmol, 5.0 eq.), and 2 mol% Rh2(4S,5R-MNOSO)4 in 1 mL toluene at −40 °C. The resulting solution was vigorously stirred at −40 °C for two days until most diazo compound was completely consumed. |
|---|
|
Conclusions
In summary, we have designed and synthesized a series of chiral Rh(II) complexes, dirhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate), dirhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate) acids and dirhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide), in which the N-sulfonyl functionalities included –NO2, –F, –CF3, –Me, –tert-Bu, –OMe and –n-dodecyl, which have been fully characterized by EA, IR, UV, NMR and optical rotation measurements. The capabilities of these Rh(II) complexes have been tested in nitrene and carbene transfer reactions. In general, Rh(II) oxazolidinecarboxylate displayed higher enantioselectivities than Rh(II) thiazolidinecarboxylate 1,1-dioxide with the same arylsulfonyl groups. The n-dodecylphenyl substituted Rh(II) complexes (e.g. Rh2(4S-DOSO)4, Rh2(4S,5R-MDOSO)4 and Rh2(4R-DOST)4) gave high asymmetric inductions of 94%, 89% and 79% ee, respectively, in the aziridination reaction of styrene. Meanwhile, the nitro-functionalized catalyst Rh2(4S,5R-MNOSO)4 gave the best results in the cyclopropanation reactions in terms of yields and enantioselectivities. Exceptional stereocontrol (>93:7 E/Z) and enantiocontrol (up to 98% ee) were achieved with various olefins, which differ in steric and electronic factors. The preliminary results are encouraging. Studies are in progress to determine the full potential of these new dirhodium complexes in organic synthesis. In the carbene transfer reactions, an additional methyl group at the 5-position endowed the 5-methyloxazolidine-4-carboxylate complexes with higher asymmetric inductions than the corresponding Rh(II) oxazolidine-4-carboxylate catalysts. We are currently pursuing the modifications of the ligands (e.g. incorporating alkyl groups at the 5-position of the heterocycle ring) and syntheses of the second generation of Rh(II) thiazolidine-4-carboxylates, and the results will be published in due course.
Experimental section
General information
All the reagents in the present work were obtained from a commercial source and used directly without further purification. The elemental analyses were performed with a Perkin-Elmer 240 elemental analyzer. HRESI-MS was performed by using a Bruker Daltonics ESI-Q-TOF maXis4G. Infrared spectra on KBr pellets were collected with a Nicolet/Nexus-670 FT-IR spectrometer in the region of 4000–400 cm−1. UV-Vis spectra were tested on a Shimadzu/UV-3600 spectrophotometer. 1H, 13C{1H} and 19F{1H} NMR spectra were recorded on a Bruker AVANCE III 400. Chemical shifts are quoted in parts per million (ppm) referenced to the appropriate solvent peak or 0 ppm for TMS. HPLC spectra were obtained on a HPLC of Agilent 1200 series. Specific rotations were measured on ADP440 + B + S.Caution! Although we have not experienced any problem in the handling of diazo compounds, extreme care should be taken when manipulating them due to their explosive nature.
Syntheses of (4S)-3-(arylsulfonyl)oxazolidine-4-carboxylic acids
(4S)-3-(Arylsulfonyl)oxazolidine-4-carboxylic acids were prepared with a similar procedure,29 as exemplified by the synthesis of (4S)-3-((4-methoxyphenyl)sulfonyl)oxazolidine-4-carboxylic acid: (S)-oxazolidine-4-carboxylic acid hydrochloride (0.50 g, 3.25 mmol) was dissolved in a water (15 mL) solution of a mixture of Na2CO3 (0.82 g, 7.8 mmol) and NaHCO3 (0.32 g, 3.90 mmol) at room temperature to give a clear solution, which was then cooled down in an ice bath. An acetone (2 mL) solution containing p-methoxybenzenesulfonyl chloride (0.84 g, 4.1 mmol) was added slowly to the above solution with stirring. The whole reaction mixture was allowed to warm to room temperature. After stirring overnight, the reaction mixture was acidified with aqueous HCl (1 M) till the pH value reached 2–3, and then extracted with EtOAc (30 ml × 3). The combined organic layer was washed with H2O (30 ml × 2), brine (50 mL) and dried over Na2SO4, and filtered. The solvent was removed by using a rotary evaporator and the crude product was obtained as a white solid which was then purified by chromatography on silica gel.:PE:HOAc = 100:50:5). 1H NMR (300 MHz, DMSO-d6) δ 8.39 (d, J = 8.5 Hz, 2H), 8.18 (d, J = 8.5 Hz, 2H), 5.18 (d, J = 6.3 Hz, 1H), 4.60 (d, J = 6.3 Hz, 1H), 4.48 (dd, J = 7.9, 5.7 Hz, 1H), 3.94 (t, J = 8.7 Hz, 1H), 3.74 (dd, J = 8.7, 5.7 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 171.02, 150.85, 143.17, 129.86, 125.22, 81.48, 69.81, 59.38. FTIR (KBr, cm−1): 3114 w, 2894 w, 1715 s, 1609 w, 1532 s, 1496 w, 1352 s, 1310 s, 1167 s, 1101 w, 1056 w, 1010 w, 997 w, 927 m, 854 m, 742 m, 688 m, 660 m, 631 m, 574 m. HRMS (ESI) ([M − H]−) Calcd for C10H9N2O7S: 301.0136; found: 301.0137.
:PE:HOAc = 100:50:5), [α]20D = −135.6 (c 3.62, acetone). 1H NMR (300 MHz, DMSO-d6) δ 8.11–7.91 (m, 2H), 7.48–7.42 (m, 2H), 5.13 (d, J = 6.6 Hz, 1H), 4.54 (d, J = 6.6 Hz, 1H), 4.40 (dd, J = 7.8, 6.0 Hz, 1H), 3.87 (t, J = 8.3 Hz, 1H), 3.70 (dd, J = 8.7, 5.9 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 171.10, 165.52 (JC–F = 249.75 Hz), 134.07, 131.40, 117.36 (JC–F = 21.75 Hz), 81.56, 69.76, 59.41. 19F NMR (282 MHz, DMSO-d6) δ 105.42. FTIR (KBr, cm−1): 3110 w, 2895 m, 1713 s, 1595 s, 1495 s, 1353 m, 1298 s, 1205 s, 1170 s, 1156 s, 1101 m, 1070 m, 1057 m, 999 m, 926 s, 839 s, 686 s, 597 s, 545 s. HRMS (ESI) ([M − H]−) Calcd for C10H9FNO5S: 274.0191; found: 274.0199.
:PE:HOAc = 20:10:1). [α]20D = −122.7 (c 5.76, acetone). 1H NMR (300 MHz, DMSO-d6) δ 8.14 (d, J = 8.1 Hz, 2H), 7.99 (d, J = 8.1 Hz, 2H), 5.17 (d, J = 6.3 Hz, 1H), 4.57 (d, J = 6.3 Hz, 1H), 4.45 (dd, J = 7.8, 6.0 Hz, 1H), 3.93 (t, J = 8.4 Hz, 1H), 3.73 (dd, J = 8.7, 6.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 170.67, 141.10, 133.27 (JC–F = 33.0 Hz), 132.79, 128.81, 123.45 (JC–F = 272.0 Hz), 80.87, 69.09, 58.74. 19F NMR (282 MHz, DMSO-d6) δ −62.43. FTIR (KBr, cm−1): 3400 br, 2896 w, 1714 s, 1433 w, 1406 w, 1354 s, 1329 s, 1298 w, 1257 w, 1170 s, 1111 m, 1064 m, 999 w, 927 m, 842 m, 717 m, 657 w, 630 w, 599 w, 570 w. HRMS (ESI) ([M − H]−) Calcd for C11H9F3NO5S: 324.0159; found: 324.0155.
:PE:HOAc = 100:100:5), [α]20D = −168.2 (c 4.30, acetone). 1H NMR (300 MHz, CDCl3) δ 7.76 (d, J = 8.2 Hz, 2H), 7.40–7.30 (m, 2H), 5.18 (d, J = 6.0 Hz, 1H), 4.72 (d, J = 6.0 Hz, 1H), 4.38 (t, J = 6.8 Hz, 1H), 3.95 (m, 2H), 2.46 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 173.44, 145.17, 134.09, 130.38, 128.03, 81.96, 69.74, 59.14, 22.09. FTIR (KBr, cm−1): 3433 br, 2895 w, 1713 s, 1599 w, 1433 w, 1351 s, 1309 w, 1294 w, 1256 w, 1166 s, 1108 w, 1170 w, 997 w, 926 w, 885 w, 814 w, 712 w, 683 m, 579 m, 554 m, 465 w. HRMS (ESI) ([M − H]−) Calcd for C11H12NO5S: 270.0442; found: 270.0440.
:PE:HOAc = 100:100:2), [α]20D = −158.5 (c 4.48, acetone). 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 5.10 (d, J = 6.3 Hz, 1H), 4.54 (d, J = 6.3 Hz, 1H), 4.36 (dd, J = 8.4, 6.0 Hz, 1H), 3.85 (t, J = 8.4 Hz, 1H), 3.70 (dd, J = 8.4, 6.0 Hz, 1H), 1.31 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 171.40, 157.43, 135.00, 128.35, 127.13, 81.74, 69.81, 59.70, 35.87, 31.69. FTIR (KBr, cm−1): 3416 w, 2897 m, 1713 s, 1597 s, 1598 w, 1497 m, 1467 w, 1443 w, 1348 s, 1307 m, 1257 s, 1157 s, 1114 m, 1069 w, 1026 w, 998 w, 926 m, 834 m, 685 m, 598 s, 554 s. HRMS (ESI) ([M − H]−) Calcd for C14H18NO5S: 312.0911; found: 312.0911.
:petroleum ester:HOAc = 4:4:1), [α]20D = −153.0 (c 7.36, acetone). 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 9.0 Hz, 2H), 7.11 (d, J = 9.0 Hz, 2H), 5.09 (d, J = 6.6 Hz, 1H), 4.52 (d, J = 6.6 Hz, 1H), 4.33 (dd, J = 7.2, 6.0 Hz, 1H), 3.84 (s, 3H), 3.82 (t, J = 7.8 Hz, 1H), 3.68 (dd, J = 8.7, 6.0 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 171.33, 163.71, 130.53, 129.10, 115.31, 81.59, 69.56, 59.56, 56.47. FTIR (KBr, cm−1): 2988 m, 1713 s, 1597 s, 1580 m, 1498 m, 1464 w, 1442 w, 1348 s, 1306 m, 1257 s, 1156 s, 1113 m, 1069 w, 1026 w, 998 w, 926 m, 834 m, 685 m, 598 s, 554 s. HRMS (ESI) ([M − H]−) Calcd for C11H14NO7S: 286.0380; found: 286.0354.
