
Nano-Fe3O4 encapsulated-silica supported boron trifluoride as a novel heterogeneous solid acid for solvent-free synthesis of arylazo-1-naphthol derivatives
Abdolhamid Bamoniri* and
Naimeh Moshtael-Arani
Department of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan 87317-51167, Iran. E-mail: bamoniri@kashanu.ac.ir
First published on 19th January 2015
Abstract
Nano-Fe3O4 encapsulated-silica supported boron trifluoride (Fe3O4@SiO2–BF3) as a new type of green heterogeneous solid acid was prepared by the immobilization of BF3·Et2O on the surface of Fe3O4@SiO2 core–shell nanoparticles and characterized by Fourier transform-infrared spectroscopy (FT-IR), X-ray diffraction (XRD), vibrating sample magnetometer (VSM), field emission-scanning electron microscope (FE-SEM), energy dispersive X-ray (EDS), and transmission electron microscope (TEM). Then, this super solid acid was used as an acidic reagent for the synthesis of aryl diazonium salts as a starting reactant, followed by its diazo coupling with 1-naphthol in a basic solvent-free medium at room temperature. The main advantages of this clean method were high yields, short reaction times, the possibility of performing it at room temperature, and no need of corrosive and toxic liquid acids and solvents. In addition, the long-term stability of aryl diazonium salts supported on Fe3O4@SiO2–BF3 magnetic nanoparticles (MNPs) at room temperature was one of the most important results of this procedure.
Introduction
In recent years, solvent-free organic reactions1 have attracted great interest because of their various advantages such as high efficiency and selectivity, easy separation and purification, mild reaction conditions, reduction in waste, simplicity in progress and handling, and benefits to the industry as well as the environment. Aromatic azo compounds constitute a very important class of organic dyes because of their widespread applications in many areas of technology and medicine. They are well-known for their use as colorants in textile industries,2 digital printing and photography.3 They are also applied as chiral receptors,4 liquid crystals,5 new glassy materials,6 chiral switches in photochemistry,7 dyes for drugs, food, cosmetic, biomedicine,8 and molecular recognition.9 The synthesis of azo dyes requires some special conditions, such as low temperature and concentrated liquid acid, in addition to high costs because it leads to the corrosion of the reactors and equipments.10 Nowadays, solid supported reagents, rather than individual reagents, have resolved these problems and improved the activity and selectivity.11 Such reagents not only simplify the purification processes, but they also help prevent the release of reaction residues into the environment.12 In this regard, nanostructured solid acids exhibit higher activity and selectivity than their corresponding bulk materials due to their particular physical and chemical properties, especially large surface to volume ratio.13 Recently, Fe3O4 MNPs has appeared as a new type of efficient catalyst support due to its low toxicity, ease of surface modification, and unique physical properties, including their high surface area and superparamagnetism.14 To prevent the aggregation of Fe3O4 nanoparticles, its surface is usually modified with a silica layer, taking advantage of the fact that they are biocompatible and hydrophilic and also because the surface silanol groups can easily react with various organic and inorganic materials to achieve certain purposes, especially in the field of catalysis.15 As a continuation of our efforts on the development of heterogeneous solid acids in organic transformations,16 we herein report the preparation and