
Micropatterned model membrane with quantitatively controlled separation of lipid phases†
Fumiko Okadaa and
Kenichi Morigaki*ab
aGraduate School of Agricultural Science, Kobe University, Rokkodaicho 1-1, Nada, Kobe 657-8501, Japan
bResearch Center for Environmental Genomics, Kobe University, Rokkodaicho 1-1, Nada, Kobe 657-8501, Japan. E-mail: morigaki@port.kobe-u.ac.jp; Fax: +81-78-803-5941
First published on 25th November 2014
Abstract
The localization of lipids and proteins in microdomains (lipid rafts) is believed to play important functional roles in the biological membrane. Herein, we report on a micropatterned model membrane that mimics lipid rafts by quantitatively controlling the spatial distribution of lipid phases. We generated a composite membrane of polymeric and fluid lipid bilayers by lithographic polymerization of diacetylene phospholipid(1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine: DiynePC). The composite membrane comprised polymer free-region (R0), partially polymerized region (R1), and fully polymerized region (R2). As a ternary mixture of saturated lipid, unsaturated lipid, and cholesterol was introduced into the voids between polymeric bilayers, liquid-ordered (Lo) and liquid-disordered (Ld) lipid phases were accumulated in R0 and R1, respectively. Local enrichment of Ld phase in R1 (and Lo phase in R0) was enhanced with a heightened coverage of polymeric bilayer in R1, supporting the premise that polymeric bilayer domains are inducing the phase separation. The pattern geometry (the area fractions of R0 and R1) also affected the enrichment due to the balance of gross Lo/Ld area fractions. Therefore, we could control the local Lo/Ld ratios by modulating the pattern geometry and polymer coverage in R1. Micropatterned model membrane with quantitatively controlled distribution of Lo/Ld phases offers a new tool to study the functional roles of lipid rafts by enabling to separate membrane-bound molecules according to their affinities to Lo and Ld phases.
1. Introduction
The cell membrane is made of a heterogeneous mixture of lipids and proteins. It is generally regarded that the lateral organization of membrane microdomains (“lipid rafts”) is closely related with the cellular functions.1–4 The lateral heterogeneity is generated by the spontaneous segregation of lipids. Bilayer membranes containing saturated lipids, unsaturated lipids, and cholesterol spontaneously separate into the liquid-ordered (Lo) and liquid-disordered (Ld) phases in certain compositional regimes.5,6 Phase separations in model membranes such as giant unilamellar vesicles (GUVs) and substrate-supported phospholipid bilayers (SPBs) have been extensively studied as models of lipid rafts.7–10 Studies using model membranes have provided important insight into the formation and physicochemical properties of lipid rafts.4In the case of SPBs, micropatterning techniques have been applied to generate arrayed patches of Lo and Ld phases in the model membranes.11–16 For example, Yoon et al. accumulated Lo and Ld phases on a silicon substrate by locally modulating the surface curvatures. Lo and Ld phases were enriched on the flat and corrugated surfaces, respectively, due to the difference in bending energy.11 Some other studies have also exploited the different bending energies of Lo and Ld phases to realize patterned accumulation.12–14 Alternatively, some studies utilized kinetic effects to realize a patterned phase separation by using photolithography and micro-fluidics.15,16 Patterned Lo/Ld phases with controlled size and spatial distribution would provide a model membrane for systematic in vitro parallel assays of the lipid-raft-related functions.
We have previously developed a methodology to create patterned Lo/Ld phases by using a composite membrane of polymeric and fluid lipid bilayers.17 The polymeric bilayer was lithographically generated from a diacetylene phospholipid by UV illumination.18 The density of polymeric bilayer domains could be locally modulated by applying varied UV doses and removing non-reacted monomers with a detergent solution (Fig. 1(A)).19,20 As a fluid bilayer containing a mixture of saturated lipid, unsaturated lipid, and cholesterol was incorporated, saturated lipid and cholesterol (Lo domains) were enriched in the polymer-free region (R0), whereas unsaturated lipid (Ld domains) was enriched in the partially polymeric region (R1) (Fig. 1(B) and (C)).17 A fluorescent phospholipid (TR-PE) was used as the marker of Ld phase.21 Selective binding of dye-conjugated cholera toxin subunit B (CTB-488) to a glycolipid (GM1) was used to detect Lo phase22 (representative fluorescence micrographs of the phase separation process are shown in ESI (Fig. S1†)). We inferred that the driving force of the patterned Lo/Ld phase separation was the local bending of fluid bilayer at the boundary with polymeric bilayer, since a slight mismatch of the thickness is expected between polymeric and fluid bilayers.17 Due to the higher energetic penalty of bending, Lo domains are expected to be excluded from the boundaries, resulting in the accumulation of Ld domains around polymeric bilayer domains.23,24
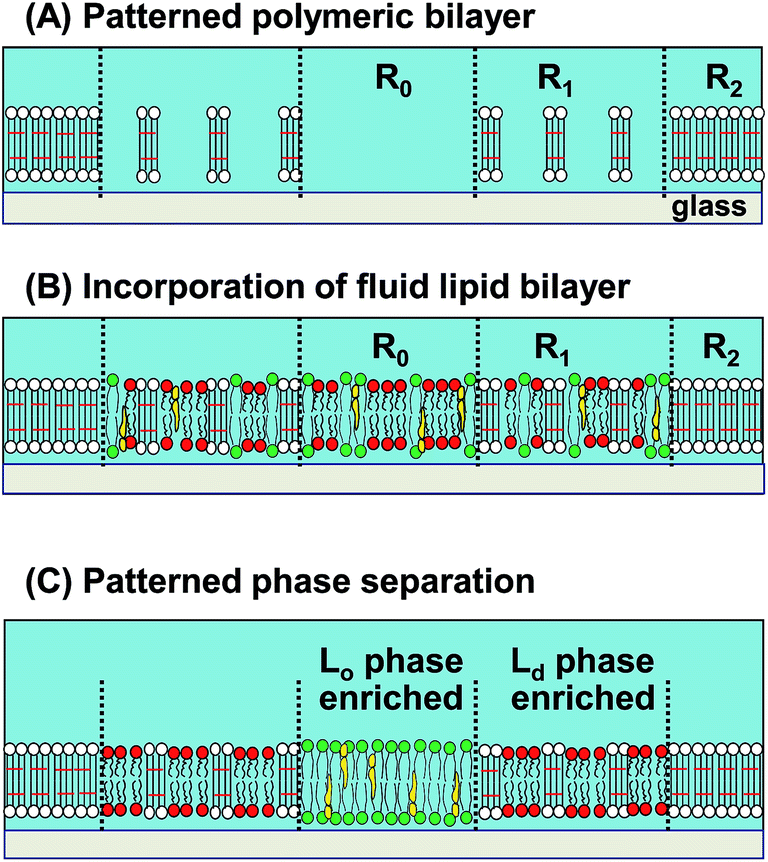 | ||
| Fig. 1 Schematic illustration of the patterned phase separation in a model membrane. (A) The patterned membrane has three regions: polymer-free region (R0), partially polymeric region (R1), and fully polymeric region (R2). As a mixed bilayer is introduced into the voids between polymeric bilayers (B), Lo and Ld phases spontaneously accumulate in R0 and R1, respectively (C). | ||
