
Selectfluor promoted NHC–oxazoline gold(I) complex catalyzed cycloaddition/oxidation reaction of enynones with alkenes†
Qin
Xu
a,
Peng
Gu
a,
Feijun
Wang
*a and
Min
Shi
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, China. E-mail: Mshi@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925137
bKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Mei-Long Road, Shanghai 200237, China
First published on 2nd September 2015
Abstract
The ortho- and meta-substituted NHC–oxazoline ligands based on a phenyl scaffold have been synthesized in a six-step pathway, and their coordination with Au(Me2S)Cl gave the corresponding NHC–gold(I) complexes in good yields. Resulting from restricted rotation between the phenyl ring and the benzimidazole ring, the ortho-substituted NHC–oxazoline gold(I) complexes exhibited as a pair of diastereomers, and their structures have been further confirmed by X-ray diffraction analysis. Moreover, combination of these gold(I) complexes with Selectfluor in situ was used to catalyze the reaction of enynone with alkenes using air as an oxidant to give naphthalene derivatives in moderate to excellent yields. Some controlled experiments were carried out to reveal the active species as a chelated NHC–oxazoline gold(I) species and the role Selecfluor played in this reaction.
Introduction
The first decade of the century has witnessed the exploration of homogeneous gold chemistry.1 Owing to the character as soft carbophilic Lewis acids towards C–C multiple bonds, these homogeneous gold complexes have been successfully applied as pivotal catalysts for enyne cyclization reactions. Among the various factors of gold-catalyzed reactions, the ligands play a decisive role in the catalytic activities. The N-heterocyclic carbene (NHC) ligands have been widely applied as a sort of metal ligands, which could connect to various main and transition metals.2 The NHC–gold complexes have showed dramatically different catalytic activities and selectivities compared to other normally used phosphine ligands3 due to the fact that the NHC ligands exhibited stronger σ-donor but weaker π-acceptor abilities.4 Thus, developing highly efficient NHC–gold catalysts is urgent and significant.In the past few years, numerous NHC–gold(I) complexes have been synthesized and successfully applied in metal-catalyzed homogeneous reactions, even for some challenging reactions in which other catalysts exhibited bad catalytic activities.5 In comparison, the synthesis and application of NHC–gold(III) complexes received less attention despite Fehlhammer's group first reporting the synthesis of NHC gold(III) complexes in 1974.6 Later, the Raubenheimer's group revealed another synthetic method of NHC–gold(III) complexes by oxidative addition of halogens to the NHC–gold(I) complexes.7 Several synthesized NHC–gold(III) complexes have been applied in gold-catalyzed reactions. Unfortunately, few of these gold(III) complexes exhibited good catalytic activities.8 Some typical examples of NHC–gold(I) and NHC–gold(III) complexes are presented in Fig. 1.9
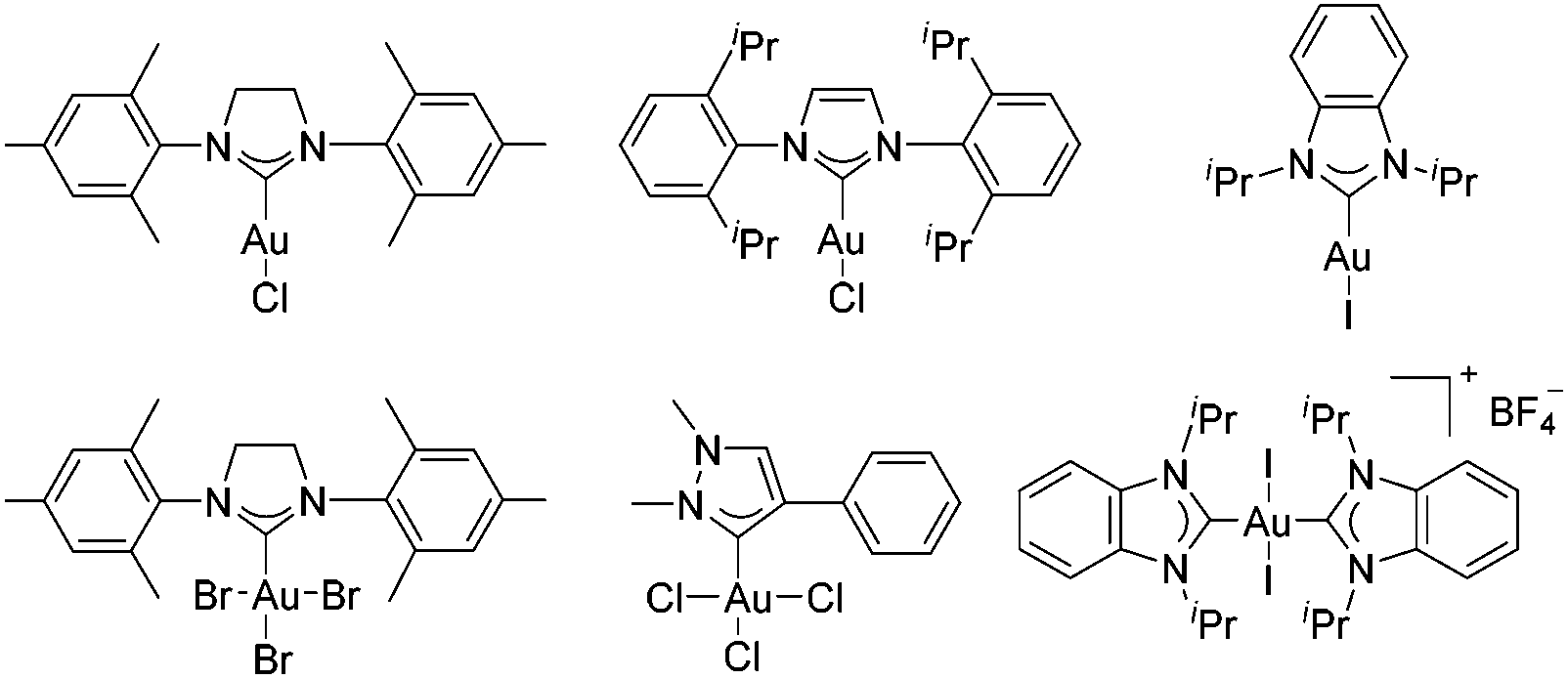 | ||
| Fig. 1 Typical NHC–gold(I) and NHC–gold(III) complexes. | ||
In consideration of the linear coordination of gold(I) complexes, the corresponding NHC ligands connected with the gold(I) atom were always designed as monodentate ligands. Even for the multiple-coordinated gold(III) atoms, the polydentate NHC ligand ligated gold(III) complexes were also rare on account of the limited synthetic methods. But for some gold-catalyzed reactions, the multiple-ligand coordinated gold complexes exhibited better catalytic activities than the traditional monodentate ligand connected gold complexes. Recently, Shi's group has developed a series of 1,2,3-triazole-coordinated phosphine-gold(I) complexes to catalyze challenging chemical transformations, achieving some excellent results.10 The Hashmi's group disclosed the synthesis of chelated bidentate PicAuCl2 complexes. This new gold(III) complex was also used in many homogeneous reactions and exhibited outstanding catalytic activities.11 Inspired by recent developments of multiple-coordinated gold complexes, we were intrigued by the synthesis of polydentate functionalized NHC ligand ligated gold(I) or gold(III) atoms to seek out more active gold catalysts.
Selectfluor has been frequently used to oxidize gold(I) species to the gold(III) species even though the specific oxidation process is still ambiguous.12 Therefore, we assumed to design bidentate NHC–oxazoline ligands A and make them ligated with gold(I) species. According to the previous literature, oxazoline substituted NHC–gold(I) complex B should be transformed to chelated NHC–oxazoline gold(III) complex C upon treatment with Selectfluor (Fig. 2). In this paper, we would like to report a novel performance of Selectfluor to make the NHC–gold(I) complex B being transformed to the ionic chelated NHC–oxazoline gold(I) complex D. This in situ generated gold(I) species were used to catalyze the reaction of enynone with alkenes using air as an oxidant to give naphthalene derivatives in moderate to excellent yields.
