DOI:
10.1039/C4RA11195H
(Paper)
RSC Adv., 2015,
5, 8070-8076
Synthesis, stability and electrical properties of new soluble pentacenes with unsaturated side groups
Received
25th September 2014
, Accepted 22nd December 2014
First published on 23rd December 2014
Abstract
A series of pentacene derivatives with unsaturated hydrocarbon substituents at the 6,13-positions as side groups have been synthesized. The effects of conjugated π-bonding throughout the side groups on the solubility, oxidative stability, crystallinity, and electrical properties of the compounds were investigated. Here we report that the solubility of pentacene can be greatly improved by the insertion of linear long side groups at the 6,13-positions: the obtained pentacene derivatives were soluble in common organic solvents. Time-dependent UV-visible spectra in chloroform solutions as well as thin films were recorded to investigate the oxidative stability of each pentacene compound under ambient conditions. The photooxidative stability of the pentacene derivatives was remarkably enhanced by increasing the electron conjugation length through the installation of unsaturated hydrocarbon substituents at the 6,13-positions. The solution-processed TFT devices based on 6,13-bis(4-pentylphenyl-ethynyl)pentacene (P5) and 6,13-bis(4-hexylphenylethynyl)pentacene (P6) showed charge transport mobilities of 2.14 × 10−2 cm2 V−1 s−1 and 3.94 × 10−2 cm2 V−1 s−1, respectively, and exhibited an on/off ratio of 105. Here we demonstrate that P5 and P6 revealed much more stable electrical performances than the parent pentacene under the oxidative environment.
Introduction
Over the last decade, intensive research and development efforts have focused on the synthesis of organic semiconducting materials and their applications in light emitting diodes (OLEDs), photovoltaic cells, sensors, radio frequency identification (RFID) tags,1 and organic thin film transistors (OTFTs). Recently, organic semiconductors have attracted more attention than inorganic semiconductors such as Si, GaAs, and ZnO, since the latter require high-temperature and high-vacuum-deposition processes for device fabrication, while the former can be more easily fabricated by solution processes such as spin-coating, inkjet printing, and screen printing. Moreover, future displays are predicted to exhibit flexibility, which can be easily achieved with organic materials. Therefore, organic semiconductors are considered to be potential candidates for TFTs.2–5
Pentacene has attracted considerable attention for its use in high-performance conjugated semiconductors.6–8 However, pentacene TFTs cannot be prepared using solution deposition processes due to their limited solubility. Thus, their fabrication requires high-vacuum deposition techniques. Furthermore, even though pentacene is quite stable under dark, its 6,13-positions are extremely sensitive to oxygen when exposed to visible light.9–12
To improve both the solubility and photooxidative stability of pentacene, various research groups are designing and synthesizing new pentacene derivatives.13–19
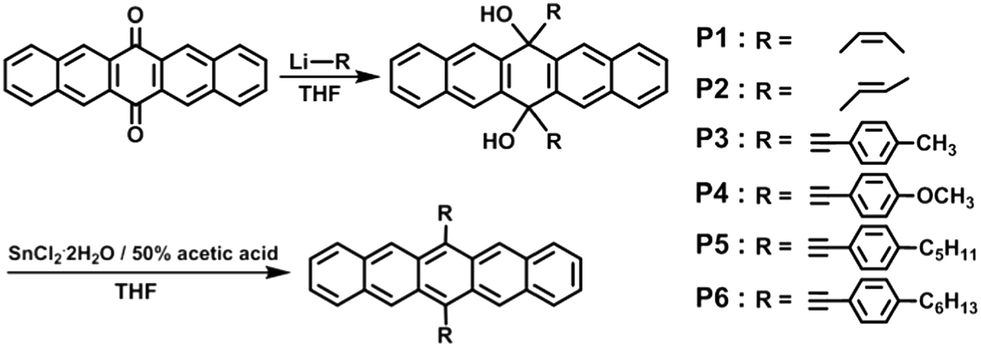
In this study, we designed new soluble and stable pentacene derivatives through the introduction of unsaturated hydrocarbon substituents into the 6,13-position of a parent pentacene: the target molecules have been synthesized by the “organolithium reaction” method, followed by reduction with SnCl2·2H2O. The resulting six compounds are 6,13-bis(Z-prop-1-enyl)pentacene (P1); 6,13-bis(E-prop-1-enyl)pentacene (P2); 6,13-bis(4-tolylethynyl)-pentacene (P3); 6,13-bis(4-methoxyphenyl-ethynyl)pentacene (P4); 6,13-bis(4-pentylphenylethynyl)pentacene (P5); and 6,13-bis(4-hexylphenylethynyl)pentacene (P6), as depicted in Scheme 1. We investigated the solubility, oxidative stability, crystallinity, and electrical properties of these pentacene derivatives for their application in solution-processable organic semiconductors for TFT fabrication.
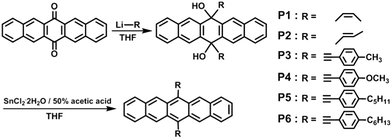 |
| | Scheme 1 Synthesis of pentacene derivatives. | |
Experimental
Materials and measurements
All reagent-grade starting materials were purchased from Aldrich and TCI. Commercially available reagents were used without further purification. All solvents were further purified prior to use by general methods. 6,13-Pentacenequinone was synthesized according to reported literature procedures.17 The structures of all isolated compounds were identified by FT-IR (Jasco, FT/IR-300E) and 1H NMR spectroscopy (Bruker, DPX 200 MHz). The 1H NMR chemical shifts are presented in the unit of δ (ppm) relative to tetramethylsilane (TMS, δ = 0) and referenced to the peaks signals corresponding to the residual non-deuterated solvent. The absorption spectra of the pentacene derivatives were measured by a UV-visible spectrophotometer (Mecasys, UV-3220). The current–voltage characteristics of the devices were measured at room temperature in dark by using a semiconductor parameter analyzer (Keithley, S4200). X-ray diffraction (XRD) measurements were obtained by using the synchrotron source at the Pohang Accelerator Laboratory (PAL) in Korea (10C1). The optical microscopy (OM) and polarized optical microscopy (POM) images were observed by using an Axioplan microscope (ZEISS).