characterization of a novel and eco-friendly magnetic solid acid Fe3O4@SiO2–BF3, as well as its utility for the synthesis of arylazo-1-naphthol dyes in a solvent-free environment at room temperature. In this method, special cold conditions are not required for the stabilization of aryl diazonium salts. The reaction easily takes place at room temperature, and the resulting diazonium salt can remain on the solid substrate for several months. To the best of our knowledge, this research is the first report about the long-term stability of aryl diazonium salts supported on the surface of MNPs and their use as the reactant for the synthesis of arylazo-1-naphthols in solvent-free conditions. The findings of this research may have implications for effective synthesis on a larger scale in dyeing and medical industries.Results and discussion
This research was performed in three stages. Initially, Fe3O4@SiO2–BF3 MNPs was synthesized and identified by FT-IR, XRD, VSM, FE-SEM, EDS, and TEM. In the second stage, aryl diazonium salts were synthesized in the presence of Fe3O4@SiO2–BF3 nanoparticles at room temperature and their structure and stability was investigated. The third stage was the diazo coupling of aryl diazonium salts supported on Fe3O4@SiO2–BF3 with basic 1-naphthol under solvent-free grinding condition.Synthesis and characterization of Fe3O4@SiO2–BF3 nanoparticles
Fe3O4@SiO2–BF3 core–shell nanoparticles, with Fe3O4 spheres as the core and silica supported BF3 as the shell, were prepared by a simple and convenient method. First, Fe3O4 nanoparticles were prepared by the chemical co-precipitation of FeCl2·4H2O and FeCl3·6H2O in an ammonium hydroxide solution. To improve the chemical stability of Fe3O4 and prevent self-aggregation, its surface engineering was successfully performed by the suitable deposition of SiO2 on Fe3O4 surface via the ammonia-catalyzed hydrolysis of tetraethylorthosilicate (TEOS). Next, the Fe3O4@SiO2 spheres served as support for the immobilization of BF3 groups by the simple stirring of Fe3O4@SiO2 and BF3·Et2O in ethanol. All three steps were carried out under sonication at room temperature (Scheme 1).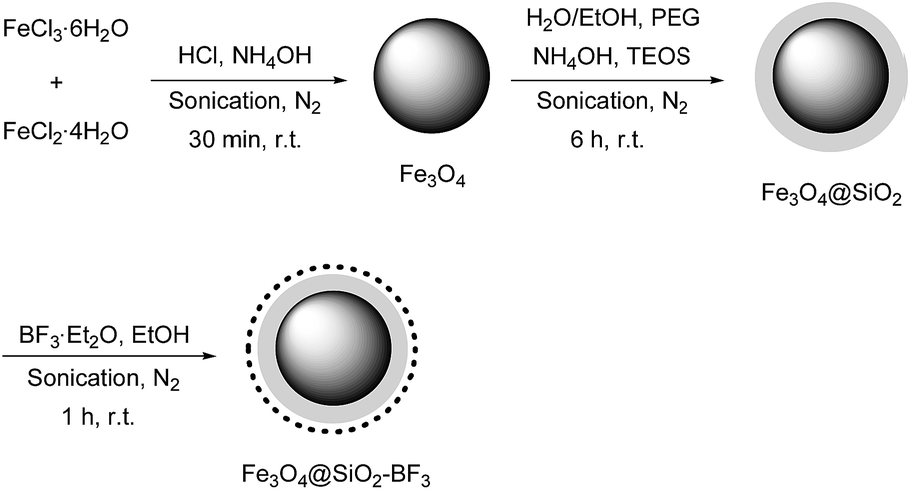 | ||
| Scheme 1 Preparation of Fe3O4@SiO2–BF3 MNPs under sonication at room temperature. | ||
To identify the molecular structures of Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–BF3 MNPs, FT-IR analysis of three mentioned samples was performed (Fig. 1). Fe3O4 was identified by a stretching vibration of the Fe–O absorption peak at 576 cm−1, O–H stretching vibration at 3429 cm−1 and O–H deformed vibration at 1625 cm−1 in Fig. 1a. The FT-IR spectrum of Fe3O4@SiO2 (Fig. 1b) displayed characteristic peaks at 1093 and 800 cm−1, corresponding to the symmetrical and asymmetrical vibrations of Si–O–Si, respectively. The weak band at 466 cm−1 corresponded to the Si–O–Fe stretching vibrations of the Fe3O4@SiO2 core–shell, overall confirming the presence of SiO2 in the sample. The successful covalent linking of the BF3·Et2O on the surface of Fe3O4@SiO2 core–shell was proved by the appearance of a new band at 1457 cm−1, which originates from the absorption of B–O (Fig. 1c). The absorption band of B–F was masked by the Si–O band. Moreover, the ethanolic OH and existing moisture in BF3·Et2O caused a broad O–H stretching band at the wavenumber of 3221 cm−1. The observation of Fe3O4 absorption peaks in both Fig. 1b and c implies that Fe3O4 MNPs do not change chemically or physically after the coating and surface modification processes. According to this information and from the reported structure of BF3–SiO2 in the literature,17 the final structure of nano Fe3O4@SiO2–BF3 is predicted and displayed in Scheme 2.