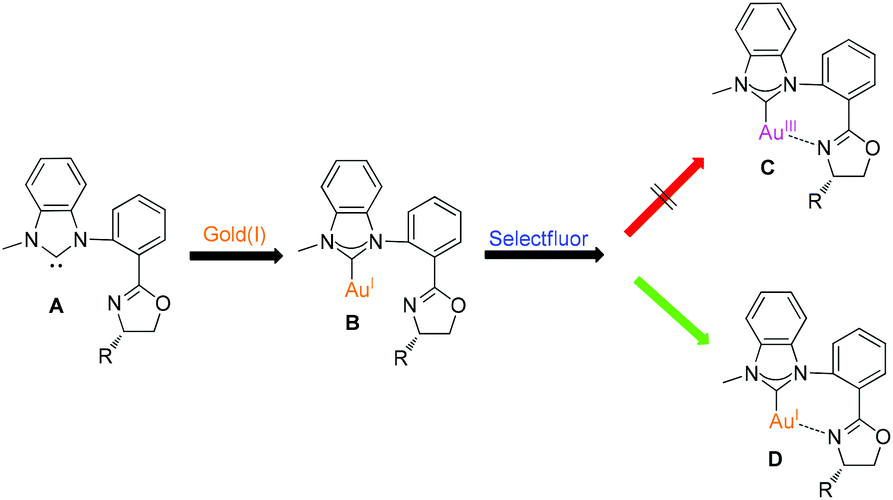 | ||
| Fig. 2 Design of NHC–oxazoline gold species. | ||
Results and discussion
Preparation of ortho-substituted NHC–oxazoline gold(I) complexes
The synthesis of ortho-substituted benzimidazolium salts based on the phenyl ring started from methyl anthranilate. Namely, compound 1 was used to react with 2-bromonitrobenzene in the presence of a DPE-phos-Pd(OAc)2 catalyst and Cs2CO3 to give the Buchwald-type coupling product 2 in quantitative yield. Then, treatment of compound 2 with Pd/C under a hydrogen atmosphere afforded the corresponding hydrogenation product 3 in 85% yield. The benzimidazole derivative 4 could be obtained by simple cyclization of compound 3 with triethyl orthoformate and a catalytic amount of TsOH at 100 °C. Consequently, compound 4 was utilized to react with two different kinds of chiral aminoalcohols promoted by Cs2CO3 in toluene to give the corresponding amide derivative 5 in good yields, which further reacted with thionyl chloride and MeONa in methanol respectively via the halogenation/cyclization reaction to give the desired benzimidazole-oxazoline product 6 in good yields. Finally, a combination of 6 with methyl iodide in acetonitrile under reflux afforded the carbene precursor 7 with high yields, which could be directly used in the next step without any further purification (Scheme 1).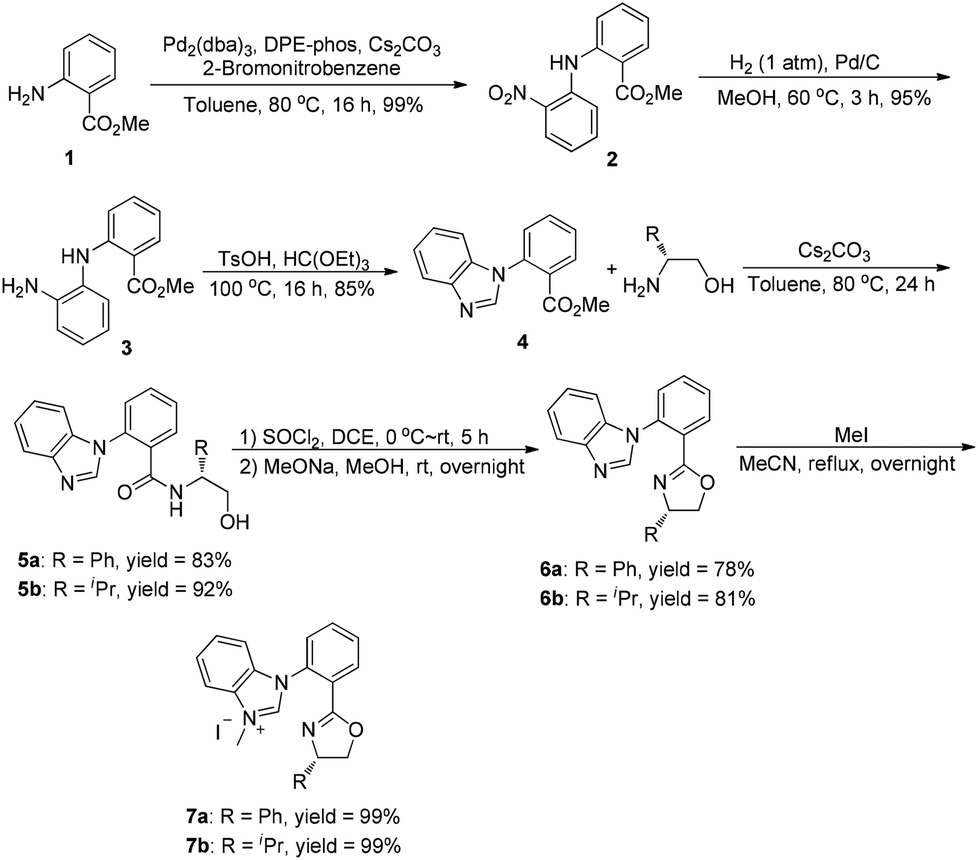 | ||
| Scheme 1 Synthesis of ortho-substituted NHC–oxazoline ligands. | ||
With the carbene precursor 7 in hand, we turned our attention to the synthesis of NHC–oxazoline gold(I) complexes. Treatment of benzimidazolium salt 7a with Au(Me2S)Cl in the presence of NaOAc in acetonitrile under reflux generated the corresponding NHC–gold(I) complex 8a in high yield (Scheme 2). 1H NMR spectra of complex 8a revealed two sets of alkyl group signals with an equal ratio. The same process was operated with the isopropyl substituted benzimidazolium salt 7b to afford the corresponding NHC–oxazoline gold(I) complex 8b in good yield as well. However, the isomeric ratio of complexes (aS, S)-8b and (aR, S)-8b was 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 on the basis of 1H NMR spectroscopic data, presumably due to the steric influence of the oxazoline group.13
:1 on the basis of 1H NMR spectroscopic data, presumably due to the steric influence of the oxazoline group.13
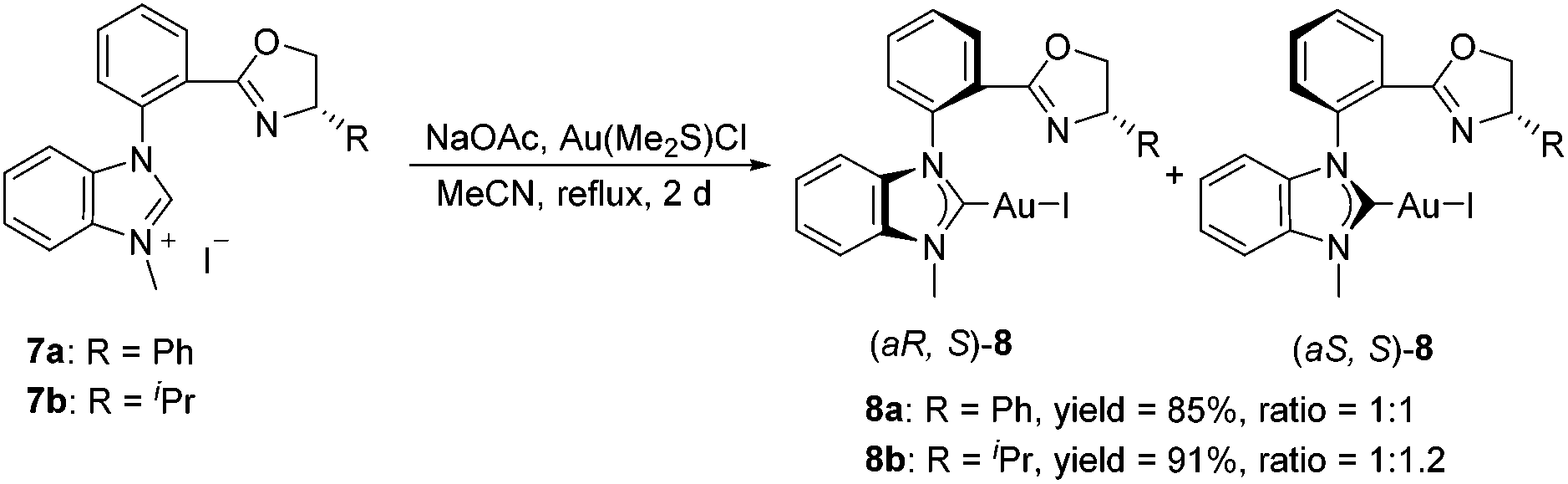 | ||
| Scheme 2 Synthesis of ortho-substituted NHC–oxazoline gold(I) complexes. | ||
The structures of complexes 8 were confirmed by the X-ray diffraction of complex 8a. A suitable single crystal of complex 8 could be obtained from the mixed solvents of dichloromethane and ethyl ether. Furthermore, the structures of two isomers, (aS, S)-8a and (aR, S)-8a, were both confirmed by the X-ray diffraction data in one crystal cell as a pair of diastereoisomer. The ORTEP drawing is shown in Fig. 3 and the CIF data are presented in the ESI.† According to the X-ray diffraction data, only the benzimidazole carbene rather than the oxazoline group is coordinated with the gold atom. When the benzimidazole group was ligated with the gold atom, the free rotation of the bond between the phenyl ring and the benzimidazole ring is blocked, causing the appearance of the axial chirality.