Fabrication of OTFT device
Bottom-gate and top-contact OTFTs prepared using pentacene derivatives as channel semiconductors were fabricated under ambient conditions. A highly n-doped silicon wafer was used as a gate electrode as well as a substrate. A silicon dioxide (SiO2) layer thermally grown to 300 nm thickness was employed as a gate dielectric. Then, 0.5 wt% and 0.1 wt% solutions of the pentacene derivatives in chloroform were used for spin-coating on HMDS coated surface and drop-casting on bare surface, respectively. The channel width/lengths of 1500 μm/40 μm were adopted. As a source/drain electrode, 60 nm Au was thermally deposited through a shadow mask.
Synthesis of 6,13-di[(Z)-prop-1-enyl]pentacene (P1)
A 2.0 M solution of n-BuLi in cyclohexane (2.0 mL, 4.0 mmol) was added dropwise to a solution of cis-1-bromo-1-propene (0.47 g, 3.9 mmol) in dry THF (20 mL) at −78 °C under a nitrogen atmosphere. The solution was stirred for 50 min. After adding 6,13-pentacenequinone (0.30 g, 1.0 mmol), the solution was stirred for 1 h at −78 °C and for an additional 7 h at room temperature. Into the solution, SnCl2·2H2O (2.0 g, 8.8 mmol) and 50% acetic acid (2.0 mL) were added. The mixture was stirred for 12 h at room temperature, and then poured into water (300 mL). To this mixture, dichloromethane (300 mL) was added and the organic layer was separated and washed with water (300 mL × 2). After removing the solvent by evaporation under reduced pressure, the residue was purified by column chromatography on silica gel using chloroform as an eluent to afford the product. Yield: 45%. IR (KBr pellet, cm−1): 3040 (aromatic = CH, st), 2950, 2814 (aliphatic CH, st), 1429, 1381 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, st). 1H NMR (CDCl3, δ in ppm): 9.22 (4H, s, ArH), 8.12–8.03 (4H, m, ArH), 7.56–7.48 (4H, m, ArH), 2.55 (6H, s, CH3).
C, st). 1H NMR (CDCl3, δ in ppm): 9.22 (4H, s, ArH), 8.12–8.03 (4H, m, ArH), 7.56–7.48 (4H, m, ArH), 2.55 (6H, s, CH3).
Synthesis of 6,13-di[(E)-prop-1-enyl]pentacene (P2)
An alkylation reaction of 6,13-pentacenequinone (0.30 g, 1.0 mmol) with trans-1-bromo-1-propene (0.47 g, 3.9 mmol) and reduction with SnCl2·2H2O was carried out following the procedure described above to afford the product. Yield: 43%. IR (KBr pellet, cm−1): 3040 (aromatic = CH, st), 2950, 2814 (aliphatic CH, st), 1427, 1379 (CC, st). 1H NMR (CDCl3, δ in ppm): 9.22 (4H, s, ArH), 8.12–8.03 (4H, m, ArH), 7.56–7.48 (4H, m, ArH), 2.62 (6H, s, CH3).
Synthesis of 6,13-bis[4-tolylethynyl]pentacene (P3)
An alkylation reaction of 6,13-pentacenequinone (0.30 g, 1.0 mmol) with 1-ethynyl-4-methylbenzene (0.34 g, 2.6 mmol) and reduction with SnCl2·2H2O was carried out following the procedure described above to afford the product. Yield: 36%. IR (KBr pellet, cm−1): 3023 (aromatic = CH, st), 2913, 2854 (aliphatic CH, st), 2183 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, st), 1508 (CC, st). 1H NMR (CDCl3, δ in ppm): 9.31 (4H, s, ArH), 8.16–8.05 (4H, m, ArH), 7.85, 7.80 (4H, d, ArH), 7.48–7.41 (4H, m, ArH), 7.39, 7.32 (4H, d, ArH), 2.49 (6H, s, Ar–CH3).
C, st), 1508 (CC, st). 1H NMR (CDCl3, δ in ppm): 9.31 (4H, s, ArH), 8.16–8.05 (4H, m, ArH), 7.85, 7.80 (4H, d, ArH), 7.48–7.41 (4H, m, ArH), 7.39, 7.32 (4H, d, ArH), 2.49 (6H, s, Ar–CH3).
Synthesis of 6,13-bis[(4-methoxyphenyl)ethynyl]pentacene (P4)
An alkylation reaction of 6,13-pentacenequinone (0.30 g, 1.0 mmol) with 4-ethynylanisole (0.39 g, 2.7 mmol) and reduction with SnCl2·2H2O was carried out following the procedure described above to afford the product. Yield: 52%. IR (KBr pellet, cm−1): 3045, 3033 (aromatic = CH, st), 2954, 2925 (aliphatic CH, st), 2183 (CC, st), 1602, 1510 (CC, st), 1247, 1027 (C–O, st). 1H NMR (CDCl3, δ in ppm): 9.31 (4H, s, ArH), 8.13–8.09 (4H, m, ArH), 7.92, 7.88 (4H, d, ArH), 7.48–7.44 (4H, m, ArH), 7.13, 7.09 (4H, d, ArH), 3.97 (6H, s, Ar–O–CH3).
Synthesis of 6,13-bis[(4-pentylphenyl)ethynyl]pentacene (P5)
An alkylation reaction of 6,13-pentacenequinone (0.30 g, 1.0 mmol) with 1-ethynyl-4-pentylbenzene (0.51 g, 2.7 mmol) and reduction with SnCl2·2H2O was carried out following the procedure described above to afford the product. Yield: 36%. IR (KBr pellet, cm−1): 3041 (aromatic = CH, st), 2919, 2850 (aliphatic CH, st), 2179 (CC, st), 1506, 1457 (CC, st). 1H NMR (CDCl3, δ in ppm): 9.15 (4H, s, ArH), 8.05–8.00 (4H, m, ArH), 7.86, 7.81 (4H, d, ArH), 7.46–7.42 (4H, m, ArH), 7.40, 7.36 (4H, d, ArH), 2.80–2.73 (4H, t, Ar–CH2–CH2), 1.81–1.76 (4H, m, Ar–CH2–CH2), 1.51–1.42 (8H, m, CH2–CH2–CH3), 1.03–0.96 (6H, t, CH3).