In the present work, we report that we can generate a micropatterned membrane with quantitatively controlled local Lo/Ld ratios. Although a number of techniques have been reported for patterning Lo/Ld phases, previous studies, including our work, have shown only qualitative separation of Lo and Ld phases. However, quantitatively controlled distribution of Lo/Ld phases and associated molecules in pre-designed patterns would be desirable for quantitatively evaluating the functional roles of lipid rafts. We established a methodology to modulate the local Lo/Ld ratios through two experimentally controllable parameters. The first parameter is the area fractions of partially polymeric and polymer-free regions in the pattern. The second parameter is the area fraction of polymeric bilayers within the partially polymeric bilayer region. Well-defined separation of Lo and Ld phases should offer a new tool to study the functional roles of lipid rafts by enabling to separate membrane-bound molecules according to their affinities to Lo and Ld phases.
2. Materials and methods
2.1. Materials
1,2-Bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine (DiynePC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), sphingomyelin (egg, chicken) (SM), cholesterol (ovine wool) (Chol), and GM1 ganglioside (brain, ovine) (GM1) were purchased from Avanti Polar Lipids (Alabaster, AL). Texas Red 1,2-dihexadecanoyl-sn-glycero-phosphoethanolamine (TR-PE) and cholera toxin subunit B-Alexa Fluor 488 conjugate (CTB-488) were purchased from Molecular Probes (Eugene, OR). Bovine serum albumin (BSA) was purchased from Sigma-Aldrich (St. Louis, MO). Sodium dodecyl sulfate (SDS), glucose oxidase, catalase, and glucose were purchased from Nacalai Tesque (Kyoto, Japan). Deionized water used in the experiments was ultrapure Milli-Q water (Millipore) with a resistance of 18.2 MΩ cm. It was used for cleaning substrates, preparing buffer solution (0.01 M sodium phosphate buffer with 0.15 M NaCl, pH 6.6 (PBS)) and all other experiments.2.2. Substrate cleaning
Microscopy cover slips (Matsunami, Osaka, Japan) were used as substrates for bilayer deposition. The substrates were cleaned in an SDS solution (0.1 M) for 20 min under sonication, rinsed with Milli-Q water, treated in a solution of NH4OH (28%)/H2O2 (30%)/H2O (0.05![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5) for 10 min at 65 °C, rinsed extensively with Milli-Q water, and then dried in a vacuum oven for 30 min at 80 °C. Before use, these substrates were further cleaned by the UV/ozone treatment for 20 min (PL16-110, Sen Lights, Toyonaka, Japan).
:1:5) for 10 min at 65 °C, rinsed extensively with Milli-Q water, and then dried in a vacuum oven for 30 min at 80 °C. Before use, these substrates were further cleaned by the UV/ozone treatment for 20 min (PL16-110, Sen Lights, Toyonaka, Japan).
2.3. Preparation of patterned polymeric bilayers
Bilayers of monomeric DiynePC were deposited onto glass substrates by the spontaneous spreading of vesicles. DiynePC powder was suspended in Milli-Q water by freezing in liquid nitrogen and thawing at 60 °C (five cycles). After the freeze-and-thaw, DiynePC suspension was homogenized by an ultrasonic homogenizer (Branson Sonifier150) at 60 °C (30 s × 2). Monomeric DiynePC suspension was applied onto a cleaned substrate on ice to immediately cool the membrane (we previously discovered that it is important to deposit monomers at a low temperature for generating homogeneous DiynePC bilayers25).Polymerization of DiynePC bilayers was conducted by UV irradiation using a mercury lamp (UVE-502SD, Ushio, Tokyo, Japan) as the light source. A closed system that comprised a water reservoir, a pump, and a cell (ca. 4 mL volume) was used. The water reservoir was depleted of oxygen by purging with argon.19 Oxygen-free water was circulated continuously by the pump through the cell where polymerization of the bilayers was conducted. The cell had two walls on the opposite sides, one being the sample (the SPB was inside the cell) and the other being a quartz window through which UV light was illuminated. Desired patterns were transferred onto the SPB in the polymerization process by illuminating the sample through a mask (a quartz slide with a patterned chromium coating) which was placed directly on the SPB. After sufficient circulation of deaerated water (typically for 15 minutes), the pump was stopped and the polymerization was started. The applied UV intensity was typically 7 mW cm−2 at 254 nm and the UV dose was varied by changing the illumination time. After the UV irradiation, non-polymerized DiynePC molecules were removed from the substrate surface by immersing in 0.1 M SDS solution at 30 °C for 30 min and rinsing with Milli-Q water extensively. The polymerized bilayers were stored in Milli-Q water in the dark at 4 °C.
The patterned membrane consisted of polymer-free region (R0), partially polymeric region (R1), and fully polymerized region (R2) (Fig. 1). These patterns were generated by the successive UV exposure of monomeric membrane using two different photomasks (100 μm squares/8 μm circles or 10 μm squares). We varied two experimental parameters to modulate the phase separation. First, the area fractions of R0 and R1 (A0 and A1: A0 + A1 = 1) were varied by changing the pattern geometries. Second, the fraction of the polymeric bilayer in R1 (p) was changed by the applied UV dose for polymerization. These parameters are schematically summarized in ESI (Fig. S2†).
