
K2S2O8-mediated metal-free direct C–H functionalization of quinones using arylboronic acids†
Andivelu
Ilangovan
*,
Ashok
Polu
 and
Gandhesiri
Satish
and
Gandhesiri
Satish
School of Chemistry Bharathidasan University, Tiruchirappalli, Tamilnadu-620024, India. E-mail: ilangovanbdu@yahoo.com
First published on 15th October 2015
Abstract
Direct C–H functionalization of quinones with arylboronic acids is achieved using K2S2O8 as the sole and efficient green catalytic system. This method provides a straightforward and sustainable way to construct arylquinones via a radical pathway in moderate to good yields under metal-free conditions.
Introduction
Arylated benzoquinone, naphthoquinone and heterocyclic ring systems form important skeletal frameworks of several biologically active compounds and natural products (Fig. 1).1a–d Unique electronic and visual properties of arylated quinones make them act as useful dyes and photoactive materials.2a,b Recent developments on C–C bond forming methods made it possible to carryout direct C–H functionalization of quinones using different aryl counter parts. Consequently, several new procedures have been reported for the synthesis of arylated quinones. Employing stannyl quinones for Stille type coupling3a or halogenated quinones for Suzuki–Miyaura type coupling3b have their own drawbacks as they require pre-functionalization of quinones. Haloquinones or stannyl quinones are hard to access due to chemo and regioselectivity issues. Heck type cross coupling fails in the case of quinones due to the fact that quinones can coordinate with Pd and usually acts as a catalyst for reoxidation of Pd(0) species4a,b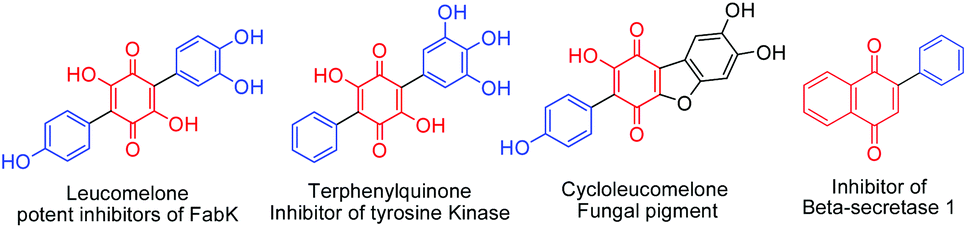 | ||
| Fig. 1 Bioactive arylquinone motifs. | ||
Direct C–H/C–H cross coupling is highly advantageous, as this avoids the involvement of functionalized starting materials. Rh(III) together with an expensive re-oxidant (AgSbF6–AgOAc) worked well for C–H/C–H coupling of quinones when used in combination with a directing group.5 Similarly, a stoichiometric quantity of Pd(OAc)2 or a catalytic quantity of Pd(OAc)2–Na2S2O8 was used for direct C–H/C–H cross coupling.6a Pd(acac)2 with a large excess of Ag2CO3 as a co-oxidant was found useful for the synthesis of arylated quinone from hydroquinone.6b Pd(TFA)2 with (NH4)2S2O8 as the co-oxidant is yet another useful catalytic system reported for arylation of quinones as well as olefins.6c Direct C–H/C–H cross coupling is mainly applicable for electron rich aryl counterparts, thus making it substrate specific5,6a,c and less attractive.
Arylation of quinones using a radical is a successful approach. Although Meerwein arylation of quinone was reported earlier,7a,b improved methods were reported only later. It requires hydroquinone or a reducing metal like copper for the homolytic decomposition of diazonium functionality to form an aryl radical. Aryl diazonium salts are very unstable, sometimes explosive above 0 °C and require highly acidic conditions for their synthesis, thus not suitable for large scale applications.7c Recently, application of metal-free methods for making C–C or C–heteroatom bonds gained the attention of chemists.8 However, methods involving metal-free generation of radicals are scarce.9 Aryl radicals obtained from phenyl hydrazine-IBX were used for arylation of naphthoquinones to obtain products in moderate to good yields.9a Different Ar2IOTf salts were also used successfully to generate aryl radicals for arylation of benzoquinone and naphthoquinone.9b The scarce availability of substituted phenyl hydrazine and Ar2IOTf salts makes these approaches of limited substrate scope and hence less attractive.