:PE:HOAc = 7.5:30:1), [α]20D = −151.1° (c 13.44, acetone). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.8 Hz, 2H), 7.31 (m, 2H), 5.16 (d, J = 5.6 Hz, 1H), 4.70 (d, J = 5.6 Hz, 1H), 4.42 (m, 2H), 2.57 (m, 1H), 1.60 (m, 4H), 1.12 (m, 15H), 0.81 (m, 5H). FTIR (KBr, cm−1): 3567 br, 2958 m, 2928 s, 2871 w, 2856 m, 1719 s, 1598 s, 1466 m, 1413 w, 1352 s, 1298 w, 1166 s, 1108 m, 1069 w, 996 w, 929 s, 678 s, 605 s, 572 w. HRMS (ESI) ([M − H]−) Calcd for C23H37NO5S: 438.2320; found: 438.2320.
Syntheses of (4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylic acids
(4S,5R)-5-Methyl-3-(arylsulfonyl)oxazolidine-4-carboxylic acids were prepared with a similar procedure,29 as exemplified by the synthesis of (4S,5R)-3-((4-fluorophenyl)sulfonyl)-5-methyloxazolidine-4-carboxylic acid: (4S,5R)-5-methyloxazolidine-4-carboxylic acid hydrochloride (0.50 g, 3.25 mmol)17 was dissolved in a water (15 mL) solution of a mixture of Na2CO3 (0.82 g, 7.8 mmol) and NaHCO3 (0.32 g, 3.90 mmol) at room temperature to give a clear solution, which was then cooled down in an ice bath. An acetone (2 mL) solution containing p-fluorobenzenesulfonyl chloride (1.58 g, 4.1 mmol) was added slowly to the above solution with stirring. The whole reaction mixture was allowed to warm to room temperature. After stirring overnight, the reaction mixture was acidified with aqueous HCl (1 M) till the pH value reached 2–3, and then extracted with EtOAc (30 ml × 3). The combined organic layer was washed with H2O (30 ml × 2), brine (50 mL) and dried over Na2SO4, and filtered. The solvent was removed by using a rotary evaporator and the crude product was obtained as a white solid which was then purified by chromatography on silica gel.:PE:HOAc = 20:20:1). [α]20D = −153.4 (c 7.08, acetone). 1H NMR (400 MHz, acetone-d6) δ 8.50 (m, 2H), 8.28 (m, 2H), 5.35 (d, J = 6.8 Hz, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.11 (m, 1H), 3.94 (d, J = 7.1 Hz, 1H), 1.16 (d, J = 6.1 Hz, 3H). 13C NMR (100 MHz, acetone-d6) δ 170.40, 151.71, 144.45, 130.42, 125.47, 81.80, 79.94, 66.04, 18.84. FTIR (KBr, cm−1): 3435 br, 3114 w, 1713 m, 1610 w, 1531 s, 1351 s, 1309 w, 1167 s, 1069 w, 927 w, 854 w, 741 m, 574 w, 468 w. HRMS (ESI) ([M − H]−) Calcd for C11H12N2O7S: 315.0292; found: 315.0288.
:PE:HOAc = 20:20:1), [α]20D = −132.6 (c 8.90, acetone). 1H NMR (400 MHz, DMSO-d6) δ 8.01 (m, 2H), 7.45 (m, 2H), 5.24 (d, J = 7.1 Hz, 1H), 4.57 (d, J = 7.1 Hz, 1H), 3.97 (m, 1H), 3.72 (d, J = 7.0 Hz, 1H), 0.98 (d, J = 6.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.42, 165.07 (d, JC–F = 252.8 Hz), 133.37 (d, J = 3.0 Hz), 131.08 (d, J = 9.7 Hz), 116.74 (d, J = 22.7 Hz), 80.60, 78.14, 65.02, 18.37. 19F NMR (282 MHz, DMSO-d6) δ −109.14. FTIR (KBr, cm−1): 3420 br, 2980 w, 1737 s, 1494 s, 1295 m, 1352 s, 1238 m, 1170 s, 1156 s, 1089 m, 1075 w, 980 m, 842 s, 680 m, 591 s, 547 s. HRMS (ESI) ([M − H]−) Calcd for C11H12FNO5S: 288.0347; found: 288.0349.
:PE:HOAc = 20:20:1). [α]20D = −132.3 (c 11.78, acetone). 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.2 Hz, 2H), 7.82 (d, J = 8.2 Hz, 2H), 5.26 (d, J = 6.5 Hz, 1H), 4.70 (d, J = 6.5 Hz, 1H), 4.12 (m, 1H), 3.84 (d, J = 7.0 Hz, 1H), 1.22 (d, J = 6.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.85, 141.31, 135.53 (q, JC–F = 33.0 Hz), 128.58, 126.74 (q, J = 3.6 Hz), 123.28 (d, J = 273.1 Hz), 81.38, 79.31, 65.22, 18.61. 19F NMR (377 MHz, CDCl3) δ −63.21. FTIR (KBr, cm−1): 3400 br, 2987 w, 1715 s, 1405 w, 1390 m, 1324 s, 1296 w, 1259 w, 1227 w, 1185 s, 1133 s, 1107 m, 1062 m, 1015 w, 983 m, 889 w, 838 m, 717 s, 649 w, 617 s, 568 m, 428 m. HRMS (ESI) ([M − H]−) Calcd for C12H12F3NO5S: 338.0316; found: 338.0311.
:PE:HOAc = 20:20:1), [α]20D = −159.7 (c 9.00, acetone). 1H NMR (400 MHz, CDCl3) δ 7.75 (m, 2H), 7.34 (m, 2H), 5.23 (d, J = 6.7 Hz, 1H), 4.67 (d, J = 6.7 Hz, 1H), 4.09 (m, 1H), 3.76 (d, J = 7.1 Hz, 1H), 2.43 (s, 1H), 1.15 (d, J = 6.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.84, 145.05, 134.04, 130.22, 128.13, 81.44, 79.02, 65.31, 21.78, 18.55. FTIR (KBr, cm−1): 3380 br, 2885 w, 1737 s, 1598 w, 1348 s, 1278 w, 1161 s, 1089 s, 1071 w, 972 m, 818 w, 705 m, 665 s, 595 s, 546 s. HRMS (ESI) ([M − H]−) Calcd for C12H15NO5S: 284.0598; found: 284.0597.
:PE:HOAc = 20:20:1), [α]20D = −122.6 (c 8.60, acetone). 1H NMR (400 MHz, CDCl3) δ 7.78 (m, 2H), 7.54 (m, 2H), 5.25 (d, J = 6.6 Hz, 1H), 5.25 (d, J = 6.6 Hz, 1H), 4.67 (d, J = 6.7 Hz, 1H), 4.08 (m, 1H), 3.77 (d, J = 7.0 Hz, 1H), 1.32 (s, 9H), 1.11 (d, J = 6.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 173.74, 158.01, 134.07, 128.05, 126.56, 81.52, 79.00, 65.25, 35.40, 31.20, 18.51. FTIR (KBr, cm−1): 3480 br, 2966 s, 1737 s, 1595 m, 1461 w, 1400 m, 1349 s, 1268 w, 1164 s, 1113 m, 1086 m, 1039 w, 980 m, 891 w, 844 m, 754 m, 637 s, 591 s, 554 w. HRMS (ESI) ([M − H]−) Calcd for C15H21NO5S: 326.1068; found: 326.1058.
:PE:HOAc = 20:20:1), [α]20D = −179.5 (c 7.80, acetone). 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.9 Hz, 2H), 6.98 (d, J = 8.9 Hz, 2H), 5.17 (d, J = 6.8 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.04 (m, 1H), 3.82 (s, 3H), 3.71 (d, J = 7.1 Hz, 1H), 1.10 (d, J = 6.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 172.86, 163.89, 130.25, 128.26, 114.73, 81.35, 78.91, 65.32, 55.85, 18.53. FTIR (KBr, cm−1): 3500 w, 2880 w, 1738 s, 1596 s, 1577 w, 1499 s, 1460 w, 1443 w, 1348 s, 1309 w, 1264 s, 1157 s, 1092 m, 1074 w, 1022 w, 980 w, 892 w, 836 m, 802 w, 673 s, 593 s, 557 s. HRMS (ESI) ([M − H]−) Calcd for C12H15NO6S: 300.0547; found: 300.0538.
:PE:HOAc = 7.5:30:1), [α]20D = −151.1° (c 13.44, acetone). 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.2 Hz, 2H), 7.32 (m, 2H), 5.28 (d, J = 6.9 Hz, 1H), 4.69 (d, J = 6.9 Hz, 1H), 4.09 (m, 1H), 3.74 (m, 1H), 2.59 (m, 1H), 1.60 (m, 4H), 1.12 (m, 18H), 0.81 (m, 5H). FTIR (KBr, cm−1): 3560 br, 2958 m, 2927 s, 2871 w, 2855 m, 1738 s, 1597 m, 1411 m, 1352 s, 1165 s, 1074 w, 982 w, 834 w, 655 w, 601 m, 575 w. HRMS (ESI) ([M − H]−) Calcd for C23H37NO5S: 438.2320; found: 438.2320.
Syntheses of (4R)-3-(arylsulfonyl)thiazolidine-4-carboxylic acids
(4R)-3-(Arylsulfonyl)thiazolidine-4-carboxylic acids were prepared with a similar procedure,29 as exemplified by the synthesis of (4R)-3-tosylthiazolidine-4-carboxylic acid: L-4-thiazolidinecarboxylic acid (1.00 g, 7.5 mmol) was dissolved in a water (30 mL) solution of a mixture of Na2CO3 (1.59 g, 15.0 mmol) and NaHCO3 (0.63 g, 7.5 mmol) at room temperature to give a clear solution, which was then cooled down in an ice bath. An acetone (10 mL) solution containing 4-methylbenzene-1-sulfonyl chloride (1.80 g, 9.4 mmol) was added slowly to the above solution with stirring. The whole reaction mixture was allowed to warm to room temperature. After stirring for another 4 h, the reaction mixture was washed with Et2O (5 mL × 5). The aqueous layer was collected, acidified with aqueous HCl (1 M) till the pH value reached around 2, extracted with EtOAc (30 ml × 3), washed with water until the pH value reached around 7, dried over anhydrous sodium sulfate, and then concentrated in a vacuum.:EtOAc:acetic acid = 30:20:2). 1H NMR (300 MHz, CDCl3) δ 8.39 (d, J = 9.0 Hz, 2H), 8.08 (d, J = 9.0 Hz, 2H), 4.97 (dd, J = 6.9, 3.6 Hz, 1H), 4.74 (d, J = 9.0 Hz, 1H), 4.41 (d, J = 9.0 Hz, 3.0 Hz, 1H), 3.32 (dd, J = 11.1, 3.6 Hz), 3.20 (dd, J = 11.1, 6.9 Hz, 1H).
:EtOAc:acetic acid = 30:20:2). 1H NMR (300 MHz, acetone-d6) δ 7.85 (m, 2H), 7.17 (m, 2H), 4.84 (dd, J = 7.2, 3.4 Hz, 1H), 4.63 (d, J = 9.3 Hz, 1H), 4.35 (d, J = 9.3 Hz, 1H), 3.21 (dd, J = 11.5, 3.4 Hz, 1H), 3.01 (dd, J = 11.5, 7.2 Hz, 1H). 19F NMR, (282 MHz, acetone-d6) δ −103.98.
