
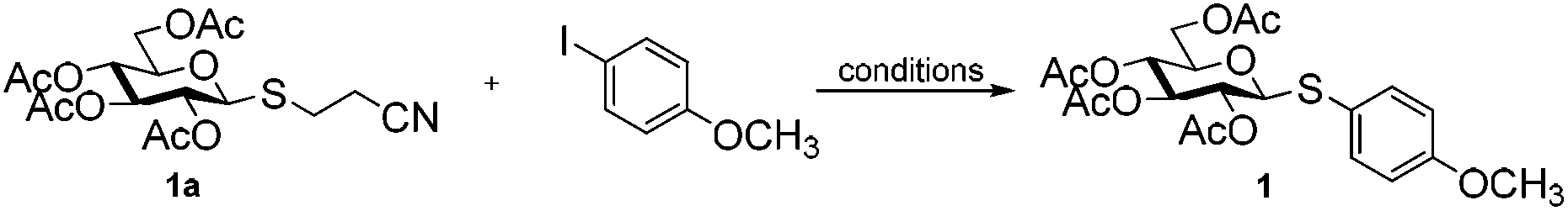
2′-Cyanoethyl thioglycosides: effective nucleophiles for synthesis of (hetero)aryl thioglycosides under the catalysis of Cu†
Xiaolong
Yuan
,
Yazhen
Kou
,
Lan
Yu
,
Zhan-Xin
Zhang
* and
Weihua
Xue
*
School of Pharmacy, Lanzhou University, Lanzhou, 730000, China. E-mail: xuewh@lzu.edu.cn; zhangzhx@lzu.edu.cn
First published on 14th October 2015
Abstract
2′-Cyanoethyl thioglycosides are designed and synthesized, which are demonstrated to function as powerful nucleophiles to a wide variety of organic iodides. The coupling reactions smoothly proceed under the catalysis of Cu to afford the corresponding (hetero)aryl thioglycosides in good to excellent yields.
Thioglycosides function as a significant type of glycosylation donors which are widely applied in the synthesis of biologically potent oligosaccharides and glycoconjugates. Furthermore, through judicious adjustments of protection groups and leaving groups, a highly efficient ‘one pot’ protocol can be realized with various thioglycosides as building blocks.1 On the other hand, owing to the stability of thioglycosidic bonds against enzymatic hydrolysis, thioglycosides have been seen as very promising candidates for the preparation of carbohydrate-based therapeutics.2 They are also employed as ligands coupled with chromatographic gel beads for binding the target proteins.3 Interestingly, the bioactive compound Afrostyraxthioside A from Afrostyrax lepidophyllus structurally features the thioglycoside unit.4 Another representative natural product carrying the C–S glycosidic bond is the clinically used antibiotic Lincomycin A (Fig. 1).5
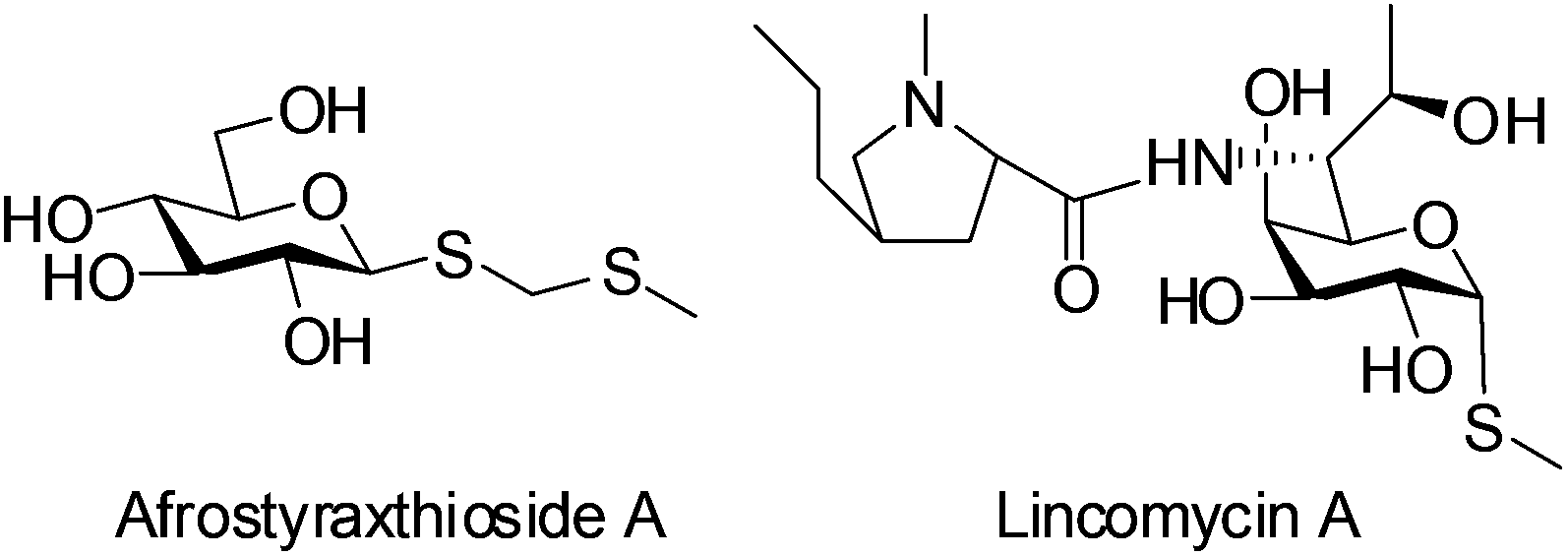 | ||
| Fig. 1 Representative thioglycoside motifs in the naturally occurring products. | ||
For these reasons, significant efforts have been made to develop general methods for the preparation of thioglycosides. One of the more widely used procedures is through the Lewis acid-promoted glycosylation of aldose pentaacetates with mercaptans6 (Scheme 1A), while the frequent formation of anomeric mixtures due to the poor stereoselectivity at the anomeric position during glycosylation process makes chromatographic process pretty cumbersome. Alternatively, a direct approach involves reactions of glycosyl halides with thiolate anions (Scheme 1B).7 But a requirement for a strong base constitutes its major disadvantage, which necessarily limits its application to those substrates with compatible