Arylboronic acids are highly stable, even in water and are readily available in a variety of substituted forms, making it a convenient precursor for building compounds of wide substrate scope. After Molina et al. demonstrated that naphthoquinones can be directly arylated using arylboronic acids in the presence of a Pd/Cu catalyst and a FeCl3 co-oxidant,10 a series of reports appeared on the application of arylboronic acids for arylation of quinones using different metal salts such as AgNO3,11a FeS,11b FeNO3,11c FeSO4,11d and Fe(acac)2,11e all in combination with a large excess of K2S2O8 as the co-oxidant. Pd(OCOCF3)2 with a large excess of 2,6-dichlorobenzoquinone (2.5 equiv.) as the co-oxidant was also reported for the diarylation of quinones using arylboronic acids.12 Analysis of methods reported so far reveals that except for few reagents (IBX and Ar2IOTf)9a,b almost all the methods reported require metal salts and a re-oxidant (Scheme 1).
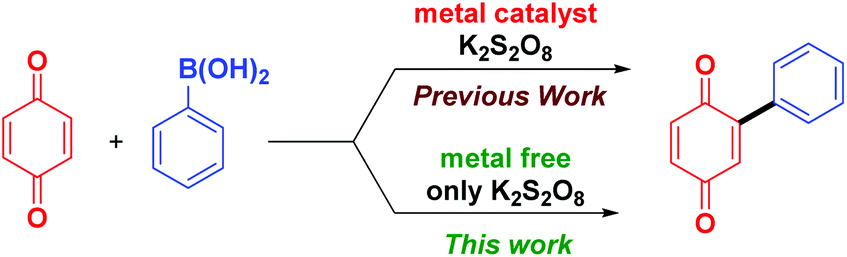 | ||
| Scheme 1 Arylation of quinones. | ||
It is well known that K2S2O8 decomposes at higher temperature to generate radicals.13a,b However, in the presence of metal salts, decomposition of K2S2O8 takes place at room temperature to generate sulphate radicals.11d,14 Thus we envisaged that an aryl radical could be generated from arylboronic acids using K2S2O8 at higher temperature and used for arylation of quinones. K2S2O8 is a versatile oxidant available at much cheaper price and can be used for the regeneration of quinone from hydroquinone. Avoiding metallic reagents is advantageous in terms of elimination of metal toxicity, cost saving and minimization of environmental pollution. Herein, we present a simple method for arylation of quinones and heterocycles using arylboronic acid under metal free conditions.
Results and discussion
Our studies began with optimization of conditions for the reaction between quinone 1a (1.0 equiv.) and arylboronic acid 2a (1.5 equiv.) in the presence of K2S2O8 as an oxidant in DCE/H2O. Various reaction parameters were studied and the results are summarized in Table 1. No reaction was observed at room temperature (rt) with 1.0 equiv. of K2S2O8, in DCE. However when the solvent system was changed to DCE/H2O, we were delighted to see the formation of 3a in traces (entry 2). Increase in temperature to 80 °C lead to formation of product 3a in 30% isolated yield (entries 1 and 3). It was concluded that temperature is the key factor for the reaction to take place. Increasing the amount of K2S2O8 to 2.0 equiv. had a beneficial effect and the product 2a was obtained in the highest yield (72%) in the shortest reaction time (entry 4). Further, increase in the quantity of K2S2O8 to 3.0 equiv. had no improvement on the yield (entry 5). However, when the quantity of K2S2O8 was increased further to 5.0 equiv. the yield of the product 3a decreased (entry 6). Similarly, increasing or decreasing the temperature from 80 °C only provided the product 3a in low yield (entries 7 and 8). Likewise, when the quantity of arylboronic acid 2a was decreased to 1.0 equiv. the yield decreased (entry 9). In the absence of K2S2O8 no desired product was formed, indicating that K2S2O8 is crucial to this reaction (entry 10). Other oxidants such as TBHP, H2O2 and Oxone as catalysts were ineffective for the reaction (entries 11–13). Among the different solvent systems, DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O furnished the highest yield (entries 14–18). Thus a systematic screening study revealed that 1 (1.0 equiv.), 2a (1.5 equiv.) and K2S2O8 (2.0 equiv.) in DCE–H2O at 80 °C are the optimum conditions for arylation.