:EtOAc:acetic acid = 10:10:1). 1H NMR (300 MHz, CDCl3) δ 8.02 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.1 Hz, 2H), 4.96 (dd, J = 6.9, 3.3 Hz, 1H); 4.71 (d, J = 9.0 Hz, 1H), 4.42 (d, J = 9.0 Hz, 1H), 3.3 (dd, J = 11.4, 3.6 Hz, 1H), 3.15 (dd, J = 11.4, 6.9 Hz, 1H). 19F NMR (282 MHz, CDCl3) δ −63.92.
:EtOAc:acetic acid = 30:20:2). 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 4.86 (dd, J = 7.2, 3.6 Hz, 1H), 4.67 (d, J = 9.3 Hz, 1H), 4.44 (d, J = 9.3 Hz, 1H), 3.25 (dd, J = 11.4, 3.6 Hz, 1H), 2.96 (dd, J = 11.4, 7.2 Hz, 1H), 2.45 (s, 3H).
:EtOAc:acetic acid = 30:20:2). 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 6.6 Hz, 2H), 7.55 (d, J = 6.6 Hz, 2H), 4.88 (dd, J = 7.2, 5.4 Hz, 1H), 4.65 (d, J = 9.3 Hz, 1H), 4.45 (d, J = 9.3 Hz, 1H), 3.28 (dd, J = 11.4, 5.4 Hz, 1H), 2.98 (dd, J = 11.4, 7.2 Hz, 1H), 1.36 (s, 9H).
:EtOAc:acetic acid = 30:20:2). 1H NMR (300 MHz, DMSO-d6) δ 7.83 (d, J = 6.9 Hz, 2H), 7.10 (d, J = 6.9 Hz, 2H), 4.81 (dd, J = 7.3, 3.6 Hz, 1H), 4.68 (d, J = 10.3 Hz, 1H), 4.32 (d, J = 10.3 Hz, 1H), 3.83 (s, 3H), 3.07 (dd, J = 11.4, 3.6 Hz, 1H), 2.78 (dd, J = 11.4, 7.3 Hz, 1H).
:EtOAc:acetic acid = 20:10:1). 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 4.90 (d, J = 4.1 Hz, 1H), 4.66 (d, J = 9.6 Hz, 1H), 4.44 (d, J = 9.6 Hz, 1H), 3.20 (d, J = 10.8 Hz, 1H), 2.87 (d, J = 4.1 Hz, 1H), 2.56 (m, 1H), 1.55 (m, 4H), 1.09 (m, 15H), 0.79 (m, 5H). FTIR (KBr, cm−1): 3513 br, 2956 m, 2926 s, 2855 m, 1731 s, 1596 m, 1458 m, 1411 w, 1351 s, 1227 w, 1165 s, 1091 w, 1009 w, 828 w, 648 w, 632 w, 595 s.
Syntheses of (4R)-3-(arylsulfonyl)thiazolidine-4-carboxylic acid 1,1-dioxides
(4R)-3-(Arylsulfonyl)thiazolidine-4-carboxylic acid 1,1-dioxides were prepared with a similar procedure, as exemplified by the synthesis of (4R)-3-tosylthiazolidine-4-carboxylic acid 1,1-dioxide: a mixture of (R)-3-tosylthiazolidine-4-carboxylic acid (2.0 g, 6.96 mmol) and the finely powdered urea–hydrogen peroxide adduct (3.27 g, 34.79 mmol) in acetonitrile (3 mL) was heated at 85 °C for 4 h. After that, water (5 mL) was added to the reaction mixture, which was then acidified with HCl (1 M) till the pH value reached around 2, extracted with EtOAc (30 mL × 3), washed with water till the pH value reached around 7, and then dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure to afford the crude product, which was purified by chromatography to deliver the pure product.:EtOAc:formic acid = 30:20:2). [α]15D = −62.2° (c 1.96, acetone). 1H NMR (300 MHz, acetone-d6): δ 8.45 (d, J = 9.0 Hz, 2H), 8.25 (d, J = 9.0 Hz, 2H), 5.39 (dd, J = 9.1, 4.1 Hz, 1H), 4.90 (d, J = 11.8 Hz, 1H), 4.37 (d, J = 11.8 Hz, 1H), 3.73 (dd, J = 13.5, 9.1 Hz, 1H), 3.63 (dd, J = 13.5, 4.1 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 168.97, 150.13, 143.01, 129.14, 124.62, 62.35, 56.91, 51.28. FTIR (KBr, cm−1): 3592 s, 3522 s, 3073 w, 3017 w, 2964 w, 2852 w, 2769 w, 2698 w, 2548 w, 1739 s, 1608 s, 1530 s, 1355 s, 1332 s, 1224 m, 1161 s, 1124 s, 1090 s, 1043 s, 967 w, 921 w, 858 w, 769 s, 682 m, 468 m, 430 m. HRMS (ESI) ([M − H]−) Calcd for C10H9N2O8S2: 348.9806; found: 348.9784.
:EtOAc:formic acid = 30:20:2). [α]15D = −58.8° (c 10.18, acetone). 1H NMR (300 MHz, acetone-d6) δ 8.05 (m, 2H), 7.39 (m, 2H), 5.28 (dd, J = 8.4, 4.5 Hz, 1H), 4.81 (d, J = 12.0 Hz, 1H), 4.32 (d, J = 12 Hz, 1H), 3.60 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.80, 165.52 (JC–F = 251.0 Hz) 134.54, 131.25, 117.07 (JC–F = 23.0 Hz), 62.82, 57.43, 50.75, 31.09. 19F NMR (377 MHz, acetone-d6) δ −106.06. FTIR (KBr, cm−1): 3554 br, 3109 s, 3023 s, 2955 s, 2601 w, 1732 s, 1625 m, 1591 s, 1494 s, 1408 s, 1333 s, 1297 s, 1232 s, 1157 s, 1089 s, 1041 m, 997 m, 915 m, 841 s, 820 s, 758 s, 727 w, 577 s, 545 s, 440 m. HRMS (ESI) ([M − H]−) Calcd for C10H9FNO6S2: 321.9861; found: 321.9841.
:EtOAc:acetic acid = 30:20:2). [α]15D = −67.7° (c 8.98, acetone). 1H NMR (300 MHz, DMSO-d6) δ 8.13 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H), 5.14 (dd, J = 9.0 Hz, 4.5 Hz, 1H), 4.96 (d, J = 12.0 Hz, 1H), 4.30 (d, J = 12.0 Hz 1H), 3.68 (dd, J = 13.8, 9.0 Hz, 1H), 3.56 (dd, J = 13.8, 4.5 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 169.32, 141.50, 133.12 (JC–F = 24.8 Hz), 126.37, 126.39, 123.26 (JC–F = 270.8 Hz), 62.40, 57.11, 51.33. 19F NMR (377 MHz, acetone-d6) δ −63.66. FTIR (KBr, cm−1): 3212 w, 2959 w, 1735 m, 1407 w, 1368 m, 1324 s, 1268 w, 1170 s, 1134 s, 1092 w, 1062 m, 1015 m, 996 w, 840 m, 756 m, 711 m, 613 s, 555 w, 431 w. HRMS (ESI) ([M − H]−) Calcd for C11H9F3NO6S2: 371.98296; found: 371.9829.
:EtOAc:acetic acid = 30:20:2). [α]15D = −47.2° (c 10.07, acetone). 1H NMR (300 MHz, acetone-d6) δ 7.84 (d, J = 8.3 Hz, 2H), 7.43 (d, J = 8.3 Hz, 2H), 5.25 (t, J = 6.4 Hz, 1H), 4.77 (d, J = 11.9 Hz, 1H), 4.30 (d, J = 11.9 Hz, 1H), 3.55 (d, J = 6.4 Hz, 2H), 2.44 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 169.21, 144.31, 134.67, 129.31, 127.45, 62.89, 57.13, 51.61, 21.18. FTIR (KBr, cm−1): 3219 s, 3025 w, 2972 w, 1760 s, 1434 w, 1332 s, 1286 w, 1269 w, 1229 w, 1190 s, 1113 m, 1088 m, 1056 w, 812 m, 762 s, 663 s, 578 s, 546 s, 466 m. HRMS (ESI) ([M − H]−) Calcd for C11H12NO6S2: 318.0112; found: 318.0100.
:EtOAc:acetic acid = 30:20:2). [α]15D = −69.9° (c 8.97, acetone). 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 5.07 (dd, J = 8.4, 4.8 Hz, 1H), 4.82 (d, J = 12.1 Hz, 1H), 4.24 (d, J = 12.1 Hz, 1H), 3.57 (dd, J = 13.8 Hz, 8.4 Hz, 1H), 3.53 (dd, J = 13.8 Hz, 4.8 Hz, 1H), 1.30 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 169.22, 156.75, 134.84, 127.29, 126.13, 62.72, 57.11, 51.21, 35.02, 30.80. FTIR (KBr, cm−1): 3435 br, 2961 s, 1747 w, 1627 w, 1596 w, 1403 w, 1339 m, 1162 s, 1110 s, 806 w, 765 m, 631 m, 583 w, 553 w, 467 m. HRMS (ESI) ([M − H]−) Calcd for C14H18NO6S2: 360.0581; found: 360.0576.
:EtOAc:acetic acid = 30:20:2). [α]15D = −66.6° (c 10.11, acetone). 1H NMR (400 MHz, DMSO-d6) δ 7.87 (d, J = 8.7 Hz, 2H), 7.14 (d, J = 8.7 Hz, 2H), 5.07 (t, J = 5.1 Hz, 1H), 4.83 (d, J = 12.3 Hz, 1H), 4.26 (d, J = 12.3 Hz, 1H), 3.87 (s, 3H), 3.57 (d, J = 5.1 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.89, 163.75, 130.37, 129.39, 115.07, 63.30, 56.26, 51.96. FTIR (KBr, cm−1): 3207 br s, 3000 s, 2942 s, 1770 m, 1596 s, 1579 s, 1500 s, 1446 m, 1264 m, 1171 s, 1105 s, 1046 s, 1019 s, 995 m, 884 m, 806 s, 779 s, 733 s, 668 s, 579 s, 553 s, 445 s. HRMS (ESI) ([M − H]−) Calcd for C11H12NO7S2: 334.0061; found: 334.0035.
:EtOAc:acetic acid = 20:10:1). [α]25D = −61.3° (c 39.20, acetone). 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.2 Hz, 2H), 7.34 (m, 2H), 5.15 (dd, J = 8.7, 3.4 Hz, 1H), 4.60 (d, J = 11.8 Hz, 1H), 4.41 (d, J = 11.8 Hz, 1H), 3.62 (dd, J = 8.7, 4.7 Hz, 1H), 3.31 (m, 1H), 2.62 (m, 1H), 1.65 (m, 4H), 1.17 (m, 15H), 0.86 (m, 5H). FTIR (KBr, cm−1): 3552 br, 2958 m, 2927 s, 2855 m, 1741 s, 1597 w, 1462 w, 1412 w, 1336 s, 1226 w, 1164 s, 1040 w, 919 w, 830 w, 770 w, 650 w, 593 s. HRMS (ESI) ([M − H]−) Calcd for C22H35NO6S2: 472.1833; found: 472.1825.