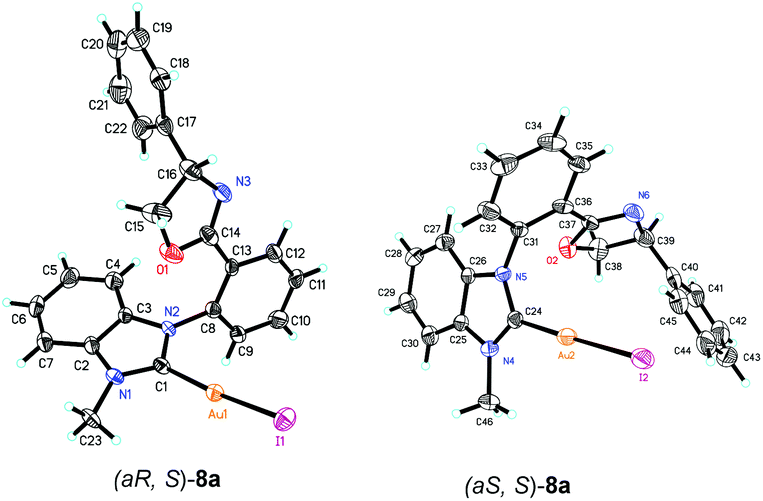 | ||
| Fig. 3 Molecular structure of complexes (aR, S)-8a and (aS, S)-8a. Ellipsoids are shown at 30% probability. Selected bond lengths [Å] and angles [°] of complexes (aR, S)-8a and (aS, S)-8a: Au1–C1 2.000(7), Au2–C24 1.999(7), N1–C1 1.321(9), N4–C24 1.328(9), O1–C14 1.336(10), N3–C14 1.247(10), N6–C37 1.282(11), C1–Au1–I1 171.9(2), C24–Au2–I2 178.1(2), C1–N1–C2 111.2(6), N1–C1–N2 107.0(6), C24–N4–C25 111.0(6), N3–C14–O1 117.8(9), N4–C24–N5 106.6(6). | ||
Preparation of meta-substituted NHC–oxazoline gold(I) complexes
According to our previous work,14 the meta-substituted benzimidazolium salts 9 could be obtained smoothly. Treatment of these carbene precursors with gold species and a base in the same method as that mentioned above afforded the corresponding NHC–oxazoline gold(I) complexes 10 in good yields (Scheme 3). The 1H NMR spectra of complexes 10 showed only one set of alkyl signals, suggesting that the structures of complexes 10 were completely different from those of NHC–gold(I) complexes 8.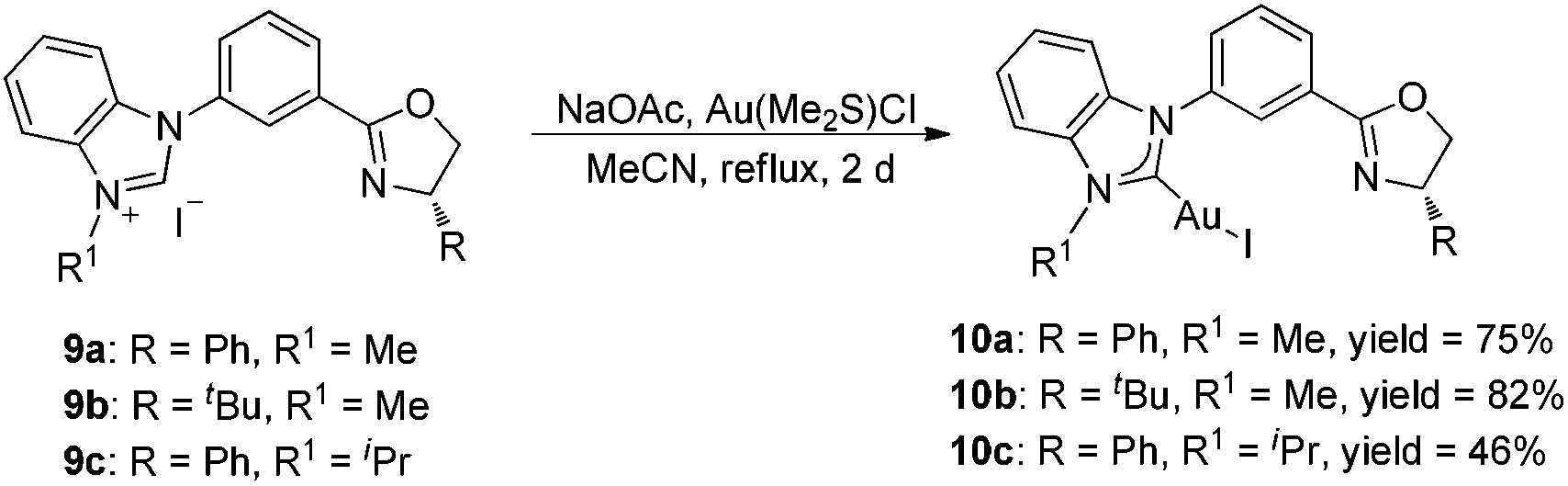 | ||
| Scheme 3 Synthesis of meta-substituted NHC–oxazoline gold(I) complexes. | ||
The accurate structures of complexes 10 were also disclosed by the X-ray diffraction of NHC–oxazoline gold(I) complex 10c. The high quality single crystal for X-ray diffraction was obtained from the mixed solvents of petroleum ether and dichloromethane. The ORTEP drawing is shown in Fig. 4 and the CIF data are summarized in the ESI.† As for the NHC–oxazoline gold(I) complexes 10c, the gold atom was only ligated with the NHC ligand, and the oxazoline group did not participate in the ligation process.