2.4. Preparation of vesicle suspensions
Two types of vesicle suspensions were prepared: (a) DOPC with TR-PE (1 mol%) (b) DOPC/SM/Chol (1:1:1) with GM1 and TR-PE (1 mol% each). Lipids dissolved in organic solvents (DOPC, SM, Chol, and TR-PE were dissolved in chloroform, and GM1 was dissolved in methanol) were mixed in a round-bottom flask, dried with nitrogen (15 min), and subsequently evaporated for at least 4 h in a vacuum desiccator. The dried lipid films were hydrated in PBS containing 3 mM CaCl2 overnight (the total lipid concentration was 1 mM). Lipid membranes were dispersed by five freeze/thaw cycles, and the suspension was extruded by using a Liposofast extruder (Avestin, Ottawa, Canada) with 100 nm polycarbonate membrane filter (10 times) and 50 nm polycarbonate filter (15 times).
2.5. Phase separation of fluid bilayer in a patterned membrane
Fluid bilayers were incorporated into the voids between polymeric bilayers in a micropatterned membrane by spontaneous spreading of vesicles.26,27 A droplet of vesicle suspensions (100 μL) was put on a petri-dish and covered with a substrate having a patterned polymeric bilayer. The substrate was incubated for 30 minutes to allow complete spreading of SPBs on the patterned membrane. Excess vesicles were rinsed off by extensively flushing the substrate surface with Milli-Q water.We first incorporated DOPC/TR-PE into the patterned membrane to estimate the area fraction of the polymeric bilayer in R1 (p). After the fluorescence microscopy observation, DOPC/TR-PE was removed by immersing the sample in 0.1 M SDS at 30 °C for 30 min and extensively rinsing with Milli-Q water. Subsequently, DOPC/SM/Chol (1:1:1) with GM1 and TR-PE (1 mol% each) was introduced into the voids and incubated at 25 °C for 1–3 days (although phase separation started immediately after the introduction of fluid bilayer, we waited long enough to complete the phase separation17). After the completion of phase separation, we observed the same positions of patterned membrane.
2.6. Fluorescence microscopy observation
Fluorescence microscopy observations were performed using an upright microscope (BX51WI, Olympus, Tokyo, Japan) equipped with a xenon lamp (UXL-75XB, Olympus), a 20× objective (NA 0.95), and a CCD camera (DP30BW, Olympus). Two types of filter sets were used: (1) excitation 470–490 nm/emission 510–550 nm (green fluorescence), (2) excitation 545–580 nm/emission >610 nm (red fluorescence). Fluorescence images were processed with the MetaMorph program (Molecular Devices, Sunnyvale, CA).3. Results
We studied Lo/Ld phase separation in the patterned membranes with defined geometry and density of polymeric bilayer domains. Fig. 2 shows the fluorescence micrographs of three patterned membranes comprising polymer-free region (R0), partially polymeric region (R1), and fully polymeric region (R2). The large squares (100 μm) contained R1 and small windows of R0 (10 μm squares or 8 μm circles). Outside of the large squares was R2. The area fractions of R0 (A0) and R1 (A1) were varied by using photomasks with different geometries (A0 and A1 are the area fractions within R0 and R1 (A0 + A1 = 1)) (R2 is not included in calculating the area fractions, because we assume that fluid bilayers are excluded from it). A0/A1 was 0.25/0.75 for the pattern (A), and 0.05/0.95 for the patterns (B) and (C), respectively. The polymeric bilayer fraction in R1 (p) was varied by changing the applied UV dose. To estimate the values of p, we first incorporated a fluid bilayer that did not separate into two phases (DOPC/TR-PE) (upper panels of Fig. 2). The fluorescence intensity of TR-PE in R0 (ITR0) was higher than that in R1 (ITR1) due to the fact that R1 was partially covered by polymeric bilayer. The values of p were estimated with the following equation, assuming that TR-PE is uniformly distributed in the fluid bilayer (ITR0 and ITR1 represent the true fluorescence intensities after subtracting the background fluorescence intensity from the measured intensities):20| p = 1 − (ITR1/ITR0). | (1) |
 | ||
| Fig. 2 Fluorescence micrographs of the phase separation in patterned bilayers having different geometries and polymeric bilayer fractions: the area fractions of R1 (A1) (shown at the top) were varied by using different photomasks upon polymerization. The polymeric bilayer fractions in R1 (p) (shown at the top) were determined by incorporating a homogeneous bilayer (DOPC/TR-PE) (upper panel). Subsequently, the fluid bilayer was exchanged with a mixed lipid bilayer (DOPC/SM/Chol/TR-PE/GM1) and the phase separation was observed using the markers of Ld and Lo phases (TR-PE and CTB-488) (middle and lower panels). The enrichment of TR-PE and CTB-488 in R1 and R0 (DTR1 and DCTB0) were estimated from the fluorescence intensities (see the text). The scale bar is 40 μm. | ||
The obtained p values are given in Fig. 2. After evaluating the p values, we removed DOPC/TR-PE with a detergent solution (0.1 M SDS, 30 min at 30 °C: this treatment did not alter polymeric bilayer domains20) and incorporated a new lipid membrane (DOPC/SM/Chol (1:1:1)) that separated into Lo and Ld phases. The membrane contained TR-PE and GM1 (1 mol% each). After incubation, TR-PE was enriched in R1, as evidenced by the higher fluorescence intensity in R1 compared with R0, in spite of the fact that there was less fluid membrane in R1 (note the inverted contrast between the upper and middle panels). Fluidity of the membrane in R1 was confirmed by the fluorescence recovery after photobleaching (FRAP) measurements (ESI, Fig. S3†). After observing the distribution of TR-PE, we added CTB-488 to detect Lo phase. CTB-488 was preferentially found in R0 (lower panels). The line profiles of fluorescence intensities confirmed the inverted accumulation of TR-PE and CTB-488 (ESI, Fig. S4†). Comparing the middle panels of (B) and (C), we note that the fluorescence intensity of TR-PE in R1 was higher for the sample having a larger p (C). Concomitantly, the fluorescence of CTB-488 in R0 was more prominent in this sample (bottom panel). These observations suggested that a higher density of polymeric bilayer in R1 enhanced the patterned separation of Lo and Ld phases. The local enrichment of TR-PE and CTB-488 was evaluated from the fluorescence intensities in R0 and R1 (ITR0, ITR1, ICTB0, ICTB1) using the following equations (the background fluorescence intensities were subtracted from the measured intensities to obtain the true fluorescence intensities of TR-PE and CTB-488):
| Enrichment of CTB-488 in R0: DCTB0 = (ICTB0/ICTB1)(1 − p). | (2) |
| Enrichment of TR-PE in R1: DTR1 = (ITR1/ITR0)/(1 − p) | (3) |
The fluorescence intensity in R1 was normalized with the area fraction of fluid bilayer (1 − p), considering the fact that the region contained less fluid bilayer due to polymeric bilayer. The obtained values of DTR1 and DCTB0 are given in Fig. 2. Enrichment of Lo and Ld phases in R0 and R1 was enhanced for a sample with a higher p value ((B) and (C)).