Synthesis of 6,13-bis[(4-hexylphenyl)ethynyl]pentacene (P6)
An alkylation reaction of 6,13-pentacenequinone (0.30 g, 1.0 mmol) with 1-ethynyl-4-hexylbenzene (0.55 g, 2.7 mmol) and reduction with SnCl2·2H2O was carried out following the procedure described above to afford the product. Yield: 42%. IR (KBr pellet, cm−1): 3027 (aromatic = CH, st), 2921, 2850 (aliphatic CH, st), 1508, 1459 (CC, st). 1H NMR (CDCl3, δ in ppm): 9.16 (4H, s, ArH), 8.04–8.00 (4H, m, ArH), 7.88, 7.03 (4H, d, ArH), 7.49–7.46 (4H, m, ArH), 7.40, 7.37 (4H, d, ArH), 2.82–2.76 (4H, t, Ar–CH2–CH2), 1.82–1.76 (4H, m, Ar–CH2–CH2), 1.51–1.42 (12H, m, CH2–CH2–CH2–CH3), 1.03–0.96 (6H, t, CH3).
Results and discussion
Synthesis and solubility
Six pentacene derivatives with unsaturated side groups were synthesized by the “organolithium reaction” method, followed by reduction with SnCl2·2H2O, as shown in Scheme 1. They contain Z-prop-1-enyl (P1), E-prop-1-enyl (P2), 4-tolylethynyl (P3), 4-methoxyphenylethynyl (P4), 4-pentylphenylethynyl (P5), and 4-hexylphenylethynyl (P6) side groups. The structures of the products obtained from each reaction step were characterized by 1H NMR and FT-IR spectroscopy. The results were in accordance with the expected formula.
The solubilities of the resulting compounds were investigated to examine potential for the solution process. Three levels of solubility were classified in a common organic solvent (e.g., chloroform) at room temperature, and the results are summarized in Table 1. Triple (+++), double (++), and single (+) plus signs were used to indicate solubility levels corresponding to more than 7 mg mL−1 (excellent solubility), between 4 and 6 mg mL−1 (good solubility), and less than 3 mg mL−1 (poor solubility), respectively. P1 and P2, which contain propenyl side groups, exhibited poorer solubility levels than P3–P6, which contain alkylphenylethynyl side groups. Specifically, P5 and P6 with p-pentylphenylethynyl and p-hexylphenylethynyl groups, respectively, exhibited the highest solubility levels. This indicates that linear long side groups may inhibit the packing of the pentacene molecules by loosening the π–π stacking interactions of the fused rings.
Table 1 Solubility, half-life times, and photophysical properties of various pentacene derivatives
| Compound |
Solubilitya |
λmax (nm) |
λonset (nm) |
Eoptg (eV) |
t1/2 (day) |
| Solution |
Film |
Solutionb |
Filmc |
| Classification: +++ (>7 mg mL−1); ++ (4–6 mg mL−1); + (<3 mg mL−1). Concentration: 2 mg mL−1. Films coated on glasses. |
| P1 |
+ |
580, 631 |
604, 656 |
650 |
1.91 |
1.5 |
0.6 |
| P2 |
+ |
580, 631 |
606, 661 |
650 |
1.91 |
1.4 |
0.8 |
| P3 |
++ |
611, 666 |
775 |
688 |
1.80 |
3.3 |
>5 |
| P4 |
++ |
614, 668 |
794 |
693 |
1.79 |
2.5 |
>5 |
| P5 |
+++ |
611, 666 |
762 |
688 |
1.80 |
2.7 |
∼5 |
| P6 |
+++ |
611, 666 |
748 |
688 |
1.80 |
2.7 |
2.9 |
Photophysical properties and oxidative stabilities
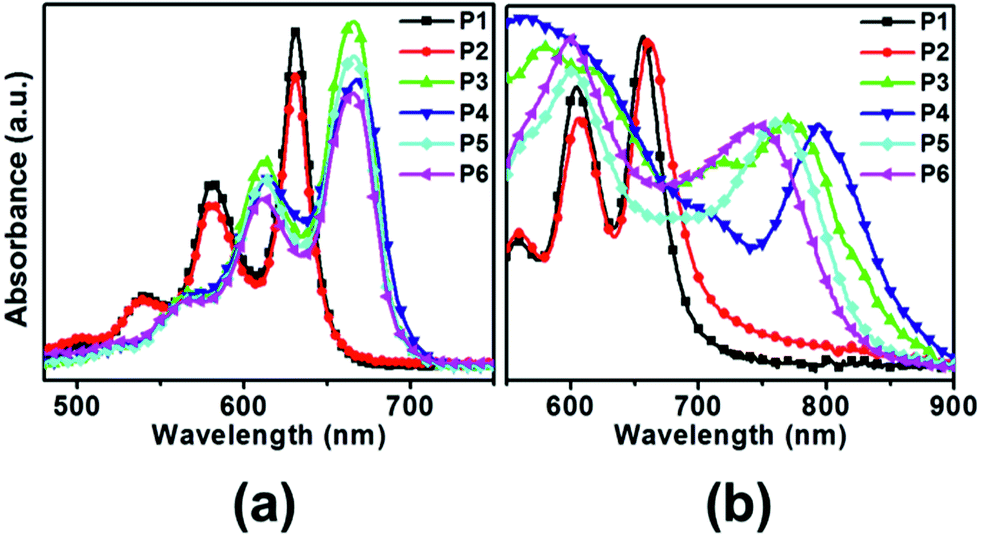
UV-vis spectra of obtained pentacene derivatives were recorded in chloroform solutions and thin films. As shown in Fig. 1, the pentacene derivatives in chloroform solutions showed characteristic vibrational finger-like absorbances similar to the parent pentacene (λmax = 577 nm) and exhibited significant red shifts. As depicted in Table 1, the pentacene derivatives show absorbances λmax in the range 580–614 nm and 631–668 nm in chloroform solutions. Interestingly, P3–P6 exhibited more red shifts than P1–P2 due to the relatively large extended π-electron delocalization in the former. Moreover, for all compounds, the film state shows greater red shifts compared with the solution state because the intermolecular interaction may increase in the dense state. The HOMO–LUMO bandgaps were calculated using the absorption onset obtained in the solution and are also included in Table 1. The all pentacene derivatives exhibited relatively low bandgaps (Eoptg = 1.79–1.91 eV) compared to the parent pentacene (Eg = 2.12 eV).