 | ||
| Fig. 1 FT-IR spectra of (a) Fe3O4, (b) Fe3O4@SiO2 and (c) Fe3O4@SiO2–BF3. | ||
 | ||
| Scheme 2 The structure of Fe3O4@SiO2–BF3 MNPs. | ||
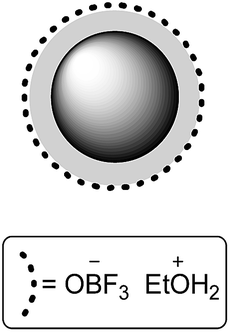
Fig. 2 shows the XRD powder diffraction patterns of three synthesized MNPs. The data for the Fe3O4 nanoparticles at 2θ of 30.22°, 35.61°, 43.25°, 53.58°, 57.30°, 62.89°, and 74.66° (Fig. 2a) correspond to the standard Fe3O4 powder diffraction data. Moreover, the relatively sharp peaks observed indicate the phase purity of Fe3O4 nanoparticles, which are consistent with the presence of a cubic inverse spinel structure of Fe3O4. The XRD pattern of the Fe3O4@SiO2 (Fig. 2b) is in good agreement with that of Fe3O4 phase, except for a broad peak around 2θ of 20°–30° corresponding to the amorphous phase of SiO2. This indicates that the MNPs obtained after the coating process are composed of Fe3O4 core and amorphous SiO2 shell. The XRD pattern of the modified Fe3O4@SiO2 with BF3·Et2O in Fig. 2c is nearly the same as in Fig. 2b, meaning that the surface modification by BF3 group has little effect on the XRD pattern of Fe3O4@SiO2 nanoparticles because of the shielding effect of Fe3O4 and SiO2 peaks. However, the changes in the intensity of peaks in spectra Fig. 2b and c, as well as the increase of noise in Fig. 2c verify the linking of BF3 on the surface of Fe3O4@SiO2 core–shell MNPs. Furthermore, the characteristic peaks of Fe3O4 are observed in three samples, thus indicating that the binding process did not induce any phase change.
 | ||
| Fig. 2 XRD patterns of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2–BF3. | ||
The magnetic properties of synthesized Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–BF3 nanoparticles were assessed by VSM at room temperature. The magnetization curve in Fig. 3 indicates magnetization as a function of the applied magnetic field. The saturation magnetization of Fe3O4@SiO2 nanoparticles in Fig. 3b was about 29.15 emu g−1, and it was reduced to 27.44 emu g−1 after supporting with BF3·Et2O (Fig. 3c). Both of these values were considerably lower than the initial saturation magnetization of Fe3O4 nanoparticles (59.2 emu g−1) in Fig. 3a. The decrease of the saturation magnetization after the surface coating of Fe3O4 confirms the presence of a diamagnetic outer shell (SiO2 or SiO2–BF3) at the surface of the Fe3O4 particles.