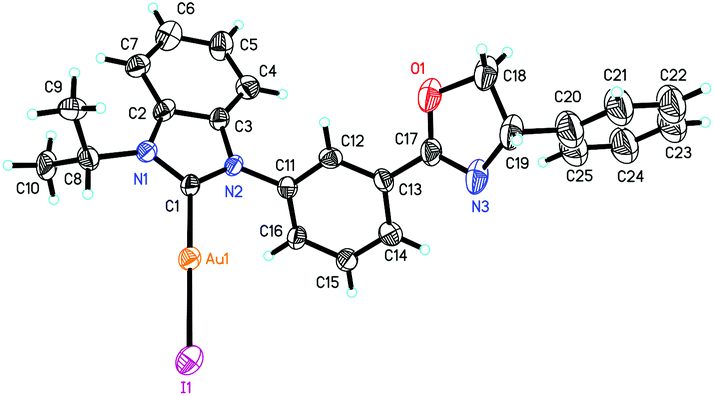 | ||
| Fig. 4 Molecular structure of complex 10c. Ellipsoids are shown at 30% probability. Selected bond lengths [Å] and angles [°] of complex 10c: Au1–C1 2.018(8), Au1–I1 2.5459(8), N1–C1 1.343(9), C17–N3 1.28(3), C17–O1 1.35(3), N1–C8 1.483(10), C1–Au1–I1 176.8(2), C1–N1–C2 109.5(6), N2–C1–N1 107.6(7), N2–C1–Au1 125.2(6), N3–C17–O1 114(2). | ||
The Selectfluor promoted NHC–oxazoline gold(I) complexes catalyzed cycloaddition/oxidation reaction
With all these NHC–oxazoline gold(I) complexes in hand, our attention turned to investigate the catalytic activities of these oxazoline-substituted NHC–gold(I) complexes in the presence of Selectfluor. The gold-catalyzed cycloaddition/oxidation reaction of enynone with alkenes, which was realized as one important kind of method to synthesize the multiple-substituted naphthalene derivatives, has already been reported widely.15 Recently, Zhu's group disclosed the in situ generated NHC–gold(III) catalyzed cycloaddition of enynone with two kinds of alkenes to afford the corresponding cyclohexane derivatives in good yields under mild conditions.16 At the end of this work, they presented the cycloaddition/oxidation reaction under an oxygen atmosphere to afford the aromatization product with moderate yield. Therefore, we decided to use our NHC–oxazoline gold(I) complexes to optimize this reaction. Initially, enynone 11a and styrene 12a were chosen as substrates for the model reaction. Treatment of these substrates with NHC–oxazoline gold(I) complex 8a (5 mol%) and Selectfluor (15 mol%) in dichloroethane at 80 °C under an air atmosphere (1.0 atm) furnished the corresponding naphthalene derivative 13a in 92% yield (Table 1, entry 1). The absence of Selectfluor significantly decreased the yield of 13a in the same reaction (Table 1, entries 2 and 3). The in situ generated NHC–gold(I) species in the presence of AgSbF6 caused the reaction system to become complex (Table 1, entry 4). The reaction could not take place only in the presence of Selectfluor (Table 1, entry 5). All the experiments disclosed the indispensability of the NHC–gold(I) complex and the Selectfluor in the reaction. Next, other NHC–oxazoline gold(I) complexes were selected to test their catalytic activities in this reaction, furnishing the naphthalene derivative 13a in 29–83% yields (Table 1, entries 6–9). The NHC–oxazoline gold(I) complex 8a functioned better than others which was confirmed as the best catalyst in this reaction.|
|
|||
|---|---|---|---|
| Entrya | Au-cat | Additive | Yieldb(%) |
| a Substrates 11a (0.1 mmol) and 12a (0.5 mmol) were stirred in DCE (2 ml) at 80 °C for 18 h using 5 mol% gold catalyst and 15 mol% Selectfluor as the catalytic system under an air atmosphere. b Isolated yield. | |||
| 1 | 8a | Select-F | 92 |
| 2 | 8a | — | 15 |
| 3 | 10a | — | Trace |
| 4 | 8a/AgSbF6 (5 mol%) | — | Complex |
| 5 | — | Select-F | No reaction |
| 6 | 8b | Select-F | 83 |
| 7 | 10a | Select-F | 63 |
| 8 | 10b | Select-F | 29 |
| 9 | 10c | Select-F | 77 |
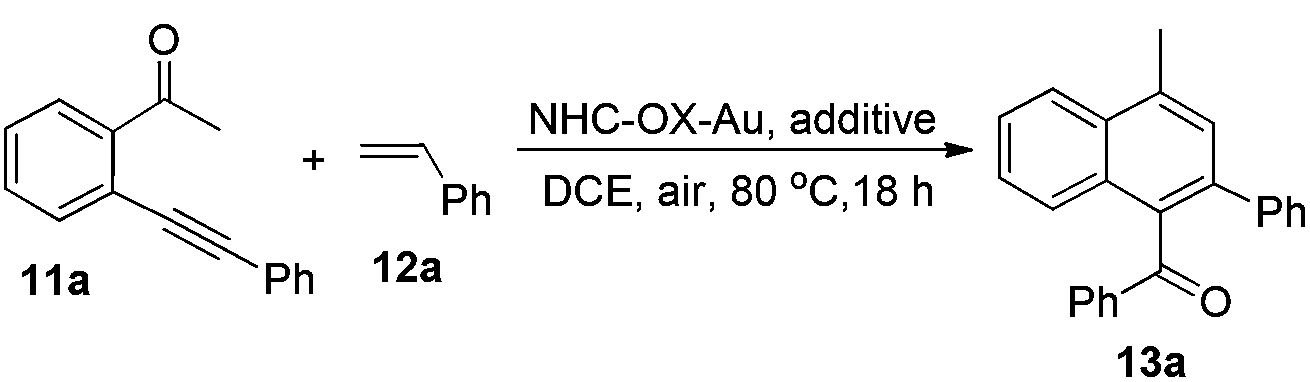
With the optimized reaction conditions in hand, the substrate scope was then examined. Some typical substrates have been chosen to be applied in this catalytic process and the results are shown in Table 2. Firstly, various styrene derivatives could successfully react with enynone 11a to give the desired products in moderate to excellent yields. This reaction was slightly sensitive to the steric hindrance of alkene. For example, the indene participated reaction could only furnish the fused-ring product 13c in 57% yield. Next several enynone derivatives were chosen to test this catalytic system. The terminal groups of the alkyne moiety were changed to n-butyl and cyclopropyl groups, which also smoothly furnished the corresponding products 13d and 13e in good yields. Finally, the substituted groups of the carbonyl moiety were examined. The highly active enynal derivative 11f could be used to react with styrene, affording the corresponding product 13f in 32% yield. The reactivity of more sterically hindered phenyl-substituted enynone 11g was poor, and its reaction should be carried out under an oxygen (1.0 atm) atmosphere for 72 h in 1,2-dichloroethane to give the desired polycyclic aromatic product 13g in 70% yield.