:H2O furnished the highest yield (entries 14–18). Thus a systematic screening study revealed that 1 (1.0 equiv.), 2a (1.5 equiv.) and K2S2O8 (2.0 equiv.) in DCE–H2O at 80 °C are the optimum conditions for arylation.
|
|
|||||
|---|---|---|---|---|---|
| S.NO | Additive | Solvent | Temp (°C) | Time | Yielda (%) |
| a Isolated yield. b No reaction. c 2a (1.0 mmol). | |||||
| 1 | K2S2O8 (1.0 equiv.) | DCE | rt | 24 h | nrb |
| 2 | K2S2O8 (1.0 equiv.) | DCE/H2O | rt | 24 h | 5 |
| 3 | K2S2O8 (1.0 equiv.) | DCE/H2O | 80 | 24 h | 30 |
| 4 | K 2 S 2 O 8 (2.0 equiv.) | DCE/H 2 O | 80 | 1 h 30 min | 72 |
| 5 | K2S2O8 (3.0 equiv.) | DCE/H2O | 80 | 1 h 10 min | 72 |
| 6 | K2S2O8 (5.0 equiv.) | DCE/H2O | 80 | 1 h | 65 |
| 7 | K2S2O8 (2.0 equiv.) | DCE/H2O | 60 | 2 h | 60 |
| 8 | K2S2O8 (2.0 equiv.) | DCE/H2O | 100 | 1 h | 64 |
| 9 | K2S2O8 (2.0 equiv.) | DCE/H2O | 80 | 2 h 30 min | 58c |
| 10 | — | DCE/H2O | 80 | 24 h | nrb |
| 11 | TBHP (2.0 equiv.) | DCE/H2O | 80 | 24 h | nrb |
| 12 | H2O2 (2.0 equiv.) | DCE/H2O | 80 | 24 h | nrb |
| 13 | Oxone (2.0 equiv.) | DCE/H2O | 80 | 24 h | nrb |
| 14 | K2S2O8 (2.0 equiv.) | H2O | 80 | 24 h | 36 |
| 15 | K2S2O8 (2.0 equiv.) | DMF/H2O | 80 | 1 h 50 min | 45 |
| 16 | K2S2O8 (2.0 equiv.) | Toluene/H2O | 80 | 24 h | 50 |
| 17 | K2S2O8 (2.0 equiv.) | DMSO/H2O | 80 | 24 h | nrb |
| 18 | K2S2O8 (2.0 equiv.) | MeOH/H2O | 80 | 24 h | Trace |
With the optimal condition in hand, the scope of quinone arylation reaction was explored on different substrates (Scheme 2). Different arylboronic acids, bearing electron donating groups 2b–2e reacted well with 1,4-benzoquinone to give the corresponding products 3ab–3ae in moderate to good yields. Similarly, arylboronic acids 2f and 2g containing substituents at the sterically crowded ortho position reacted fast to furnish the products 3af and 3ag in moderate yield. Arylboronic acids, 2h–2j bearing halogen groups were well tolerated and offered 3ah–3aj in moderate to good yields. Next, the effect of the substituent on quinone was investigated. Mono substituted benzoquinone such as 2-methylcyclohexa-2,5-diene-1,4-dione (1b) produced a regio isomeric mixture of arylated quinones 3bka and 3bkb in 72% overall yield. Similarly, more sterically hindered 2,6-dimethoxybenzoquinone (1c) reacted successfully with different aryl boronic acids 2a, 2b, 2h, and 2j to provide the corresponding products 3cl–3co in good yields. However, 2,6-dimethylbenzoquinone (1d) and 2,5-dichlorobenzoquinone (1e) delivered the desired products 3dp, 3dq (inseparable mixture along with starting material) and 3er respectively in low yield. The (4-cyanophenyl)boronic acid (2s) containing a strong electron withdrawing –CN group on reaction with simple benzoquinone (1a) failed to produce the desired product 3as. This may be due to the unstable nature of the cyano aryl radical.11a Similarly, 2,6-dihydroxybenzoquinone (1f) and t-butylbenzoquinone (1g) on treatment with phenylboronic acid (2a) also failed to produce the expected arylated products 3t and 3u, even after a prolonged reaction time.