To evaluate the effects of polymeric bilayer on the phase separation, we measured the enrichment of TR-PE and CTB-488 (DTR1 and DCTB0) in samples with systematically varied p (we generated patterned samples with varied UV doses to obtain different p values). The two pattern geometries shown in Fig. 2 (A1 = 0.75 and A1 = 0.95) were used. The plot of DTR1 versus p is summarized in Fig. 3(A). DTR1 increased with the p value. For low p values, DTR1 was close to 1, as expected, and increased gradually with p. The increase was more prominent for higher p values. In the case of the membrane with A1 = 0.95, DTR1 increased very steeply as the p value exceeded 0.7 (Fig. 3(A)). The plot of DCTB1 versus p also shows that more CTB-488 molecules are localized in R0 for a higher p value (Fig. 3(B)). The data for CTB-488 were rather scattered, presumably due to the effects of non-specific adsorption, although we suppressed it by applying a blocking agent (BSA). These results clearly show that the patterned phase separation is positively correlated with the amount of polymeric bilayer domains.
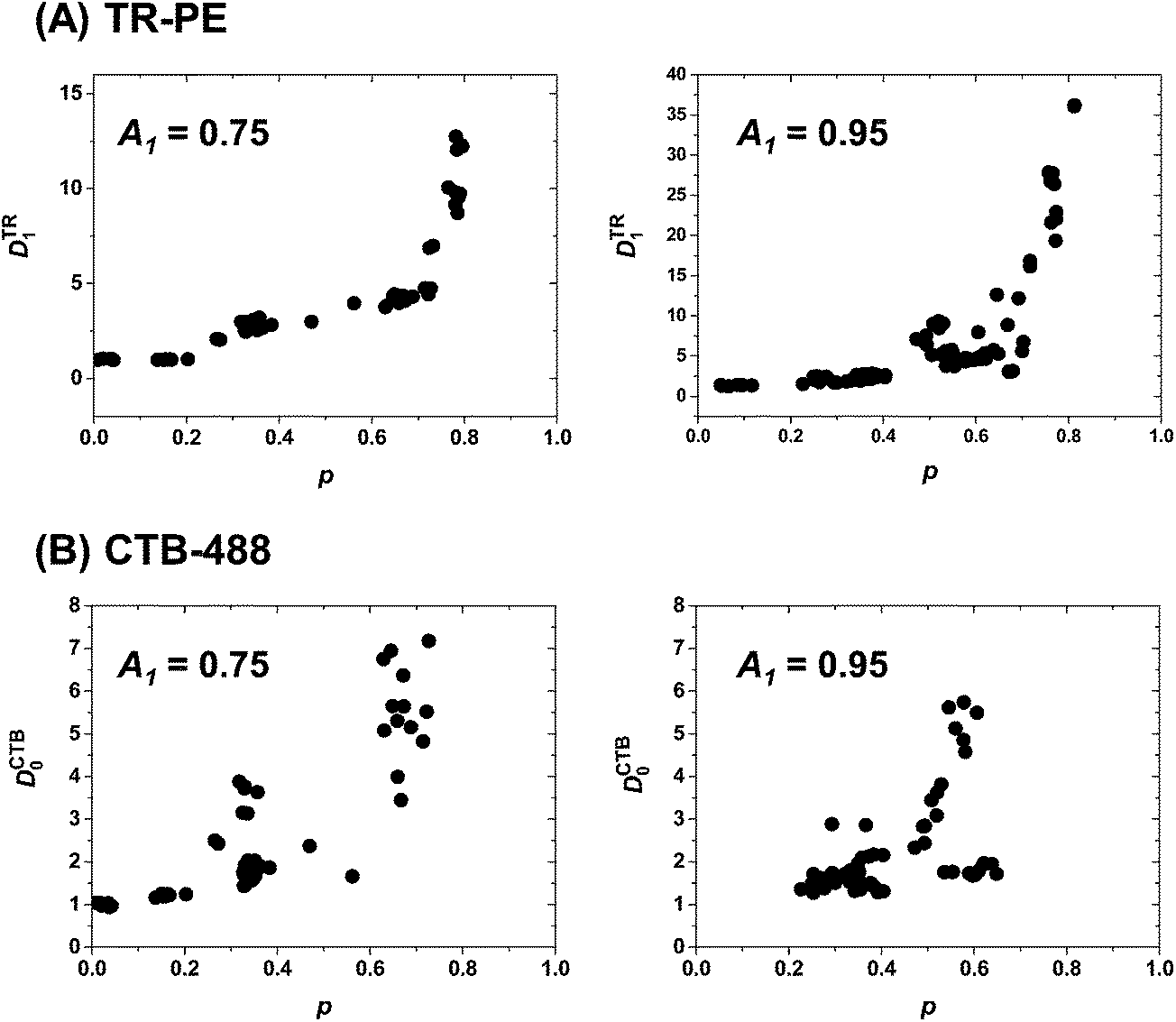 | ||
| Fig. 3 Enrichment of TR-PE in R1 (DTR1) (A) and CTB-488 in R0 (DCTB0) (B) in patterned samples with varied p. The two pattern geometries in Fig. 2 (A1 = 0.75 and 0.95) were used. Each data point represents the evaluation from a single fluorescence micrograph. Results from at least four independent samples were compiled. | ||
Localization of TR-PE and CTB-488 in the patterned membranes reflects the accumulation of Ld and Lo phases in R1 and R0, respectively. We estimated the occupied area fractions of Ld phase in R0 and R1 (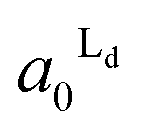 and
and 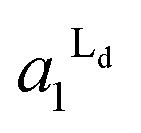 ) from the observed enrichment of TR-PE in R1, assuming the following two conditions. First, we assumed that the gross area fractions of Lo and Ld phases were 0.5 (equal area of the two phases) for the lipid composition used (DOPC/SM/Chol = 1:1:1), as previously estimated by the atomic force microscopy observations.28 Second, we assumed that the area fraction of Ld phase was proportional to the fluorescence intensity of TR-PE, since TR-PE was predominantly partitioned in the Ld phase. By applying these boundary conditions to the experimentally obtained enrichment of TR-PE in R1 (DTR1 in Fig. 3(A)), we could calculate the area fractions of Lo/Ld in R0 and R1 (eqn (4)–(6)).