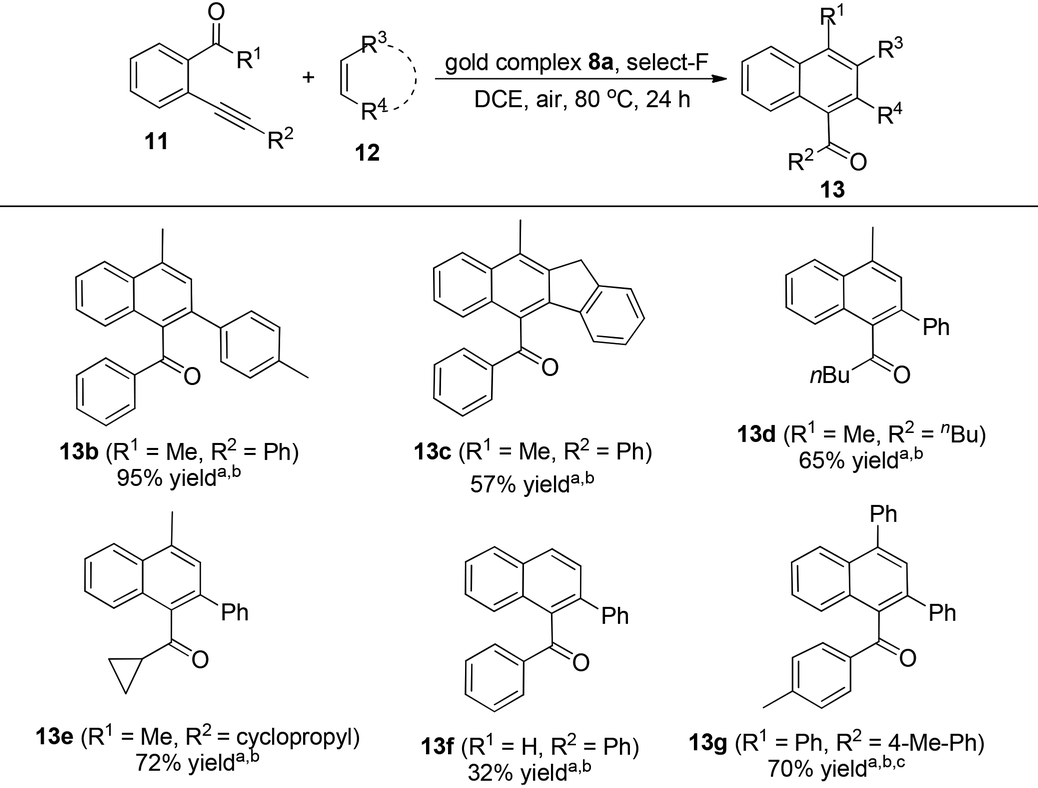
Next, we turned our attention to reveal the accurate structure of NHC–oxazoline gold species. Unfortunately, the NHC–oxazoline gold complex 14 could not be purified by column chromatography or upon recrystallization because they are quite labile and unstable. Firstly, the 19F NMR spectrum of the active gold species showed the existence of a BF4− anion which indicated that the active species probably was an ionic complex (see the ESI†). Next, we started to investigate the coordination state of the oxazoline group through its 1H NMR spectrum because the chemical shift of the Ha and Ha′ atoms ligated with chiral carbon in oxazoline moved to a low field when the oxazoline group connected with the metal atom.17 After combination of complex 8a with 2.0 equivalent of Selectfluor in CDCl3, the mixture was stirred at room temperature for 5 min and then its 1H NMR spectrum was directly measured. The typical double signal sets of complex 8a vanished since in the alkyl area, only one group of alkyl signals could be identified. Besides, the chemical shift of the Ha′′ atom ligated with the chiral carbon atom in the oxazoline moiety moved from the 5.1 ppm to the 5.9 ppm (Fig. 5) which strongly supported the generation of a chelated structure. As for meta-substituted NHC–gold(I) complexes 10, the corresponding Au(I) species was also generated. However, the proof of the generation of chelated NHC–oxazoline gold(I) complexes could not be observed. The 1H NMR spectra of the catalytic species generated by combination of complex 10a and Selectfluor only gave complex mixtures and could not detect any useful information. We assumed that the long distance between the NHC ligand and the oxazoline ligand probably led to the complex coordination mode of active species. This might explain the low catalytic activities of complexes 10 in the reaction.
 | ||
| Fig. 5 The 1H NMR spectra of complex 8a and complex 8a after adding Selectfluor. | ||
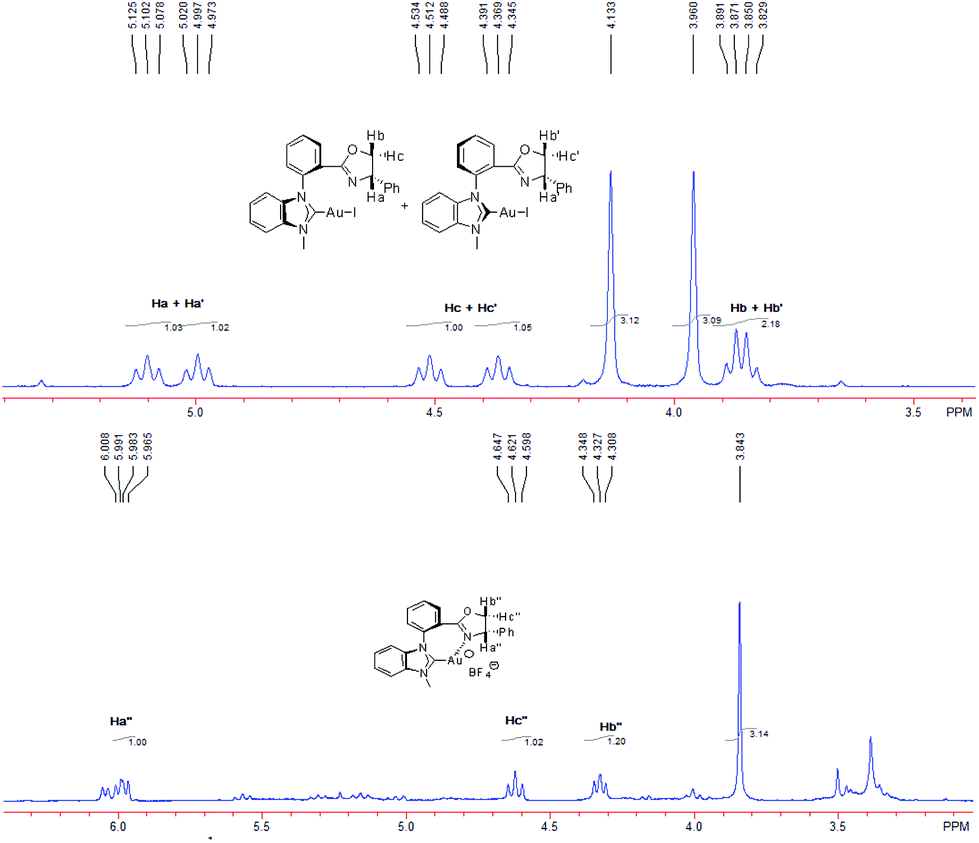
Some control experiments were also carried out to verify our hypothesis (Scheme 4). The chelated NHC–oxazoline gold(I) complex 14 could be also synthesized through the common anion-exchange process. The NHC–gold(I) complex 8a and 1.1 equivalent of AgBF4 were stirred in dichloromethane at room temperature for 0.5 h. After that, the mixture was filtered through the celite pad twice to remove AgI and an excess amount of AgBF4 to furnish gold complex 14 with some inseparable impurities. The high ESI-Mass detection of gold complex 14 showed that the generated gold species’ m/z was 550.1196 as a divalent species (z = 2), which indicated that the chelated NHC–oxazoline gold(I) complex 14 probably was formed as a binuclear structure. Considering of the ESI-Mass result and the coordination mode of the gold(I) atom, we assumed that complex 14 might be connected by two ligands and the gold(I) atoms end to end just like the coordination mode of Hoveyda group's bidentate NHC–oxygen Cu or Ag complexes.18 The axial chirality of complex 14 could not be identified at the present stage. The synthesized crude complex 14 generated from complex 8a and AgBF4 was also used as the catalyst in the same reaction under the standard reaction conditions, furnishing the corresponding product 13a in 90% yield. The bad performance of the in situ generated gold(I) complex shown in Table 1, entry 4 might be attributed to the disturbance of the active silver(I) salt to this reaction.19 Meanwhile, the combination of synthesized NHC–oxazoline gold(I) species 14 with two equivalents of tetrabutylammonium iodide (TBAI) in acetone at room temperature reversibly furnished the NHC–gold(I) complex 8a, which was confirmed by its NMR and ESI-Mass spectra. All these experiments strongly supported our hypothesis.
 | ||
| Scheme 4 The controlled experiments. | ||
In Scheme 5, we proposed a plausible mechanism according to Zhu's previous work and our findings. Firstly, the diastereoisomer of complexes (aR, S)-8a and (aS, S)-8a could be transformed to the chelated NHC–oxazoline gold(I) species 14, but the practical axial chirality of chelated Au(I) species could not be identified at the present stage. In this specific reaction system, the Selectfluor probably oxidized the iodine atom preferentially to generate the ionic NHC–oxazoline gold(I) complexes instead of oxidizing the gold(I) species to gold(III) species. Then the Au(I) species could be coordinated with the triple bond of the alkyne moiety along with the dissociation of the gold(I) atom and the oxazoline group. The nucleophilic attack of carbonyl oxygen to the alkyne moiety then took place to form intermediate II. The intermediate II could undergo a Diels–Alder reaction with styrene to afford intermediate III. The ligated gold atom could be dissociated through a β-metal elimination process to give the cyclic o-quinodimethane IV. The product 13a could be obtained after an aromatization process of intermediate IV in the presence of air atmosphere. The dissociative ionic gold species V could coordinate with another enynone molecule to complete the catalytic cycle.