Syntheses of dirhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate)
Dirhodium tetrakis((4S)-3-(arylsulfonyl)oxazolidine-4-carboxylate) was prepared with a similar procedure,31a as exemplified by the synthesis of dirhodium tetrakis((S)-3-tosyloxazolidine-4-carboxylate): Na4Rh2(CO3)4 (122.6 mg, 0.228 mmol) and (4S)-3-tosyloxazolidine-4-carboxylic acid (495 mg, 1.824 mmol) were added to water (12 mL). The mixture was stirred at 95 °C for 4 h, during which time the catalyst color changed from blue to green and the insoluble sticky matter clotted on the stir bar. The reaction mixture was cooled to room temperature and added to DCM (20 mL). The mixture was then washed sequentially with saturated NaHCO3 and brine, dried over anhydrous Na2SO4, and then concentrated in vacuo. The crude reaction mixture was purified on silica using ether to afford the product as a green solid.:PE = 1:1). 1H NMR (300 MHz, acetone-d6) δ 8.42 (J = 8.7 Hz, 2H), 8.14 (d, J = 8.7 Hz, 2H), 5.10 (d, J = 5.6 Hz, 1H), 4.52 (d, J = 5.6 Hz, 1H), 4.38 (dd, J = 7.5, 6.0 Hz, 1H), 4.02 (dd, J = 8.7, 7.5 Hz, 1H), 3.57 (dd, J = 8.7, 6.0 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ 188.89, 150.55, 143.94, 129.19, 124.46, 80.88, 70.30, 59.52. Anal. Calcd for C44H46N8O31Rh2S4 (Rh2(NOSO)4·H2O·EtOAc): C, 34.83%; H, 3.06%; N, 7.39%; S, 8.45%; found: C, 34.49%; H, 3.108%; N, 7.34%; S, 8.26%. FTIR (KBr, cm−1): 3451 br, 3106 w, 2888 w, 1612 s, 1531 s, 1477 w, 1419 s, 1352 s, 1312 m, 1225 w, 1169 s, 1090 m, 1068 m, 1011 m, 937 m, 857 m, 739 s, 685 m, 626 s, 578 m, 455 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 593.5, 312.1; 447.5, 147.4
:PE = 1:1). 1H NMR (300 MHz, CDCl3) δ 7.83 (dd, J = 8.6, 5.0 Hz, 2H), 7.18 (t, J = 8.4 Hz, 2H), 5.00 (d, J = 5.4 Hz, 1H), 4.67 (d, J = 5.4 Hz, 1H), 4.12 (m, 1H), 3.93 (t, J = 8.4 Hz, 1H), 3.71 (m, 1H). 13C NMR (100 MHz, acetone-d6) δ 188.94, 165.40 (JC–F = 251.0 Hz), 134.55, 130.73, 116.41 (JC–F = 23.0 Hz), 80.98, 70.02, 59.62. 19F NMR (282 MHz, CDCl3) δ −104.07. Anal. Calcd for C40H38F4N4O21Rh2S4 (Rh2(FLSO)4·H2O): C, 36.38%; H, 2.90%; N, 4.24%; S, 9.71%; found: C, 36.36%; H, 3.10%; N, 4.31%; S, 9.22%. FTIR (KBr, cm−1): 3473 br, 3107 w, 3074 w, 2888 w, 1612 s, 1592 s, 1494 s, 1526 s, 1420 s, 1355 w, 1239 m, 1171 s, 1155 s, 1090 w, 1068 m, 1011 m, 935 m, 841 s, 764 w, 739 w, 677 s, 598 s, 548 s, 483 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 597.5, 322.1; 449.5, 145.3.
:PE = 1:1). 1H NMR (300 MHz, CDCl3) δ 7.94 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 8.1 Hz, 2H), 5.00 (d, J = 5.7 Hz, 1H), 4.70 (d, J = 5.2 Hz, 1H), 4.15 (m, 1H), 3.96 (t, J = 8.0 Hz, 1H), 3.75 (m, 1H). 13C NMR (100 MHz, acetone-d6) δ 188.86, 142.36, 133.93 (JC–F = 33.0 Hz), 128.60, 126.50 (JC–F = 4.0 Hz), 123.61 (JC–F = 270.0 Hz), 80.96, 70.12, 59.55. 19F NMR (282 MHz, CDCl3) δ −63.98. Anal. Calcd for C44H40F12N4O22Rh2S4 (Rh2(TFSO)4·2H2O): C, 34.34%; H, 2.62%; N, 3.64%; S, 8.33%; found: C, 34.33%; H, 2.54%; N, 3.69%; S, 8.33%. FTIR (KBr, cm−1): 3466 br, 2893 w, 1613 m 1421 versus 1407 m, 1359 m, 1324 s, 1172 s, 1133 s, 1109 w, 1063 s, 1015 m, 936 s, 714 m, 625 m, 601 w, 429 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 594.5, 308.8; 447.5, 138.3.
:PE = 2:1). 1H NMR (300 MHz, CDCl3) δ 7.68 (d, J = 6.6 Hz, 2H), 7.28 (d, J = 6.6 Hz, 2H), 4.99 (d, J = 5.5 Hz, 0H), 4.64 (d, J = 5.7 Hz, 1H), 4.13 (t, J = 7.2 Hz, 1H), 3.85 (t, J = 8.1 Hz, 1H), 3.68 (t, J = 6.9 Hz, 1H), 2.39 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ 188.94, 144.14, 135.20, 129.81, 127.74, 81.06, 69.91, 59.68, 20.56. Anal. Calcd for C50H61N4O23.5Rh2S4 (Rh2(MESO)4·0.5H2O·1.5EtOAc): C, 42.05%; H, 4.365%; N, 4.59%; S, 8.332%; found: C, 42.05%; H, 4.31%; N, 3.92%; S, 8.98%. FTIR (KBr, cm−1): 3434 br, 2955 m, 2924 s, 2853 m, 1736 w, 1735 w, 1705 w, 1612 s, 1420 s, 1353 s, 1220 w, 1164 s, 1091 m, 1068 m, 1001 m, 934 m, 815 m, 764 w, 739 w, 705 w, 669 s, 598 s, 548 m, 470 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 594.5, 273.8; 446.5, 128.7.
:PE = 1:1) . 1H NMR (300 MHz, CDCl3) δ 7.70 (d, J = 8.5 Hz, 2H), 7.49 (d, J = 8.5 Hz, 2H), 4.99 (d, J = 5.5 Hz, 1H), 4.64 (d, J = 5.5 Hz, 1H), 4.12 (t, J = 7.0 Hz, 1H), 3.86 (t, J = 8.0 Hz, 1H), 3.71 (dd, d, J = 8.4, 6.6 Hz, 1H), 1.32 (s, 9H). 13C NMR (100 MHz, acetone-d6) δ 188.96, 156.87, 135.21, 127.65, 126.24, 81.10, 69.91, 59.70, 34.85, 30.39. Anal. Calcd for C56H76N4O22Rh2S4 (Rh2(TBSO)4·2H2O): C, 45.10%; H, 5.14%; N, 3.76%; S, 8.60%; found: C, 44.78%; H, 5.38%; N, 3.72%; S, 8.30%. FTIR (KBr, cm−1): 3486 br, 3068 w, 2964 s, 2905 w, 2872 w, 1802 w, 1611 s, 1465 w, 1419 s, 1352 s, 1269 m, 1269 m, 1198 s, 1167 m, 1085 m, 1069 m, 1011 m, 935 m, 841 m, 753 m, 641 s, 593 s, 544 m, 486 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 594.5, 303.7; 446.5, 144.0.
:PE = 3:1). 1H NMR (300 MHz, CDCl3) δ 7.73 (d, J = 8.9 Hz, 2H), 6.94 (d, J = 8.9 Hz, 2H), 5.02 (d, J = 5.6 Hz, 1H), 4.63 (d, J = 5.6 Hz, 1H), 4.14 (dd, J = 11.1, 7.2 Hz, 1H), 3.89 (dd, J = 11.1, 9.0 Hz, 1H), 3.83 (s, 3H), 3.72 (dd, J = 9.0, 7.2 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ 189.01, 163.51, 129.93, 129.67, 114.39, 81.05, 69.88, 59.72, 55.27. Anal. Calcd for C48H63N4O29.5Rh2S4 (Rh2(MOSO)4·3.5H2O·EtOAc): C, 38.38%; H, 4.23%; N, 3.73%; S, 8.54%; found: C, 38.14%; H, 3.95%; N, 4.20%; S, 8.55%. FTIR (KBr, cm−1): 3537 br, 2916 w, 1596 s, 1499 m, 1418 s, 1350 m, 1308 m, 1263 m, 1156 s, 1133 w, 1092 w, 1067 w, 1020 w, 933 w, 836 w, 677 m, 560 s, 578 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 594.5, 275.4; 449.5, 137.3.
:PE = 1:3), [α]20D = −237.6° (c 1.01, acetone). 1H NMR (400 MHz, CDCl3) δ 7.73 (m, 2H), 7.28 (m, 2H), 5.05 (d, J = 5.0 Hz, 1H), 4.72 (d, J = 5.0 Hz, 1H), 4.21 (m, 1H), 3.85 (m, 2H), 2.64 (m, 1H), 1.60 (m, 4H), 1.15 (m, 15H), 0.85 (m, 5H). Anal. Calcd for C88H136F12N4O20Rh2S4 (Rh2(4S-DOSO)4): C, 55.51%; H, 7.20%; N, 2.94%; S, 6.74%; found: C, 54.93%; H, 7.41%; N, 2.81%; S, 6.62%. FTIR (KBr, cm−1): 3448 br, 2958 m, 2927 s, 2871 w, 2855 m, 1610 s, 1421 s, 1354 s, 1166 s, 1092 w, 1070 w, 936 m, 765 w, 657 w, 606 m, 577 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 596.0, 301.6; 448.0, 144.0.
Syntheses of dirhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate)
Dirhodium tetrakis((4S,5R)-5-methyl-3-(arylsulfonyl)oxazolidine-4-carboxylate) was prepared with a similar procedure,31a as exemplified by the synthesis of dirhodium tetrakis((4S,5R)-5-methyl-3-tosyloxazolidine-4-carboxylate): Na4Rh2(CO3)4 (61.5 mg, 0.114 mmol), and (4S,5R)-5-methyl-3-tosyloxazolidine-4-carboxylic acid (285 mg, 0.912 mmol) were added to water (6 mL). The mixture was stirred at 95 °C for 4 h, during which time the catalyst color changed from blue to green and the insoluble sticky matter clotted on the stir bar. The reaction was cooled to room temperature and added to DCM (20 mL). The mixture was then washed sequentially with saturated NaHCO3 and brine, dried over anhydrous Na2SO4, and then concentrated in vacuo. The crude reaction mixture was purified on silica using ether to afford the product as a green solid.:PE = 1:1). 1H NMR (400 MHz, acetone-d6) δ 8.44 (m, 2H), 8.44 (m, 2H), 5.16 (d, J = 6.0 Hz, 1H), 4.48 (d, J = 6.0 Hz, 1H), 3.82 (m, 1H), 3.75 (d, J = 7.4 Hz, 1H), 1.09 (d, J = 5.9 Hz, 3H). 13C NMR (100 MHz, acetone-d6) δ 189.51, 151.56, 145.00, 130.20, 125.44, 81.43, 80.38, 67.08, 18.59. Anal. Calcd for C45H48N8O29.5Rh2S4 (Rh2(4S,5R-MNOSO)4·H2O·0.25EtOAc): C, 35.87%; H, 3.21%; N, 7.44%; S, 8.51%; found: C, 35.58%; H, 3.37%; N, 7.37%; S, 8.10%. FTIR (KBr, cm−1): 3453 br, 3106 w, 1613 s, 1532 s, 1417 m, 1351 s, 1311 w, 1163 s, 1068 w, 856 w, 738 s, 685 m, 625 s, 579 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 596.0, 353.3; 449.0, 171.4.