) from the observed enrichment of TR-PE in R1, assuming the following two conditions. First, we assumed that the gross area fractions of Lo and Ld phases were 0.5 (equal area of the two phases) for the lipid composition used (DOPC/SM/Chol = 1:1:1), as previously estimated by the atomic force microscopy observations.28 Second, we assumed that the area fraction of Ld phase was proportional to the fluorescence intensity of TR-PE, since TR-PE was predominantly partitioned in the Ld phase. By applying these boundary conditions to the experimentally obtained enrichment of TR-PE in R1 (DTR1 in Fig. 3(A)), we could calculate the area fractions of Lo/Ld in R0 and R1 (eqn (4)–(6)).
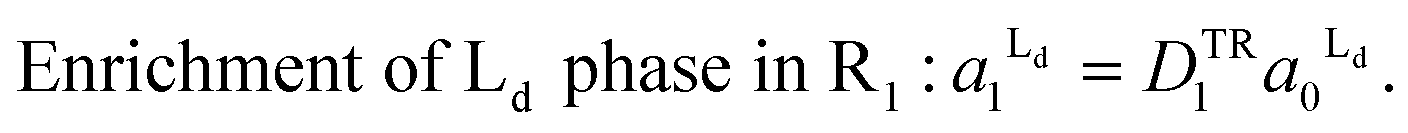 | (4) |
The total areas of Ld domains after the phase separation should be equal to the gross area of Ld phase:
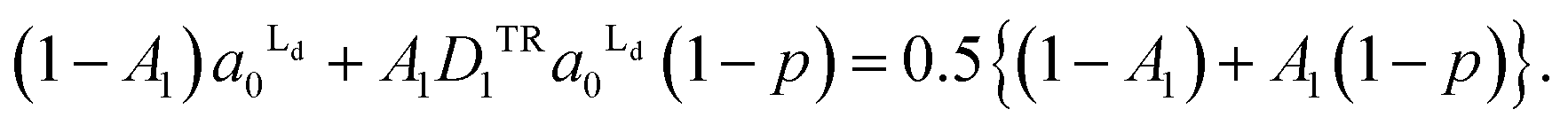 | (5) |
From the eqn (4) and (5), the area fraction of Ld phase in R0 can be calculated as follows:
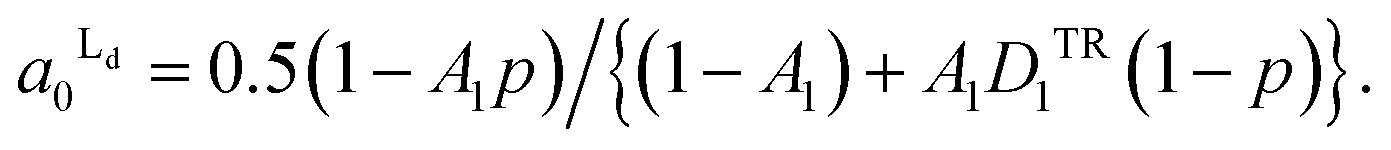 | (6) |
The estimated area fractions are shown in Fig. 4. For A1 = 0.75, 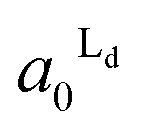 decreased and
decreased and 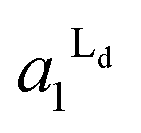 increased progressively with p, indicating enrichment of Lo and Ld phases in R0 and R1, respectively. On the other hand, in the case of A1 = 0.95,
increased progressively with p, indicating enrichment of Lo and Ld phases in R0 and R1, respectively. On the other hand, in the case of A1 = 0.95,  decreased with p, whereas
decreased with p, whereas 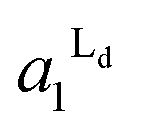 increased only slightly, indicating that R0 consisted mostly of Lo phase, whereas R1 remained a mixture of Lo and Ld phases. This asymmetric enrichment can be understood by considering the fact that R0 is much smaller compared with that of R1 (A0 = 0.05 and A1 = 0.95). As a consequence, a part of the Lo phase should have remained in R1, even if R0 was highly enriched with Lo phase.
increased only slightly, indicating that R0 consisted mostly of Lo phase, whereas R1 remained a mixture of Lo and Ld phases. This asymmetric enrichment can be understood by considering the fact that R0 is much smaller compared with that of R1 (A0 = 0.05 and A1 = 0.95). As a consequence, a part of the Lo phase should have remained in R1, even if R0 was highly enriched with Lo phase.