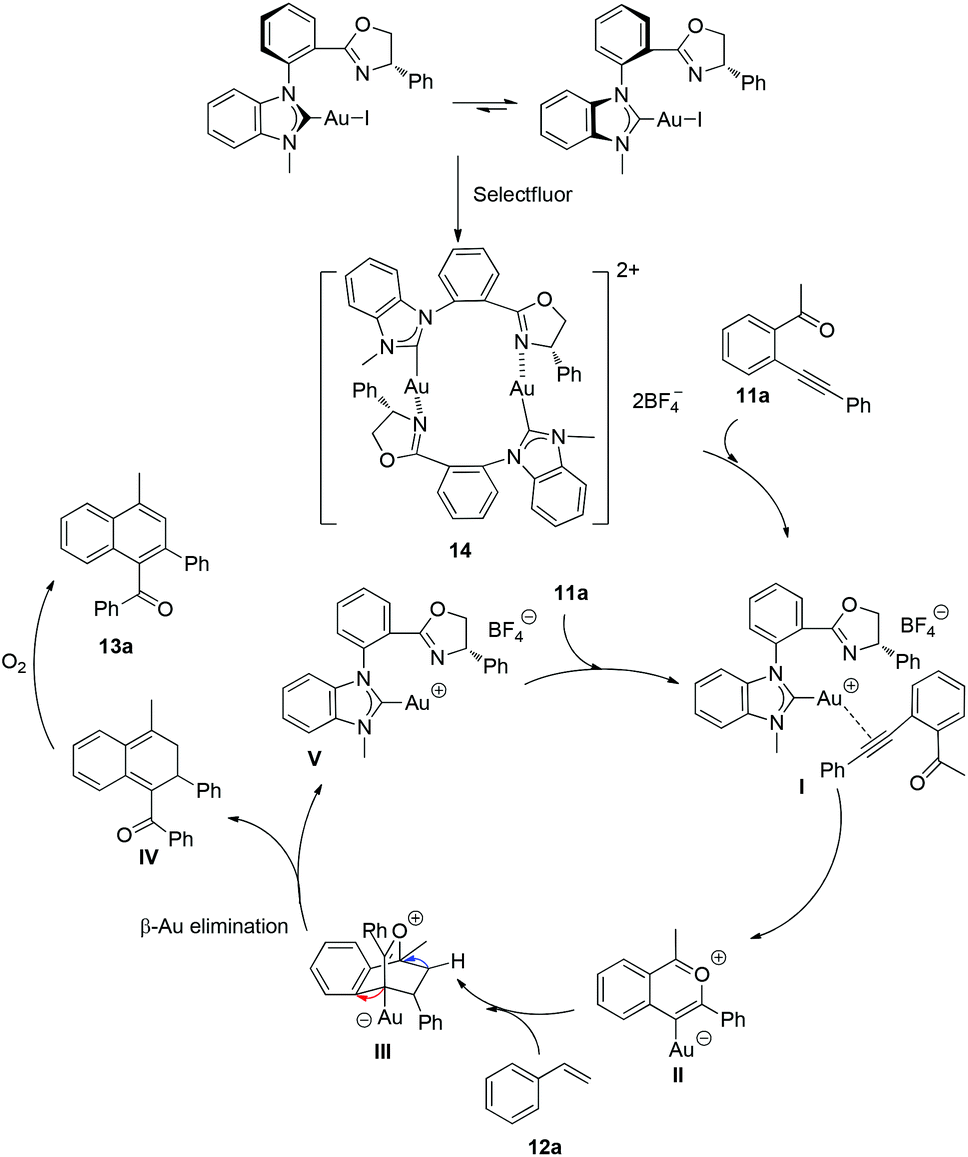 | ||
| Scheme 5 The plausible mechanism for gold-catalyzed cycloaddition/oxidation reaction. | ||
Conclusions
In summary, we have synthesized a series of new ortho- and meta-substituted NHC–oxazoline ligands based on a phenyl scaffold in a six-step pathway. These ligands could be used to coordinate with Au(Me2S)Cl by a simple deprotonation process in the presence of NaOAc. The ortho-substituted NHC–oxazoline gold(I) complexes have been shown as a pair of diastereoisomers due to the appearance of axial chirality. The meta-substituted NHC–oxazoline gold(I) complexes have been exhibited as a single structure. Both structures of the two kinds of NHC–oxazoline gold(I) complexes have been confirmed by X-ray diffraction analyses and the oxazoline group did not connect with the gold atom. These NHC–oxazoline gold(I) complexes could be combined with Selectfluor in situ to catalyze the cycloaddition/oxidation reaction of enynone with alkenes using air as an oxidant to give the naphthalene derivatives in good to excellent yields. In this reaction, a novel function of the Selectfluor reagent in gold-catalyzed reactions was disclosed. The Selectfluor preferentially oxidized the iodine atom to give the ionic chelated binuclear NHC–oxazoline gold(I) complexes instead of oxidizing the gold(I) complexes to gold(III) complexes on the basis of the NMR spectra and mass spectroscopy analysis of complex 14. Some controlled experiments were also carried out to verify our hypothesis. In any event, the role of Selectfluor should be still largely as an oxidant. More investigations on these highly active ionic chelated NHC–oxazoline gold(I) complexes are on-going in our laboratory.Experimental
General
Dichloromethane and acetonitrile were freshly distilled from calcium hydride. Toluene was distilled from sodium (Na) under an argon (Ar) atmosphere. Melting points were measured on a Yanagimoto micro melting apparatus and uncorrected. NMR spectra were recorded with a Varian Mercury Vx or Bruker spectrometer at 400 MHz (1H NMR), 100 or 125 MHz (13C NMR) and 376 MHz (19F NMR) in CDCl3, respectively. Chemical shifts were reported in ppm down field from internal TMS. Optical rotations were determined at 589 nm (sodium D line) using a Perkin Elmer 341 MC Polarimeter. [α]D values are given with units of 10 cm2 deg−1 g−1. Mass spectra were recorded on the HP-5989 instrument by EI/ESI methods. Infrared spectra were recorded on a Perkin-Elmer PE-983 spectrometer with absorption in cm−1. Satisfactory CHN microanalyses were obtained by using a Carlo-Erba 1106 analyzer. X-ray diffraction analysis was performed by using a Bruker Smart-1000 or Bruker SMART APEXIIX-ray diffractometer. Commercially obtained reagents were used without further purification. All reactions were monitored by TLC with Huanghai GF254 silica gel coated plates. Flash column chromatography was carried out using 300–400 mesh silica gel at increased pressure.It is a known compound (0.54 g, 99%),201H NMR (400 MHz, CDCl3, TMS) δ 11.14 (s, 1H), 8.17 (dd, J = 1.6, 8.4 Hz, 1H), 8.04 (dd, J = 1.6, 7.6 Hz, 1H), 7.62–7.60 (m, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.47–7.42 (m, 2H), 7.07–7.03 (m, 1H), 6.96–6.92 (m, 1H), 3.97 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 167.5, 142.3, 139.0, 137.3, 134.7, 133.4, 132.0, 126.6, 121.8, 119.9, 119.0, 118.6, 118.5, 52.3.
It is a known compound (0.46 g, 95%),201H NMR (400 MHz, CDCl3, TMS) δ 8.96 (s, 1H), 7.95 (dd, J = 1.6, 8.0 Hz, 1H), 7.28–7.24 (m, 1H), 7.14–7.06 (m, 2H), 6.82–6.75 (m, 2H), 6.70–6.61 (m, 2H), 3.91 (s, 3H), 3.81 (br, 2H). 13C NMR (100 MHz, CDCl3) δ 169.1, 149.5, 143.3, 134.4, 131.4, 127.7, 127.1, 125.9, 118.8, 116.4, 115.9, 113.7, 110.9, 51.7.
It is a known compound (0.64 g, 85%),211H NMR (400 MHz, CDCl3, TMS) δ 8.11–8.09 (m, 1H), 8.00 (s, 1H), 7.87 (d, J = 7.2 Hz, 1H), 7.74–7.69 (m, 1H), 7.62–7.58 (m, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.33–7.25 (m, 2H), 7.16 (d, J = 7.6 Hz, 1H), 3.48 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 143.1, 135.1, 135.0, 133.2, 131.9, 129.0, 128.32, 128.28, 123.5, 122.4, 120.3, 109.7, 52.3.