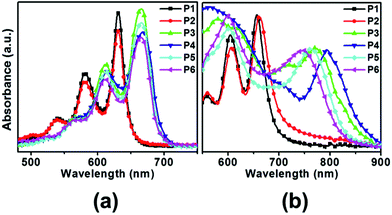 |
| | Fig. 1 UV-vis spectra of various pentacene derivatives obtained in chloroform solutions (a) and in thin films (b). | |
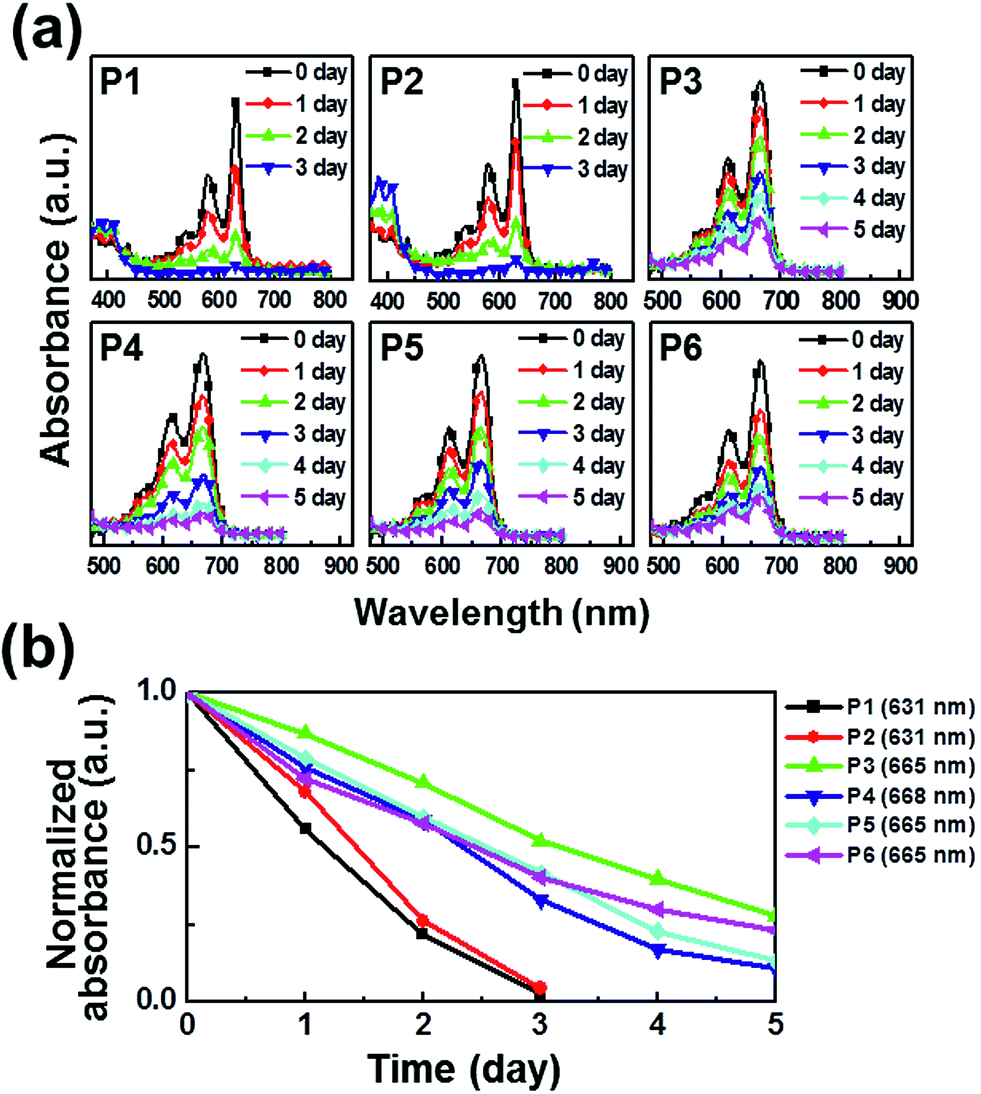
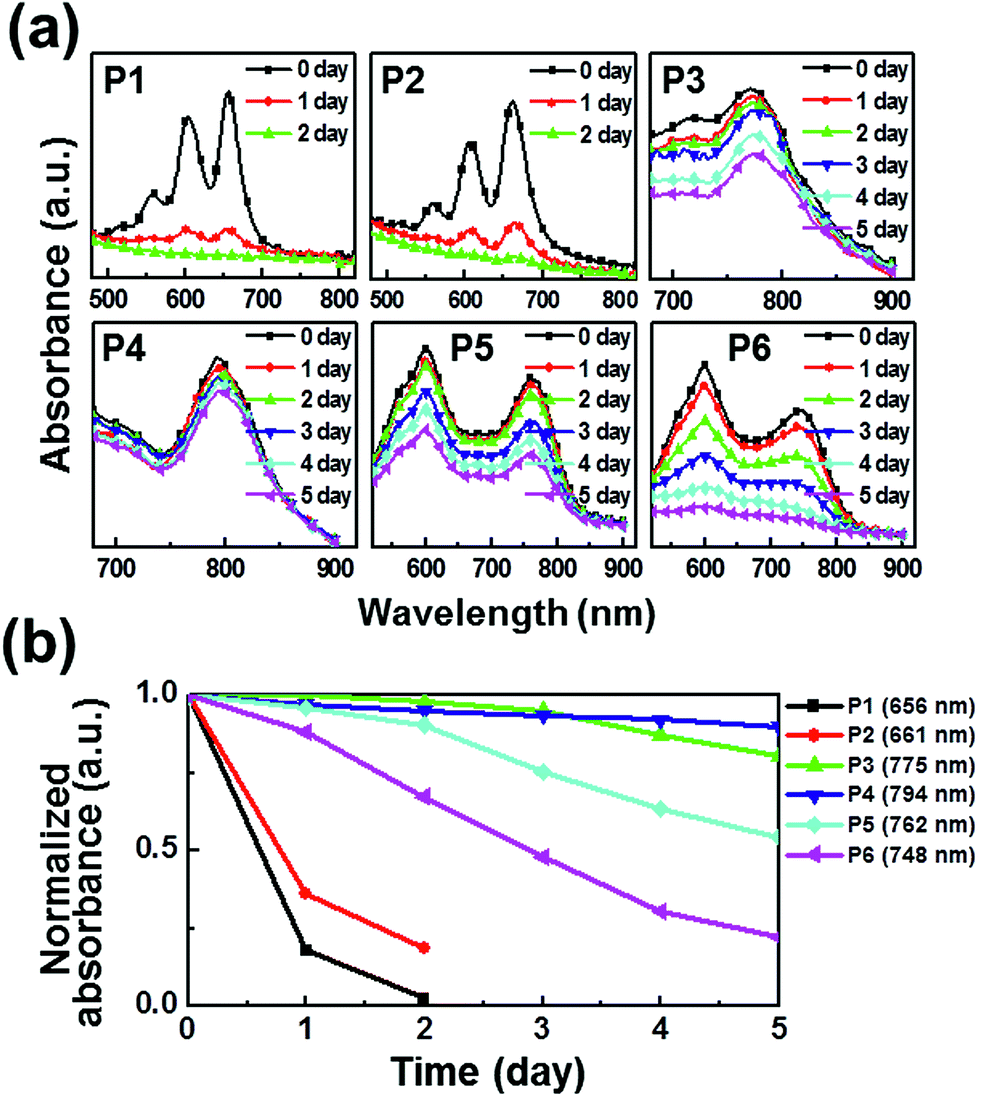
The photooxidative stability of pentacene derivatives has been investigated under ambient conditions. The time-dependent UV-vis spectra were measured to estimate their oxidative stabilities in solutions (Fig. 2) and thin films (Fig. 3). Solutions of pentacene derivatives in chloroform gradually became colorless and the peak intensities of λmax in the UV-vis spectra decreased depending upon the exposure period under ambient atmosphere, light, and temperature. In Table 1, the pentacene derivatives exhibited remarkably long half-life times (t1/2 = 1–3 day) in solution compared with the parent pentacene (t1/2 = ∼3 min).16 Interestingly, the derivatives containing alkylated phenylethynyl groups (P3–P6) exhibited greater oxidative stabilities than those containing E/Z-propenyl groups (P1 and P2). In Fig. 2b and 3b, the λmax peaks of the latter molecules almost disappeared after 3 days in the solutions and 2 days in the films, whereas those of the former molecules remained till 5 days. We infer that the steric hindrance of the bulkier phenyl groups against attacking of oxygen atoms prevents the oxidation of the 6,13-positions of P3–P6, and the extended π-electron delocalization thermodynamically stabilizes the molecular structures. The half-life times determined in films are also included in Table 1. Notably, P3–P5 presented longer half-life times (≥5 day) than the values obtained in solution, whereas P1 and P2 exhibited relatively shorter half-life times (<1 day). Meanwhile, P6 shows the half-life time in the film similar to that in the solution.