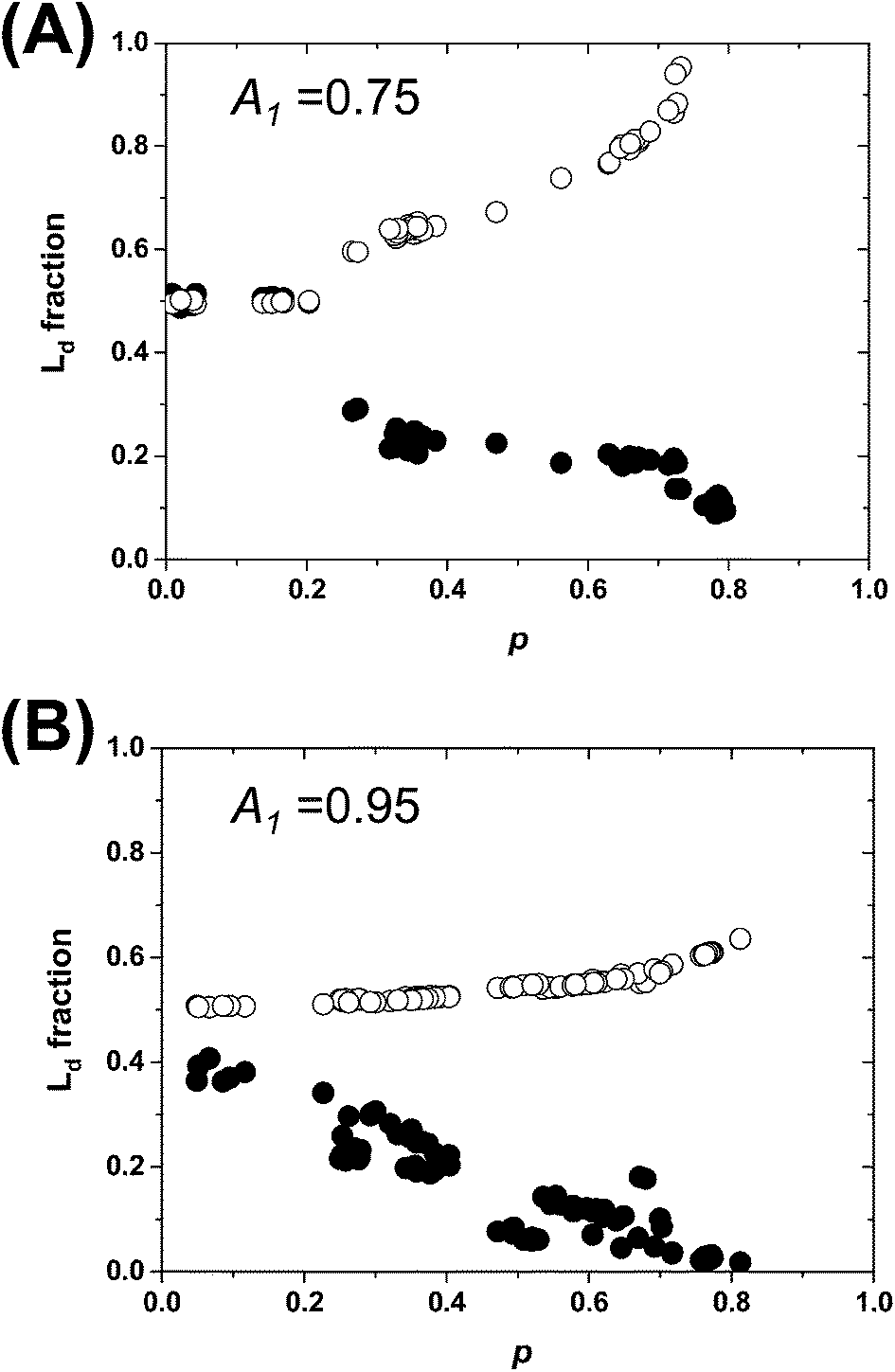 | ||
Fig. 4 The area fractions of Ld phase in R0 and R1 were estimated from the enrichment of TR-PE in Fig. 3(A). We assumed that the gross area fractions of Lo and Ld phases were 0.5 for the lipid composition used. Open circles: Ld phase in R1 (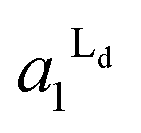 ); filled circles: Ld phase in R0 ( ); filled circles: Ld phase in R0 (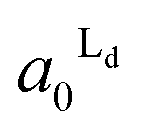 ). ). | ||
The results in Fig. 4 demonstrate that we can quantitatively control the local Lo/Ld ratios in a patterned membrane by the pattern geometry (area fractions of R0 and R1) and the polymeric bilayer fraction in the partially polymeric region. This feature enables to create an array of model membranes with varied local Lo/Ld ratios. Fig. 5 shows a patterned membrane that has four regions with different polymeric bilayer coverages, a polymer free region (a), two partially polymeric regions (b and c), and a fully polymeric region (d). By incorporating DOPC/TR-PE, we could estimate the area fractions of polymeric bilayers in (b) and (c) to be 0.02 and 0.48, respectively (Fig. 5(A)). Subsequently, we incorporated DOPC/SM/Chol (1:1:1) containing GM1 and TR-PE (1 mol% each), and observed that TR-PE and CTB-488 were distributed in the three regions ((a)–(c)) according to the densities of polymeric bilayer domains. TR-PE was most accumulated in the region (c) where the density of polymeric bilayers was highest (except for the fully polymerized region (d)) (Fig. 5(B)), whereas CTB-488 was most accumulated in the polymer-free region (a) (Fig. 5(C)) (enrichment of TR-PE at the boundaries between the regions (b) and (d) was caused presumably by the partial polymerization at the boundary of these regions). From the fluorescence intensities of TR-PE, we evaluated the fractions of Ld phase in each region to be 0.24 (a), 0.34 (b), and 0.96 (c) (the Lo/Ld ratio could not be quantified in the fully polymeric region).