Synthesis of ortho NHC–oxazoline gold(I) complex 8
Compound 7 (0.4 mmol), NaOAc (66 mg, 0.8 mmol) and Au(Me2S)Cl (118 mg, 0.4 mmol) were added to a dry flask under argon, then refluxed in MeCN (20 mL) for 2 days. The volatiles were removed under reduced pressure and the residue was purified by silica gel flash column chromatography (eluent: petroleum ether/EtOAc = 2:1) to give products 8.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1].
It is a white solid (199 mg, 85%). M.p. 210.2–212.0 °C. [α]20D −29.3 (c 0.75, CH2Cl2). 1H NMR (400 MHz, CDCl3, TMS) δ 8.20 (t, J = 8.4 Hz, 2H), 7.74–7.59 (m, 6H), 7.48–7.29 (m, 9H), 7.15–7.09 (m, 7H), 6.64 (d, J = 6.0 Hz, 2H), 5.08 (t, J = 9.2 Hz, 1H), 4.98 (t, J = 9.2 Hz, 1H), 4.49 (t, J = 8.8 Hz, 1H), 4.35 (t, J = 8.8 Hz, 1H), 4.11 (s, 3H), 3.94 (s, 3H), 3.86 (d, J = 8.0, 1H), 3.82 (d, J = 8.4, 1H). 13C NMR (100 MHz, CDCl3) δ 188.9 (188.8), 161.5 (161.2), 141.6 (141.3), 135.5 (134.9), 133.2 (133.0), 132.6 (132.5), 132.4, 131.7 (131.5), 130.3 (130.2), 129.7 (129.2), 128.6 (128.4), 127.5 (127.2), 126.9 (126.2), 126.1 (125.7), 124.8 (124.5), 111.9 (111.8), 111.2 (111.0), 74.0 (73.99), 70.2 (69.9), 34.9 (34.6). IR (KBr) ν 2924, 2853, 1644, 1601, 1504, 1455, 1387, 1358, 1311, 1263, 1243, 1204, 1096, 1057, 1035, 954, 895, 787, 699, 661 cm−1. MS (ESI) m/z: 678.0 (M+ + H). HRMS (ESI) Calcd for C23H20N3OAuI requires: 678.0317. Found: 678.0303.
:1.2].
It is a yellow solid (201 mg, 91%). M.p. 156.0–157.6 °C. [α]20D −58.1 (c 0.33, CH2Cl2). 1H NMR (400 MHz, CDCl3, TMS) δ 8.10 (d, J = 7.2 Hz, 1.83H), 7.71–7.42 (m, 9.2H), 7.39–7.31 (m, 1.83H), 7.11 (d, J = 8.0 Hz, 1H), 7.03 (d, J = 8.0 Hz, 0.83H), 4.15–4.12 (m, 6.5H), 3.92 (t, J = 8.8 Hz, 0.83H), 3.75–3.58 (m, 3.8H), 1.58–1.51 (m, 1H), 1.19–1.12 (m, 0.85H), 0.69 (d, J = 6.8 Hz, 6H), 0.57 (d, J = 6.8 Hz, 2.5H), 0.44 (d, J = 6.8 Hz, 2.5H). 13C NMR (100 MHz, CDCl3) δ 188.6 (188.4), 160.0 (159.5), 135.3 (135.2), 134.8 (134.7), 133.1 (132.9), 132.1 (131.9), 131.3 (131.1), 130.1 (129.5), 129.1, 126.4 (125.8), 124.8 (124.6), 124.5 (124.4), 111.9 (111.8), 110.94 (110.89), 73.1 (72.7), 70.3 (69.9), 34.9 (34.8), 32.9 (32.7), 18.74 (18.67), 18.5 (18.1). IR (KBr) ν 2960, 2924, 1651, 1453, 1391, 1261, 1102, 1022, 1009, 913, 801, 752, 670 cm−1. MS (ESI) m/z: 644.0 (M+ + H). HRMS (ESI) Calcd for C20H22N3OAuI requires: 644.0473. Found: 644.0460.
:1) to give products 10.
:1].
It is a white solid (199 mg, 85%). M.p. 210.2–212.0 °C. [α]20D −29.3 (c 0.75, CH2Cl2). 1H NMR (400 MHz, CDCl3, TMS) δ 8.20 (t, J = 8.4 Hz, 2H), 7.74–7.59 (m, 6H), 7.48–7.29 (m, 9H), 7.15–7.09 (m, 7H), 6.64 (d, J = 6.0 Hz, 2H), 5.08 (t, J = 9.2 Hz, 1H), 4.98 (t, J = 9.2 Hz, 1H), 4.49 (t, J = 8.8 Hz, 1H), 4.35 (t, J = 8.8 Hz, 1H), 4.11 (s, 3H), 3.94 (s, 3H), 3.86 (d, J = 8.0, 1H), 3.82 (d, J = 8.4, 1H). 13C NMR (100 MHz, CDCl3) δ 188.9 (188.8), 161.5 (161.2), 141.6 (141.3), 135.5 (134.9), 133.2 (133.0), 132.6 (132.5), 132.4, 131.7 (131.5), 130.3 (130.2), 129.7 (129.2), 128.6 (128.4), 127.5 (127.2), 126.9 (126.2), 126.1 (125.7), 124.8 (124.5), 111.9 (111.8), 111.2 (111.0), 74.0 (73.99), 70.2 (69.9), 34.9 (34.6). IR (KBr) ν 2924, 2853, 1644, 1601, 1504, 1455, 1387, 1358, 1311, 1263, 1243, 1204, 1096, 1057, 1035, 954, 895, 787, 699, 661 cm−1. MS (ESI) m/z: 678.0 (M+ + H). HRMS (ESI) Calcd for C23H20N3OAuI requires: 678.0317. Found: 678.0303.
:1.2].
It is a yellow solid (201 mg, 91%). M.p. 156.0–157.6 °C. [α]20D −58.1 (c 0.33, CH2Cl2). 1H NMR (400 MHz, CDCl3, TMS) δ 8.10 (d, J = 7.2 Hz, 1.83H), 7.71–7.42 (m, 9.2H), 7.39–7.31 (m, 1.83H), 7.11 (d, J = 8.0 Hz, 1H), 7.03 (d, J = 8.0 Hz, 0.83H), 4.15–4.12 (m, 6.5H), 3.92 (t, J = 8.8 Hz, 0.83H), 3.75–3.58 (m, 3.8H), 1.58–1.51 (m, 1H), 1.19–1.12 (m, 0.85H), 0.69 (d, J = 6.8 Hz, 6H), 0.57 (d, J = 6.8 Hz, 2.5H), 0.44 (d, J = 6.8 Hz, 2.5H). 13C NMR (100 MHz, CDCl3) δ 188.6 (188.4), 160.0 (159.5), 135.3 (135.2), 134.8 (134.7), 133.1 (132.9), 132.1 (131.9), 131.3 (131.1), 130.1 (129.5), 129.1, 126.4 (125.8), 124.8 (124.6), 124.5 (124.4), 111.9 (111.8), 110.94 (110.89), 73.1 (72.7), 70.3 (69.9), 34.9 (34.8), 32.9 (32.7), 18.74 (18.67), 18.5 (18.1). IR (KBr) ν 2960, 2924, 1651, 1453, 1391, 1261, 1102, 1022, 1009, 913, 801, 752, 670 cm−1. MS (ESI) m/z: 644.0 (M+ + H). HRMS (ESI) Calcd for C20H22N3OAuI requires: 644.0473. Found: 644.0460.
:1) to give products 10.
Acknowledgements
We thank the Shanghai Municipal Committee of Science and Technology (11JC1402600), the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21372250, 21372075, 21121062, 21302203, 20732008 and 21572052).Notes and references
- For some reviews, see: (a) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (b) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (c) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (d) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (e) R. A. Widenhoefer, Chem. – Eur. J., 2008, 14, 5382 CrossRef CAS PubMed; (f) S. Sengupta and X. Shi, ChemCatChem, 2010, 2, 609 CrossRef CAS PubMed; (g) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC.