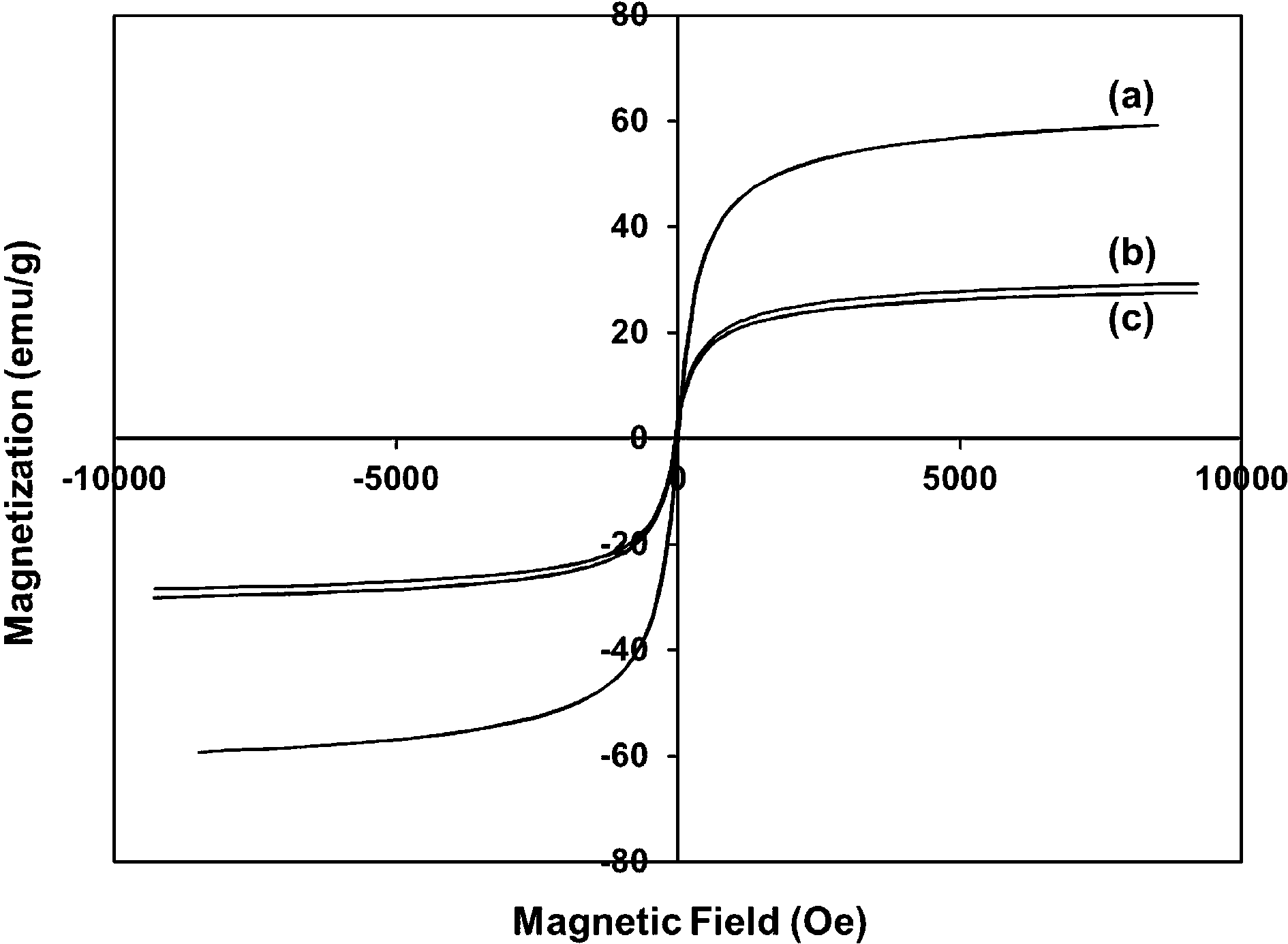 | ||
| Fig. 3 Magnetization curves of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2–BF3. | ||
The high magnification FE-SEM images of the purified MNPs are displayed in Fig. 4a–c. These images clearly showed the surface morphology of the three types of synthesized MNPs with a nearly spherical shape. The elemental components of three MNPs were characterized by EDS analysis. Fig. 4d shows the EDS of Fe3O4 nanoparticles; the particles contain Fe and O elements. The presence of Fe, O and Si elements in Fig. 4e verified the coating of the Fe3O4 core by SiO2 shell. The appearance of two new signals related to F and C elements in Fig. 4f confirmed the supporting of BF3·Et2O on Fe3O4@SiO2 core–shell nanoparticles, according to Scheme 2.
 | ||
| Fig. 4 FE-SEM images of (a) Fe3O4, (b) Fe3O4@SiO2, (c) Fe3O4@SiO2–BF3 and EDS spectra of (d) Fe3O4, (e) Fe3O4@SiO2, (f) Fe3O4@SiO2–BF3. | ||
TEM images of Fe3O4 and Fe3O4@SiO2–BF3 are displayed in Fig. 5. As demonstrated in Fig. 5a, Fe3O4 nanoparticles have a spherical morphology. In Fig. 5b, two regions with different electron densities can be distinguished, confirming that the Fe3O4 nanoparticles were successfully coated with a thin layer of a different phase. However, it can be observed that the sample is nearly in a core–shell structure. An electron dense region (black colour), which corresponds to the Fe3O4 cores, and a less dense or more translucent region (ash colour) surround these cores, which corresponds to the SiO2–BF3 shell. From the size distribution histograms, the average size of 9 nm for Fe3O4 and 13 nm for Fe3O4@SiO2–BF3 nanoparticles could be estimated.
 | ||
| Fig. 5 TEM images of (a) Fe3O4, and (b) Fe3O4@SiO2–BF3 nanoparticles. | ||
Finally, Fe3O4@SiO2–BF3 was identified by using the techniques described above, and applied for the synthesis of arylazo-1-naphthol derivatives.