:PE = 1:1). 1H NMR (400 MHz, acetone-d6) δ 7.92 (m, 2H), 7.37 (m, 2H), 5.12 (d, J = 6.3 Hz, 1H), 4.49 (d, J = 6.3 Hz, 1H), 3.79 (m, 1H), 3.58 (d, J = 7.4 Hz, 1H), 1.01 (d, J = 6.0 Hz, 3H). 13C NMR (100 MHz, acetone-d6) δ 189.61, 166.35 (d, JC–F = 253.0 Hz), 135.55 (d, JC–F = 3.1 Hz), 131.78 (d, JC–F = 9.6 Hz), 117.32 (d, JC–F = 22.8 Hz), 81.58, 80.17, 67.18, 18.68. 19F NMR (377 MHz, acetone-d6) δ −103.85. Anal. Calcd for C48H56F4N4O24Rh2S4 (Rh2(4S,5R-MFLSO)4·2H2O·EtOAc): C, 38.87%; H, 3.81%; N, 3.78%; S, 8.65%; found: C, 38.76%; H, 3.70%; N, 4.21%; S, 8.16%. FTIR (KBr, cm−1): 3448 br, 2979 w, 1612 s, 1592 m, 1493 m, 1417 s, 1352 s, 1295 w, 1240 m, 1169 m, 1155 s, 1090 m, 869 m, 771 m, 709 w, 677 m, 598 s, 548 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 591.5, 243.4; 449.5, 122.1.
:PE = 1:1). 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.2 Hz, 2H), 5.10 (d, J = 6.2 Hz, 1H), 4.76 (d, J = 6.2 Hz, 1H), 3.99 (m, 1H), 3.65 (d, J = 7.3 Hz, 1H), 1.02 (d, J = 5.9 Hz, 3H).13C NMR (101 MHz, CDCl3) δ 189.26, 140.83, 135.31 (q, JC–F = 33.0 Hz), 126.70 (d, JC–F = 3.2 Hz), 123.24 (d, JC–F = 273.0 Hz), 81.26, 79.65, 76.91, 66.29, 18.26. 19F NMR (377 MHz, CDCl3) δ −63.21. Anal. Calcd for C50H52F12N4O23Rh2S4 (Rh2(4S,5R-MFLSO)4·2H2O·0.5EtOAc): C, 36.64%; H, 3.20%; N, 3.42%; S, 7.83%; found: C, 36.60%; H, 3.41%; N, 3.34%; S, 7.37%. FTIR (KBr, cm−1): 3462 br, 2981 w, 1614 s, 1418 m, 1406 w, 1324 s, 1253 w, 1167 s, 1134 s, 1063 m, 1016 w, 901 w, 844 m, 771 m, 714 m, 624 s, 601 w, 428 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 595.5, 320.4; 448.5, 157.9.
:PE = 1:1). 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.9 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 5.06 (d, J = 5.9 Hz, 1H), 4.65 (d, J = 5.9 Hz, 1H), 4.04–3.83 (m, 1H), 3.62 (d, J = 7.3 Hz, 1H), 2.38 (s, 3H), 1.02 (d, J = 5.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 189.52, 144.44, 134.63, 130.07, 128.09, 81.22, 79.43, 66.40, 21.74, 18.30. Anal. Calcd for C50H66N4O24Rh2S4 (Rh2(4S,5R-MMESO)4·3H2O·0.5EtOAc): C, 41.67%; H, 4.62%; N, 3.89%; S, 8.90%; found: C, 41.62%; H, 4.52%; N, 3.78%; S, 8.43%. FTIR (KBr, cm−1): 3448 br, 2879 w, 1612 s, 1418 s, 1350 s, 1251 w, 1161 s, 1090 m, 815 w, 771 m, 705 s, 599 s, 550 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 597.0, 336.6; 448.0, 180.5.
:PE = 1:1). 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 5.09 (d, J = 5.7 Hz, 1H), 4.65 (d, J = 5.7 Hz, 1H), 3.97 (m, 1H), 3.62 (d, J = 7.1 Hz, 1H), 1.26 (s, 9H), 0.97 (d, J = 5.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 189.45, 157.32, 134.64, 127.97, 126.35, 81.28, 79.43, 66.29, 35.31, 31.18, 18.18. Anal. Calcd for C62H88N4O23Rh2S4 (Rh2(4S,5R-MTBSO)4·2H2O·0.5EtOAc): C, 46.79%; H, 5.57%; N, 3.52%; S, 8.06%; found: C, 46.66%; H, 5.44%; N, 3.62%; S, 7.64%. FTIR (KBr, cm−1):, 3500 br, 2965 s, 1613 s, 1417 s, 1351 s, 1267 w, 1163 s, 1113 m, 1086 s, 1014 w, 984 w, 842 m, 771 m, 754 m, 640 s, 594 m, 542 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 596.5, 308.7; 449.0, 159.6.
:PE = 1:1). 1H NMR (400 MHz, acetone-d6) δ 7.77 (d, J = 8.6 Hz, 2H), 7.10 (d, J = 8.6 Hz, 2H), 5.08 (d, J = 6.2 Hz, 1H), 4.48 (d, J = 6.2 Hz, 1H), 3.89 (s, 3H), 3.79 (m, 1H), 3.52 (d, J = 7.4 Hz, 1H), 0.99 (d, J = 5.8 Hz, 3H). 13C NMR (100 MHz, acetone-d6) δ 189.73, 164.47, 130.95, 130.70, 115.29, 81.67, 80.09, 67.30, 56.26, 18.77. Anal. Calcd for C50.4H64.8N4O27.2Rh2S4 (Rh2(4S,5R-MMOSO)4·2H2O·0.6EtOAc): C, 40.47%; H, 4.37%; N, 3.75%; S, 8.57%; found: C, 40.36%; H, 4.32%; N, 3.85%; S, 8.14%. FTIR (KBr, cm−1): 3517 br, 2977 w, 1612 s, 1596 s, 1498 m, 1416 m, 1347 m, 1309 w, 1156 s, 1092 w, 984 w, 836 w, 771 w, 877 m, 600 m, 558 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 597.5, 309.2; 448.5, 146.7.
:PE = 1:3), [α]20D = −259.8° (c 1.27, acetone). 1H NMR (400 MHz, CDCl3) δ 7.75 (m, 2H), 7.30 (m, 2H), 5.15 (d, J = 6.0 Hz, 1H), 4.69 (d, J = 6.0 Hz, 1H), 3.97 (m, 1H), 3.59 (m, 1H), 2.64 (m, 1H), 1.63 (m, 4H), 0.99 (m, 23H). Anal. Calcd for C92H144N4O20Rh2S4 (Rh2(4S,5R-MDOSO)4): C, 56.37%; H, 7.40%; N, 2.86%; S, 6.54%; found: C, 55.79%; H, 7.73%; N, 2.79%; S, 6.28%. FTIR (KBr, cm−1): 3467 br, 2958 m, 2928 s, 2871 w, 2856 m, 1611 s, 1466 w, 1458 w, 1419 s, 1178 s, 1161 s, 1078 m, 985 w, 770 m, 677 w, 655 m, 605 s, 577 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 596.0, 353.3; 449.0, 171.4.
Syntheses of dirhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide)
Dirhodium tetrakis((4R)-3-(arylsulfonyl)thiazolidine-4-carboxylate 1,1-dioxide) was prepared with a similar procedure,31a as exemplified by the synthesis of dirhodium tetrakis((4R)-3-tosylthiazolidine-4-carboxylate 1,1-dioxide): Na4Rh2(CO3)4 (122.6 mg, 0.228 mmol) and (4R)-3-tosylthiazolidine-4-carboxylic acid 1, 1-dioxide (582.4 mg, 1.824 mmol) were added to water (12 mL). The mixture was stirred at 95 °C for 4 h, during which time the catalyst color changed from blue to green and the insoluble sticky matter clotted on the stir bar. The reaction was cooled to room temperature and added to EtOAc (10 mL) with stirring, and then extracted with EtOAc (30 mL × 3). The combined organic layer was washed sequentially with saturated NaHCO3 and brine, dried over anhydrous sodium sulfate, and then concentrated in vacuo. The crude reaction mixture was purified on silica using ether to afford the product as a green solid.:PE = 1:1) . 1H NMR (300 MHz, acetone-d6): δ 8.46 (m, 2H), 8.19 (m, 2H), 5.12 (dd, J = 8.40, 4.5 Hz, 1H), 4.71 (d, J = 12.0 Hz, 1H), 4.11 (d, J = 12.0 Hz, 1H), 3.52 (dd, J = 13.5, 8.4 Hz, 1H), 3.40 (dd, J = 13.5, 4.5 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ 186.99, 150.78, 143.70, 129.37, 124.56, 62.30, 57.79, 52.03. Anal. Calcd for C44H46N8O35Rh2S8 (Rh2(NOST)4·H2O·EtOAc): C, 30.92%; H, 2.71%; N, 6.56%; S 15.01%; found: C, 30.53%; H, 2.94%; N, 6.55%; S, 14.72%. FTIR (KBr, cm−1): 3543 br, 3107 m, 3020 m, 2963 m, 2871 w, 1624 s, 1532 s, 1478 w, 1414 s, 1351 m, 1226 m, 1265 s, 1089 m, 1044 m, 1008 m, 917 w, 895 w, 856 s, 822 w, 765 s, 745 s, 684 m, 619 s, 566 m, 461 m, 433 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 595, 281.2; 450.5, 131.1.
:PE = 1:1). 1H NMR (300 MHz, acetone-d6): δ 7.96 (m, 2H), 7.39 (t, J = 8.6 Hz, 2H), 5.00 (m, 1H), 4.63 (m, 1H), 4.07 (m, 1H), 3.39 (m, 2H). 13C NMR (100 MHz, acetone-d6) δ 187.02, 165.61 (JC–F = 251 Hz), 164.35, 134.61, 116.53 (JC–F = 23 Hz), 62.55, 57.79, 52.11. 19F NMR (377 MHz, acetone-d6) δ −105.93. Anal. Calcd for C44H48F4N4O28Rh2S8 (Rh2(FLST)4·2H2O·EtOAc): C, 32.64%; H, 2.99%; N, 3.46%; S 15.84%; found: C, 32.67%; H, 3.26%; N, 3.62%; S, 15.63%. FTIR (KBr, cm−1): 3423 br, 3108 w, 3019 w, 2964 w, 1624 s, 1591 s, 1494 m, 1413 s, 1297 w, 1237 m, 1156 s, 1089 w, 1006 w, 841 m, 759 m, 671 m, 576 versus 545 m, 471 versus 447 w. Vis (acetone solution, λ (nm), ε (M−1 cm−1)): 593.5, 278.3; 449.5, 133.3.
