
Pincer CNC bis-N-heterocyclic carbenes: robust ligands for palladium-catalysed Suzuki–Miyaura arylation of bromoanthracene and related substrates†
V. M.
Zende
a,
C.
Schulzke
b and
A. R.
Kapdi
*a
aDepartment of Chemistry, Institute of Chemical Technology, Matunga, Mumbai-400019, India. E-mail: ar.kapdi@ictmumbai.edu.in
bInstitute fur Biochemie, Ernst-Moritz-Arndt Universität Greifswald, Felix-Hausdorff-Straße 4, D-17487 Greifswald, Germany
First published on 2nd September 2015
Abstract
Novel pincer-type N-heterocyclic carbene ligands have been synthesised and characterised by FAB-MS and single crystal X-ray analysis. The ligands have proved to be highly active towards palladium-catalysed Suzuki–Miyaura cross-coupling of bromoanthracene and related substrates allowing the efficient and easy access of molecules for possible OLED applications.
Introduction
Palladium-catalysed cross-coupling reactions have emerged in the last few decades as the most powerful C–C bond forming processes with applications extending from simple arenes to complex biological targets.1 This has been achieved over the years via the employment of highly active σ-donor ligands such as bulky electron-rich phosphines or N-heterocyclic carbenes.2 The latter have found a great number of applications3 not just as ligands in metal-mediated processes4 but also as useful organocatalysts.5Since the first reports of their isolation by Arduengo,6 N-heterocyclic carbenes have gained enormous interest in terms of their organometallic chemistry and versatile nature. Important contributions from the group of Herrmann7 and Nolan8 have pointed towards better σ-donating properties of the NHCs over the phosphine donor ligands, making them all more useful for further catalytic applications. Nolan and Organ have in recent years led the way for the extensive application of NHCs in palladium-catalyzed cross-coupling reactions, either as added ligands or coordinated Pd-complexes.9 Besides this, NHCs have also found a wide variety of applications that have been highlighted in several recent reviews.10
Imidazole-based N-heterocyclic carbenes4a,6 have over the years proved to be effective ligands for a large number of C–C and C–N bond forming reactions due to their intrinsic properties of forming thermally stable metal complexes11 either in solution or in the preformed state. The thermal stability can be further enhanced by the introduction of pincer-type chelation12 which has already proved to be successful in the phosphine analogues. In the literature several research groups have reported on the synthesis of palladium complexes of CNC pincer-type carbene ligands13 and their application towards catalysing cross-coupling reactions.14 In order to achieve even better applicability and sustainability of this chemistry, developing more robust and highly active pincer-type CNC carbene ligands is requisite.
Palladium-catalyzed Suzuki–Miyaura cross-coupling15 is one such reaction that has shown rapid growth owing to its use as a sharpening tool for newer catalytic systems including pincer-type NHC ligands. Besides serving as a powerful methodology for the synthesis of simple molecules, it has also played a significant role in the development of functional molecules with varied applications. One of the areas that has benefitted immensely is the field of Organic Light Emitting Diodes (OLEDs)16 which are most commonly synthesized using Suzuki–Miyaura cross-coupling reactions.17 Key features of OLEDs that contribute towards the fabrication of full-color OLED displays involve high red, green and blue electroluminescence possessing good thermal properties and longer lifetimes.18 This might be achieved by incorporating organic molecules that specifically help enhance these properties for better OLED displays.
Anthracene as a structural motif has found wide OLED application due to the unique blend of properties such as chemical and electron-rich structure, and a low electronic band gap exhibiting strong blue electroluminescence.17 The incorporation of anthracene into the OLED main structure has helped solve problems associated with the formation of films with good optical quality. Several other advantages are also associated with the presence of the anthracene moiety with palladium-catalyzed Suzuki–Miyaura cross-coupling reactions providing the synthetic pathway for developing robust OLEDs with enhanced qualities. Although these routes have provided researchers with the possibility of obtaining OLEDs, they suffer from a number of problems such as low reactivity and high concentration of the employed Pd catalyst.17a–e Even today a general catalytic system is not available for accessing such synthetically important molecules routinely.
To address this issue we herein report the synthesis of novel and particularly robust pincer bis-carbene ligands which were characterized by NMR spectroscopic techniques plus X-ray crystallographic analysis of a single crystal obtained for one of the ligands. Thermogravimetric analysis of the ligands suggests exceptional thermal stability. The bis-carbene ligands were later employed for the development of an efficient protocol for the palladium-catalyzed Suzuki–Miyaura cross-coupling of 9-bromoanthracene and related structures with an overall catalyst concentration that is as low as 0.1 mol%. Synthesis of 9,10-substituted anthracene was also achieved under milder reaction conditions in good to high yields.
Results and discussion
Synthesis and characterization of pincer bis-carbene NHC ligands
The synthesis of carbene precursors 2,6-bis(1-phenylimidazolium-3-yl)pyridine dibromide (1A), 2,6-bis(1-(p-tolyl)imidazolium-3-yl)pyridine dibromide (1B) and 2,6-bis(1-(o-tolyl)imidazolium-3-yl)pyridine dibromide (1C) was carried out in two simple steps in good to high yields. First step of the synthesis involved the N-arylation of imidazoles via Cu-catalyzed coupling with arylboronic acids which was achieved by following a literature protocol developed by Xie and co-workers.19N-Phenylimidazole, N-(4-tolyl)imidazole and N-(2-tolyl)imidazole were obtained in yields of 79%, 70% and 56% respectively from the corresponding arylboronic acids. Having synthesized the substituted imidazoles the next step was the quarternization with 2,6-dibromopyridine using a slightly modified protocol of the report published by Sakamoto and co-workers.13c The reaction of N-arylimidazoles with 2,6-dibromopyridine was carried out under microwave radiation for 15 minutes at 100 psi pressure and 200 °C temperature under solvent-free conditions (Scheme 1). This resulted in the formation of the carbene precursors in good yields.
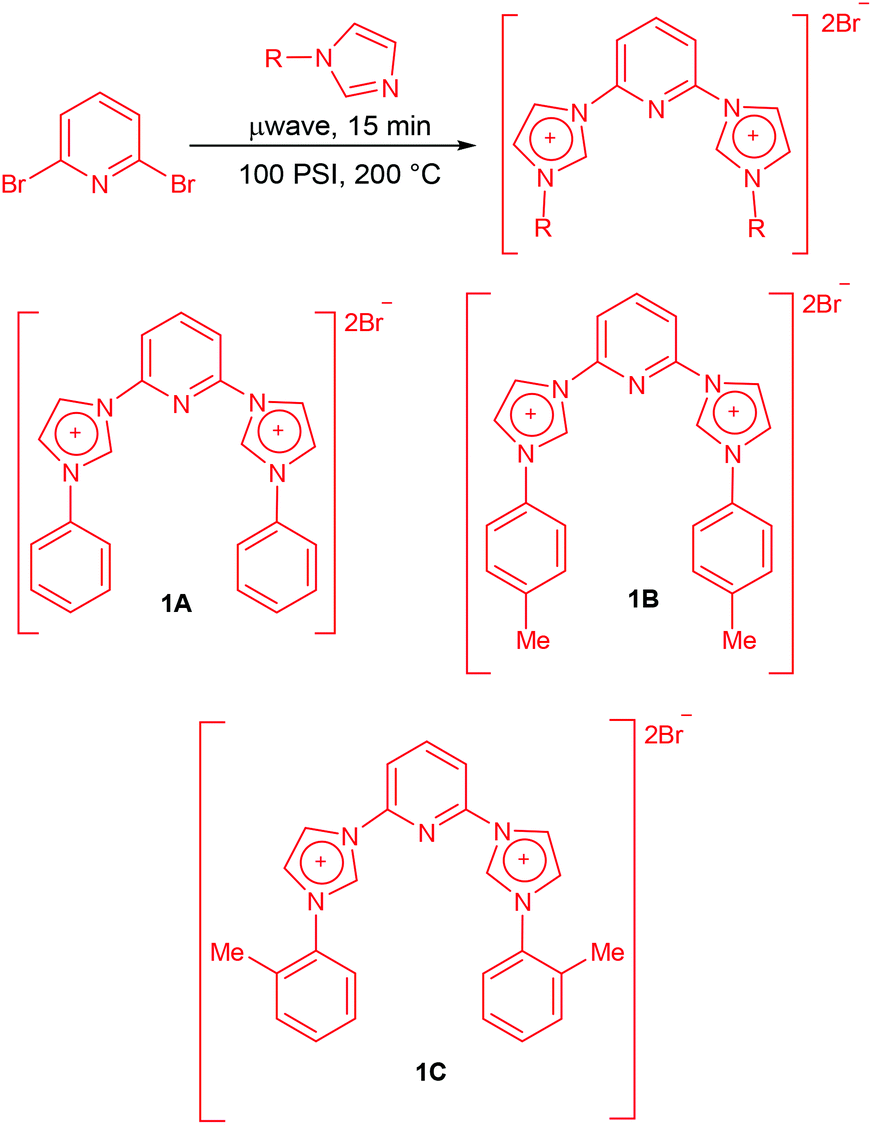 | ||
| Scheme 1 Synthesis of novel N-heterocyclic carbene ligands. | ||
The bis-carbene precursors thus obtained were characterized by spectroscopic techniques. The formation of the bis-carbene precursors was evident from the 1H and 13C NMR spectra showing characteristic peaks for N–CH–N (carbene proton) as represented in Table 1. Small differences were found in the case of the 13C values, however, 1H NMR values show appreciable differences suggesting the influence of substituent based electronics on the N–CH–N linkage.
| Bis-carbene precursor | 1A | 1B | 1C |
|---|---|---|---|
| 1H-δ value for N–CH–N (ppm) | 10.9 | 10.8 | 10.7 |
| 13C-δ value for N–CH–N (ppm) | 145.7 | 145.7 | 145.7 |
Next we carried out fast atom bombardment analysis of the ligands which revealed the following details in support of the proposed structures. Fast Atom Bombardment Mass Spectrometry (FAB-MS)20 is a highly versatile and relatively mild analytical technique optimal for the analysis of ligand and complex salts. Accordingly, in all three cases the fragment showing the loss of both the bromide anions was observed along with the presence of a mono-brominated fragment (lower in abundance). Fragmentation patterns also revealed the presence of N-arylimidazoles (in some cases assuming base peak intensity). A representative FAB spectrum with the fragmentation pattern is shown below for 3,3′-(pyridine-2,6-diyl)bis(1-(p-tolyl)-1H-imidazol-3-ium) bromide (1B) (see Fig. 1).