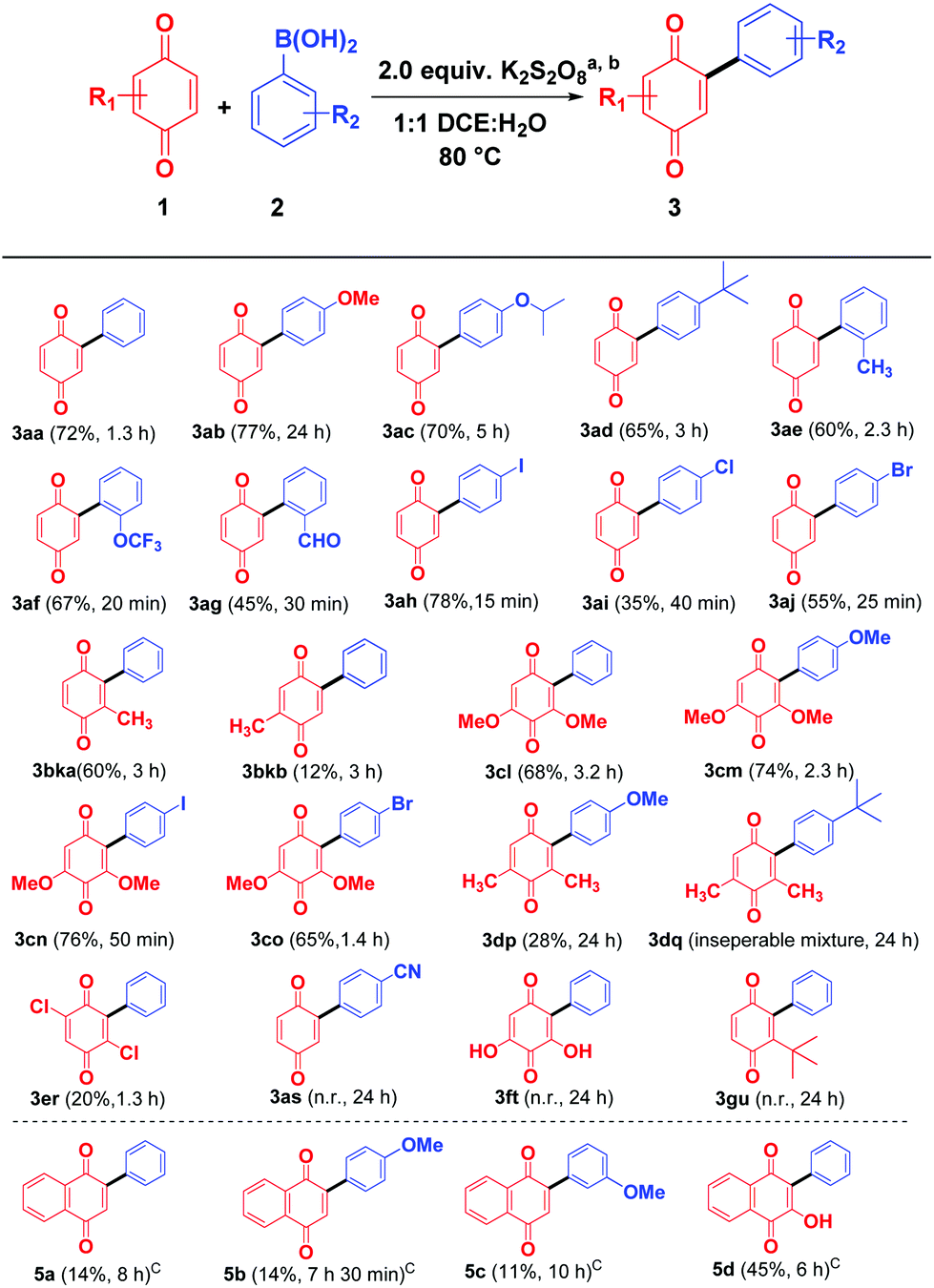 | ||
| Scheme 2 Scope of quinone and boronic acid coupling partners. aReaction conditions: 1 (1.0 mmol), 2 (1.5 mmol), K2S2O8 (2.0 equiv.), CH2Cl2–H2O (1:1, v/v, 4 mL) at 80 °C; bisolated yields; cthe reaction was performed in the presence of Et3N (10.0 equiv.) at 80 °C in CH2Cl2–H2O (1:1, v/v, 4 mL). | ||
As a part of the study we next examined the reactivity of 1,4 naphthoquinone (4a) and its derivatives. In the case of 1,4 naphthoquinone (4a), the desired product 5a could not be obtained under the optimum reaction conditions, however in the presence of triethylamine (TEA, 10.0 equiv.) the expected product 5a was obtained albeit in low yield (14%). In the presence of diisopropylamine the product 5a was formed only in 5% yield, whereas in the presence of inorganic bases such as K2CO3 and NaOH the yield was decreased to a trace. Similarly, 4-methoxyphenylboronic acid (2b) and 3-methoxy phenylboronic acid (2l) furnished the expected products 5b and 5c in 14% and 11% respectively. Interestingly, the hydroxyl group in 2-hydroxy naphthoquinone (4b) tolerated the reaction conditions well and the arylated quinone 5d was obtained in moderate yield (45%).