Synthesis and characterization of aryl diazonium salts
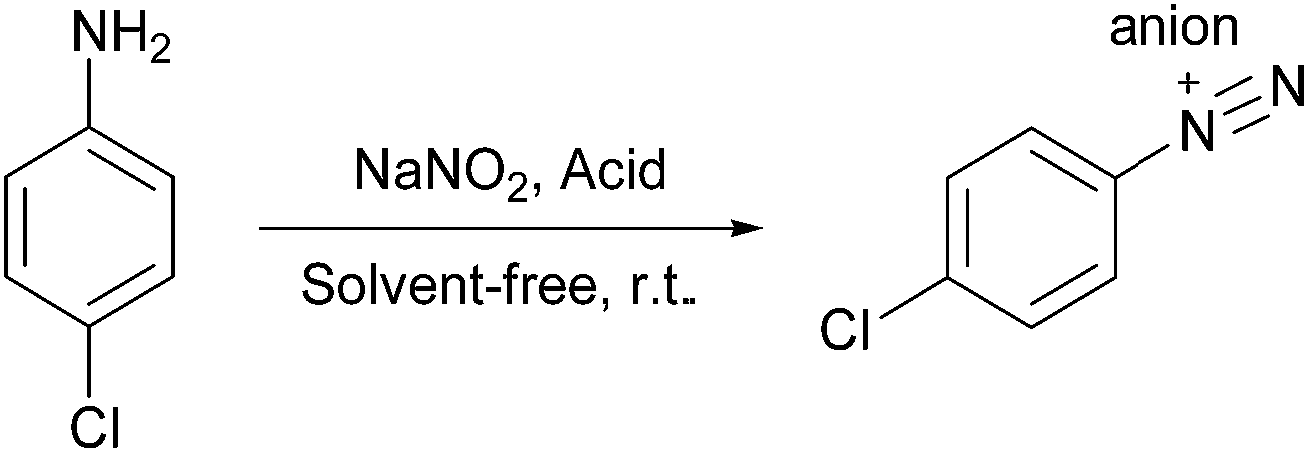
Aryl diazonium salts are usually synthesized in the presence of a liquid acid dissolved in water at low temperatures between 0 and 5 °C because temperatures above 5 °C generally promote phenol formation in aqueous media. Thus, the synthesis of aryl diazonium salts has limitations and drawbacks such as the control and maintenance of the low-temperature, use of toxic liquid acids that are incompatible with environment, and most importantly the instability of aryl diazonium salts at room temperature. We tried to resolve these problems by the development of a green and simple procedure for the synthesis of aryl diazonium salts followed by their diazo coupling with 1-naphthol.First, to evaluate the efficiency of Fe3O4@SiO2–BF3 MNPs in the diazotization reaction, an initial optimization of the type and amount of acidic reagent was performed via the diazotization of 4-chloroaniline as a model substrate. A range of parameters, such as the stability of 4-chlorophenyl diazonium salt at room temperature, the reaction time of diazotization, and the yield of the resulting 4-chlorophenylazo-1-naphthol are reported in Table 1.
|
||||
|---|---|---|---|---|
| Entry | Acid (wt%) | Stability at r.t. | Time of diazotization | Yielda (%) |
| a The yields refer to the total isolated yield of 2-(4-chlorophenylazo)-1-naphthol and 4-(4-chlorophenylazo)-1-naphthol after adding fresh 4-chlorophenyl diazonium salt into basic 1-naphthol.b Silica phosphoric acid.c Silica chloride. | ||||
| 1 | SPAb (10) | ∼2 days | 35 min | 57 |
| 2 | SCc (10) | ∼2 days | 30 min | 61 |
| 3 | Fe3O4@SiO2 | — | No reaction | — |
| 4 | BF3·Et2O | — | No reaction | — |
| 5 | Fe3O4@SiO2–BF3 (5) | >13 months | 12 s | 89 |
| 6 | Fe3O4@SiO2–BF3 (10) | >13 months | 6 s | 95 |
| 7 | Fe3O4@SiO2–BF3 (15) | >13 months | 8 s | 90 |
| 8 | Fe3O4@SiO2–BF3 (20) | >13 months | 8 s | 87 |
Solid acids, such as silica phosphoric acid and silica chloride, gave 4-chlorophenylazo-1-naphthol with modest yields: 57% and 61%, respectively (Table 1, entries 1 and 2). The stability of aryl diazonium salt supported on the above mentioned solid acids was at maximum after 2 days. Fe3O4@SiO2 and BF3·Et2O sources were also tested individually. There was no reaction in the presence of neither Fe3O4@SiO2 nor BF3·Et2O (Table 1, entries 3 and 4), while Fe3O4@SiO2–BF3 with different loadings afforded improved yields: from 87% to 95% for 4-chlorophenylazo-1-naphthol (Table 1, entries 5–8). These results clearly indicate that the presence of Brönsted acid sites on the solid acid surface is essential for promoting diazotization. Moreover, the rate of diazotization in the presence of Fe3O4@SiO2–BF3 MNPs was considerably higher than silica phosphoric acid and silica chloride.