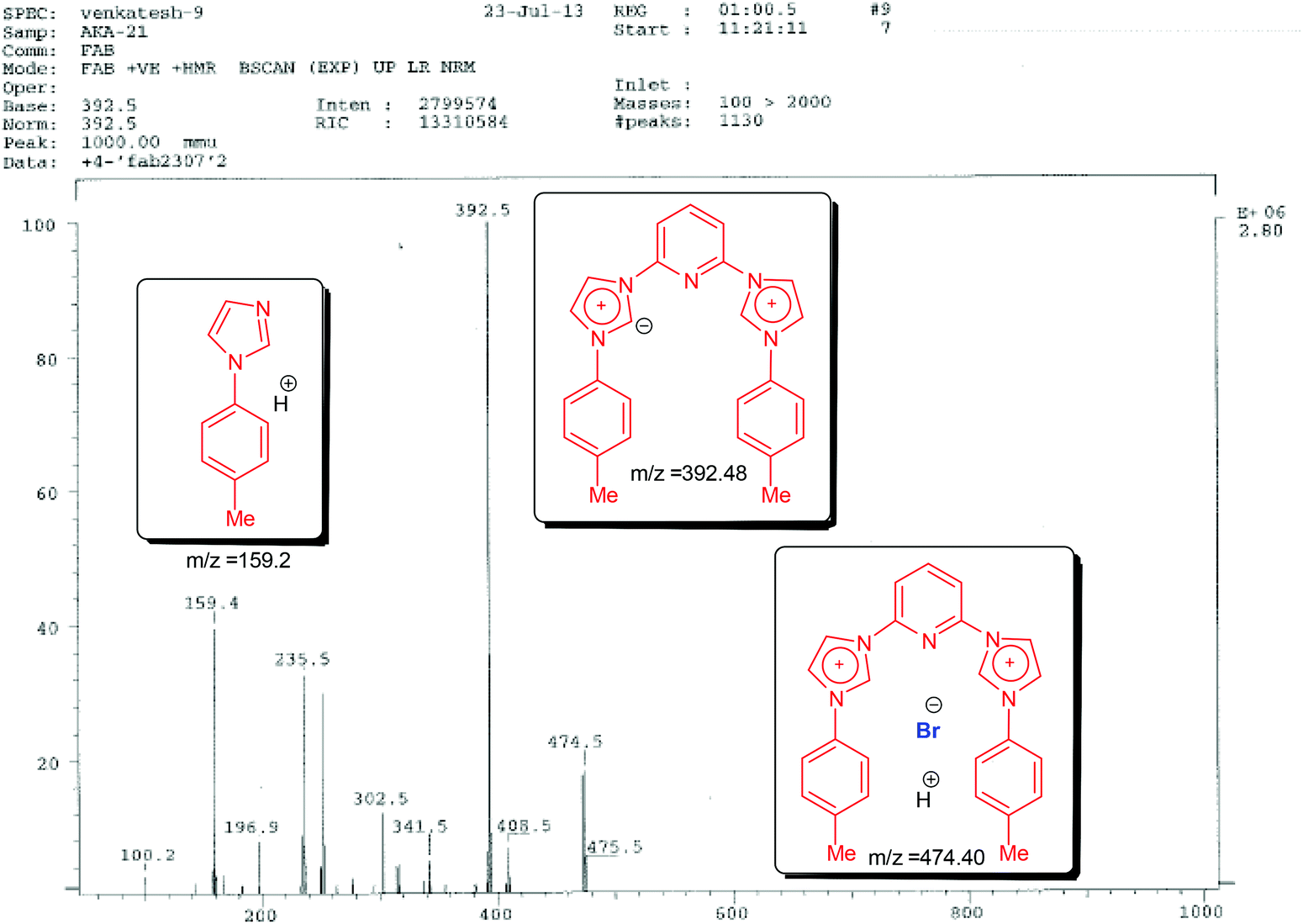 | ||
| Fig. 1 Fast atom bombardment analysis for ligand 1B. | ||
Further proof for the molecular structures of the bis-carbene precursors was provided by growing crystals and obtaining conclusive evidence by X-ray structural analysis. Several attempts to crystallize the precursors failed. However, in the case of ligand 1B it was possible to obtain the X-ray crystal structure from single crystals grown from MeOH.211B crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the large heterocyclic bis-imidazolium cation cocrystallized with two water molecules and two bromides in the asymmetric unit.
with the large heterocyclic bis-imidazolium cation cocrystallized with two water molecules and two bromides in the asymmetric unit.
The two imidazolium rings and the central pyridine are almost perfectly coplanar with the torsion angles of only 3.7° and 0.8° between pyridine and the two imidazolium moieties on both sides. The torsion angles between the phenyl substituents and the imidazolium rings, they are connected to, are a bit larger with 18.0° and 22.7°. In addition the N–C bonds connecting pyridine with the imidazolium rings (N2–C11; N4–C15) and the imidazolium with the phenyl rings (N1–C5; N5–C19) are slightly shorter than typical C–N single bonds with bond lengths ranging from 1.424 to 1.435 Å (1.47 Å is the sum of covalent radii). All this suggests that to some extent a more than usually delocalized π-system is present.
This is further supported by comparing it to the closest related crystallographically characterised molecule in which a pyridine binds an imidazolium ring which binds a mesityl substituent.22 There, both the torsion angles are considerably larger than in the present case which is for the imidazolium mesityl connection (74.6°) certainly due to steric restraints caused by the mesityl's bulkiness. For the second connection between imidazolium and pyridine (29.1°), however, there are no steric effects which could be responsible, therefore it has to be based on the electronic structure, which apparently is different from the one found here. Interestingly, despite having been measured at even lower temperature all bond lengths in the known structure are longer than the ones found here. In the imidazolium rings all C–N bonds between the nitrogen atoms and the C2 groups are considerably larger (1.383–1.392 Å) than those of the “single” carbon atoms (C10, C18) ranging from 1.327 to 1.345 Å. This suggests a tendency to a certain localization of double bond character within the imidazolium rings. No such effect was found for the two phenyl substituents or the pyridine ring. With the exception of a particularly short bond between C6 and C7 of one phenyl ring with 1.358 Å the C–C bonds range from 1.374 to 1.398 Å and the C–N bonds are 1.321 and 1.331 Å long respectively and all are evenly distributed throughout the moieties with no apparent pattern.
Finally we performed thermal stability studies on the bis-carbene precursors 1A–C and were compared using thermal gravimetric analysis/differential scanning calorimetry (see the ESI† for graphs). A trend observed for all ligands 1A–C suggests first a loss of bromine atoms. The thermal stability of all the bis-carbene precursors was found to be very high in the range of 400 °C or above (Fig. 2).
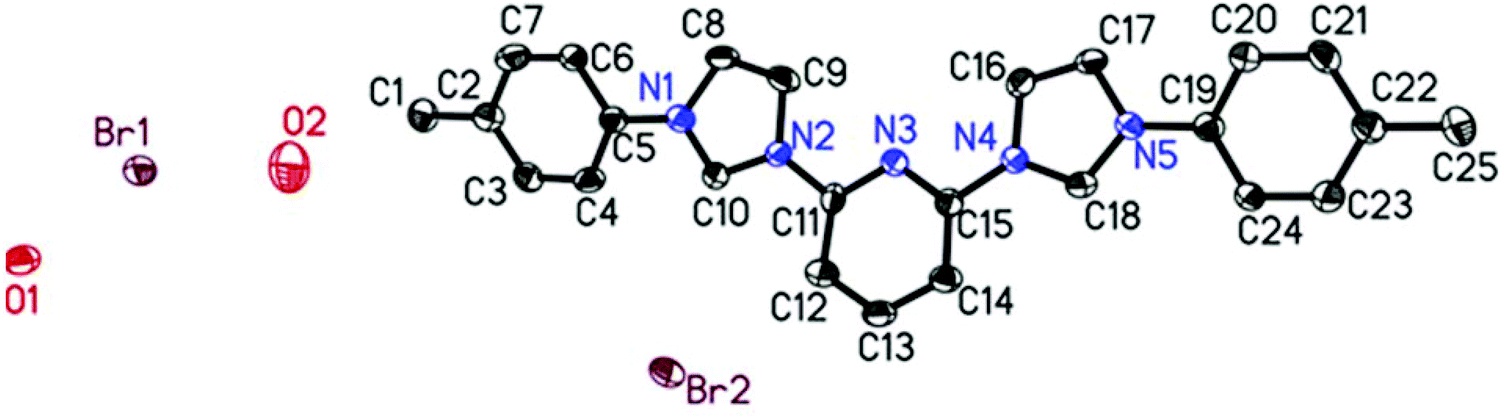 | ||
| Fig. 2 Single crystal X-ray structure of N-heterocyclic carbene ligand 1B (H atoms removed for clarity). Ellipsoids are shown with 50% probability. | ||
Palladium-catalyzed Suzuki–Miyaura arylation of anthracene and related substrates