In the absence of TEA, while 2-methoxy naphthoquinone (4c) failed to undergo reaction with phenylboronic acid (2a, Scheme 3, eqn (1), 5e), it underwent reaction with 4-methoxy boronic acid (2b, Scheme 3, eqn (1)) successfully albeit in low yield 5f. Surprisingly, in the presence of TEA, the –OMe, –OCOCH3 and –OCOPh naphthoquinones 4c–e underwent a C–O bond cleavage reaction to give the corresponding arylated naphthoquinones 5a and 5d in low yields (eqn (3)–(5), Scheme 3). It is interesting to note that in the absence of Et3N no C–O bond cleavage took place indicating that a base plays a vital role in C–O bond cleavage (eqn (1) and (2), Scheme 3). To the best of our knowledge, it is for the first time that C–O bond cleavage of naphthoquinone derivatives such as 4c, 4d, and 4e was observed in the presence of K2S2O8 and TEA.
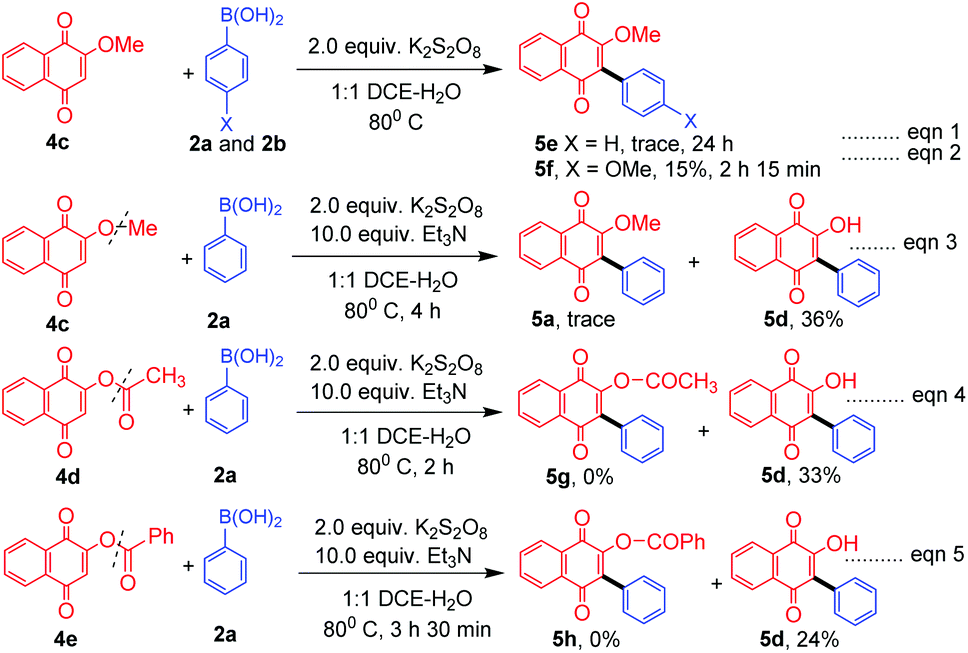 | ||
| Scheme 3 K2S2O8–Et3N mediated C–O bond cleavage of naphthoquinones. | ||