functional groups. Besides, we must emphasize that some arenethiols are expensive or inaccessible, so general preparation is not easy to furnish their thioglycoconjugates. To overcome the problems mentioned above, Messaoudi and his co-workers have recently developed the metal-catalyzed coupling reaction of glycosyl thiols with (hetero)aryl halides (Scheme 1C and D).8,9 Despite the good yields and the broad substrate scope, a major drawback of these procedures for large-scale synthesis is the use of the expensive palladium catalyst. Moreover, another serious drawback of using nickel salts for coupling reaction is their toxicity, which limits their potential applications in the large-scale industrial processes.10 Additionally, in my opinion, glycosyl thiols do not seem to be the sulfuration reagent of choice for these couplings since mercaptans are usually prone to oxidation upon exposure to air. Therefore, there is a need for alternative protocols which address the shortcomings of currently available methods. The cyanoethyl group is a useful protecting group for carboxylic acid11 and thioalcohols,12 and its removal with concomitant formation of a thioanion can be easily achieved by β-elimination under the effect of a base. Against this background, we surmised that if the protecting group was installed at the anomeric position of sugar residues, producing 2′-cyanoethyl thioglycosides, substitution reactions of glycosyl thioanions generated in situ with electrophiles readily took place to furnish glycosyl sulfides. Meanwhile, recent great development in transition metal-catalyzed carbon–sulfur bond formation enables the coupling reaction of (hetero)aryl halides with sulfurating reagents.13 Among these metal catalysts, copper aroused our attention in part because many of the Cu-catalyzed reactions proceed under very mild conditions to afford (hetero)aryl ethers.14 In general, copper catalysts also tolerate a wide variety of functional groups and are therefore particularly suitable for the synthesis of complex molecules having versatile bioactivities. Moreover, compared to noble-metal catalysts, copper-based methods have appreciable economic advantages. With these considerations in mind, we expected that 2′-cyanoethyl thioglycosides would serve as effective nucleophiles, which enabled the formation of (hetero)aryl thioglycosides under the catalysis of Cu (Scheme 1E). Herein we would like to report our efforts to put this conception to practice.