Once the ligands were characterised, their activity towards catalysing palladium-catalysed Suzuki–Miyaura cross-coupling of bromoanthracenes and related substrates was analysed. Initially, we tested the bis-carbene precursors for the arylation of 9-bromoanthracene with different arylboronic acids. At the outset of these studies, the bis-carbene precursors were screened first for the arylation reaction at 0.1 mol% catalyst loading in a toluene/H2O system at 80 °C (Scheme 2 – isolated yields). It was observed that the bis-carbene precursor 1A (88%) furnished the cross-coupled product in higher yield than the other two (1B-60% and 1C-50%). The possibility of Pd(OAc)2 catalysing the coupling reaction without an added ligand at low catalyst loading is well documented in the literature.23 Therefore, to test the validity of the result obtained with ligand 1A, we performed a ligand-free reaction under identical conditions. The ligand-free reaction provided the desired product only in poor yield (30%) emphasising the bis-carbene precursor enhanced activity in combination with Pd(OAc)2 in the present study.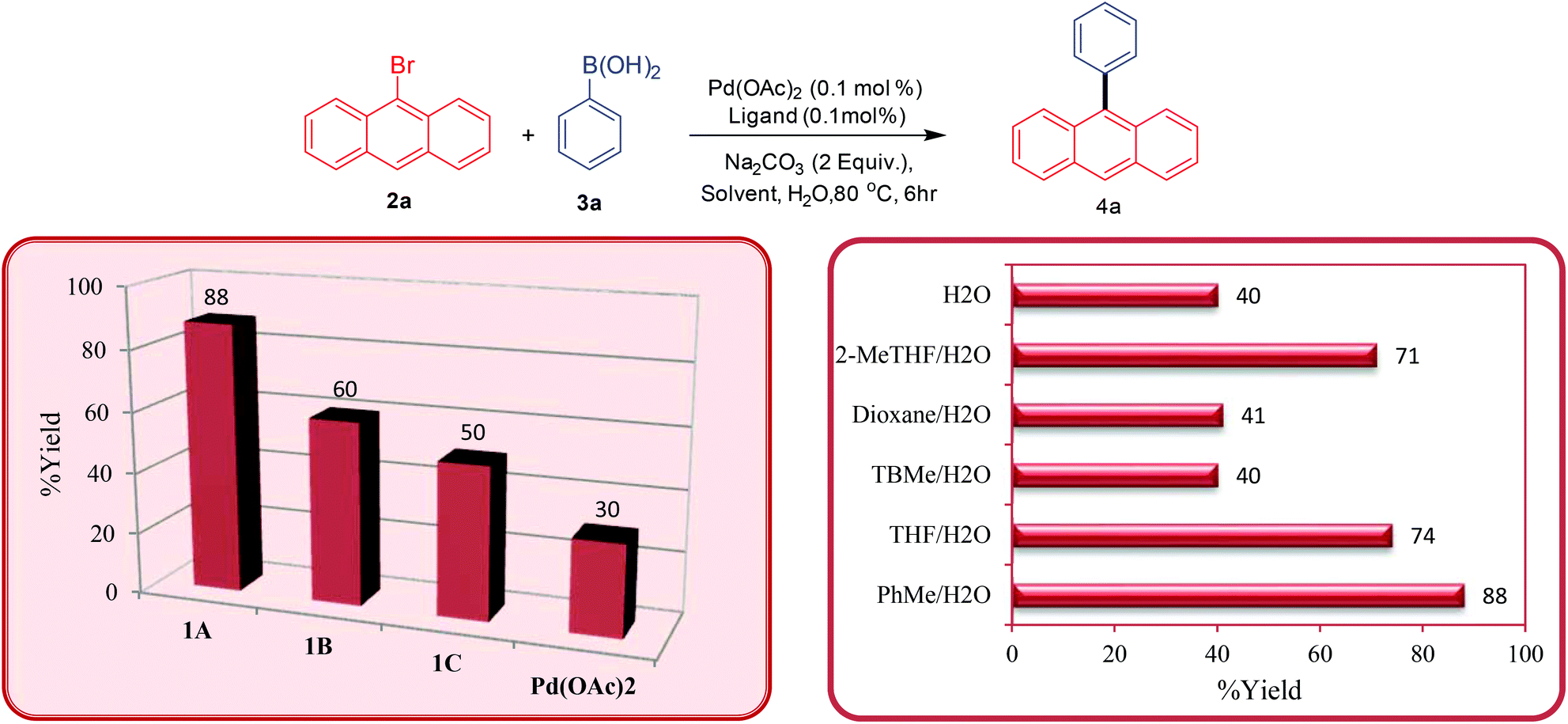 | ||
| Scheme 2 Screening studies for arylation of 9-bromoanthracene using NHCs 1A–C. | ||
The employment of traditional phosphine ligands such as PPh3 or P(o-Tol)3 (either 1.0 equiv. wrt Pd or 2.0 equiv.) also failed to enhance the reactivity compared to NHC ligands (see the complete screening table in the ESI†).
In order to optimise the conditions, the effect of solvent on the catalytic reaction was investigated. The reaction when performed in neat water gave poor yields of the product due to the lower solubility of the starting substrates. A combination of a variety of organic solvents with water was employed, however, the combination of the PhMe/H2O (88%) solvent system proved to be the most effective in catalysing the cross-coupling reaction, although decent yields were also obtained for the THF/H2O (74%) and the 2-MeTHF/H2O systems (71%). The combination of PhMe/H2O was identified as the solvent of choice for all further tests and applications.
The next step was to utilise the catalytic conditions developed towards the synthesis of various 9-(hetero)aryl anthracenes (Scheme 3). A variety of aryl and heteroaryl boronic acids were employed as the coupling partners. The reactivity observed for aryl boronic acids with electron-donating and electron-withdrawing substituents was comparable and no specific substituent effect was observed. The cross-coupling with one heteroaryl boronic acid (furan-2-boronic acid) also proceeded with good yields.
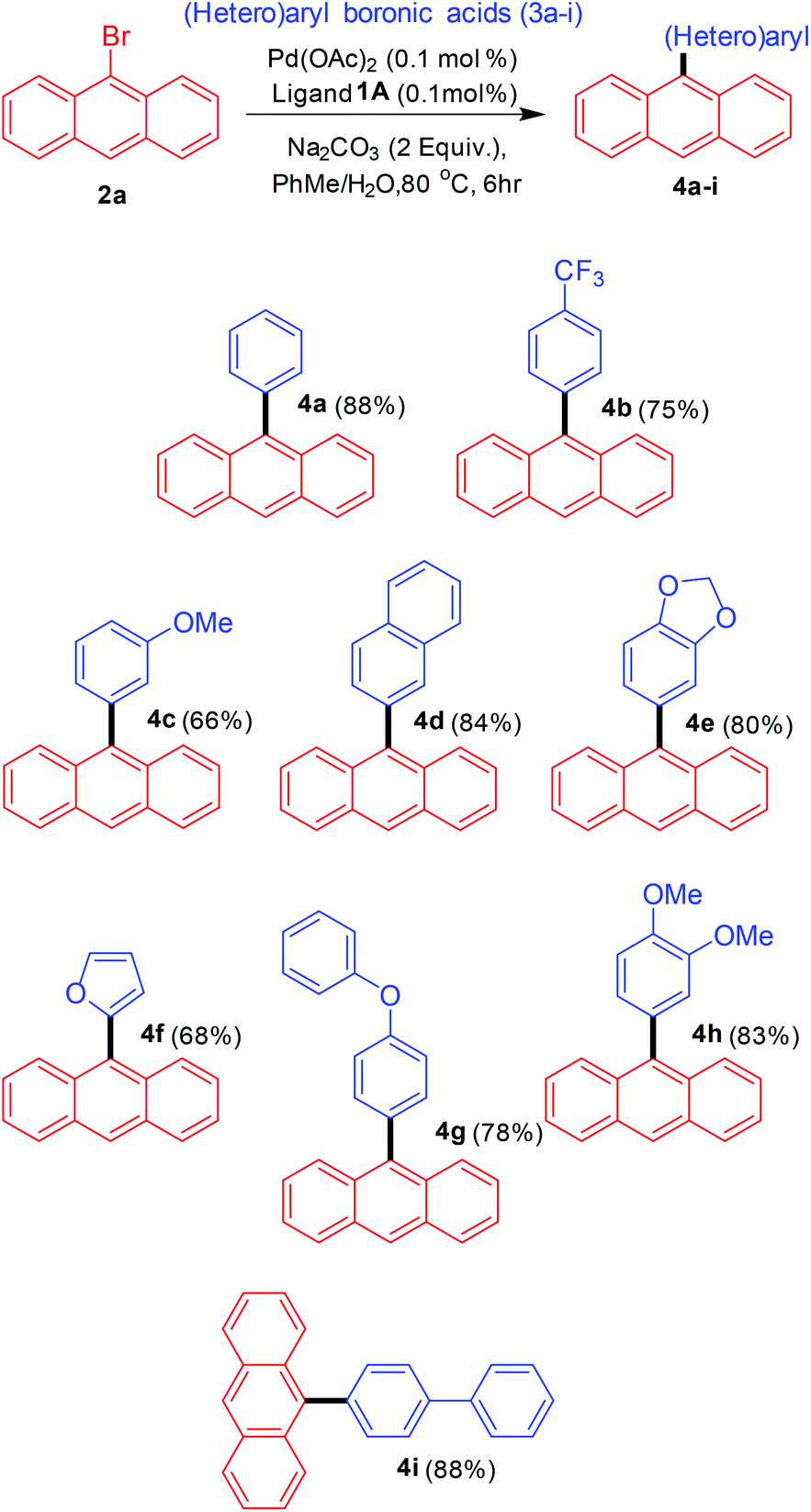 | ||
| Scheme 3 Substrate scope for the arylation of 9-bromoanthracene. | ||
Besides characterising the products obtained by general techniques, the crystalline nature of some of these molecules allowed them to be subjected to single crystal X-ray analysis (Fig. 3). 4e crystallised in the monoclinic space group C2/c with one molecule in the asymmetric unit. The torsion angle between the anthracene and the benzodioxole part is 74.4°.24 No apparent π–π stacking was found in the crystal lattice. Although ends of the aromatic rings approach the typical distance of in between 3 and 4 Å there are no real overlays of aromatic rings for such interactions. Despite the presence of two oxygen atoms in the molecule no hydrogen bonding was observed. The aromatic bond lengths range from 1.350 to 1.441 Å with the longer bonds located in the anthracene moiety. The bond connecting anthracene and benzodioxole is 1.497 Å long.