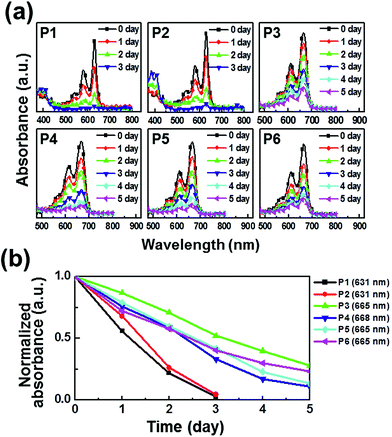 |
| | Fig. 2 Photooxidation stability in chloroform solutions: (a) time-dependent UV-vis spectra under ambient condition; (b) linear fits for the time-dependent λmax. | |
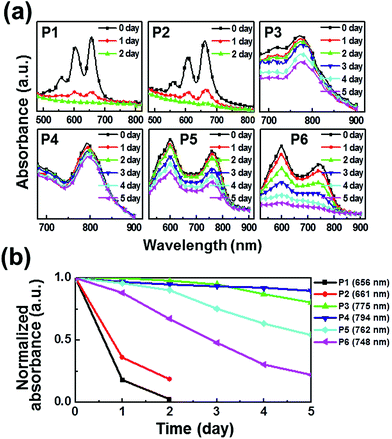 |
| | Fig. 3 Photooxidation stability in thin films: (a) time-dependent UV-vis spectra under ambient condition; (b) linear fits for the time-dependent λmax. | |
XRD studies and TFT performances
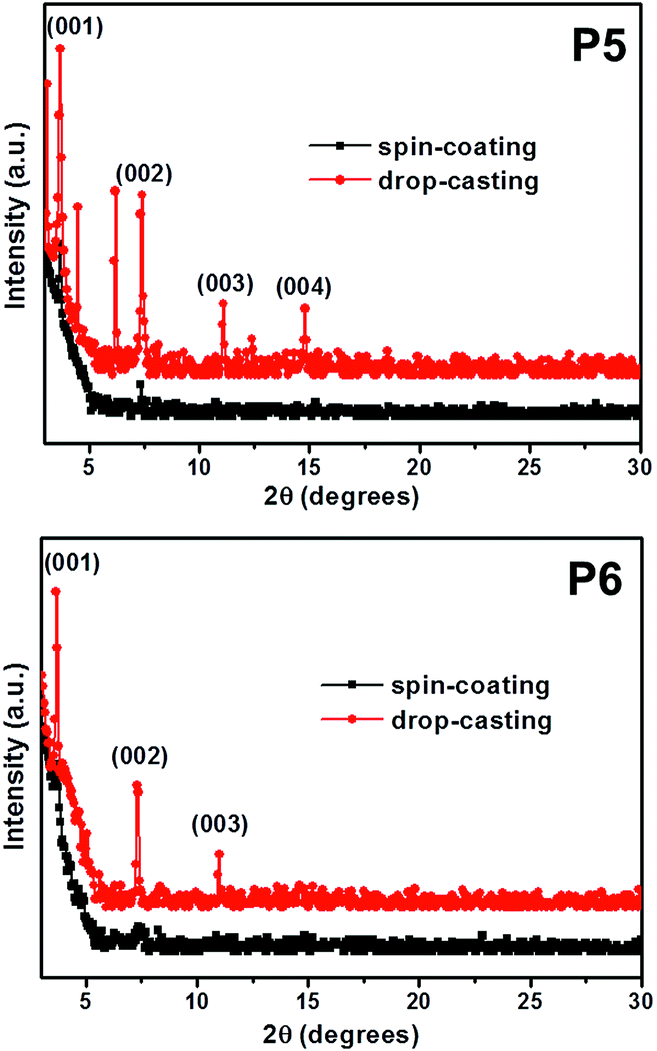
Fig. 4 depicts the out-of-plane XRD patterns of P5 and P6 thin-films deposited by spin-coating or drop-casting methods. The drop-casted films show several strong reflection peaks, whereas the spin-coated films exhibit only very weak reflection peaks. This indicates that the degree of crystallinity of films obtained via drop-casting are much higher than the films obtained via spin-coating. We suggest that the molecular stacking dynamics of these pentacene derivatives strongly correlate with the rate of solvent drying during film fabrication. That is, the spin-coating process with a faster drying rate does not provide enough time to form π–π stacks with the P5 and P6 molecules, whereas the drop-casting process with the slower drying rate does. In the XRD patterns for the drop-casted films, P6 showed only the (001) reflection peaks, while P5 exhibited the (001) reflection peaks as well as additional Bragg peaks. This implies that the crystalline structure of P5 may be more complex than that of P6. Consistent with this X-ray result, we observed that the drop-casted films by POM, P5 and P6, exhibited different optical textures at ambient temperature as shown in Fig. 5.