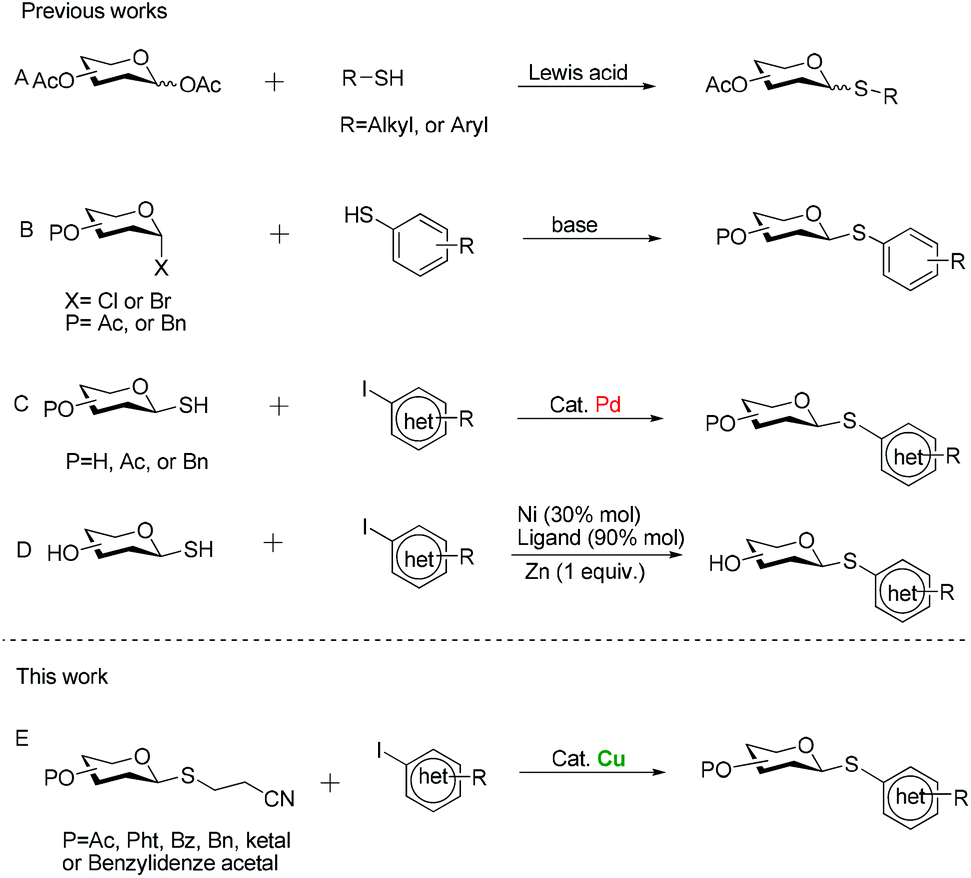 | ||
| Scheme 1 Synthetic approaches of (hetero)aryl thioglycosides. | ||
A set of 2′-cyanoethyl thioglycoside 1a–g from cheap starting materials using the simple procedures (see the ESI†) were prepared, which differed in the pattern of protecting groups (Fig. 2). These nitrile compounds were isolated as colorless oils in satisfactory yields following chromatography over silica gel. They were demonstrated to be stable towards air or moisture at room temperature and the samples have been stored for three months on the bench top with no detectable decomposition or anomerization. Their configuration was determined from the coupling constant of the anomeric proton signal in 1H NMR spectrum. Previous reports about thiofuranosides were scarce. It should be noted that the data (J1,2 = 3.6 Hz) from 1H NMR, in fairly good agreement with the reported value,15 showed that thiomannofuranoside 1f was the β-anomer.