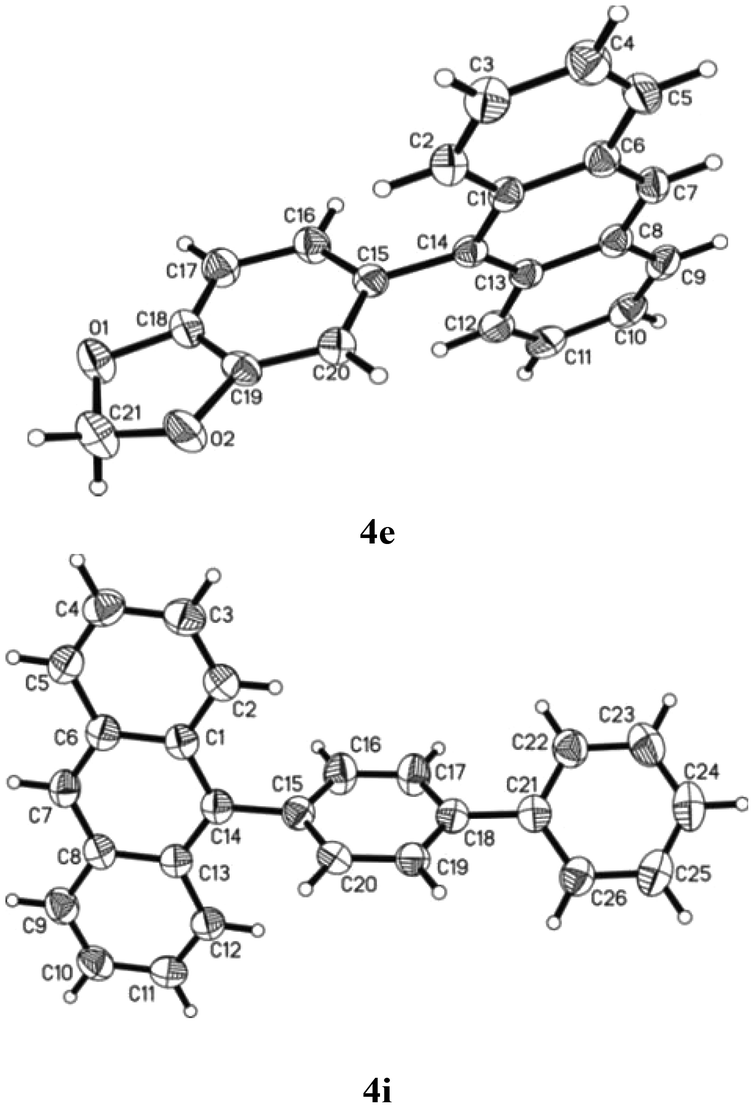 | ||
| Fig. 3 Single crystal X-ray analyses of 4e and 4i. Ellipsoids are shown with 50% probability. | ||
Similarly, 4i crystallised in the monoclinic space group P21/n with one molecule in the asymmetric unit.25 The torsion angle between the anthracene part and the first phenyl ring is 76.0°. The torsion angle between anthracene and the second, i.e. outer, phenyl ring is 38.6°. Hence, the torsion angle between the two phenyl rings is 37.5°. Again there are no π–π interactions in the crystal lattice. It appears, however, as if there was an interaction of the anthracene pi-system with the anthracene hydrogen atoms of another molecule arranged perpendicular to the first anthracene. This could be the reason for the residual electron density found in a distance of 1.18 Å to C7 which might be based on an elongated actual C–H bond. The aromatic bond lengths are in between 1.350 and 1.437 Å and the single bonds connecting anthracene and the first, phenyl of biphenyl part as well as that of the first phenyl with the second in the same biphenyl system equal 1.485 and 1.493 Å, respectively. All this is perfectly consistent with the previously discussed structure.
With an apparently powerful methodology in hand we explored the possibility of arylating other bromoanthracene related substrates such as 1-bromopyrene, 3-bromo-N-phenylcarbazole, 5-bromoacenaphthene, 9-bromophenanthrene and 4-bromotriphenylamine under Suzuki–Miyaura cross-coupling conditions using the bis-carbene precursor 1A (Scheme 4). Most of the coupled products were obtained in competitive yields under mild conditions. The reactivity, though, was found to be somehow lower than for 9-bromoanthracene. These results still reconfirm the claim for the development of a general catalytic system for accessing a wide variety of substituted arenes.
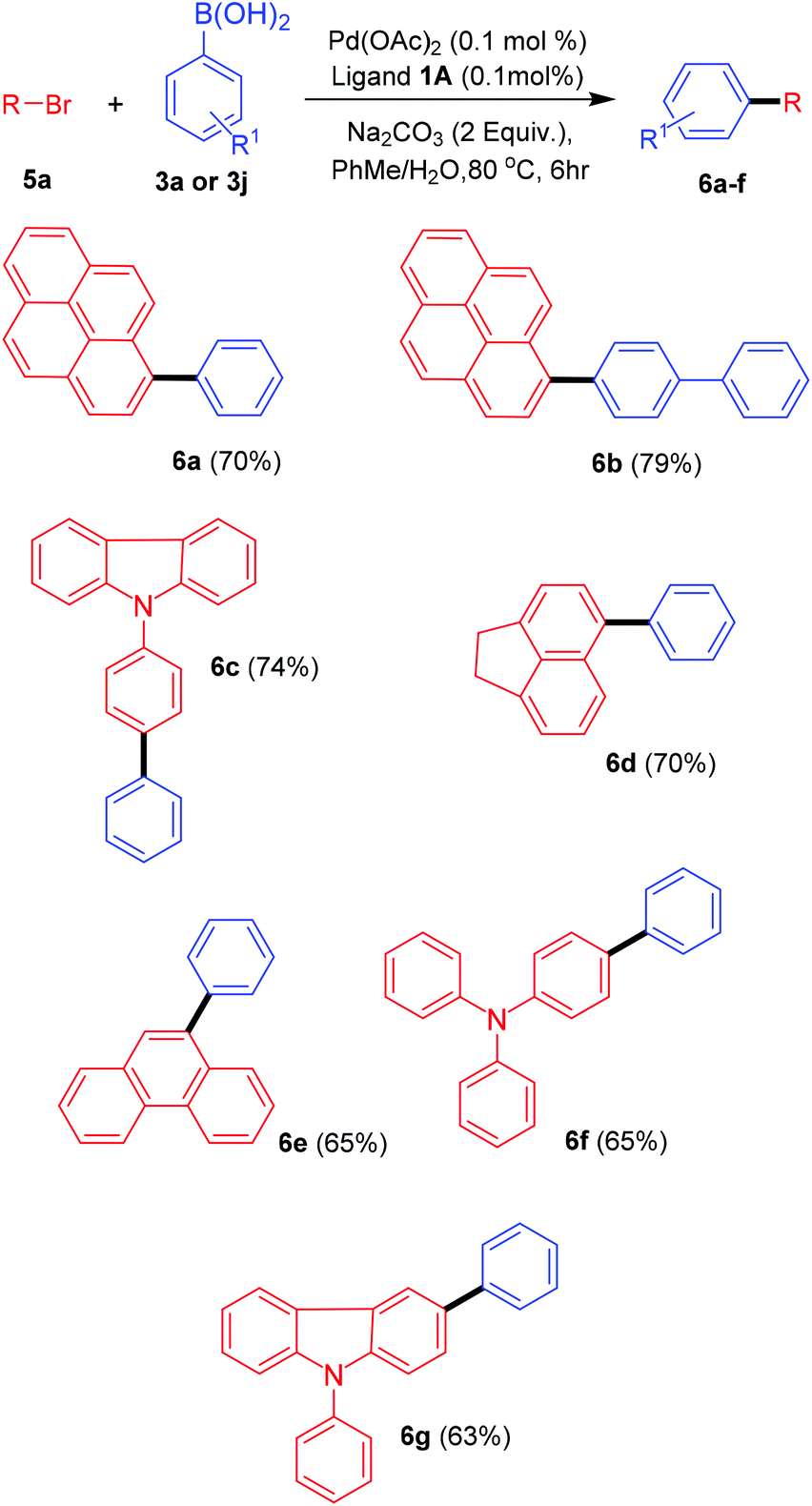 | ||
| Scheme 4 Suzuki–Miyaura arylation of related substrates. | ||
The 9,10-diarylated anthracene structural motif has found many applications in a variety of fields such as OLEDs and anthracene-based dyes.26 These molecules generally exhibit strong electroluminescence due to the promising host and hole-transporting properties. In the case of their use as anthracene-based dyes the placement of the aryl groups at the 9 and 10 positions imparts a slight twist in the structure that is beneficial in suppressing the aggregation and electron combination for obtaining dyes with better functional properties. Most of the catalytic systems employed for accessing these molecules suffer from low reactivity at relatively higher concentrations of Pd.17 For addressing this issue, the diarylation of 9,10-dibromoanthracene was undertaken at only 0.1 mol% catalyst loading in the PhMe/H2O solvent system (Scheme 5).
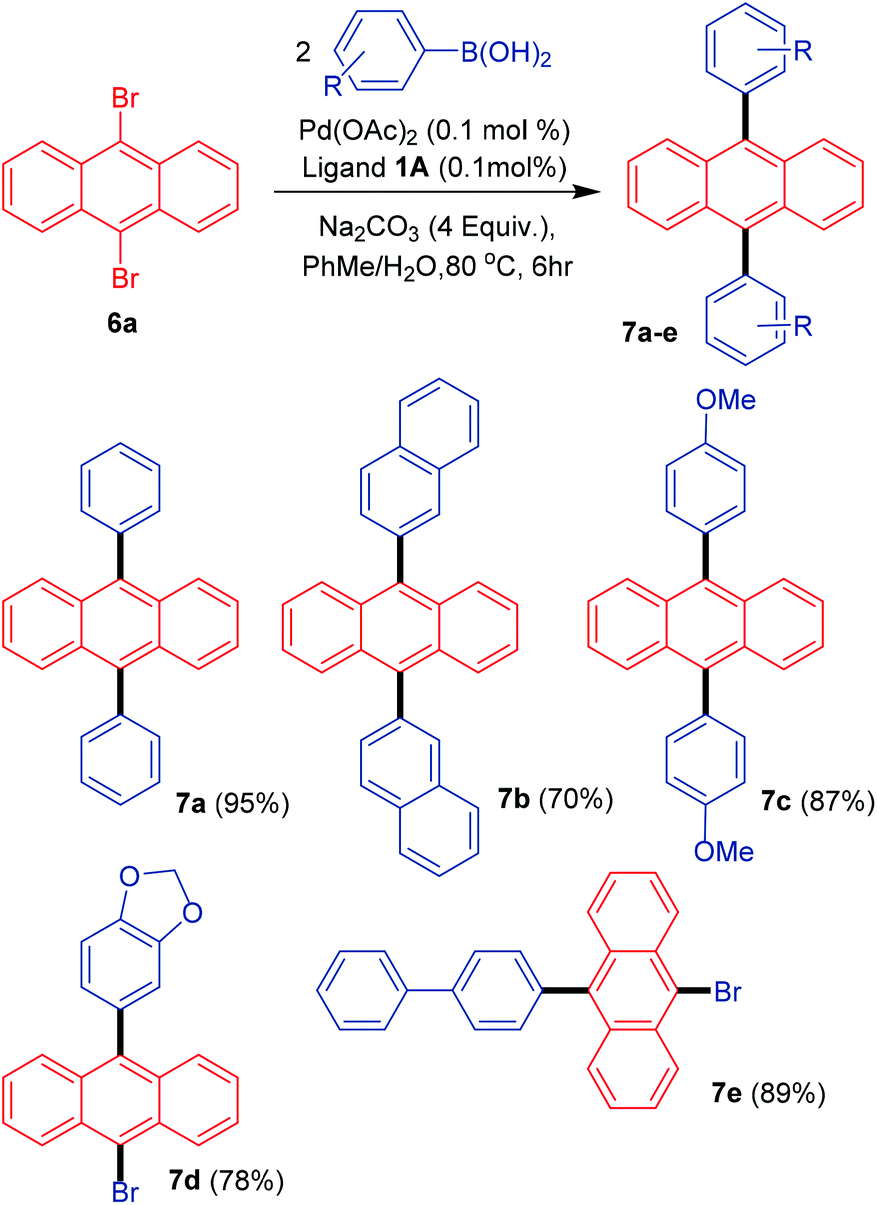 | ||
| Scheme 5 Diarylation of 9,10-dibromoanthracene. | ||
The incorporation of different arene groups helps enhance the conjugation in the anthracene moiety with the reactivity also depending on the steric effect of the aryl boronic acids. Phenyl boronic acid helped furnish the product in excellent yield (7a-95%). Similar observations were made in the case of 4-methoxyphenyl boronic acids providing the diarylated product in excellent yield. 2-Naphthylboronic acid, however, gave decent yields of the diarylated product possibly due to steric effects. Interesting results were obtained in the case of methylenedioxy and 4-biphenyl boronic acids for which, rather than furnishing the desired diarylated product, complete mono-selectivity was observed with products 7d and 7e obtained in good to excellent yields respectively.