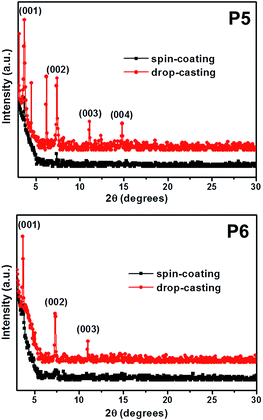 |
| | Fig. 4 Synchrotron radiation X-ray diffraction patterns of P5and P6 on bare Si/SiO2 substrates. | |
 |
| | Fig. 5 Optical microscopy (OM) and polarized optical microscopy (POM) images of drop-casted films for P5 (a) and P6 (b). | |
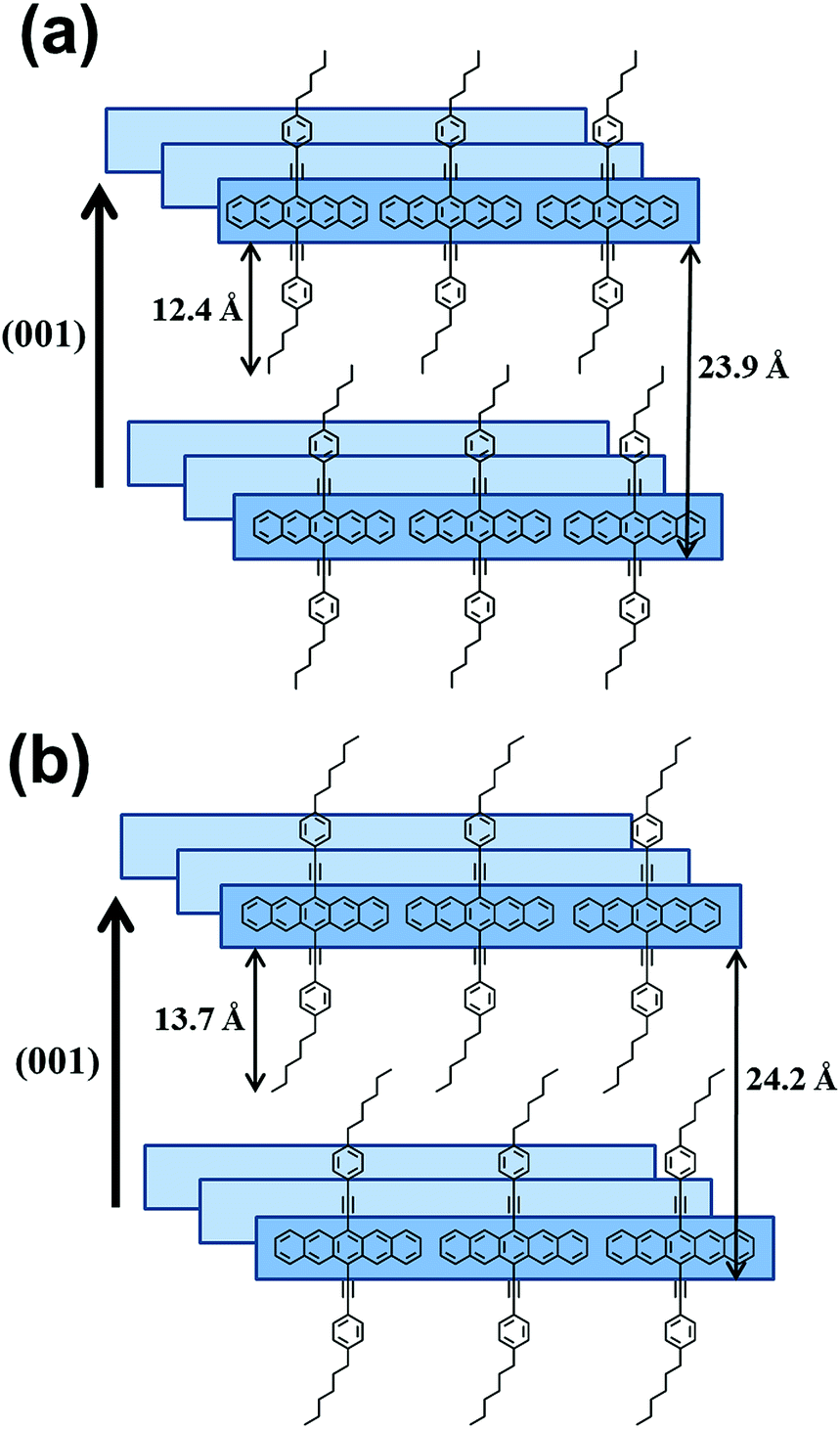
The d-values for the (001) peaks for P5 and P6 were calculated to be 23.9 Å and 24.2 Å, respectively, and the contour lengths of the side groups for P5 and P6 were estimated to be 12.4 Å and 13.7 Å, respectively. These values are indicative of the intercalation of side groups into the layer spacings. Based on these results, we conclude that the long axes of the P5 and P6 molecules are perpendicular to the surface and that the molecular axes of pentacenes stack cofacially, as proposed in Fig. 6. In case of P5 and P6, a linear long n-alkylated phenylethynyl side group can discourage the herringbone packing arrangement and promote the cofacial π-stacked packing arrangement. Meanwhile, P1–P4, which contain a short vinyl side group, might form imperfect herringbone stacks and thus somewhat distort the cofacial stacks. These results are in agreement with those of previous studies that patacene derivatives with bulky side silyl groups (TIPS- and TES-PENs) promote the cofacial π-stacked packing arrangement instead of the herringbone packing arrangement.14
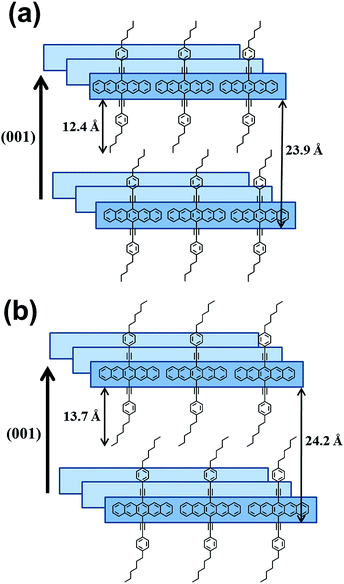 |