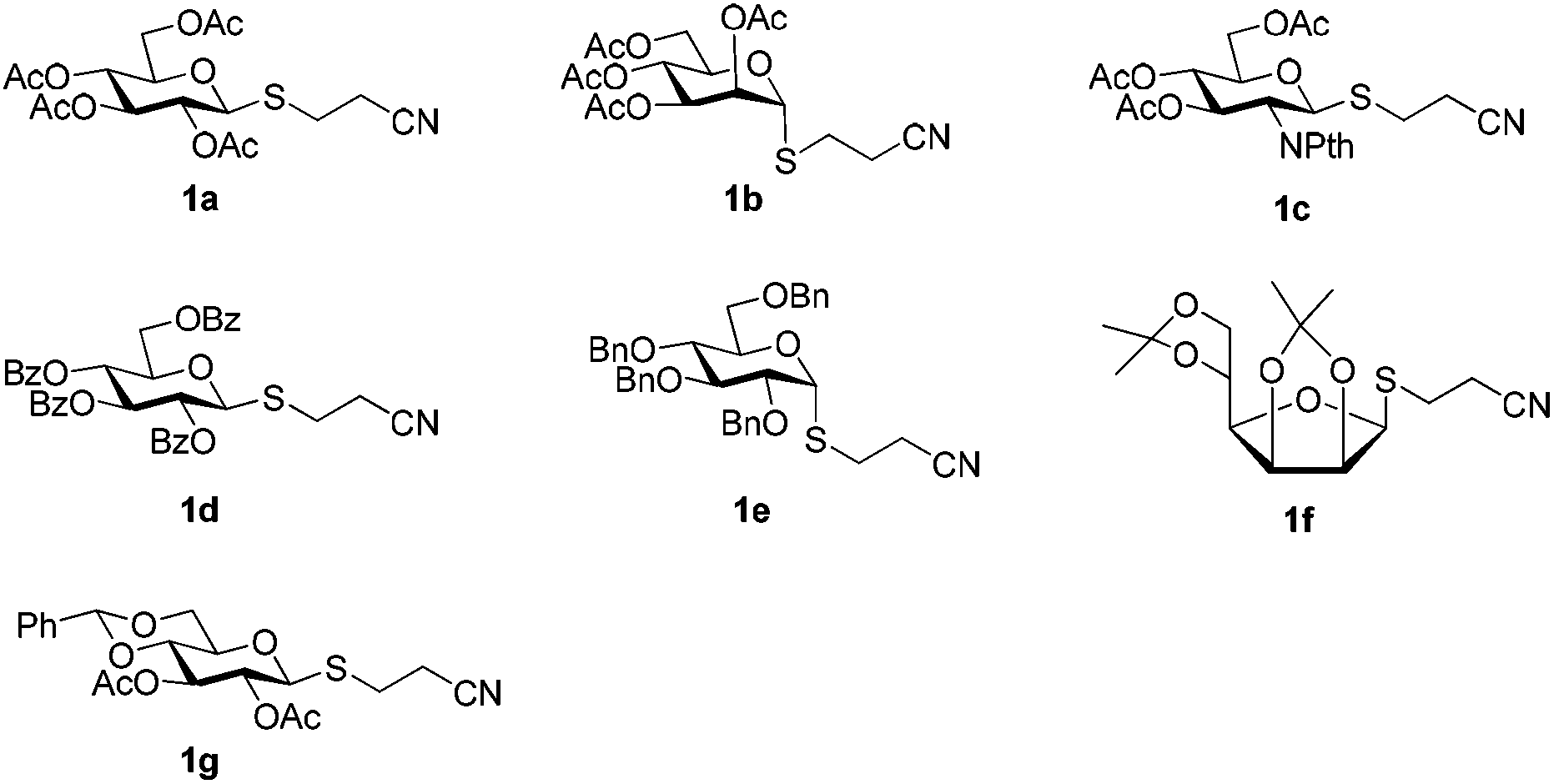 | ||
| Fig. 2 Differentially protected 2′-cyanoethyl thioglycosides in this study. | ||
The arylation conditions were optimized starting from the reaction of thioglycoside 1a with p-iodoanisole (1.2 equiv.) in refluxing 1,4-dioxane (2 ml) with various copper catalysts (10 mol%) in the presence of Cs2CO3 (2 equiv.), as summarized in Table 1. After 24 h, it was observed that CuCl was the best catalyst, providing the desired product 1 in a 44% isolated yield (entry 7). CuI, Cu2O and CuBr were also effective, albeit affording the products with slightly diminished yields (entries 1, 2 and 6). Other catalysts such as IPr-CuCl, Me2S·CuBr, and (CH3CN)4CuPF6, were found to be essentially ineffective (entries 3–5). Further inspection of the reaction conditions revealed that solvents also strongly influenced the reactivity. In CH3CN the yield reached 54% (entry 8), while CH3NO3, THF and DME were less efficacious and even led to no reactions (entries 9–11). Then, we screened the bases in CH3CN using CuCl as the catalyst (entries 12–15). Cs2CO3 is superior to other bases such as DBU, DABCO, TEA, and K2CO3. To achieve better results a series of familiar ligands was evaluated. To our excitement, the addition of L1 to the reaction system led to a higher yield (entry 16). This improvement encouraged us to test combinations of CuCl and other ligands. L2 proved to be an excellent ligand in the Cu-catalyzed coupling, which gave product 1 in 98% yield (entry 17), whereas other ligands afforded inferior