In another comparative study (Table 1, entries 5–8), the effect of Fe3O4@SiO2 loading by BF3·Et2O on the acidic performance of Fe3O4@SiO2–BF3 was investigated. Although the time of diazotization and the stability of aryl diazonium salt supported on Fe3O4@SiO2–BF3 nanoparticles with different loadings in the same conditions were approximately identical, but 10 wt% Fe3O4@SiO2–BF3 resulted in the highest yield of the corresponding azo dye (Table 1, entry 6). In conclusion, 10 wt% Fe3O4@SiO2–BF3 was selected as the most ideal acid to be used for the synthesis of arylazo-1-naphthol dyes among those listed in Table 1.
In addition, with more investigations it was found that aryl diazonium salts supported on Fe3O4@SiO2–BF3 nanoparticles underwent no significant changes at room temperature for several months. To explore the reason of this unusual stability, the FT-IR spectrum of 4-chlorophenyl diazonium salt was studied (Fig. 6).
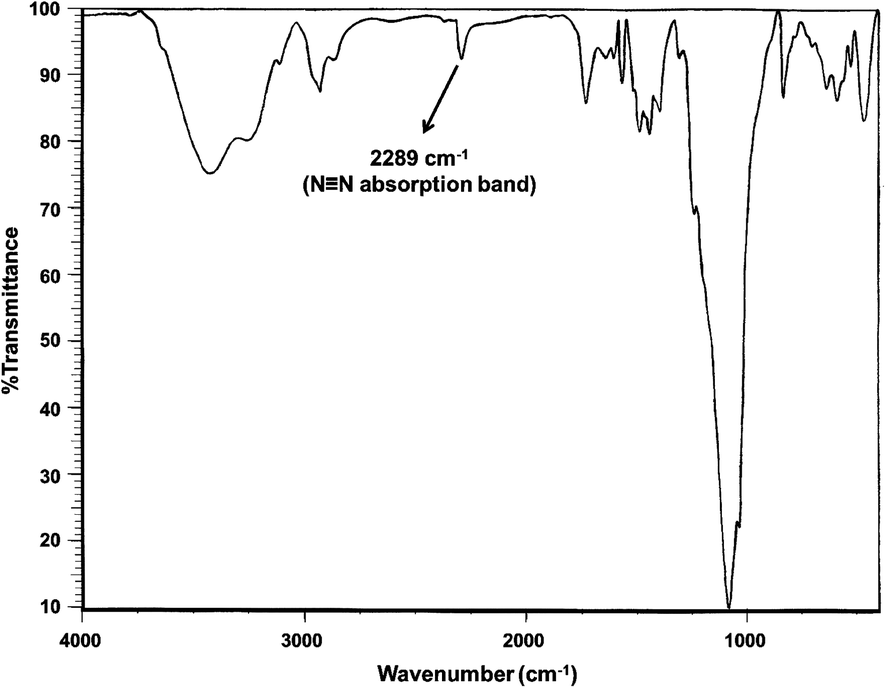 | ||
| Fig. 6 FT-IR spectrum of 4-chlorophenyl diazonium salt supported on Fe3O4@SiO2–BF3 MNPs. | ||
In this spectrum, the ethanolic OH and the existing moisture in BF3·Et2O caused a broad O–H stretching band at the wavenumber of 3419 cm−1. The appearance of a new band at 2289 cm−1 clearly demonstrated N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration and verified the formation of 4-chlorophenyl diazonium salt. The absorption bands of B–O and Si–O vibrations were observed at 1442 and 1085 cm−1, respectively. Aromatic C–H bending vibrations, C–Cl and Fe–O stretching bands were revealed at 829, 634, and 586 cm−1, respectively.
N stretching vibration and verified the formation of 4-chlorophenyl diazonium salt. The absorption bands of B–O and Si–O vibrations were observed at 1442 and 1085 cm−1, respectively. Aromatic C–H bending vibrations, C–Cl and Fe–O stretching bands were revealed at 829, 634, and 586 cm−1, respectively.