The bis-arylation of anthracene at positions 9 and 10 leads to a slight twist in the structure; a property that could be very useful in developing more efficient OLEDs as well as fluorescent dyes.26b This twist is evident from the X-ray single crystal structure obtained for the phenyl substituted diaryl anthracene 7a (Fig. 4).277a crystallized in the monoclinic space group C2/c with half the molecule in the asymmetric unit. The structure is identical with the ones published before.28 As the whole molecule is generated by symmetry operation the phenyl-substituents are necessarily perfectly coplanar. Both are in a torsion angle with the anthracene moiety of 113.0°. Neither π–π nor C–H-π interactions were found in the crystal lattice. The found bond lengths range from 1.357 to 1.436 Å for the aromatic moieties with the longer bonds found in the anthracene part and the single bond between the anthracene and phenyl substituent being 1.490 Å long, again consistent with the previously discussed and reported28 structures.
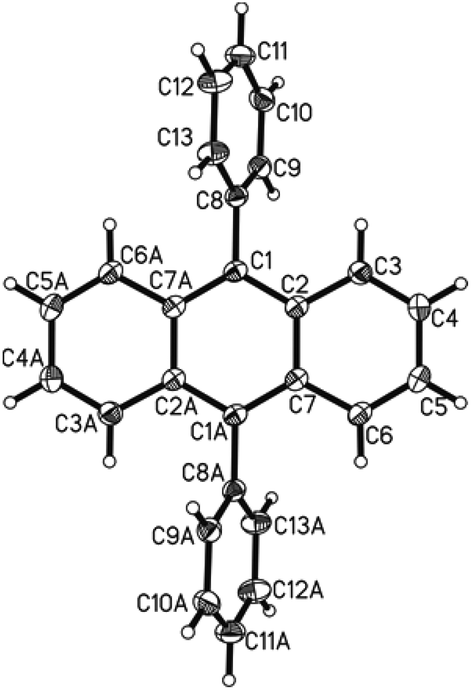 | ||
| Fig. 4 Single crystal X-ray analysis of 7a. Ellipsoids are shown with 50% probability. | ||
Finally, we turned our attention towards synthesizing differently substituted 9,10-diarylated anthracenes. Differences in the electronic and steric properties of the 9,10-diarylated anthracenes (different aryl or heteroaryl groups) have been utilized for the development of OLEDs with improved features such as stability and efficiency resulting in maintaining color.16–19 The strategy employed for achieving this outcome was the scale-up synthesis of 9-phenylanthracene followed by bromination with N-bromosuccinimide to provide 9-bromo-10-phenylanthracene17g that was able to undergo a further Suzuki–Miyaura cross-coupling with a different aryl boronic acid (Scheme 6).
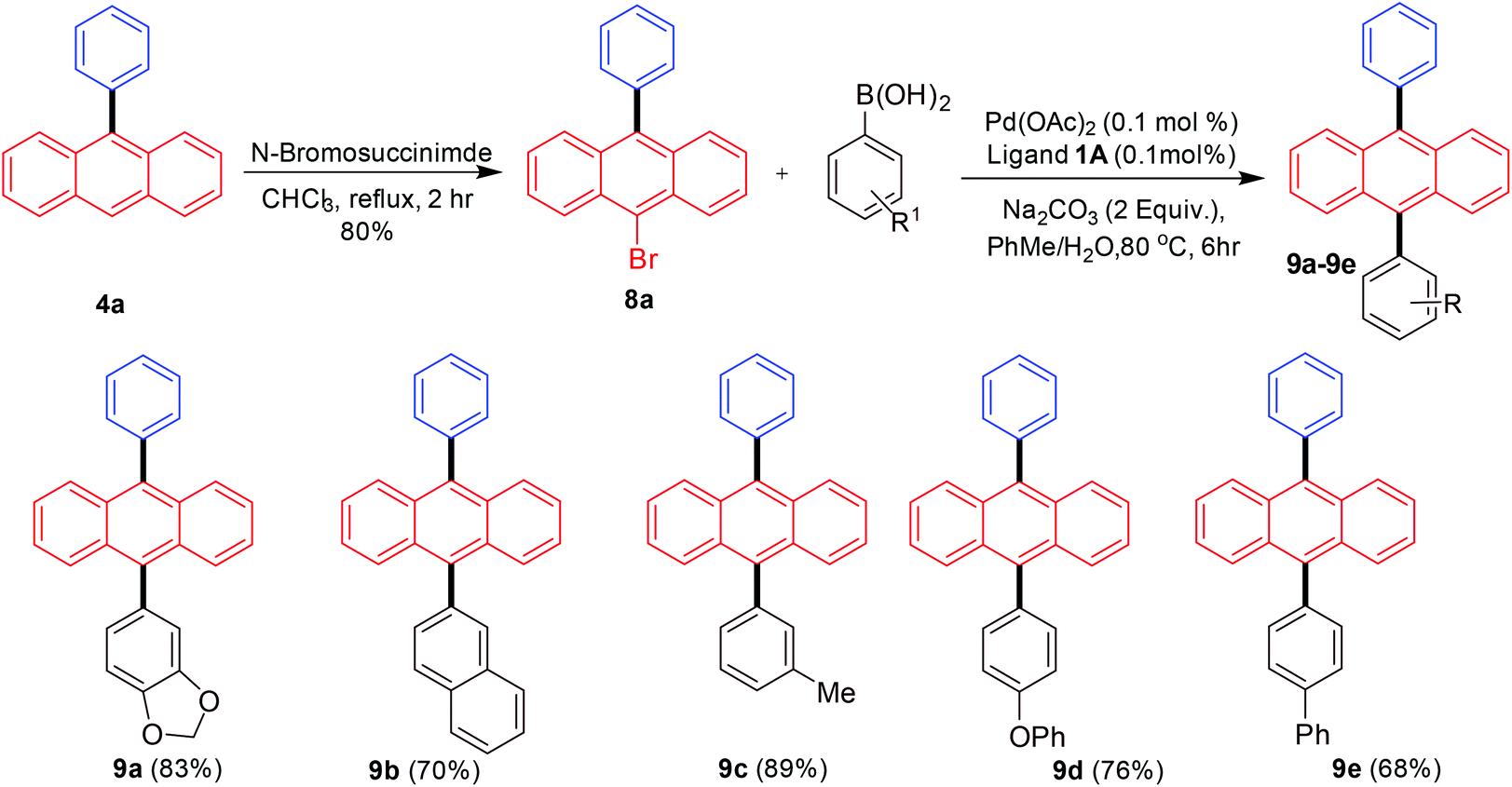 | ||
| Scheme 6 Diarylated anthracene synthesis via Suzuki–Miyaura arylation of 9-bromo-10-phenylanthracene. | ||
The initial synthesis of 9-phenylanthracene from 9-bromoanthracene was scaled-up to 10 mmol under palladium-catalysed conditions. This in the next step was brominated selectively on the anthracene moiety giving 80% yield of 9-bromo-10-phenylanthracene as a white solid. The product was further subjected to Suzuki–Miyaura coupling reactions with arylboronic acids to synthesize a variety of 9,10-diarylated anthracenes useful for OLED applications. In all cases the bis-arylated products were obtained in good to excellent yields.
UV-studies on cross-coupled products 7a–e and 9a–e
The cross-coupled products obtained were further subjected to ultra-violet visible spectroscopic analysis. Initially, their UV-absorbance spectra were obtained in dichloromethane as the solvent (giving better absorbance than other solvents). For the series 7a–e and 9a–e the highest absorbance value range was obtained for 7e (360–400 nm) while most others showed absorbance values in the range of (350–390 nm).A similar trend was observed when emission spectra were recorded for each of the coupled products in dichloromethane as the solvent. A bathochromic shift of around 50–60 nm with respect to the absorption wavelengths was observed in most cases making these molecules excellent candidates for possible OLED applications (Fig. 5).