:PE = 1:1). 1H NMR (300 MHz, acetone-d6): δ 8.14 (d, J = 9.0 Hz, 6.0 Hz, 2H), 7.98 (d, J = 9.0 Hz, 2H), 5.05 (m, 1H), 4.68 (d, J = 12.0 Hz, 1H), 4.15 (d, J = 12.0 Hz, 1H), 3.41 (m, 2H). 13C NMR (100 MHz, acetone-d6) δ 187.03, 142.11, 134.26 (JC–F = 33 Hz), 128.75, 126.55, 123.62 (JC–F = 271 Hz), 62.40, 57.77, 51.92. 19F NMR (377 MHz, acetone-d6) δ −63.49. Anal. Calcd for C60H68F12N4O32Rh2S8 (Rh2(TFST)4·4EtOAc): C, 35.20%; H, 3.35%; N, 2.74%; found: C, 35.20%; H, 3.58%; N, 3.25%. FTIR (KBr, cm−1): 3542 br, 3020 w, 2966 m, 2872 w, 1624 s, 1408 s, 1324 s, 1270 w, 1225 w 1168 versus 1136 s, 1111 s, 1090 w, 1063 m, 1043 w, 1012 m, 917 w, 843 m, 763 m, 712 m, 635 s, 579 m, 553 m, 465 m, 432 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 593.5, 306.9; 450.5, 141.3.
:PE = 1:1). 1H NMR (300 MHz, acetone-d6): δ 7.77 (d, J = 9.0 Hz, 2H), 7.42 (d, J = 9.0 Hz, 2H), 4.91 (dd, J = 8.4, 5.1 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 4.02 (d, J = 12.0 Hz, 1H), 3.35 (dd, J = 13.2, 5.1 Hz, 1H), 3.26 (dd, J = 13.2, 8.4 Hz, 1H), 2.46 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ 186.89, 163.84, 130.18, 129.66, 114.52, 62.81, 57.81, 55.42, 52.03. Anal. Calcd for C48H58N4O27Rh2S8 (Rh2(MEST)4·H2O·EtOAc): C, 36.37%; H, 3.69%; N, 3.53%; S 16.18%; found: C, 36.11%; H, 3.76%; N, 3.79%; S, 15.78%. FTIR (KBr, cm−1): 3499 br, 3018 m, 2961 m, 1623 s, 1598 s, 1494 w, 1414 s, 1335 s 1224 w, 1161 s, 1090 m, 1041 m, 1004 w, 916 w, 867 w, 816 w, 757 w, 666 s, 612 w, 576 m, 547 m, 467 m, 436 m. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 589.5, 247.7; 450.5, 120.5.
:PE = 1:1). 1H NMR (300 MHz, acetone-d6): δ 7.84 (d, J = 9.0 Hz, 2H), 7.64 (d, J = 9.0 Hz, 2H), 4.88 (dd, J = 8.4, 5.1 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 4.06 (d, J = 12 Hz, 1H), 3.35 (dd, J = 13.5, 5.1 Hz, 1H), 3.22 (dd, J = 13.5, 8.4 Hz, 1H), 1.36 (s, 9H). 13C NMR (100 MHz, acetone-d6) δ 186.95, 157.40, 135.32, 127.75, 126.34, 62.80, 57.80, 51.81, 34.95. Anal. Calcd for C56H76N4O26Rh2S8 (Rh2(TBST)4·3H2O): C, 39.53%; H, 4.62%; N, 3.29%; S 15.07%; found: C, 39.73%; H, 4.67%; N, 3.32%; S, 14.69%. FTIR (KBr, cm−1): 3349 br, 2964 m, 2871 m, 1625 s, 1595 s, 1462 w, 1412 m, 1336 s, 1268 w, 1220 m, 1164 m, 1134 m, 1113 m, 1087 w, 1040 w, 840 w, 770 m, 637 m, 579 m, 553 m, 464 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 591.5, 278.8; 451, 129.7.
:PE = 1:1).1H NMR (400 MHz, acetone-d6): δ 7.85 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 4.94 (dd, J = 8.8, 4.8 Hz, 1H), 4.59 (d, J = 12.0 Hz, 1H), 4.04 (d, J = 12.0 Hz, 1H), 3.38 (dd, J = 13.2, 4.8 Hz, 1H), 3.29 (dd, J = 13.2, 8.8 Hz, 1H), 2.96 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ 186.89, 163.80, 130.18, 129.66, 114.52, 62.81, 57.81, 55.42, 52.03. Anal. Calcd for C44H48N4O28Rh2S8 (Rh2(MOST)4·4H2O·EtOAc): C, 33.85%; H, 3.79%; N, 3.29%; S 15.06%; found: C, 33.78%; H, 3.58%; N, 3.42%; S, 15.32%. FTIR (KBr, cm−1): 3513 br, 3102 w, 3016 w, 2951 w, 2844 w, 1622 s, 1595 s, 1499 m, 1414 s, 1335 s, 1264 s, 1225 w, 1157 s, 1092 m, 1020 m, 869 w, 837 w, 805 w, 758 w, 671 m, 614 w, 579 m, 557 m, 466 w, 434 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 592.5, 271.9; 452.5, 118.0.
:PE = 1:2). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.1 Hz, 2H), 7.34 (m, 2H), 4.93 (s, 1H), 4.39 (m, 1H), 3.61 (d, J = 10.8 Hz, 1H), 3.43 (s, 1H), 3.08 (s, 1H), 2.59 (m, 1H), 1.58 (m, 4H), 1.18 (m, 15H), 0.84 (m, 5H). FTIR (KBr, cm−1): 3447 br, 2957 m, 2927 s, 2855 m, 1622 s, 1462 w, 1413 m, 1338 s, 1224 w, 1161 s, 1040 w, 854 w, 589 w. UV-vis (acetone solution, λ (nm), ε (M−1 cm−1)): 596.0, 301.6; 448.0, 144.0.
General procedure for catalytic aziridination
To 2 mL of DCM were added sequentially MgO (29 mg, 0.72 mmol, 2.4 eq.), PhI(OAc)2 (152 mg, 0.45 mmol, 1.5 eq.), styrene (47 mg, 0.45 mmol, 1.5 eq.), NsNH2 (61 mg, 0.30 mmol, 1.0 eq.) and 2 mol% catalyst (the following data are related to Rh2(4S-DOSO)4-catalyzed reactions). The suspension was stirred vigorously overnight until complete consumption of most starting material was indicated by TLC. The solvent was removed in a vacuum, and the residue was subjected to flash silica chromatography to afford the pure 1-((4-nitrophenyl)sulfonyl)-2-phenylaziridine (1) as a white solid (78 mg, 86%). Rf = 0.35 (EtOAc:PE = 1:5). 1H NMR (400 MHz, CDCl3) δ 8.36 (m, 2H), 8.17 (m, 2H), 7.30 (m, 3H), 7.20 (m, 2H), 3.88 (dd, J = 7.2, 4.6 Hz, 1H), 3.10 (d, J = 7.2 Hz, 1H), 2.49 (d, J = 4.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 150.83, 144.10, 134.32, 129.36, 128.93, 126.63, 124.54, 42.05, 36.75. 94% ee (AD-H, flow rate 0.8 ml min−1, 10% i-PrOH in hexane), tR = 33.62 min (major), tR = 37.64 min (minor); [α]30D = +24.2° (c 0.80, acetone); HRMS (ESI) ([M + Na]+) Calcd for C14H12O4N2S: 327.0410; found: 327.0399.
General procedure for catalytic cyclopropanation
A solution of (E)-methyl 2-diazo-4-phenylbut-3-enoatemethyl (40.4 mg, 0.2 mmol, 1.0 eq.) in 1 mL toluene was added slowly to the solution of olefin (1.0 mmol, 5.0 eq.), and 2 mol% catalyst (the following data are related to Rh2(4S,5R-MNOSO)4-catalyzed reactions) in 1 mL toluene at −40 °C. The resulting solution was vigorously stirred at −40 °C for two days until most diazo compound was completely consumed. The solvent was removed in a vacuum, and the residue was dissolved in deuterium solvents and analyzed by 1H NMR to determine the ratio of diastereomers (typically, only E isomer was observed). Flash silica chromatography of the crude residue afforded the pure cyclopropanecarboxylate product (1S,2S)-methyl 2-phenyl-1-((E)-styryl)cyclopropanecarboxylate (2a) as a white solid (53 mg, 96%).:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.21 (m, 4H), 7.13 (m, 6H), 6.33 (d, J = 16.0 Hz, 1H), 6.11 (d, J = 16.0 Hz, 1H), 3.75 (s, 3H), 2.99 (dd, J = 9.1, 7.4 Hz, 1H), 2.01 (ddd, J = 9.1, 5.0, 0.5 Hz, 1H), 1.81 (dd, J = 7.4, 5.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 174.37, 137.27, 135.71, 133.28, 129.30, 128.58, 128.20, 127.54, 126.98, 126.44, 124.29, 52.65, 35.18, 33.49, 18.82. 98% ee (OJ-H, flow rate 1.0 ml min−1, 1% i-PrOH in hexanes), tR = 19.56 min (major), tR = 26.68 min (minor); [α]30D = −150.6° (c 1.16, acetone); HRMS (APCI) ([M + H]+) Calcd for C19H18O2: 279.1380; found: 279.1382.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.09 (m, 9H), 6.13 (m, 2H), 3.79 (s, 3H), 2.93 (t, J = 8.3 Hz, 1H), 2.26 (s, 3H), 2.06 (dd, J = 9.1, 5.0 Hz, 1H), 1.87 (dd, J = 7.5, 5.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 174.47, 138.83, 137.44, 134.27, 131.38, 129.80, 128.49, 127.36, 127.29, 126.28, 125.72, 123.74, 52.66, 35.16, 32.06, 19.84, 19.08. 90% ee (OD-H, flow rate 0.8 ml min−1, 1% i-PrOH in hexane), tR = 10.15 min (minor), tR = 11.67 min (major); [α]30D = −29.8° (c 1.90, acetone); HRMS (APCI) ([M + H]+) Calcd for C20H20O2: 293.1536; found: 293.1537.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.19 (m, 5H), 7.00 (m, 4H), 6.34 (d, J = 16.0 Hz, 1H), 6.13 (d, J = 16.0 Hz, 1H), 3.74 (s, 3H), 2.95 (dd, J = 9.1, 7.4 Hz, 1H), 2.25 (s, 3H), 1.98 (dd, J = 9.1, 5.0 Hz, 1H), 1.77 (dd, J = 7.4, 5.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 174.43, 137.34, 136.53, 133.15, 132.59, 129.13, 128.92, 128.57, 127.49, 126.47, 124.43, 52.61, 35.03, 33.46, 21.24, 18.89. 88% ee (OJ-H, flow rate 0.8 ml min−1, 1% i-PrOH in hexane), tR = 19.43 min (major), tR = 31.11 min (minor); [α]30D = −99.7° (c 2.17, acetone); HRMS (APCI) ([M + H]+) Calcd for C20H20O2: 293.1536; found: 293.1536.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.22 (m, 7H), 6.96 (m, 2H), 6.37 (d, J = 16.0 Hz, 1H), 6.18 (d, J = 16.0 Hz, 1H), 3.80 (s, 3H), 3.03 (dd, J = 8.9, 7.5 Hz, 1H), 2.07 (dd, J = 8.9, 5.2 Hz, 1H), 1.82 (dd, J = 7.5, 5.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 174.18, 161.92 (d, JC–F = 245.3 Hz), 137.05, 133.31, 131.44 (d, JC–F = 3.1 Hz), 130.76 (d, JC–F = 8.0 Hz), 128.62, 127.65, 126.38, 123.97, 115.07 (d, JC–F = 21.4 Hz), 52.64, 34.36, 33.34, 18.84. 19F NMR (377 MHz, CDCl3) δ −115.75. 