results (entries 18–21). Finally, we found that the yields of product 1 were apparently decreased in the case of the reaction temperature below the boiling point or a slightly lower catalyst loading (5 mol%). In addition, under the optimized reaction conditions (entry 17) a control experiment showed that no product was formed when p-bromoanisole instead of p-iodoanisole was used.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cu salts | Ligand | Base | Solvent | Yieldsb (%) |
a Reaction conditions: 1a (0.1 mmol), p-iodoanisole (0.12 mmol), base (0.2 mmol), Cu salt (10 mol%), and ligand (10 mol%) in refluxing solvent (2 mL) within 24 h.
b Isolated yields.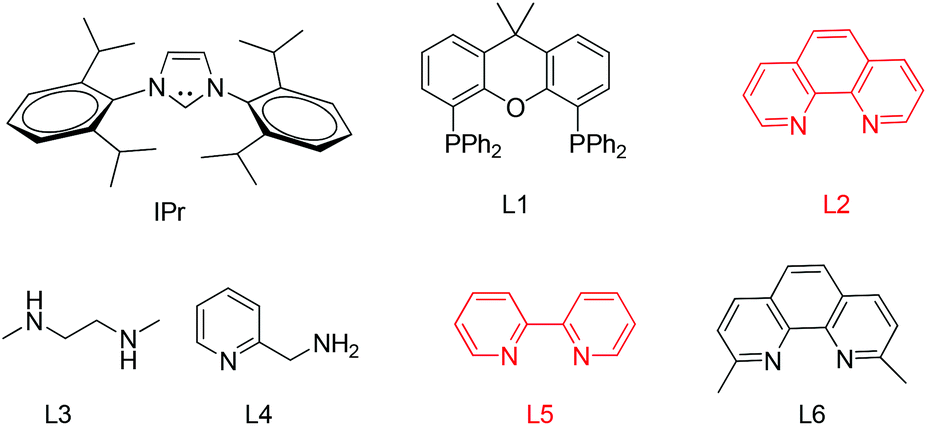
|
|||||
| 1 | CuI | — | Cs2CO3 | Dioxane | 21 |
| 2 | Cu2O | — | Cs2CO3 | Dioxane | 16 |
| 3 | IPr-CuCl | — | Cs2CO3 | Dioxane | 0 |
| 4 | Me2S·CuBr | — | Cs2CO3 | Dioxane | 0 |
| 5 | (CH3CN)4CuPF6 | — | Cs2CO3 | Dioxane | 0 |
| 6 | CuBr | — | Cs2CO3 | Dioxane | 25 |
| 7 | CuCl | — | Cs2CO3 | Dioxane | 44 |
| 8 | CuCl | — | Cs2CO3 | CH3CN | 54 |
| 9 | CuCl | — | Cs2CO3 | CH3NO2 | 0 |
| 10 | CuCl | — | Cs2CO3 | THF | 12 |
| 11 | CuCl | — | Cs2CO3 | DME | 27 |
| 12 | CuCl | — | DBU | CH3CN | 16 |
| 13 | CuCl | — | DABCO | CH3CN | 11 |
| 14 | CuCl | — | TEA | CH3CN | 6 |
| 15 | CuCl | — | K2CO3 | CH3CN | 37 |
| 16 | CuCl | L1 | Cs2CO3 | CH3CN | 87 |
| 17 | CuCl | L2 | Cs2CO3 | CH3CN | 98 |
| 18 | CuCl | L3 | Cs2CO3 | CH3CN | 51 |
| 19 | CuCl | L4 | Cs2CO3 | CH3CN | 53 |
| 20 | CuCl | L5 | Cs2CO3 | CH3CN | 31 |