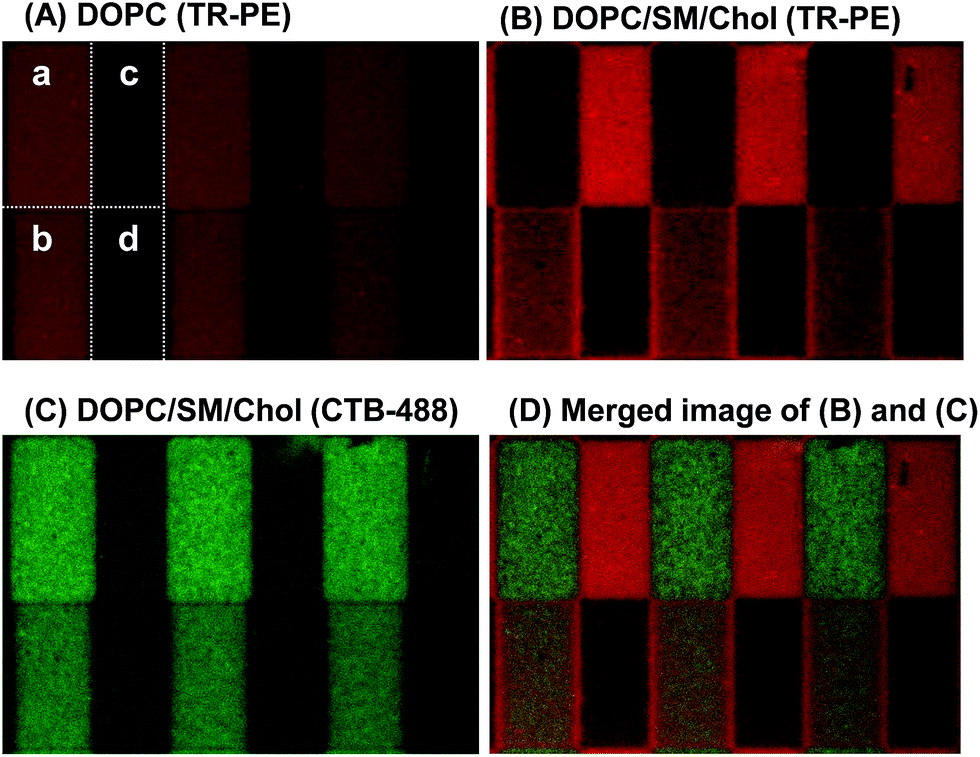 | ||
| Fig. 5 Phase separation in a patterned membrane with four different regions: the bilayer consisted of (a) polymer-free, (b) (c) partially polymeric (p = 0.02 and 0.49), and (d) polymeric regions. The patterned bilayer was first filled with a homogeneous bilayer (DOPC) to determine p (A). Subsequently, the fluid bilayer was exchanged with a mixed lipid bilayer (DOPC/SM/Chol/TR-PE/GM1) and the phase separation was observed ((B)–(D)). The size of each region was 20 × 10 μm. | ||
4. Discussion
The local Lo/Ld ratios in the micropatterned membrane could be modulated by the pattern geometry and polymer density. Localization of molecules associated with Lo and Ld phases in R0 and R1 was enhanced with a higher coverage of polymeric bilayer. This result supports the premise that patterned phase separation is induced by the accumulation of Ld domains around polymeric bilayer domains (ESI, Fig. S5†). We made a model assuming that the amount of Ld domains accumulated around polymeric bilayer domains was proportional to the area fraction of polymeric bilayers in R1 (p). The model could qualitatively reproduce the experimentally observed increase of DTR1 with p (ESI, Fig. S6†). The consistency is a further support of the premise that we can control the Lo/Ld distributions by tuning the local area fractions of polymeric bilayer.An important feature of the present micropatterning approach is the fact that polymeric and fluid bilayers are forming a continuous, two-dimensional composite membrane. Therefore, the phase separation is induced by the structural element (polymeric bilayer domain) embedded within the membrane. It is in contrast with other approaches which generally utilize the interactions of the membrane with the substrate surface for patterning Lo and Ld phases.11–16 The fact that the present approach does not rely on the interaction with the substrate should allow us to construct a model membrane on a wider variety of substrates. In the future, it may be possible to detach the membrane from the substrate with a hydrophilic polymer cushion and suspend it in a similar fashion as black lipid membranes.29,30
There are also some technical limitations at present. The phase separation takes quite a long time to complete (several hours to several days). The rate is presumably limited by the slow diffusion of lipid domains on the glass substrate. It has been reported that lateral diffusion of large domains is hindered due to the frictional drag on the substrate.31 Another important factor to be considered is the effect of embedded polymeric bilayers on the diffusion of membrane-bound molecules. Our previous studies have suggested that the lateral diffusion coefficients of lipids decreased proportionally with the polymer fraction.20 The retarded diffusion may affect the distribution of membrane-bound molecules by the kinetic effects. These technical hurdles must be mitigated by optimizing the pattern geometry and the amount of polymeric bilayer. It is also important to note that information on the gross area fractions of Lo and Ld phases for the lipid composition used is needed to determine the Lo and Ld fractions in R0 and R1 from the experimentally observed distributions of marker molecules (e.g. TR-PE) (Fig. 4). The Lo/Ld fractions have been mostly determined from the microscopic observation of giant vesicles.22,32 Since the Lo/Ld fractions may slightly vary for SPBs and giant vesicles due to the presence (or absence) of the solid support, the total area fractions should be evaluated using an SPB. The effects of polymeric bilayer domains on the phase behaviors of lipid membranes should be evaluated, as well.
In summary, a patterned composite membrane of polymeric and fluid bilayers can quantitatively control the local distribution of membrane-bound molecules according to their affinities to Lo and Ld phases. By changing the pattern geometry (R0/R1 area fractions) and polymeric bilayer coverage in R1, we can modulate the local Lo/Ld ratios with a designed pattern. A potential application of the patterned membrane should be to measure the partitioning of membrane-bound proteins to Lo and Ld phases. It is commonly conceived that the association of proteins with lipid rafts is playing important functional roles.33,34 Therefore, quantitative evaluation of protein partitioning into lipid rafts is an important issue. Conventionally, enrichment in detergent-resistant membranes (DRMs) was used to evaluate the association of proteins with lipid rafts.35,36 A more quantitative approach was recently developed by the microscopic observation of giant vesicles.37,38 Micropatterned model membrane with controlled distribution of Lo/Ld phases provides new possibilities to gauge the association of proteins to lipid rafts. Since patterned membranes are amenable to parallel analyses, it should significantly facilitate the determination process. Furthermore, we can construct an array of model membranes with multiple Lo/Ld ratios, as shown in Fig. 5. Such membranes may find various biomedical applications, including the separation of membrane bound molecules in combination with an electrophoretic or fluidic devices.