90% ee (OJ-H, flow rate 0.5 ml min−1, 1% i-PrOH in hexane), tR = 40.48 min (major), tR = 52.35 min (minor); [α]30D = −106.5° (c 1.80, acetone); HRMS (APCI) ([M + H]+) Calcd for C19H17O2F: 297.1285; found: 297.1289.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.34 (m, 2H), 7.21 (m, 5H), 6.99 (m, 2H), 6.35 (d, J = 16.0 Hz, 1H), 6.11 (d, J = 16.0 Hz, 1H), 3.75 (s, 3H), 2.93 (dd, J = 9.1, 7.3 Hz, 1H), 2.02 (ddd, J = 9.1, 5.2, 0.6 Hz, 1H), 1.77 (dd, J = 7.3, 5.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 174.03, 136.90, 134.85, 133.69, 131.25, 130.86, 128.64, 127.73, 126.43, 123.69, 120.87, 52.69, 34.30, 33.74, 18.74. 90% ee (OJ-H, flow rate 0.8 ml min−1, 3% i-PrOH in hexane), tR = 19.78 min (major), tR = 25.26 min (minor); [α]30D = −97.3° (c 2.00, acetone); HRMS (APCI) ([M + H]+) Calcd for C19H17O2Br: 357.0485; found: 357.0473.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.18 (m, 5H), 7.05 (m, 2H), 6.75 (m, 2H), 6.32 (d, J = 16.0 Hz, 1H), 6.15 (d, J = 16.0 Hz, 1H), 3.74 (s, 3H), 3.71 (s, 3H), 2.95 (dd, J = 9.1, 7.4 Hz, 1H), 1.99 (ddd, J = 9.1, 5.0, 0.5 Hz, 1H), 1.75 (dd, J = 7.4, 5.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 174.36, 158.59, 137.28, 133.00, 130.27, 128.55, 127.66, 127.46, 126.40, 124.41, 113.61, 55.33, 52.54, 34.78, 33.32, 18.94. 94% ee (OJ-H, flow rate 1.0 ml min−1, 1% i-PrOH in hexane), tR = 39.10 min (major), tR = 78.01 min (minor); [α]30D = −113.7° (c 1.80, acetone); HRMS (APCI) ([M + H]+) Calcd for C20H20O3: 309.1485; found: 309.1491.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.38 (m, 2H), 7.30 (m, 2H), 7.22 (m, 1H), 6.63 (d, J = 16.0 Hz, 1H), 6.31 (d, J = 16.0 Hz, 1H), 3.68 (s, 3H), 1.63 (m, 1H), 1.58 (m, 1H), 1.29 (m, 6H), 1.10 (dd, J = 6.6, 4.1 Hz, 1H), 0.84 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 175.21, 137.30, 131.91, 128.74, 127.57, 126.47, 124.90, 52.37, 31.92, 31.82, 30.75, 28.03, 22.56, 19.62, 14.25. 92% ee (OJ-H, flow rate 0.8 ml min−1, 1% i-PrOH in hexane), tR = 9.14 min (major), tR = 9.81 min (minor); [α]30D = −102.5° (c 1.47, acetone); HRMS (APCI) ([M + H]+) Calcd for C17H22O2: 259.1693; found: 259.1695.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.42 (m, 4H), 7.17 (m, 11H), 6.45 (d, J = 16.1 Hz, 0H), 6.18 (d, J = 16.1 Hz, 1H), 3.40 (s, 3H), 2.62 (d, J = 5.4 Hz, 1H), 2.05 (d, J = 5.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 171.40, 142.37, 140.97, 137.51, 131.10, 130.14, 128.98, 128.57, 128.54, 128.50, 127.38, 127.05, 127.01, 126.87, 126.35, 52.06, 47.34, 39.09, 22.79. 96% ee (OJ-H, flow rate 1.0 ml min−1, 5% i-PrOH in hexane), tR = 16.45 min (minor), tR = 28.54 min (major); [α]30D = −137.7° (c 1.68, acetone); HRMS (APCI) ([M + H]+) Calcd for C25H22O2: 355.1693; found: 355.1679.
:PE = 1:20). 1H NMR (400 MHz, CDCl3) δ 7.24 (m, 10H), 6.69 (d, J = 16.5 Hz, 1H), 5.85 (d, J = 16.5 Hz, 1H), 3.76 (s, 3H), 3.07 (d, J = 9.8 Hz, 1H), 2.28 (m, 1H), 1.19 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 174.79, 137.88, 135.05, 134.78, 131.10, 128.65, 128.47, 127.45, 126.84, 126.19, 122.32, 52.57, 36.45, 32.94, 28.01, 11.39. 90% ee (OD-H, flow rate 0.8 ml min−1, 1% i-PrOH in hexane), tR = 10.05 min (minor), tR = 10.67 min (major); [α]30D = −38.1° (c 1.60, acetone); HRMS (APCI) ([M + H]+) Calcd for C20H20O2: 293.1536; found: 293.1532.
X-ray crystallography
The diffraction data were collected with a Agilent Technologies SuperNova X-ray diffractometer system for 4S,5R-MNOSO at 293 K and an Oxford Gemini S Ultra diffractometer equipped with Cukα radiation (λ = 1.54178 Å) for (R)-3-((4-methoxyphenyl)sulfonyl)thiazolidine-4-carboxylic acid 1,1-dioxide (4R-MOST) at 299 K. Using Olex2,37a the structures were solved with the XS37b structure solution program using direct methods. 4S,5R-MNOSO was refined with the ShelXL37b refinement package and 4R-MOST was refined with the XL37b refinement package using least squares minimization. Anisotropical thermal factors were assigned to all of the non-hydrogen atoms. The positions of the hydrogen atoms were generated geometrically, assigned isotropic thermal parameters, and allowed to ride on their respective parent atoms before the final cycle of least-squares refinement. A summary of the crystal structure refinement data is shown in Table S1,† and selected bond angles and distances are listed in Table S2.† Crystallographic data for the structure reported in this paper have been deposited in the Cambridge Crystallographic Data Center with CCDC reference numbers 1055407 (4S,5R-MNOSO) and 1038152 (4R-MOST).Acknowledgements
This work was supported by the 973 Program (2012CB821701) and the NSFC Projects (21373278, 21102186, 91222201, 21173272, 21450110063), the NSF of Guangdong Province (S2013030013474) and the RFDP of Higher Education of China (20120171130006).Notes and references
- (a) H. M. L. Davies, Eur. J. Org. Chem., 1999, 2459 CrossRef CAS; (b) J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545 CrossRef CAS PubMed.
- (a) D. J. Timmons and M. P. Doyle, J. Organomet. Chem., 2001, 617–618, 98 CrossRef CAS; (b) M. P. Doyle, J. Org. Chem., 2006, 71, 9253 CrossRef CAS PubMed.
- P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905 CrossRef PubMed.
- G. Pons, V. Dequirez and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384 CrossRef PubMed.
- J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed.
- (a) M. Kennedy, M. A. McKervey, A. R. Maguire and G. H. P. Roos, J. Chem. Soc., Chem. Commun., 1990, 361–362 RSC; (b) T. Ye, C. F. García and M. A. McKervey, J. Chem. Soc., Perkin Trans. 1, 1995, 1373–1379 RSC; (c) C. F. García, M. A. McKervey and T. Ye, Chem. Commun., 1996, 1465–1466 RSC; (d) M. P. Doyle, Q.-L. Zhou, C. Charnsangavej, M. A. Longoria, M. A. McKervey and C. F. García, Tetrahedron Lett., 1996, 37, 4129–4132 CrossRef CAS.
- (a) H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, J. Am. Chem. Soc., 1996, 118, 6897 CrossRef CAS; (b) H. M. L. Davies, P. R. Bruzinski and M. J. Fall, Tetrahedron Lett., 1996, 37, 4133 CrossRef CAS; (c) H. M. L. Davies, D. G. Stafford, B. D. Doan and J. H. Houser, J. Am. Chem. Soc., 1998, 120, 3326 CrossRef CAS; (d) L. Deng, A. J. Giessert, O. O. Gerlitz, X. Dai, S. T. Driver and H. M. L. Davies, J. Am. Chem. Soc., 2005, 127, 1342 CrossRef CAS PubMed; (e) Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 440 CrossRef CAS PubMed; (f) J. F. Briones, J. Hansen, K. I. Hardcastle, I. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 17211 CrossRef CAS PubMed; (g) H. Wang, D. M. Guptill, A. Varela-Alvarez, D. G. Musaev and H. M. L. Davies, Chem. Sci., 2013, 4, 2844 RSC; (h) B. T. Parr, S. A. Green and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 4716 CrossRef CAS PubMed; (i) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 14516 CrossRef CAS PubMed.
- (a) H. M. L. Davies and T. Hansen, J. Am. Chem. Soc., 1997, 119, 9075 CrossRef CAS; (b) H. M. L. Davies, T. Hansen, D. W. Hopper and S. A. Panaro, J. Am. Chem. Soc., 1999, 121, 6506 CrossRef; (c) H. M. L. Davies, T. Hansen and M. R. Churchill, J. Am. Chem. Soc., 2000, 122, 3063 CrossRef CAS; (d) H. M. L. Davies and P. Ren, J. Am. Chem. Soc., 2001, 123, 2070 CrossRef CAS; (e) H. M. L. Davies and C. Venkataramani, Angew. Chem., Int. Ed., 2002, 41, 2197 CrossRef CAS; (f) H. M. L. Davies and Q. Jin, J. Am. Chem. Soc., 2004, 126, 10862 CrossRef CAS PubMed; (g) H. M. L. Davies and A. M. Walji, Angew. Chem., Int. Ed., 2005, 44, 1733 CrossRef CAS PubMed; (h) H. M. L. Davies and J. R. Manning, J. Am. Chem. Soc., 2006, 128, 1060 CrossRef CAS PubMed; (i) H. M. L. Davies, X. Dai and M. S. Long, J. Am. Chem. Soc., 2006, 128, 2485 CrossRef CAS PubMed; (j) Z. Li and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 396 CrossRef CAS PubMed; (k) J. H. Hansen, T. M. Gregg, S. R. Ovalles, Y. Lian, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 5076 CrossRef CAS PubMed; (l) Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 11940 CrossRef CAS PubMed; (m) H. M. L. Davies and C. Venkataramani, Org. Lett., 2003, 5, 1403 CrossRef CAS PubMed.
- H. T. Bonge, M. Kaboli and T. Hansen, Tetrahedron Lett., 2010, 51, 5375 CrossRef CAS PubMed.