| 21 | CuCl | L6 | Cs2CO3 | CH3CN | 0 |
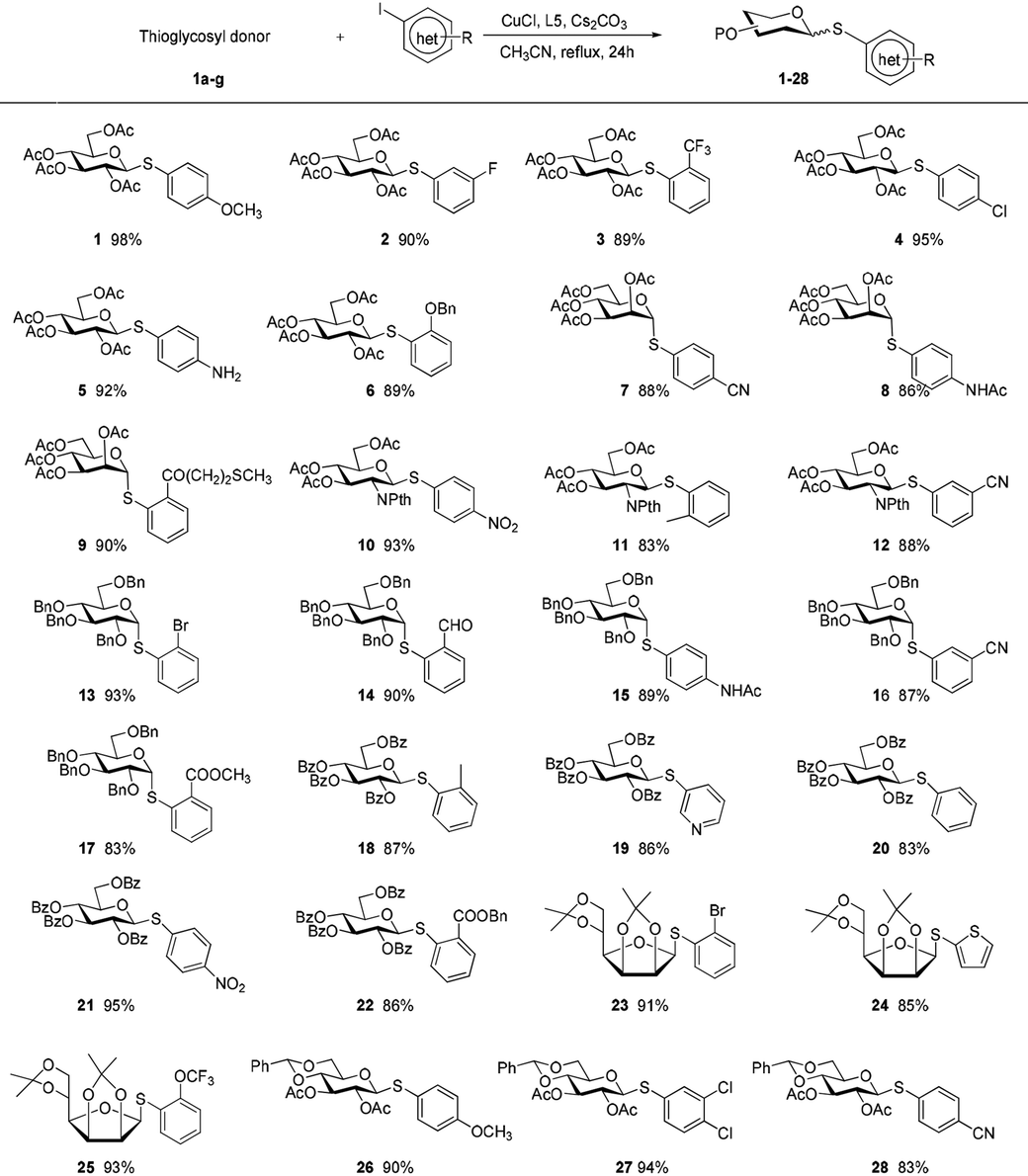
The aldose derivatives 1a–g were used to investigate the reaction scope by means of aglycons comprising a wide variety of functionalized organic iodides. As shown in Table 2, in all cases the desired products 1–28 were obtained in good to excellent isolated yields (83–95%). The reaction conditions were sufficiently mild to be compatible with a wide range of functional groups including esters, olefins, acetals, ethers, halides, amides, and ketals. Interestingly, free amino group was tolerated, allowing for the late stage conjugation or modification. Substituents at different positions on benzene rings did not obviously affect the efficiency. Especially in the case of substrates having a sterically bulky substituent at the ortho position, the corresponding products were efficiently formed. Bromo-substituted iodoarenes can act as promising difunctionalized substrates because product 13 with a relatively inert bromo group on the benzene ring in the current arylation process holds great potential as a coupling partner for following reactions. This method could be extended to heterocyclic iodides to efficiently synthesize heteroaryl thioglycosides. At present, the preparation of the differentially protected monosaccharide moieties remains the basic requirement in the synthesis of complex glycoconjugates. These differentially protected 2′-cyanoethyl thioglycosides displayed high reactivity and were smoothly coupled with partners. Our procedure not only applied to