According to this information, the structure of aryl diazonium salts supported on MNPs was predicted. Scheme 3 reveals that in this theoretical structure, the aryl diazonium cations are located on the surface of negatively charged particles called as Fe3O4-silica trifluoroborate nanoparticles. Thus, the presence of bulky anions and charge–charge interactions between nitrogen and boron atoms are the possible reasons for the unusual stability of these salts.
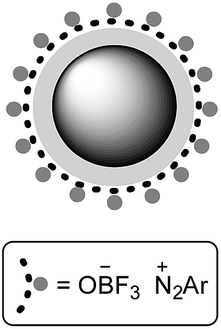 | ||
| Scheme 3 The probable structure of aryl diazonium salt supported on Fe3O4@SiO2–BF3 nanoparticles. | ||
As a result, the high conversions of aniline derivatives to aryl diazonium salts and their long-term stability showed that the Fe3O4@SiO2–BF3 has strong and sufficient acidic sites, which are responsible for the excellent performance in the synthesis of aryl diazonium salts.
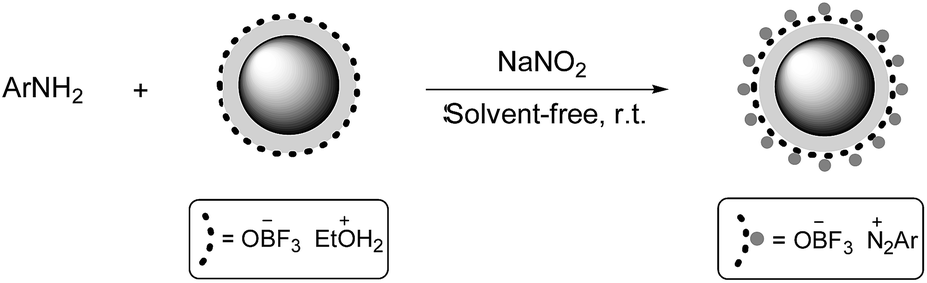
After the optimization of the conditions, aniline derivatives, including electron-withdrawing and electron-donating substituents, were ground with NaNO2 and Fe3O4@SiO2–BF3 nanoparticles. Aryl diazonium salts were obtained in a very short time with an excellent conversion at room temperature (Scheme 4). Indeed, due to the high acidic strength of Fe3O4@SiO2–BF3 nanoparticles, the reaction occurred so rapidly that the substituent type could not affect the time of diazotization.
 | ||
| Scheme 4 Diazotization of aromatic amines in the presence of Fe3O4@SiO2–BF3 nanoparticles. | ||
It is important to note that Fe3O4@SiO2–BF3 in diazotization reaction acts as a two-function reagent, such that on one side, its acidic protons convert the nitrite anions (NO2−) in NaNO2 to nitrosonium cations (NO+) to promote diazotization, while on the other side, its bulky anions facilitate the stability of aryl diazonium cations.