- (a) S. Kitagaki, M. Anada, O. Kataoka, K. Matsuno, C. Umeda, N. Watanabe and S. Hashimoto, J. Am. Chem. Soc., 1999, 121, 1417 CrossRef CAS; (b) H. Tsutsui, Y. Yamaguchi, S. Kitagaki, S. Nakamura, M. Anada and S. Hashimoto, Tetrahedron: Asymmetry, 2003, 14, 817 CrossRef CAS; (c) M. Anada, M. Tanaka, T. Washio, M. Yamawaki, T. Abe and S. Hashimoto, Org. Lett., 2007, 9, 4559 CrossRef CAS PubMed; (d) M. Yamawaki, H. Tsutsui, S. Kitagaki, M. Anada and S. Hashimoto, Tetrahedron Lett., 2002, 43, 9561 CrossRef CAS; (e) N. Shimada, M. Anada, S. Nakamura, H. Nambu, H. Tsutsui and S. Hashimoto, Org. Lett., 2008, 10, 3603 CrossRef CAS PubMed; (f) N. Shimada, T. Oohara, J. Krishnamurthi, H. Nambu and S. Hashimot, Org. Lett., 2011, 13, 6284 CrossRef CAS PubMed; (g) J. Krishnamurthi, H. Nambu, K. Takeda, M. Anada, A. Yamano and S. Hashimoto, Org. Biomol. Chem., 2013, 11, 5374 RSC; (h) T. Goto, K. Takea, N. Shimada, H. Nambu, M. Anada, M. Shiro, K. Ando and S. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 6803–6808 CrossRef CAS PubMed; (i) T. Goto, K. Takeda, M. Anada, K. Ando and S. Hashimoto, Tetrahedron Lett., 2011, 52, 4200 CrossRef CAS PubMed.
- (a) R. P. Reddy, G. H. Lee and H. M. L. Davies, Org. Lett., 2006, 8, 3437 CrossRef CAS PubMed; (b) R. P. Reddy and H. M. L. Davies, Org. Lett., 2006, 8, 5013 CrossRef CAS PubMed; (c) R. P. Reddy and H. M. L. Davies, J. Am. Chem. Soc., 2007, 129, 10312 CrossRef CAS PubMed; (d) B. D. Schwartz, J. R. Denton, Y. Lian, H. M. L. Davies and C. M. Williams, J. Am. Chem. Soc., 2009, 131, 8329 CrossRef CAS PubMed; (e) H. Wang, D. M. Guptill, A. Varela-Alvarez, D. G. Musaev and H. M. L. Davies, Chem. Sci., 2013, 4, 2844 RSC; (f) J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802 CrossRef CAS PubMed; (g) L. Lu, H. Wang and H. M. L. Davies, Org. Lett., 2014, 16, 3036 CrossRef PubMed.
- (a) P. Müller, Y. Allenbach and E. Robert, Tetrahedron: Asymmetry, 2003, 14, 779 CrossRef; (b) P. Müller, G. Bernardinelli, Y. F. Allenbach, M. Ferri and H. D. Flack, Org. Lett., 2004, 6, 1725 CrossRef PubMed; (c) P. Müller and A. Ghanem, Org. Lett., 2004, 6, 4347 CrossRef PubMed; (d) A. Ghanem, M. G. Gardiner, R. M. Williamson and P. Müller, Chem. – Eur. J., 2010, 16, 3291 CrossRef CAS PubMed; (e) C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641 CrossRef CAS PubMed; (f) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343 CrossRef CAS PubMed.
- (a) D. Marcoux and A. B. Charette, Angew. Chem., Int. Ed., 2008, 47, 10155 CrossRef CAS PubMed; (b) D. Marcoux, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 6970 CrossRef CAS PubMed.
- (a) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034 CrossRef CAS PubMed; (b) N. Grimster, L. Zhang and V. V. Fokin, J. Am. Chem. Soc., 2010, 132, 2510 CrossRef CAS PubMed; (c) M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870 CrossRef CAS PubMed; (d) M. Zibinsky and V. V. Fokin, Angew. Chem., Int. Ed., 2013, 52, 1507 CrossRef CAS PubMed; (e) S. W. Kwok, L. Zhang, N. P. Grimster and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 3452 CrossRef CAS PubMed.
- B. T. Parr and H. M. L. Davies, Angew. Chem., Int. Ed., 2013, 52, 10044 CrossRef CAS PubMed.
- (a) C. Qin, V. Boyarskikh, J. H. Hansen, K. I. Hardcastle, D. G. Musaev and H. M. L. Davies, J. Am. Chem. Soc., 2011, 133, 19198 CrossRef CAS PubMed; (b) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 14516 CrossRef CAS PubMed; (c) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 9792 CrossRef CAS PubMed; (d) D. M. Guptill and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 17718 CrossRef CAS PubMed.
- (a) M. Kim, J. Kwak and S. Chang, Angew. Chem., Int. Ed., 2009, 48, 8935 CrossRef CAS PubMed; (b) H. J. Kim, M. Kim and S. Chang, Org. Lett., 2011, 13, 2368 CrossRef CAS PubMed; (c) J. Kwak, M. Kim and S. Chang, J. Am. Chem. Soc., 2011, 133, 3780 CrossRef CAS PubMed.
- C.-Y. Zhou, J.-C. Wang, J. Wei, Z.-J. Xu, Z. Guo, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2012, 51, 11376 CrossRef CAS PubMed.
- (a) C. G. Espino, P. M. When, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935 CrossRef CAS; (b) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed; (c) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 9220 CrossRef CAS PubMed.
- (a) A. DeAngelis, O. Dmitrenko, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 7230 CrossRef CAS PubMed; (b) R. Panish, S. R. Chintala, D. T. Boruta, Y. Fang, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2013, 135, 9283 CrossRef CAS PubMed.
- (a) S. Chuprakov, F. W. Huang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757 CrossRef CAS PubMed; (b) B. Chattopadhyay and V. Gevorgyan, Org. Lett., 2011, 13, 3746 CrossRef CAS PubMed; (c) Y. Shi and V. Gevorgyan, Org. Lett., 2013, 15, 5394 CrossRef CAS PubMed.
- (a) H. Qiu, M. Li, L.-Q. Jiang, F.-P. Lv, L. Zan, C.-W. Zhai, M. P. Doyle and W.-H. Hu, Nat. Chem., 2012, 4, 733 CrossRef CAS PubMed; (b) C. Jing, D. Xing, Y. Qian, T. Shi, Y. Zhao and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 9289 CrossRef CAS PubMed; (c) D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 13356 CrossRef CAS PubMed; (d) X. Xu, X. Guo, X. Han, L. Yang and W. Hu, Org. Chem. Front., 2014, 1, 181 RSC.
- W.-B. Zhang, S.-D. Xiu and C.-Y. Li, Org. Chem. Front., 2015, 2, 47 RSC.
- W. A. J. Starmans, L. Thijs and B. Zwanenburg, Tetrahedron, 1998, 54, 629 CrossRef CAS.
- R. T. Buck, D. M. Coe, M. J. Drysdale, L. Ferris, D. Haigh, C. J. Moody, N. D. Pearson and J. B. Sanghera, Tetrahedron: Asymmetry, 2003, 14, 791 CrossRef CAS.
- (a) M. P. Doyle and R. J. Pieters, J. Am. Chem. Soc., 1991, 113, 1423 CrossRef CAS; (b) M. P. Doyle, A. van Oeveren, L. J. Westrum, M. N. Protopopova and T. W. Clayton Jr., J. Am. Chem. Soc., 1991, 113, 8982 CrossRef CAS; (c) M. N. Protopopova and M. P. Doyle, J. Am. Chem. Soc., 1992, 114, 2755 CrossRef CAS; (d) M. P. Doyle, M. Protopopova, P. Müller, D. Ena and E. A. Shapiro, J. Am. Chem. Soc., 1994, 116, 8492 CrossRef CAS.
- (a) M. P. Doyle, I. M. Phillips and W. Hu, J. Am. Chem. Soc., 2001, 123, 5366 CrossRef CAS; (b) X. Xu, X. Xu, P. Y. Zavalij and M. P. Doyle, Chem. Commun., 2013, 49, 2762 RSC.
- (a) M. P. Doyle, A. B. Dyatkin, G. H. P. Roos, F. Cañas, D. A. Pierson and A. van Oeveren, J. Am. Chem. Soc., 1994, 116, 4507 CrossRef CAS; (b) M. P. Doyle, A. V. Kalinin and D. G. Ene, J. Am. Chem. Soc., 1996, 118, 8837 CrossRef CAS; (c) M. P. Doyle, Y. Wang, P. Ghorbani and E. Bappert, Org. Lett., 2005, 7, 5035 CrossRef CAS PubMed.
- (a) M. Falorni, S. Conti, G. Giacomelli, S. Cossu and F. Soccolini, Tetrahedron: Asymmetry, 1995, 6, 287 CrossRef CAS; (b) M. R. Vishnumaya and V. K. Singh, J. Org. Chem., 2009, 74, 4289 CrossRef CAS PubMed.
- (a) D. Fattori, C. Rossi, C. I. Fincham, M. Berettoni, F. Calvani, F. Catrambone, P. Felicetti, M. Gensini, R. Terracciano, M. Altamura, A. Bressan, S. Giuliani, C. A. Maggi, S. Meini, C. Valenti and L. Quartara, J. Med. Chem., 2006, 49, 3602 CrossRef CAS PubMed; (b) K. N. Vijayadas, H. C. Davis, A. S. Kotmale, R. L. Gawada, V. G. Puranik, P. R. Rajamohanan and G. J. Sanjayan, Chem. Commun., 2012, 48, 9747 RSC.
- (a) J. Wu, Y. Chen and J. S. Panek, Org. Lett., 2010, 12, 2112 CrossRef CAS PubMed; (b) H. J. Callot and F. Metz, Tetrahedron, 1985, 41, 4495 CrossRef CAS.
- S. A. Johnson, H. R. Hunt and H. M. Neumann, Inorg. Chem., 1963, 2, 960 CrossRef CAS.
- D. C. Wynne, M. M. Olmstead and P. G. Jessop, J. Am. Chem. Soc., 2000, 122, 7638 CrossRef CAS.
- P. Müller, C. Baud, Y. Jacquier, M. Moran and I. Nägeli, J. Phys. Org. Chem., 1996, 9, 341 CrossRef.
- (a) D. A. Evans, M. M. Faul, M. T. Bilodeau, B. A. Anderson and D. M. Barnes, J. Am. Chem. Soc., 1993, 115, 5328 CrossRef CAS; (b) Z. Li, K. R. Conser and E. N. Jacobsen, J. Am. Chem. Soc., 1993, 115, 5326 CrossRef CAS; (c) S. Taylor, J. Gullick, P. McMorn, D. Bethell, P. C. B. Page, F. E. Hancock, F. King and G. J. Hutchings, J. Chem. Soc., Perkin Trans. 2, 2001, 1724 RSC.
- (a) E. J. Corey and T. G. Gant, Tetrahedron Lett., 1994, 35, 5373 CrossRef CAS; (b) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the ligands and Rh(II) complexes; CIF files giving crystallographic data. CCDC 1055407 and 1038152. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00110b |
| This journal is © the Partner Organisations 2015 |