Acknowledgements
This work was supported by Grant-in-Aid for Scientific research from Japan Society for the Promotion of Science (#21651060, #22360022, #25286062). We thank Ms Akane Nagao for the FRAP measurements.References
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 CrossRef CAS PubMed.
- K. Simons and D. Toomre, Nat. Rev. Mol. Cell Biol., 2000, 1, 31–41 CrossRef CAS PubMed.
- K. Jacobson, O. G. Mouritsen and R. G. W. Anderson, Nat. Cell Biol., 2007, 9, 7–14 CrossRef CAS PubMed.
- D. Lingwood and K. Simons, Science, 2010, 327, 46–50 CrossRef CAS PubMed.
- D. Marsh, Biochim. Biophys. Acta, 2009, 1788, 2114–2123 CrossRef CAS PubMed.
- G. W. Feigenson, Biochim. Biophys. Acta, 2009, 1788, 47–52 CrossRef CAS PubMed.
- C. Dietrich, L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson and E. Gratton, Biophys. J., 2001, 80, 1417–1428 CrossRef CAS.
- S. L. Veatch and S. L. Keller, Biophys. J., 2003, 85, 3074–3083 CrossRef CAS.
- T. Baumgart, S. T. Hess and W. W. Webb, Nature, 2003, 425, 821–824 CrossRef CAS PubMed.
- J. M. Crane and L. K. Tamm, Biophys. J., 2004, 86, 2965–2979 CrossRef CAS.
- T.-Y. Yoon, C. Jeong, S.-W. Lee, J. H. Kim, M. C. Choi, S.-J. Kim, M. W. Kim and S.-D. Lee, Nat. Mater., 2006, 5, 281–285 CrossRef CAS PubMed.
- M. O. Ogunyankin, A. Torres, F. Yaghmaie and M. L. Longo, Langmuir, 2012, 28, 7107–7113 CrossRef CAS PubMed.
- F. Roder, O. Birkholz, O. Beutel, D. Paterok and J. Piehler, J. Am. Chem. Soc., 2013, 135, 1189–1192 CrossRef CAS PubMed.
- E. L. Kendall, V. N. Ngassam, S. F. Gilmore, C. J. Brinker and A. N. Parikh, J. Am. Chem. Soc., 2013, 135, 15718–15721 CrossRef CAS PubMed.
- A. R. Sapuri-Butti, Q. Li, J. T. Groves and A. N. Parikh, Langmuir, 2006, 22, 5374–5384 CrossRef CAS PubMed.
- L. Chao and S. Daniel, J. Am. Chem. Soc., 2011, 133, 15635–15643 CrossRef CAS PubMed.
- T. Okazaki, Y. Tatsu and K. Morigaki, Langmuir, 2010, 26, 4126–4129 CrossRef CAS PubMed.
- K. Morigaki, T. Baumgart, A. Offenhäusser and W. Knoll, Angew. Chem., Int. Ed., 2001, 40, 172–174 CrossRef CAS.
- K. Morigaki, K. Kiyosue and T. Taguchi, Langmuir, 2004, 20, 7729–7735 CrossRef CAS PubMed.
- T. Okazaki, T. Inaba, Y. Tatsu, R. Tero, T. Urisu and K. Morigaki, Langmuir, 2009, 25, 345–351 CrossRef CAS PubMed.
- T. Baumgart, G. Hunt, E. R. Farkas, W. W. Webb and G. W. Feigenson, Biochim. Biophys. Acta, 2007, 1768, 2182–2194 CrossRef CAS PubMed.
- N. Kahya, D. Scherfeld, K. Bacia, B. Poolman and P. Schwille, J. Biol. Chem., 2003, 278, 28109–28115 CrossRef CAS PubMed.
- P. I. Kuzmin, S. A. Akimov, Y. A. Chizmadzhev, J. Zimmerberg and F. S. Cohen, Biophys. J., 2005, 88, 1120–1133 CrossRef CAS PubMed.
- E. Evans and W. Rawicz, Phys. Rev. Lett., 1990, 64, 2094–2097 CrossRef CAS.
- K. Morigaki, H. Schönherr and T. Okazaki, Langmuir, 2007, 23, 12254–12260 CrossRef CAS PubMed.
- R. P. Richter, R. Berat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- T. Okazaki, K. Morigaki and T. Taguchi, Biophys. J., 2006, 91, 1757–1766 CrossRef CAS PubMed.
- S. Chiantia, N. Kahya, J. Ries and P. Schwille, Biophys. J., 2006, 90, 4500–4508 CrossRef CAS PubMed.
- S. R. Garg, J. Luedtke, K. Jordan, R. Naumann and A. Christoph, Biophys. J., 2007, 92, 1263–1270 CrossRef CAS PubMed.
- A. Orth, L. Johannes, W. Römer and C. Steinem, ChemPhysChem, 2012, 13, 108–114 CrossRef CAS PubMed.
- E. Sackmann, Science, 1996, 271, 43–48 CAS.
- S. L. Veatch and S. Keller, Phys. Rev. Lett., 2005, 94, 148101 CrossRef.
- I. Levental, M. Grzybek and K. Simons, Biochemistry, 2010, 49, 6305–6316 CrossRef CAS PubMed.
- I. Levental, D. Lingwood, M. Grzybek and Ü. Coskun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22050–22054 CrossRef CAS PubMed.
- S. Mayor and F. R. Maxfield, Mol. Biol. Cell, 1995, 6, 929–944 CrossRef CAS.
- D. A. Brown and E. London, J. Biol. Chem., 2000, 275, 17221–17224 CrossRef CAS PubMed.
- N. Kahya, D. A. Brown and P. Schwille, Biochemistry, 2005, 44, 7479–7489 CrossRef CAS PubMed.
- T. Baumgart, A. T. Hammond, P. Sengupta, S. T. Hess, D. A. Holowka, B. A. Baird and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3165–3170 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09981h |
| This journal is © The Royal Society of Chemistry 2015 |