pyranosides but also worked favorably with furanosides. Moreover, 1H NMR data for these coupling products showed the absolute configuration at the anomeric positions was retained without any epimerization. It should also be noted that their unprotected counterparts were fairly difficult to obtain as cyano-based linkers were prone to cleavage, hydrolysis, or reduction under the standard conditions to remove the protecting groups from sugar rings.
|
Conclusions
In summary, we have developed a new method for preparing (hetero)aryl thioglycosides under the catalysis of Cu with designed 2′-cyanoethyl thioglycosides as effective sulfuration reagents. These nucleophiles provide good conversion of a wide range of substrates and they exhibit remarkably high functional group tolerance. Research on further uses for this procedure is ongoing in our laboratory and will be reported in due course.Acknowledgements
We gratefully acknowledge the financial support from the National Science Foundation of China (21402075) and the Fundamental Research Funds for the Central Universities, Lanzhou University (lzujbky-2015-62 and lzujbky-2015-313).Notes and references
- For selective reference, see: Z. Zhang and C.-H. J. J. Wong, Am. Chem. Soc., 1999, 121, 734 CrossRef CAS . For recent review on thioglycosides, see: G. Lian, X. Zhan and B. Yu, Carbohydr. Res., 2015, 403, 13 CrossRef PubMed.
- Z. J. Witczak, Curr. Med. Chem., 1999, 6, 165 CAS.
- C. Orgeret, E. Seillier, C. Gautier, J. Defaye and H. Driguez, Carbohydr. Res., 1992, 224, 29 CrossRef CAS.