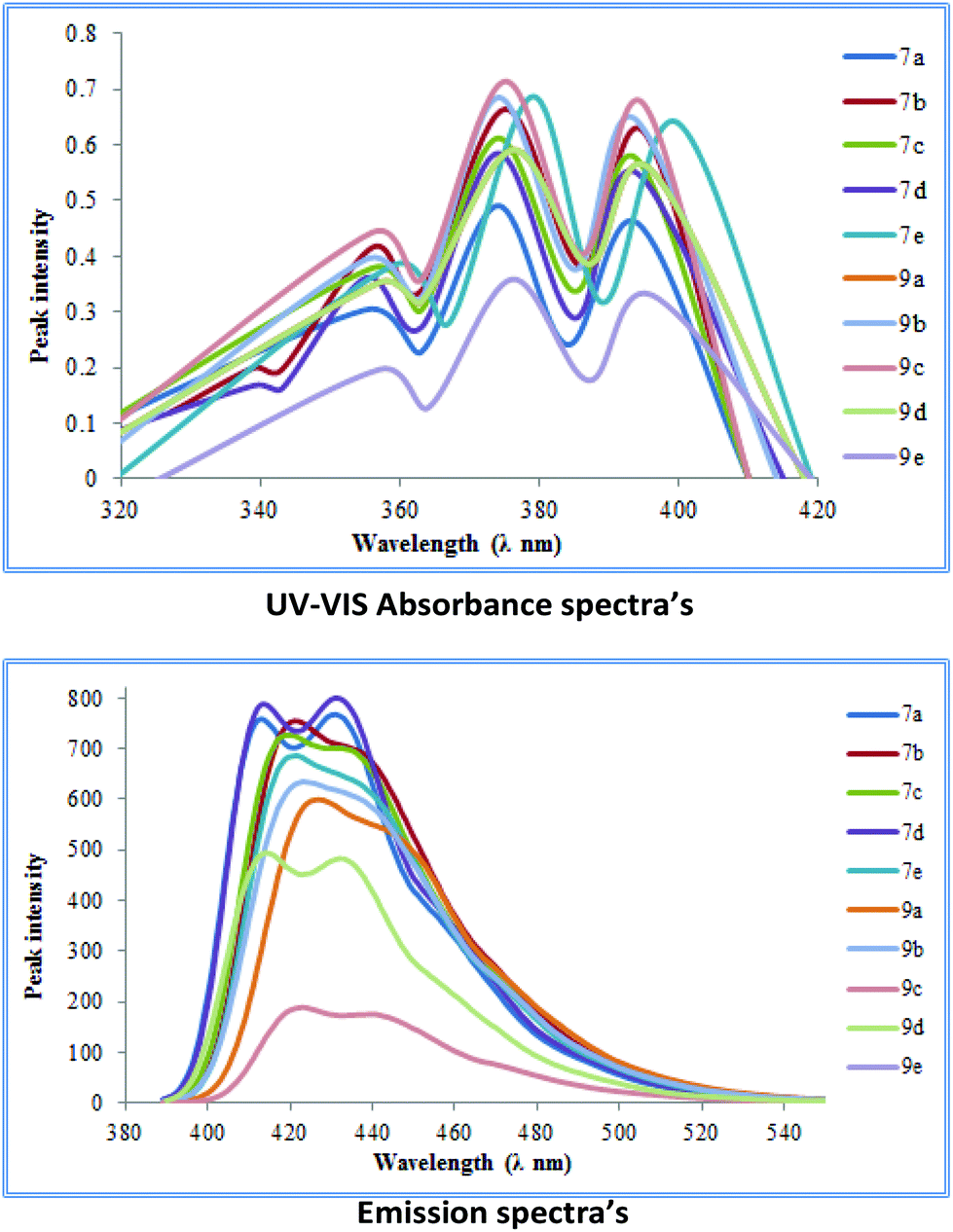 | ||
| Fig. 5 UV-Vis absorbance and emission studies of coupled products. | ||
Mechanistic studies
Promising results obtained with the bis-carbene precursor 1A towards the Suzuki–Miyaura coupling of anthracene and related substrates (vide supra), prompted us to further analyse the catalytic system for possible nanoparticle/heterogeneous or homogeneous pathways followed by the Pd species. Initially, to assess the nature of the Pd species in the catalytic reactions mercury-drop tests were performed on the Suzuki–Miyaura arylation of 9-bromoanthracene with phenyl boronic acid.29 For comparison purposes this test was also carried out for the ligand-free catalytic system, as Pd(OAc)2 under these conditions is known to catalyse coupling reactions via nanoparticle formation. To our surprise in both cases (with and without ligand) a complete retardation of the reaction was observed, thus pointing either towards the presence of a homogeneous catalytic system which is getting poisoned by the addition of mercury or active Pd nanoparticles formed under the conditions which could be sequestered by the added mercury. Furthermore, addition of catalyst poison such as carbon disulfide (CS2) also resulted in a similar observation.30 These results however, cannot completely refute the possibility for the presence of an active molecular catalyst (homotopic catalyst) (Scheme 7).31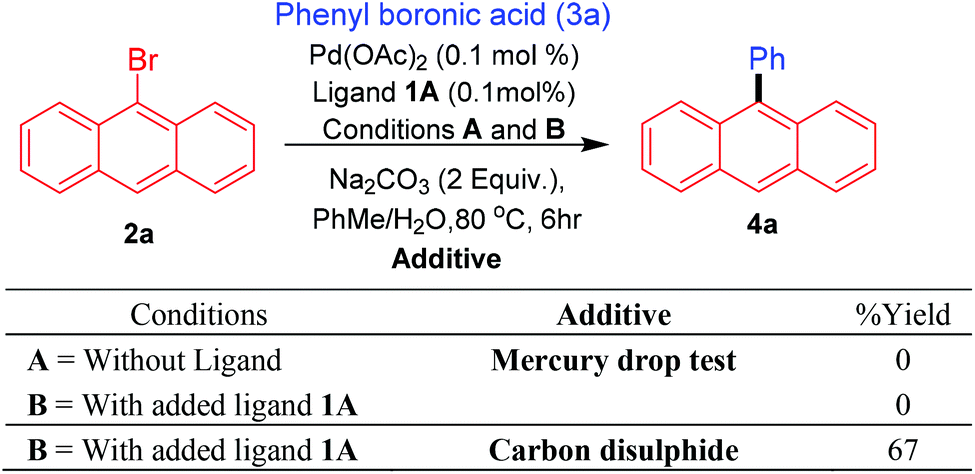 | ||
| Scheme 7 Homogeneity test for arylation of 9-bromoanthracene. | ||
In the literature the complexation of pincer NCN-type bis-carbene precursors is typically described as being carried out at elevated temperatures of 160 °C.13c,d The needed use of higher temperatures has been attributed to the hindered nature and the positioning of both the carbene functional moieties. It therefore seems unlikely that under the catalytic conditions developed for the arylation of 9-bromoanthracene using ligand 1A, coordination of palladium could take place with both of the carbenes present in the ligand. Based on recent evidence, the possibility of a mono-PdNHC coordinated species32 seems more probable and further studies will be required to ascertain such assumption.
Conclusion
In conclusion we have synthesised three novel pincer-type CNC N-heterocyclic carbene ligands which have been characterised by several techniques such as electrospray ionisation-mass spectrometry as well as single crystal X-ray analysis. The ligands were successfully employed for palladium-catalysed Suzuki–Miyaura arylation of bromoanthracene and related substrates with a view of developing a general and robust protocol for accessing molecules capable of exhibiting properties useful for OLED applications. This was achieved at a very low catalyst loading of 0.1 mol% Pd/L. Besides monoarylation, diarylated as well as differently substituted anthracenes were also synthesised to demonstrate the powerful nature of the catalytic system.Experimental section
General remarks
All catalytic reactions were conducted under an inert atmosphere of N2 on a Schlenk line. TLC analysis was performed on Merck 5554 aluminium backed silica gel plates and compounds visualized by ultraviolet light (254 nm), phosphomolybdic acid solution (5% in EtOH), or 1% ninhydrin in EtOH. Aryl boronic acids and other chemicals were obtained from commercial sources, and were used without further purification. Yields refer to isolated compounds, estimated to be >95% pure as determined by 1H-NMR. NMR data (1H, 13C) were recorded on Bruker 300, 400 and 500 MHz spectrometers. Chemical shifts are reported in parts per million downfield from an internal tetramethylsilane reference. Coupling constants (J values) are reported in hertz (Hz). LC-MS was performed on an Agilent VL mass-spectrometer. Elemental analysis was performed using a Thermo-Fischer Scientific instrument. UV-studies were performed on UV-2450 Shimadzu UV-VIS spectrophotometer in dichloromethane as the solvent at a concentration of 5 × 10−5 M. Synthesis of ligands was performed in a microwave synthesiser CEM Discover at 200 °C and 100 psi pressure. HRMS was performed for all the samples on a Bruker MAXIS IMPACT small molecules mass spectrometer.X-ray structural analysis
Suitable single crystals of 1B, 4e, 4i and 7a were mounted on a thin glass fibre coated with paraffin oil. X-ray single-crystal structural data were collected at low temperature (170 K) using a STOE-IPDS 2 T diffractometer equipped with a normal-focus, 2.4 kW, sealed-tube X-ray source with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The program XArea was used for integration of diffraction profiles; numerical absorption correction was made with the programs X-shape and X-red32; all from STOE©2010. The structures were solved by SIR92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and refined by full-matrix least-squares methods using SHELXL-2013.34 The non-hydrogen atoms were refined anisotropically. Unless otherwise stated, the hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to 1.5 Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. In 1B all four water protons were located and refined freely. All calculations were carried out using SHELX-201334 and the WinGX GUI, Ver2013.2.35 Crystallographic data are summarized in Table 2.