With these impressive results, we further focused on application of the same oxidant system for arylation of different heterocycles such as pyridine (6a), 4-acetyl pyridine (6b), quinoline (6c) and bipyridine (6d) (Scheme 4). Among the different heterocycles, quinoline (6c) was successful in undergoing reaction with the aryl radical to give 7c in moderate yield. While bipyridine (6d) produced a low yield of the product 7d, pyridine (6a) and 4-acetyl pyridine (6b) produced only a trace amount of the products 7a and 7b. Further attempts to enhance the yield for the formation of expected products 7a and 7b were not successful. This may be due to the salt formation of the pyridinium ring system during the course of the reaction.
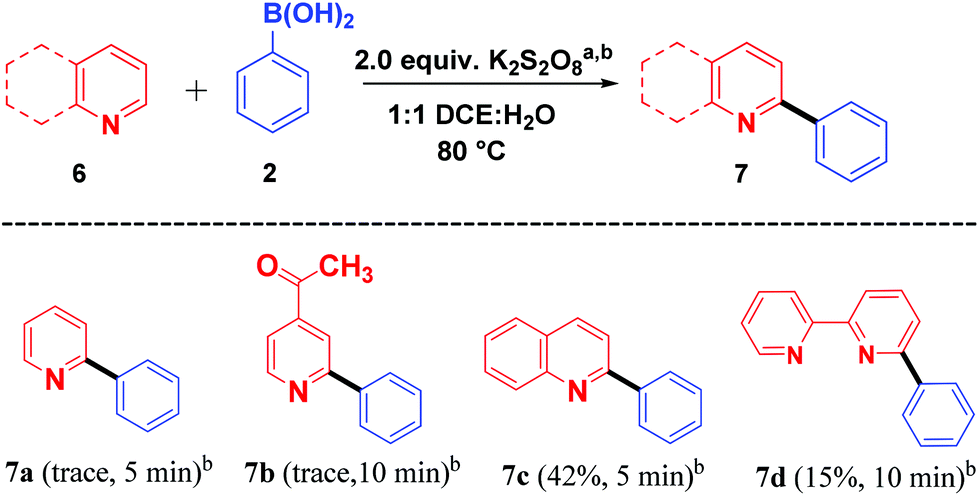 | ||
| Scheme 4 Scope of heterocycles and boronic acid coupling. aReaction conditions: 1 (1.0 mmol), 2 (1.5 mmol), K2S2O8 (2.0 equiv.), CH2Cl2–H2O (1:1, v/v, 4 mL) at 80 °C. bIsolated yields. | ||
To demonstrate the synthetic utility of the present method, a gram-scale experiment was carried out, under the optimal reaction conditions, on benzoquinone (1a, 3.0 g) using phenylboronic acid (2a, Scheme 5). The arylated product 3aa was obtained in 61% isolated yield (3.1 g), which shows that the present method can be easily adopted for the large scale preparations.