- A. N. Ngane, M. Lavault, D. Séraphin, A. Landreau and P. Richomme, Carbohydr. Res., 2006, 341, 2799 CrossRef CAS PubMed.
- R. R. Herr and M. E. Bergy, Antimicrob. Agents Chemother., 1962, 560 Search PubMed.
- For selective examples, please see: (a) S. K. Das, J. Roy, K. A. Reddy and C. Abbineni, Carbohydr. Res., 2003, 338, 2237 CrossRef CAS; (b) G. Agnihotri, P. Tiwari and A. K. Misra, Carbohydr. Res., 2005, 340, 1393 CrossRef CAS PubMed; (c) X. Li, L. Huang, X. Hu and X. Huang, Org. Biomol. Chem., 2009, 7, 117 RSC; (d) S. K. Das and N. Roy, Carbohydr. Res., 1996, 296, 275 CrossRef CAS.
- (a) M. Blanc-Muesser, J. Defaye and H. Driguez, Carbohydr. Res., 1978, 67, 305 CrossRef CAS; (b) M. Apparu, M. Blanc-Muesser, J. Defaye and H. Driguez, Can. J. Chem., 1981, 59, 314 CrossRef CAS.
- (a) E. Brachet, J.-D. Brion, S. Messaoudi and M. Alami, Adv. Synth. Catal., 2013, 355, 477 CrossRef CAS PubMed; (b) E. Brachet, J.-D. Brion, M. Alami and S. Messaoudi, Adv. Synth. Catal., 2013, 355, 2627 CrossRef CAS PubMed; (c) A. Bruneau, M. Roche, A. Hamze, J.-D. Brion, M. Alami and S. Messaoudi, Chem. – Eur. J., 2015, 21, 8375 CrossRef CAS PubMed.
- E. Brachet, J.-D. Brion, M. Alami and S. Messaoudi, Chem. – Eur. J., 2013, 19, 15276 CrossRef CAS PubMed.
- (a) D. G. Barceloux, J. Toxicol., Clin. Toxicol., 1999, 37, 239 CrossRef CAS PubMed; (b) N. B. Aquino, M. B. Sevigny, J. Sabangan and M. C. Louie, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2012, 30, 189 CrossRef CAS PubMed; (c) J. Zhao, X. Shi, V. Castranova and M. Ding, J. Environ. Pathol., Toxicol. Oncol., 2009, 28, 177 CrossRef CAS.
- P. K. Misra, S. A. N. Hashmi, W. Haq and S. B. Katti, Tetrahedron Lett., 1989, 30, 3569 CrossRef CAS.
- Y. Ohtsuka, S. Niitsuma, H. Tadokoro, T. Hayashi and T. Oish, J. Org. Chem., 1984, 49, 2326 CrossRef CAS.
- For reviews, see: (a) C. C. Eichman and J. P. Stmbuli, Molecule, 2011, 16, 590 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed.
- For selective references, see: (a) C. G. Bates, R. K. Gujadhur and D. Venkataraman, Org. Lett., 2002, 4, 2803 CrossRef CAS PubMed; (b) S. Jammi, S. Sakthivel, L. Rout, T. Mukherjee, S. Mandal, R. Mitra, P. Saha and T. Punniyamurthy, J. Org. Chem., 2009, 74, 1971 CrossRef CAS PubMed; (c) P. F. Larsson, A. Correa, M. Carril, P. O. Norrby and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5691 CrossRef CAS PubMed; (d) H. Xu, Y. Zhao, T. Feng and Y. Feng, J. Org. Chem., 2012, 77, 2878 CrossRef CAS PubMed; (e) Y. Li, J. Pu and X. Jiang, Org. Lett., 2014, 16, 1212 CrossRef PubMed.
- A. Fürstner, Liebigs Ann. Chem., 1993, 1211 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of substrates, characterization data. See DOI: 10.1039/c5qo00276a |
| This journal is © the Partner Organisations 2015 |