| | Fig. 6 Proposed molecular structures in drop-casted films for P5 (a) and P6 (b). | |
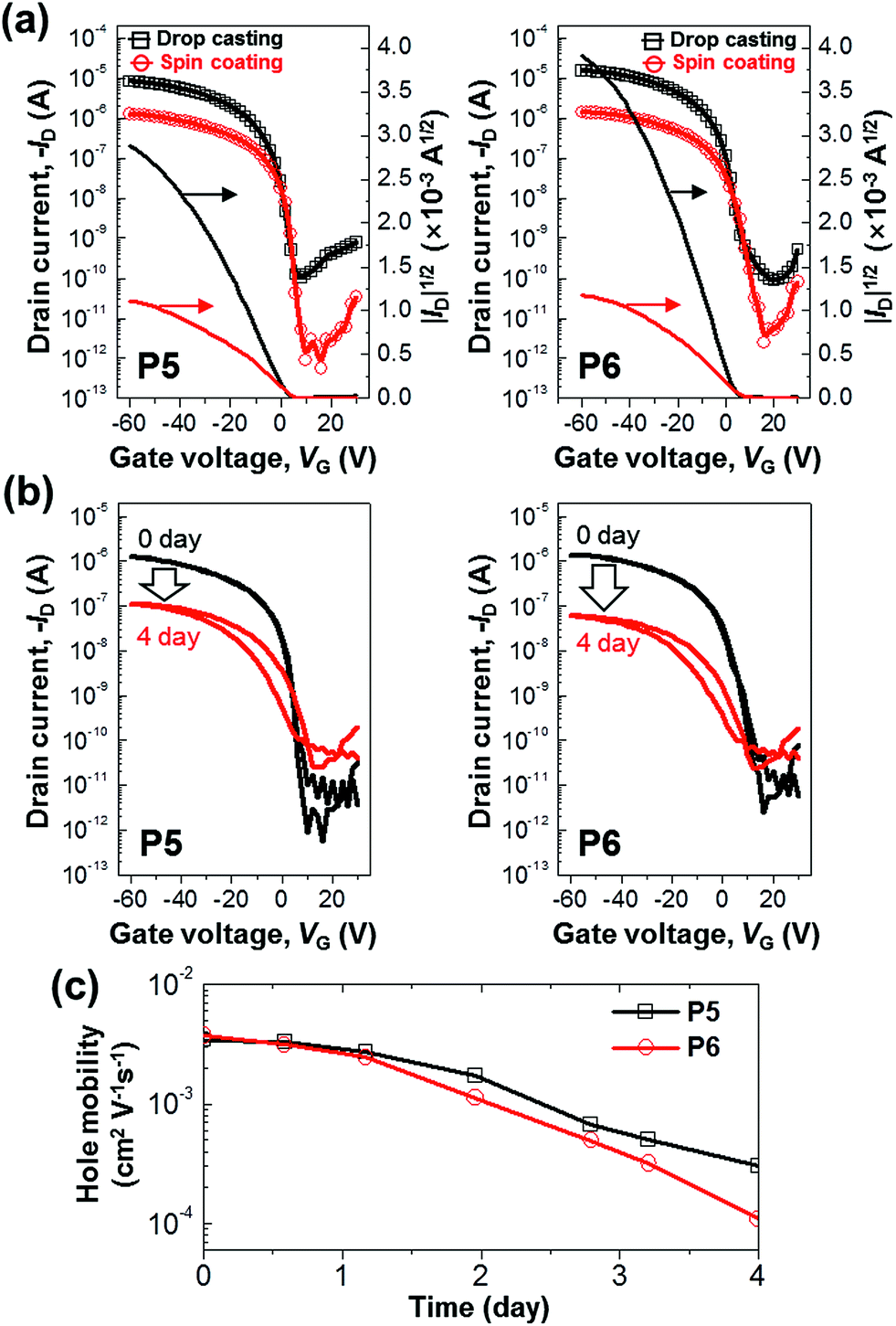
The electrical characteristics of P5 and P6 were measured by using top-contact bottom-gate OTFTs. Fig. 7a shows the typical transfer curves (ID–VG) of P5 and P6 OTFTs, whose quantitative performances are summarized in Table 2. The devices exhibited clearly hole channel-switching characteristics. The field-effect hole mobilities for drop-casted and spin-coated devices were ∼10−2 cm2 V−1 s−1 and ∼10−3 cm2 V−1 s−1, respectively. This indicates that the field-effect hole mobilities were higher in drop-casted devices than in spin-coated devices. It is attributed to the more effective π–π stacking of molecules in drop-casted film than in spin-coated film, aforementioned in Fig. 4. As adopted the drop-casted film, the P5-based devices displayed a field-effect mobility of 2.14 × 10−2 cm2 V−1 s−1 and an on/off ratio of 105, and the P6-based devices exhibited a field-effect mobility of 3.94 × 10−2 cm2 V−1 s−1 and an on/off ratio of 105. Unlike P5 and P6, P1–P4 revealed insignificant FET activities in the solution process. It is presumed that the efficient overlap of π-orbitals in P5 and P6 causes relatively superior device properties in OTFTs, compared with P1–P4. Besides, the formers form relatively effective packing structure, aforementioned in Fig. 6.