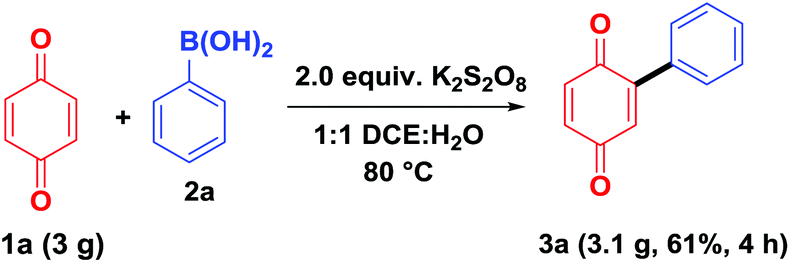 | ||
| Scheme 5 Gram-scale synthesis of aryl benzoquinone. | ||
To study the reaction mechanism, a radical scavenger effect was investigated by adding TEMPO in the reaction between benzoquinone (1a) and phenylboronic acid (2a). Obviously, the yield decreased from 72% to 12% when 2.0 equiv. of TEMPO was added. This result shows that the reaction takes place through a radical mechanism and complies with the reported literature.10 Based on these results, a suitable mechanism is proposed (Scheme 6). Initially, K2S2O8 thermally decomposes to form a sulfate radical (SO4−˙),13a,b which in turn reacts with arylboronic acid 2a to give an aryl radical A.13c,14 Further, aryl radical A reacts with quinone 1a to give B, which on re-oxidation afforded the desired product 3a (Scheme 6).
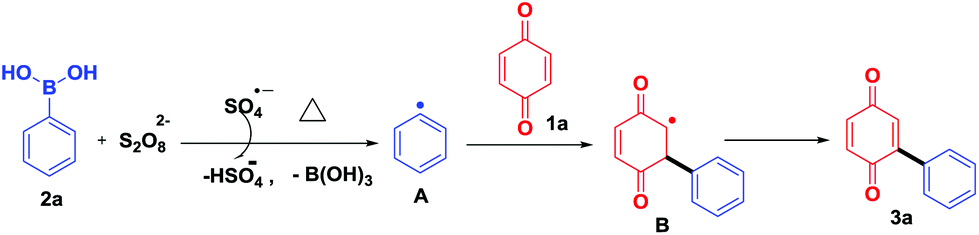 | ||
| Scheme 6 A plausible reaction mechanism. | ||
Conclusion
In summary, the present study shows that aryl radicals could be generated from arylboronic acid under metal free conditions by means of K2S2O8 and used for C–H functionalization of quinones and heterocycles. The radical pathway involved in the reaction was established by studying the reaction in the presence of a radical scavenger. The present method avoids metals and ligands. K2S2O8 is inexpensive, environmentally benign, and found to show high efficiency to generate radicals at high temperature. This method is simple, scalable, and does not require any pre-functionalization of quinones. Further study on applications of this reaction is underway in our laboratory.Acknowledgements
AP and GS thank UGC, New Delhi, for the award of BSR-RFSMS and senior research fellowship respectively. We thank DST-FIST for the use of instrument facility at the School of Chemistry, Bharathidasan University.References
- (a) C.-J. Zheng, M.-J. Sohn and W.-G. Kim, J. Antibiot., 2006, 59, 808–812 CrossRef CAS PubMed; (b) C. Puder, K. Wagner, R. Vettermann, R. Hauptmann and O. Potterat, J. Nat. Prod., 2005, 68, 323–326 CrossRef CAS PubMed; (c) J. Hiort, K. Maksimenka, M. Reichert, S. P. Ottstadt, W. H. Lin, V. Wray, K. Steube, K. Schaumann, H. Weber, P. Proksch, R. Ebel, W. E. G. Muller and G. Bringmann, J. Nat. Prod., 2004, 67, 1532–1543 CrossRef CAS PubMed; (d) D. Wang, G. Bingyang, L. Liang, J. Shan and D. Yuqiang, J. Org. Chem., 2014, 79, 8607–8613 CrossRef CAS PubMed.
- (a) W. A. Cramer and A. R. Crofts, in Photosynthesis, ed. U. Govindjee, Academic Press, New York, 1982, vol. 1, pp. 387–467 Search PubMed; (b) T. Bechtold, in Handbook of Natural Colorants, ed. T. Bechtold and R. Mussak, Wiley, New York, 2009, pp. 151–182 Search PubMed.
- (a) L. S. Liebeskind and S. W. Riesinger, J. Org. Chem., 1993, 58, 408–413 CrossRef CAS; (b) O. A. Akrawi, A. Khan, P. Tamas, V. Alexander and L. Peter, Tetrahedron, 2013, 69, 9013–9024 CrossRef CAS.