33 and refined by full-matrix least-squares methods using SHELXL-2013.34 The non-hydrogen atoms were refined anisotropically. Unless otherwise stated, the hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to 1.5 Ueq of their pivot atoms for terminal sp3 carbon atoms and 1.2 times for all other carbon atoms. In 1B all four water protons were located and refined freely. All calculations were carried out using SHELX-201334 and the WinGX GUI, Ver2013.2.35 Crystallographic data are summarized in Table 2.
| Parameters | 1B | 4e | 4i | 7a |
|---|---|---|---|---|
| Empirical formula | C25H27Br2N5O2 | C21H14O2 | C26H18 | C26H18 |
| CCDC number | 1412157 | 1412158 | 1412159 | 14121160 |
| Formula weight | 589.33 | 298.32 | 330.40 | 330.40 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P |
C2/c | P21/n | C2/c |
| a, Å | 8.8674(18) | 23.657(5) | 10.441(2) | 10.596(2) |
| b, Å | 9.3784(19) | 7.2556(15) | 7.4965(15) | 13.501(3) |
| c, Å | 16.618(3) | 18.859(4) | 22.577(5) | 12.120(2) |
| α, ° | 82.99(3) | 90 | 90 | 90 |
| β, ° | 87.24(3) | 111.31(3) | 94.06(3) | 90.38(3) |
| γ, ° | 62.65(3) | 90 | 90 | 90 |
| V, Å3 | 1218.3(5) | 3015.8(12) | 1762.7(6) | 1733.7(6) |
| Z | 2 | 8 | 4 | 4 |
| T, K | 170 | 170 | 170 | 170 |
| μ, mm−1 | 3.360 | 0.084 | 0.070 | 0.072 |
| D calcd, g cm−3 | 1.607 | 1.314 | 1.245 | 1.266 |
| F (000) | 596 | 1248 | 696 | 696 |
| Unique reflections | 4006 | 2990 | 3740 | 2345 |
| Measured reflections | 7716 | 11988 |
14359 |
9382 |
| R int | 0.0659 | 0.0727 | 0.0547 | 0.0711 |
| GOF on F2 | 1.019 | 0.929 | 1.014 | 1.053 |
| R 1[I > 2σ(I)] | 0.0541 | 0.0448 | 0.0478 | 0.0447 |
| R w[I > 2σ(I)] | 0.1482 | 0.1102 | 0.1200 | 0.1214 |
| Δρ max/min [e Å−3] | 1.132/−0.909 | 0.281/−0.188 | 0.596/−0.169 | 0.342/−0.294 |
Synthesis of substituted imidazoles19
To arylboronic acid (5 mmol) in 15 ml MeOH in an open air atmosphere, imidazole (6 mmol) with CuI (5 mol%) was added and the resulting mixture was vigorously stirred and kept for 4 h under reflux. The reaction mixture was later concentrated under vacuum. The crude product was successfully purified by column chromatography using petroleum ether–ethyl acetate as the eluent to give N-arylimidazoles.Representative procedure for the synthesis of pincer-type NHC ligands
A mixture of 2,6-dibromopyridine (0.237 g, 1.0 mmol) and phenylimidazole (0.568 g, 4.0 mmol) was allowed to react in a sealed tube in a microwave (CEM Discover) at 200 °C, 150 psi, 100 W for 15 min. The reaction mixture was cooled and dissolved in ethanol (2.0 mL). Regeneration of the solid was carried out by adding hexane. The resulting precipitate was separated by decantation and washed with CHCl3/Et2O (10 mL) and dried in a vacuum (0.462 g, 88%).Ligand 1A: 1H NMR (300 MHz, CDCl3) δ 10.96 (s, 2H), 9.20–9.18 (m, 2H), 8.77–8.72 (m, 3H), 8.53–8.51 (m, 2H), 8.02 (d, J = 7.5 Hz, 4H), 7.78–7.67 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 145.71, 145.34, 135.44, 134.99, 130.90, 130.69, 123.0, 122.91, 120.71, 115.47. FAB-MS (m/z) = 365 (M+ − 2Br−). Anal. Calcd for C23H19Br2N5: C, 52.59; H, 3.65; Br, 30.43; N, 13.33. Found: C, 52.49; H, 3.58; Br, 30.32; N, 13.26.
Ligand 1B: 1H NMR (300 MHz, CDCl3) δ 10.89 (s, 2H), 9.18–9.16 (m, 2H), 8.74–8.70 (m, 3H), 8.49 (d, J = 7.5 Hz, 2H), 7.90 (d, J = 7.8 Hz, 4H), 7.57 (d, J = 7.7 Hz, 4H), 2.45 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 145.71, 140.84, 135.12, 132.63, 130.99, 122.56, 120.64, 115.34, 21.14. FAB-MS (m/z) = 393 (M+ − 2Br−). Anal. Calcd for C25H23Br2N5: C, 54.27; H, 4.19; Br, 28.88; N, 12.66. Found: C, 54.22; H, 4.10; Br, 28.76; N, 12.58.
Ligand 1C: 1H NMR (300 MHz, CDCl3) δ 10.73 (s, 2H), 9.18–9.16 (m, 2H), 8.72 (t, J = 7.4 Hz, 1H), 8.49–8.47 (m, 4H), 7.72 (d, J = 7.8 Hz, 2H), 7.64–7.52 (m, 6H), 2.36 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 145.74, 137.17, 134.51, 134.11, 132.17, 131.58, 127.89, 127.19, 125.63, 120.10, 115.21, 17.60. FAB-MS (m/z) = 393 (M+ − 2Br−). Anal. Calcd for C25H23Br2N5: C, 54.27; H, 4.19; Br, 28.88; N, 12.66. Found: C, 54.18; H, 4.13; Br, 28.81; N, 12.55.
Representative procedure for Suzuki–Miyaura cross coupling of 9-bromo anthracene
A mixture of 9-bromoanthracene (1 mmol), arylboronic acid (1.2 mmol), 2 mL of 0.1 mol% catalytic solution (stock solution prepared by dissolving ligand 1A (1.0 mol%) and Pd(OAc)2 (1.0 mol%) in 20 mL dry PhMe) and 1.0 mL Na2CO3 (2 M) was allowed to react in a Schlenk flask under a N2 atmosphere at 80 °C for 6 h. The reaction mixture was cooled to room temperature. The organic layer was then extracted with dichloromethane (2 × 10 mL) and dried over anhydrous Na2SO4, and then concentrated under vacuum to provide the crude product which was further purified by SiO2 column chromatography using hexane as an eluent to give the desired product.Representative procedure for Suzuki–Miyaura cross coupling of related substrates
A mixture of bromoarene (1 mmol), arylboronic acid (1.2 mmol), 2 mL of 0.1 mol% catalytic solution (stock solution prepared by dissolving ligand 1A (1.0 mol%) and Pd(OAc)2 (1.0 mol%) in 20 mL dry PhMe) and 1.0 mL Na2CO3 (2 M) was allowed to react in a Schlenk flask under a N2 atmosphere at 80 °C for 12 h. The reaction mixture was cooled to room temperature. The organic layer was then extracted with dichloromethane (2 × 10 mL) and dried over anhydrous Na2SO4, and then concentrated under vacuum to provide the crude product which was further purified by SiO2 column chromatography using hexane as an eluent to give the desired product.Representative procedure for diarylation via Suzuki–Miyaura cross-coupling of 9,10-dibromoanthracene
A mixture of 9,10-dibromoanthracene (1 mmol), arylboronic acid (2.2 mmol), 2 mL of 0.1 mol% catalytic solution (stock solution prepared by dissolving ligand 1A (1.0 mol%) and Pd(OAc)2 (1.0 mol%) in 20 mL dry PhMe) and 1.0 mL Na2CO3 (2 M) was allowed to react in a Schlenk flask under a N2 atmosphere at 80 °C for 12 h. The reaction mixture was cooled to room temperature. The organic layer was then extracted with dichloromethane (2 × 10 mL) and dried over anhydrous Na2SO4, and then concentrated under vacuum to provide the crude product which was further purified by SiO2 column chromatography using hexane as an eluent to give the desired product.Scale up reaction for preparation of 9-phenylanthracene
A mixture of 9-bromoanthracene (1.285 g, 5.0 mmol), phenylboronic acid (0.731 g, 6.0 mmol), ligand 1A (0.026 g, 0.1 mol%), Pd(OAc)2 (0.011 g, 0.1 mol%) and Na2CO3 (1.06 g, 2 M in 5.0 mL water) in PhMe (10 mL) was allowed to react in a Schlenk flask under a N2 atmosphere at 80 °C for 6 h. The organic layer was extracted with dichloromethane and dried over anhydrous Na2SO4 and the concentrated crude product was purified by SiO2 column chromatography using hexane as an eluent to give the desired product (0.914 g, 72%).Preparation of 9-bromo-10-phenylanthracene17g
In 10 ml CHCl3, 9-phenylanthracene (0.25 g, 1 mmol) and N-bromosuccinimide (0.196 g, 1.1 mmol) were added and heated to 60 °C for 2 h. After cooling the reaction mixture to room temperature the solution was concentrated in vacuo. The remaining residue was re-dissolved in acetone and added in methanol. The resulting precipitate was collected by filtration and washed with methanol and dried in a vacuum to yield the product as a yellow solid (0.28 g, 85%).Representative procedure for arylation via Suzuki–Miyaura cross-coupling of 9-bromo-10-phenylanthracene
A mixture of 9-bromo-10-phenylanthracene (1 mmol), arylboronic acid (2.2 mmol), 2 mL of 0.1 mol% catalytic solution (stock solution prepared by dissolving ligand 1A (1.0 mol%) and Pd(OAc)2 (1.0 mol%) in 20 mL dry PhMe) and 1.0 mL Na2CO3 (2 M) was allowed to react in a Schlenk flask under a N2 atmosphere at 80 °C for 12 h. The reaction mixture was cooled to room temperature. The organic layer was then extracted with dichloromethane (2 × 10 mL) and dried over anhydrous Na2SO4, and then concentrated under vacuum to provide the crude product which was further purified by SiO2 column chromatography using hexane as an eluent to give the desired product.Catalyst poisoning experiments
Acknowledgements
A.R.K. would like to acknowledge the Department of Science and Technology for the DST Inspire Faculty award (IFA12-CH-22). Alexander von Humboldt Foundation is also thanked for the equipment grant to A. R. K. The authors would also like to thank University Grants Commission India for UGC-SAP fellowship for V.Z and UGC-FRP faculty award for A.R.K.References
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (b) K. C. Majumdar and B. Sinha, Synthesis, 2013, 1271 CrossRef CAS; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- Palladium-Catalysed Coupling Reactions: Practical Aspects and Future Developments, ed. A. Molnar, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) K. Öfele, J. Organomet. Chem., 1968, 12, 42 CrossRef; (b) H. W. Wanzlick and H. J. Schonherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141 CrossRef CAS PubMed.
- (a) W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS PubMed; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (c) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551 RSC; (d) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) M. Fevre, J. Pinaud, Y. Gnanou, J. Viqnolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC.
- (a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS; (b) A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913 CrossRef CAS.
- (a) W. A. Herrmann, P. W. Roesky, M. Elison, G. R. J. Artus and K. Ofele, Organometallics, 1995, 14, 1085 CrossRef CAS; (b) W. A. Herrmann, C. Kocher, L. J. Goosen and G. R. J. Artus, Chem. – Eur. J., 1996, 2, 772 CrossRef CAS PubMed; (c) W. A. Herrmann, K. Ofele, M. Elison, F. E. Kuhn and P. W. Roesky, J. Organomet. Chem., 1994, 480, C7 CrossRef CAS; (d) W. A. Herrmann, G. M. Lobmaier and M. Elison, J. Organomet. Chem., 1996, 520, 231 CrossRef CAS.