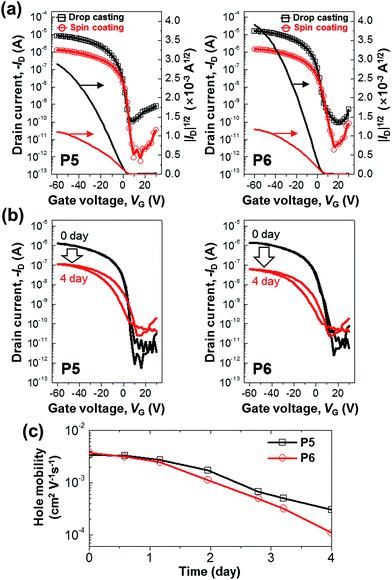 |
| | Fig. 7 (a) Transfer characteristics of top-contact bottom-gate OTFTs based on P5 and P6 films using drop casting (black square) or spin coating (red circle); gate electrode and dielectric: n-doped Si and 300 nm-thick SiO2, respectively; source/drain electrode: Au (width: 1500 μm, length: 40 μm). Photooxidation experiments for spin-coated devices: (b) representative time-dependent transfer characteristics of the OTFTs exposed to ambient air and 60 W fluorescent light; (c) dependence of field-effect hole mobilities on the exposure period. | |
Table 2 Field-effect transistor characteristics of selected pentacene derivativesa
| Compound |
Fabrication method |
Exposure periodb (day) |
Mobility (cm2 V−1 s−1) |
On/off ratio |
| Top-contact bottom-gate OTFTs; gate electrode and dielectric: n-doped Si and 300 nm-thick SiO2, respectively; source/drain electrode: Au (width: 1500 μm, length: 40 μm). OTFTs were exposed to ambient air and 60 W fluorescent light; 0 day stands for an as-prepared device. |
| P5 |
Spin-coating |
0 |
3.43 × 10−3 |
∼106 |
| 4 |
3.04 × 10−4 |
∼104 |
| Drop-casting |
0 |
2.14 × 10−2 |
∼105 |
| P6 |
Spin-coating |
0 |
3.72 × 10−3 |
∼106 |
| 4 |
1.09 × 10−4 |
∼104 |
| Drop-casting |
0 |
3.94 × 10−2 |
∼105 |
Finally, the oxidative stability of the OTFT devices based on the P5 and P6 was evaluated. Fig. 7b and c show the time-dependent transfer characteristics of the OTFTs exposed for 4 days to ambient air and 60 W fluorescent light. In Table 2, compared P5-device with P6-device, the mobility is slightly higher in the P6 than in the P5 before the exposure to ambient air and light, but the on-current and field-effect mobilities were less decreased in the P5 than in the P6 under oxidative environment. This means that the electrical performance was less diminished in P5-device than in P6-device. This result is consistent with the aforementioned tendency of time-dependent UV-vis absorption spectra of P5 and P6 thin films under oxidative environment. It is noteworthy that only the structural difference between the two compounds is very minor: P5 has a pentyl end group and P6 has a hexyl one.
Conclusions
Six new pentacene derivatives P1–P6 were synthesized and characterized. The resulting compounds were soluble in common organic solvents such as chloroform, dichloromethane, THF, and toluene. We assume that linear unsaturated side groups may inhibit the packing of molecules by loosening the π–π stacking interactions of the fused rings. The photooxidation stability for the pentacene derivatives were considerably increased by introducing propenyl (P1 and P2) as well as alkylated phenylethynyl (P3–P6) groups at the 6,13-positions. We assume that the unsaturated side groups may wave aside an attack by the oxygen atoms at the 6,13-positions through the steric hindrance and may thermodynamically stabilize the molecule through the extended π-electron delocalization. Further, we found that the morphology and crystallinity of these molecules, which are affected by the casting method, are the predominant source of the different field-effect mobilities. In case of the solution process, the OTFT devices of P5 and P6 (R = pentyl- or hexyl-phenylethynyl) showed the superior charge transport properties whereas those of P1–P4 exhibited the inferior properties. Here we demonstrate that P5 and P6 revealed much more stable electrical performances than the parent pentacene under oxidative environment. Finally, we conclude that the installation of unsaturated side groups into pentacene derivatives tended to much more improve oxidative stability rather than charge mobility. Especially, by using linear long unsaturated side groups, as exemplified by P5 and P6, the level of conjugation is improved and results in remarkable oxidative stability with superior electrical performances.
Acknowledgements
This research was supported by the Basic Science Research Program (no. NRF-2013R1A1A2012471) through the National Research Foundation of Korea (NRF), funded by the Ministry of Education Science and Technology.
Notes and references
- P. F. Baude, D. A. Ender, M. A. Haase, T. W. Kelley, D. V. Muyres and S. D. Theiss, Appl. Phys. Lett., 2003, 82, 3964 CrossRef CAS PubMed.
- G. Horowitz, R. Hajlaoui, R. Bourguiga and M. Hajlaoui, Synth. Met., 1999, 101, 401 CrossRef CAS.
- H. E. Katz, Z. N. Bao and S. L. Gilat, Acc. Chem. Res., 2001, 34, 359 CrossRef CAS PubMed.
- M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS PubMed.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99 CrossRef CAS.
- H. Klauk, M. Halik, U. Zschieschang, G. Schmid, W. Radlik and W. Weber, J. Appl. Phys., 2002, 92, 5259 CrossRef CAS PubMed.
- W. H. Lee, J. Park, S. H. Sim, S. Lim, K. S. Kim, B. H. Hong and K. Cho, J. Am. Chem. Soc., 2011, 133, 4447 CrossRef CAS PubMed.
- H. H. Choi, W. H. Lee and K. Cho, Adv. Funct. Mater., 2012, 22, 4833 CrossRef CAS.
- H. Yamada, Y. Yamashita, M. Kikuchi, H. Watanabe, T. Okujima, H. Uno, T. Ogawa, K. Ohara and N. Ono, Chem.–Eur. J., 2005, 11, 6212 CrossRef CAS PubMed.
- F. De Angelis, M. Gaspari, A. Procopio, G. Cuda and E. D. Fabrizio, Chem. Phys. Lett., 2009, 468, 193 CrossRef CAS PubMed.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980 CrossRef CAS.
- L. Li, K. Meise-Gresch, L. Jiang, C. Du, W. Wang, H. Fuchs and L. Chi, Adv. Mater., 2012, 24, 3053 CrossRef CAS PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482 CrossRef CAS.
- J. E. Anthony, D. L. Eaton and S. R. Parkin, Org. Lett., 2002, 4, 15 CrossRef CAS PubMed.
- C. D. Sheraw, T. N. Jackson, D. L. Eaton and J. E. Anthony, Adv. Mater., 2003, 15, 2009 CrossRef CAS.
- B. S. Ong, Y. N. Li, Y. L. Wu, P. Liu, Z. Prostran and S. Gardner, Chem. Mater., 2007, 19, 418 CrossRef.
- E-J. Choi, H.-G. Kim, J.-H. Park and J.-H. Kim, J. Inf. Disp., 2009, 10, 121 CrossRef.
- N. Vets, M. Smet and W. Dehaen, Tetrahedron Lett., 2004, 45, 7287 CrossRef CAS PubMed.
- S. Subramanian, S. K. Park, S. R. Parkin, V. Podzorov, T. N. Jackson and J. E. Anthony, J. Am. Chem. Soc., 2008, 130, 2706 CrossRef CAS PubMed.
Footnote |
| † The authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.