- (a) T. W. Lyons, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 4455–4464 CrossRef CAS PubMed; (b) D. Nattawan and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 5732–5735 CrossRef PubMed.
- Y. Moon, Y. Jeong, D. Kook and S. Hong, Org. Biomol. Chem., 2015, 13, 3918–3923 CAS.
- (a) T. Itahara, J. Org. Chem., 1985, 50, 5546–5550 CrossRef CAS; (b) S. Zhijie, S. Yang, Y. Huang, Y. Cheng, S. Feijie and Y. Jingsong, Chem. Commun., 2014, 50, 13914–13916 RSC; (c) Z. She, Y. Shi, Y. Huang, Y. Cheng, F. Song and J. You, Chem. Commun., 2014, 50, 13914–13916 RSC.
- (a) J. F. Bagli and Ph. Lecuyer, Can. J. Chem., 1961, 39, 1037–10480 CrossRef CAS; (b) A. Honraedt, L. C. François, E. L. Grognec, V. Fernandez and F. F. Xavier, J. Org. Chem., 2013, 78, 4604–4609 CrossRef CAS PubMed; (c) M. R. Heinrich, Chem. – Eur. J., 2009, 15, 820–833 CrossRef CAS PubMed.
- For a review see: (a) C.-L. Sun and G. -J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed; (b) A. Bhunia, Y. Santhivardhana Reddy and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC; For reference see: (c) D. Chandan, E. Lindstedt and B. Olofsson, Org. Lett., 2015, 17, 4554–4557 CrossRef PubMed; (d) L. Bering and A. P. Antonchick, Org. Lett., 2015, 17, 3134–3337 CrossRef CAS PubMed; (e) A. Ilangovan and G. Satish, J. Org. Chem., 2014, 79, 4984–4991 CrossRef CAS PubMed; (f) G. Satish, P. Ashok, R. Thangeswaran and A. Ilangovan, J. Org. Chem., 2015, 80, 5167–5175 CrossRef CAS PubMed.
- (a) P. Pravin, N. Abhay and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331–2336 CrossRef PubMed; (b) D. Wang, G. Bingyang, L. Liang, J. Shan and D. Yuqiang, J. Org. Chem., 2014, 79, 8607–8613 CrossRef CAS PubMed.
- M. T. Molina, N. Cristina, M. Ana and A. G. Csaky, Org. Lett., 2009, 11, 4938–4941 CrossRef CAS PubMed.
- (a) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, D. B. Matthew and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS PubMed; (b) J. Wang, S. Wang, G. Wang, J. Zhang and X.-Q. Yu, Chem. Commun., 2012, 48, 11769–11771 RSC; (c) D. Arghya, M. Srimanta, M. Arun, D. Uttam and D. Maiti, Eur. J. Org. Chem., 2013, 5251–5256 Search PubMed; (d) K. Komeyama, T. Kashihara and K. Takaki, Tetrahedron Lett., 2013, 54, 1084–1086 CrossRef CAS; (e) P. S. Parvinder, A. Sravan Kumar, Y. Mahipal, V. P. Singh and R. A. Vishwakarma, J. Org. Chem., 2013, 78, 2639–2648 CrossRef PubMed.
- S. E. Walker, J. A. Jordan-Hore, D. G. Johnson, S. A. Macgregor and A. –L. Lee, Angew. Chem., Int. Ed., 2014, 53, 13876–13879 CrossRef CAS PubMed.
- (a) P. Bartlett and J. D. Cotman, J. Am. Chem. Soc., 1949, 71, 1419–1422 CrossRef CAS; (b) I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055–3059 CrossRef CAS; (c) M. Srimanta, S. Maity, R. Sujoy, A. Soumitra and D. Maiti, Org. Lett., 2012, 14, 1736–1739 CrossRef PubMed.
- A. Modak, R. Sujoy and D. Maiti, J. Org. Chem., 2015, 80, 296–303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectral data and copies of spectra. See DOI: 10.1039/c5qo00246j |
| This journal is © the Partner Organisations 2015 |