- I. Huang, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 2370 CrossRef.
- (a) E. A. Kantchev, C. J. Obrien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (b) C. Valente, M. Pompeo, M. Sayah and M. G. Organ, Org. Process Res. Dev., 2014, 18, 180 CrossRef CAS; (c) N. Marion, R. S. Ramon and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448 CrossRef CAS PubMed; (d) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC; (e) S. P. Nolan, in N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley, New York, 2014 Search PubMed; (f) S. P. Nolan, in N-Heterocyclic Carbenes in Synthesis, Wiley, New York, 2006 Search PubMed.
- (a) W. D. Jones, J. Am. Chem. Soc., 2009, 131, 15075 CrossRef CAS PubMed; (b) M. W. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (c) A. Schimdt, S. Wiechmann and T. Freese, ARKIVOC, 2013, 424 Search PubMed.
- (a) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032 RSC; (b) E. Levin, E. Ivry, C. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607 CrossRef CAS PubMed.
- (a) J. Schwarz, V. P. W. Böhm, M. G. Gardiner, M. Grosche, W. A. Herrmann, W. Hieringer and G. Raudaschl-Sieber, Chem. – Eur. J., 2000, 6, 1773 CrossRef CAS; (b) W. A. Herrmann, C.-P. Reisinger and M. Spiegler, J. Organomet. Chem., 1998, 557, 93 CrossRef CAS; (c) W. A. Herrmann, M. Elison, J. Fischer, C. Köcher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371 CrossRef CAS PubMed; (d) D. Morales-Morales, R. Redón, C. Yung and C. M. Jensen, Chem. Commun., 2000, 1619 RSC; (e) D. Morales-Morales, C. Grause, K. Kasaoka, R. Redón, R. E. Cramer and C. M. Jensen, Inorg. Chim. Acta, 2000, 300–302, 958 CrossRef CAS; (f) M. Ohff, A. Ohff, M. E. van der Boom and D. Milstein, J. Am. Chem. Soc., 1997, 119, 11687 CrossRef CAS; (g) I. P. Beletskaya, A. V. Chuchurjukin, H. P. Dijkstra, G. P. M. van Klink and G. van Koten, Tetrahedron Lett., 2000, 41, 1075 CrossRef CAS; (h) D. E. Bergbreiter, P. L. Osburn and Y. S. Liu, J. Am. Chem. Soc., 1999, 121, 9531 CrossRef CAS.
- (a) E. Peris, J. A. Loch, J. Mata and R. H. Crabtree, Chem. Commun., 2000, 201 Search PubMed; (b) H. V. Hunyh and C. S. Lee, Dalton Trans., 2013, 42, 6803 RSC; (c) K. Inamoto, J. H. Kuroda, K. Hiroya, Y. Noda, M. Watanabe and T. Sakamoto, Organometallics, 2006, 25, 3095 CrossRef CAS; (d) J. A. Loch, M. Albrecht, E. Peris, J. Mata, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 700 CrossRef CAS.
- (a) S. Grundemann, M. Albrecht, J. A. Loch, J. W. Faller and R. H. Crabtree, Organometallics, 2001, 20, 5485 CrossRef; (b) F. Churruca, R. SanMartin, B. Ines, I. Tellitu and E. Dominguez, Adv. Synth. Catal., 2006, 348, 1836 CrossRef CAS PubMed; (c) G. Urgoitia, R. SanMartin, M. T. Herrero and E. Dominguez, Green Chem., 2011, 13, 2161 RSC.
- (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (b) A. Suzuki and Y. Yamamoto, Chem. Lett., 2011, 40, 894 CrossRef CAS; (c) A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC; (d) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC; (e) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (f) F. A. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef; (g) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS PubMed; (h) A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. Elshof and G. Rothenberg, Chem. – Eur. J., 2007, 13, 6908 CrossRef CAS PubMed; (i) J. D. Sellars and P. G. Steel, Chem. Soc. Rev., 2011, 40, 5170 RSC; (j) A. Suzuki, in Metal-Catalyzed Cross-Coupling Reactions, VCH, Weinheim, 1998 Search PubMed; (k) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (l) A. Zapf, in Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS PubMed; (b) J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS PubMed.
- (a) J. N. Moorthy, P. Venkatakrishnan, P. Natarajan, D. F. Huang and T. J. Chow, J. Am. Chem. Soc., 2008, 130, 17320 CrossRef CAS PubMed; (b) L. Wang, W. Y. Wong, M. F. Lin, W. K. Wong, K. W. Cheah, H. L. Tam and C. H. Chen, J. Mater. Chem., 2008, 18, 4529 RSC; (c) M. S. Gong, H. S. Lee and Y. M. Jeon, J. Mater. Chem., 2010, 20, 10735 RSC; (d) A. Thangthong, D. Meunmart, N. Prachumrak, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2011, 47, 7122 RSC; (e) N. Prachumrak, S. Namuangruk, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2013, 3825 CrossRef CAS PubMed; (f) B. Chen, G. Yu, X. Li, Y. Ding, C. Wang, Z. Liu and Y. Xie, J. Mater. Chem. C, 2013, 1, 7409 RSC; (g) J. Y. Hu, Y. J. Pu, Y. Yamashita, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, J. Mater. Chem. C, 2013, 1, 3871 RSC; (h) S. R. S. Sarsah, M. R. Lutz, M. Zeller, D. S. Crumrine and D. P. Becker, J. Org. Chem., 2013, 78, 2051 CrossRef CAS PubMed.
- J. Zhang, B. Xu, J. Chen, S. Ma, Y. Dong, L. Wang, B. Li, L. Ye and W. Tian, Adv. Mater., 2014, 26, 739 CrossRef PubMed.
- J. B. Lan, L. Chen, X. Q. Yu, J. S. You and R. G. Xie, Chem. Commun., 2004, 188 RSC.
- D. H. Williams, C. Bradley, C. Bojesen, S. Santikarn and L. C. E. Taylor, J. Am. Chem. Soc., 1981, 103, 5700 CrossRef CAS.
- CCDC number for 1B is 1412157.
- B. R. M. Lake and C. E. Willans, Organometallics, 2014, 33, 2027 CrossRef CAS.
- For reports on the influence of lower concentrations of Pd under ligand-free conditions see: (a) Q. Yao, E. P. Kinney and Z. Yang, J. Org. Chem., 2003, 68, 7528 CrossRef CAS PubMed; (b) A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS PubMed.
- CCDC number for 4f is 1412158.
- CCDC number for 4j is 1412159.
- (a) Y. H. Niu, B. Chen, T. D. Kim, M. S. Liu and A. K. Y. Jen, Appl. Phys. Lett., 2004, 85, 5433 CrossRef CAS PubMed; (b) H. Li, Y. Yang, Y. Hou, R. Tang, T. Duan, J. Chen, H. Wang, H. Han, T. Peng, X. Chen, Q. Li and Z. Li, ACS Sustainable Chem. Eng., 2014, 2, 1776 CrossRef CAS.
- CCDC number for 4j is 1412160.
- (a) Y. Fujiwara, R. Ozawa, D. Onuma, K. Suzuki, K. Yoza and K. Kobayashi, J. Org. Chem., 2013, 78, 2206–2212 CrossRef CAS PubMed; (b) M. Munakata, L. P. Wu, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga, T. Ohta and H. Konaka, Inorg. Chem., 2003, 42, 2553–2558 CrossRef CAS PubMed; (c) J. M. Adams and S. Ramdas, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 679–683 CrossRef.
- (a) G. M. Whitesides, M. Hackett, R. L. Brainard, J. P. P. M. Lavallaye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 4, 1819 CrossRef CAS; (b) R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- S. M. Khake, V. Soni, R. G. Gonnade and B. Punji, Dalton Trans., 2014, 43, 16084 RSC.
- in Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, ed. K. J. Szabo and O. F. Wendt, Wiley-VCH, Weinheim, ch. 2, 2014 Search PubMed.
- For a comprehensive review on single- and double-site Pd(II)–N-heterocyclic carbene complexes see: S. Budagumpi, R. A. Haque and A. S. Salman, Coord. Chem. Rev., 2012, 256, 1787 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- (a) M. Xu, X. Li, Z. Sun and T. Tu, Chem. Commun., 2013, 49, 11539 RSC; (b) S. Sithebe and R. S. Robinson, Beilstein J. Org. Chem., 2014, 10, 1107 CrossRef PubMed; (c) R. K. Mohamed, S. Mondal, B. Gold, C. J. Evoniuk, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 6335 CrossRef CAS PubMed; (d) K. Kotayama, M. Hirotsu, I. Kinoshita and Y. Teki, Dalton Trans., 2014, 43, 13384 RSC; (e) R. Sivasakthikumaran, M. Nandakumar and A. K. Mohanakrishnan, J. Org. Chem., 2012, 77, 9053–9071 CrossRef CAS PubMed; (f) P. E. Maligres, S. W. Krska and P. G. Dormer, J. Org. Chem., 2012, 77, 7646 CrossRef CAS PubMed; (g) Y. Sugihara, H. Takeda and J. Nakayama, Eur. J. Org. Chem., 1999, 597 CrossRef CAS; (h) J. L. Serrano, J. Pérez, L. García, G. Sánchez, J. García, P. Lozano, V. Zende and A. R. Kapdi, Organometallics, 2015, 34, 522 CrossRef CAS; (i) O. Kobayashi, D. Uraguchi and T. Yamakawa, Org. Lett., 2009, 11, 2679 CrossRef CAS PubMed; (j) S. S. Babu, M. J. Hollamby, J. Aimi, H. Ozawa, A. Saeki, S. Seki, K. Kobayashi, K. Hagiwara, M. Yoshizawa, H. Möhwald and T. Nakanishi, Nat. Commun., 2013, 4, 1969 Search PubMed; (k) W. Lee, Y. Kang and P. H. Lee, J. Org. Chem., 2008, 73, 4326 CrossRef CAS PubMed; (l) M. Periasamy, M. Beesu and M. Shanmugaraja, Synthesis, 2014, 2913 Search PubMed; (m) W. Schlenk, E. Bergmann, B. Benedikt, O. Blum, C. Bresiewicz, I. Rodloff, J. Appenrodt, K. Ehninger, H. Ender and R. Israel, Justus Liebigs Ann. Chem., 1928, 463, 98 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1412157–1412160. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00236b |
| This journal is © the Partner Organisations 2015 |