- For some reviews, see: (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (b) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (c) V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (d) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed; (e) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef PubMed; (f) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed; (g) F. Wang, L. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef CAS PubMed.
- (a) P. Klahn and S. F. Kirsch, ChemCatChem, 2011, 11, 694 Search PubMed; (b) D. Benitez, N. D. Shapiro, E. Tkatchouk, Y. Wang, W. A. Goddard III and F. D. Toste, Nat. Chem., 2009, 1, 482 CrossRef CAS PubMed; (c) S. Lopez, E. Herrero-Gomez, P. Perez-Galan, C. Nieto-Oberhuber and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 6029 CrossRef CAS PubMed.
- (a) M. T. Lee and C. H. Hu, Organometallics, 2004, 23, 976 CrossRef CAS; (b) J. Huang, L. Jafarpour, A. C. Hillier, E. D. Stevens and S. P. Nolan, Organometallics, 2001, 20, 2878 CrossRef CAS.
- (a) G. Ciancaleoni, L. Belpassi, D. Zuccaccia, F. Tarantelli and P. Belanzoni, ACS Catal., 2015, 5, 803 CrossRef CAS; (b) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC; (c) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778 CrossRef CAS PubMed; (d) P. de Fremont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551 CrossRef CAS PubMed; (e) A. S. K. Hashmi, D. Riedel, M. Rudolph, F. Rominger and T. Oeser, Chem. – Eur. J., 2012, 18, 3827 CrossRef CAS PubMed; (f) R. Manzano, F. Rominger and A. S. K. Hashmi, Organometallics, 2013, 32, 2199 CrossRef CAS.
- W. P. Fehlhammer and K. Bartel, Angew. Chem., Int. Ed. Engl., 1974, 13, 599 Search PubMed.
- P. Kuhlkamp, H. G. Raubenheimer, J. S. Field and M. Desmet, J. Organomet. Chem., 1998, 552, 69 CrossRef CAS.
- (a) M. Hoffmann, J. Weibel, P. de Frémont, P. Pale and A. Blanc, Org. Lett., 2014, 16, 908 CrossRef CAS PubMed; (b) J. P. Reeds, A. C. Whitwood, M. P. Healy and I. J. S. Fairlamb, Organometallics, 2013, 32, 3108 CrossRef CAS; (c) H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591 CrossRef CAS.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3651 CrossRef PubMed.
- (a) Q. Y. Wang, S. E. Motika, N. G. Akhmedov, J. L. Peterson and X. D. Shi, Angew. Chem., Int. Ed., 2014, 53, 5418 CrossRef CAS PubMed; (b) Q. Y. Wang, S. Aparaj, N. G. Akhmedov, J. L. Peterson and X. D. Shi, Org. Lett., 2012, 14, 1334 CrossRef CAS PubMed; (c) L. P. Liu and G. B. Hammond, Org. Lett., 2010, 12, 4640 CrossRef CAS PubMed; (d) H. F. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. D. Shi, J. Am. Chem. Soc., 2009, 131, 12100 CrossRef CAS PubMed.
- (a) N. D. Shapiro, Y. Shi and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 11654 CrossRef CAS PubMed; (b) N. Marien, B. Brigou, B. Pinter, F. De Proft and G. Verniest, Org. Lett., 2015, 17, 270 CrossRef CAS PubMed; (c) D. Li, W. Rao, G. L. Tay, B. J. Ayers and P. W. H. Chan, J. Org. Chem., 2014, 79, 11301 CrossRef CAS PubMed; (d) M. Garzon and P. W. Davies, Org. Lett., 2014, 16, 4850 CrossRef CAS PubMed; (e) M. W. Johnson, A. G. Di Pasquale, R. G. Bergman and F. D. Toste, Organometallics, 2014, 33, 4169 CrossRef CAS PubMed; (f) E. P. A. Talbot, M. Richardson, J. M. McKenna and F. D. Toste, Adv. Synth. Catal., 2014, 356, 687 CrossRef CAS PubMed; (g) Y. Zhang, B. Feng and C. Zhu, Org. Biomol. Chem., 2012, 10, 9137 RSC.
- (a) J. Ma, H. Jiang and S. Zhu, Org. Lett., 2014, 16, 4472 CrossRef CAS PubMed; (b) G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112 CrossRef CAS PubMed; (c) E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard III and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293 CrossRef CAS PubMed.
- (a) F. Wang, S. Li, M. Qu, M. Zhao, L. Liu and M. Shi, Chem. Commun., 2011, 47, 12813 RSC; (b) F. Wang, S. Li, M. Qu, M. Zhao, L. Liu and M. Shi, Beilstein. J. Org. Chem., 2012, 8, 726 CrossRef CAS PubMed.
- P. Gu, Q. Xu and M. Shi, Synlett, 2013, 1255 CAS.
- (a) N. Asao, K. Takahashi, S. Lee, T. Kasahara and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 12650 CrossRef CAS PubMed; (b) N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921 CrossRef CAS PubMed; (c) N. Asao, H. Aikawa and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 7458 CrossRef CAS PubMed; (d) N. Asao, K. Sato and Y. Yamamoto, J. Org. Chem., 2005, 70, 3682 CrossRef CAS PubMed; (e) N. Asao and H. Aikawa, J. Org. Chem., 2006, 71, 5249 CrossRef CAS PubMed; (f) S. Zhu, Z. Zhang, X. Huang, H. Jiang and Z. Guo, Chem. – Eur. J., 2013, 19, 4695 CrossRef CAS PubMed.
- S. Zhu, H. Huang, Z. Zhang, T. Ma and H. Jiang, J. Org. Chem., 2014, 79, 6113 CrossRef CAS PubMed.
- Selected papers, see: (a) Y. Imai, W. Zhang, T. Kida, Y. Nakatsuji and I. Ikeda, Tetrahedron Lett., 1997, 38, 2681 CrossRef CAS; (b) F. Wang, G. Yang, Y. J. Zhang and W. Zhang, Tetrahedron, 2008, 64, 9413 CrossRef CAS PubMed; (c) F. Wang, Y. J. Zhang, H. Wei, J. Zhang and W. Zhang, Tetrahedron Lett., 2007, 48, 4083 CrossRef CAS PubMed; (d) F. Wang, Y. Zhang, G. Yang and W. Zhang, Tetrahedron Lett., 2007, 48, 4179 CrossRef CAS PubMed.
- (a) J. J. Van Veldhuizen, S. B. Garber, J. S. Kinsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 4954 CrossRef CAS PubMed; (b) A. O. Larsen, W. Leu, C. N. Oberhuber, J. E. Campbell and A. H. Hoveyda, J. Am. Chem. Soc., 2004, 126, 11130 CrossRef CAS PubMed; (c) M. K. A. Brown, T. L. May, C. A. Baxter and A. H. Hoveyda, Angew. Chem., Int. Ed., 2007, 46, 1097 CrossRef CAS PubMed; (d) B. Jung and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 1490 CrossRef CAS PubMed.
- (a) R. X. Liao, T. M. Ma and S. F. Zhu, Org. Lett., 2014, 16, 4412 CrossRef PubMed; (b) V. Reddy, A. S. Jadhav and R. V. Anand, Org. Biomol. Chem., 2015, 13, 3732 RSC; (c) T. Godet, C. Vaxelaire, C. Michel, A. Milet and P. Belmont, Chem. – Eur. J., 2007, 13, 5632 CrossRef CAS PubMed.
- R. A. Bunce and J. E. Schammerhorn, J. Heterocycl. Chem., 2006, 43, 1031 CrossRef CAS PubMed.
- L. A. Crawford, M. Ieva, H. McNab and S. Parsons, Dalton Trans., 2010, 39, 7147 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: DETAILS. CCDC 973292 for 8a and CCDC 950237 for 10c. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00155b |
| This journal is © the Partner Organisations 2015 |