Synthesis of arylazo-1-naphthol dyes
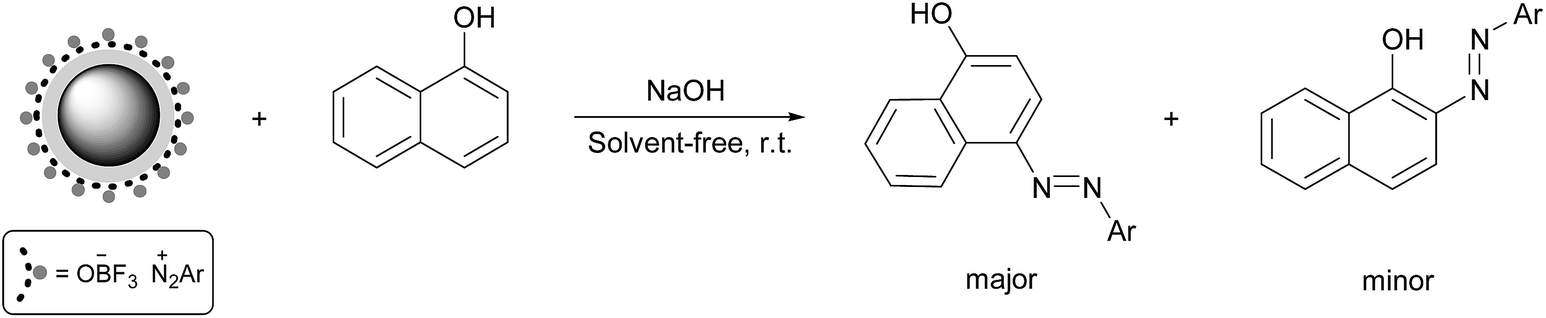
The most common synthetic route to arylazo-1-naphthol compounds involves the coupling of aryl diazonium salts with 1-naphthol in a basic solution. Using water for the preparation of 1-naphthoxide salt appears to be necessary; however, similar to the previous step, it causes the formation of some phenoxide salts and its attack to diazonium salt to form phenolic azo dyes. To prevent this problem, the reaction was performed under solvent-free conditions toward green chemistry. Thus, we ground solid 1-naphthol with some NaOH in a mortar. The moisture absorbed by the reaction mixture during the grinding appears to be sufficient for the formation of a homogeneous mixture. Moreover, the higher concentrations of reactants in the absence of a solvent at room temperature usually leads to a more favourable kinetics than in solution.18In this reaction, the diazo coupling of aryl diazonium salts with 1-naphtoxide led to two products, in which 4-arylazo-1-naphthol derivatives were the major products and 2-arylazo-1-naphthol derivatives were the minor products. The resulting two main products were easily separated by a flash column chromatography and identified by 1H-NMR and FT-IR spectroscopic methods. The molar ratios of these two dyes were determined from the integral intensities of OH/NH signals in 1H-NMR spectrum (ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 4-arylazo-1-naphthol to 2-arylazo-1-naphthol). The total yields of two products are shown in Table 2.
:1 for 4-arylazo-1-naphthol to 2-arylazo-1-naphthol). The total yields of two products are shown in Table 2.
|
|||||
|---|---|---|---|---|---|
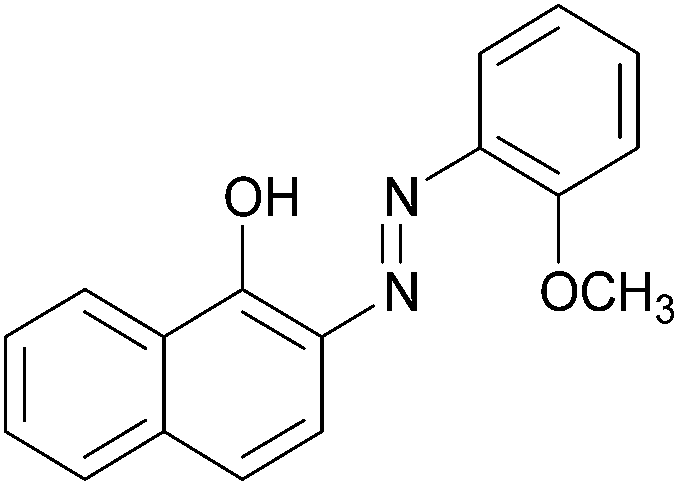
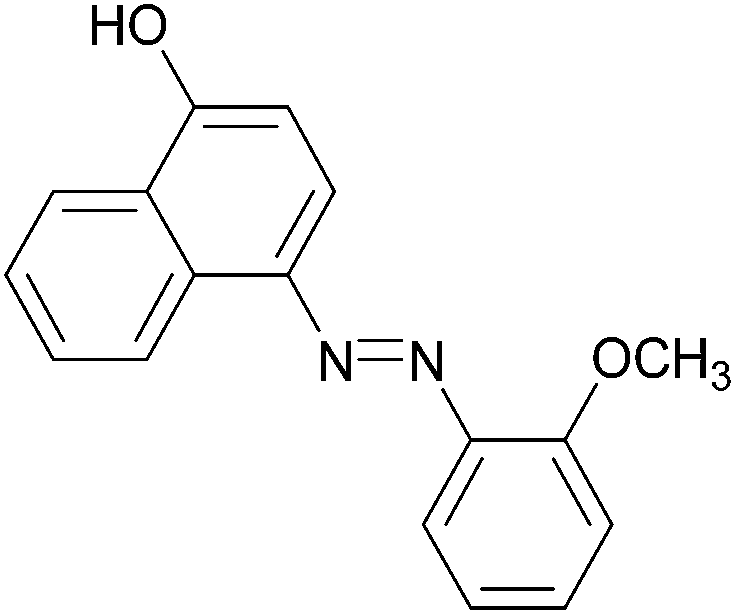
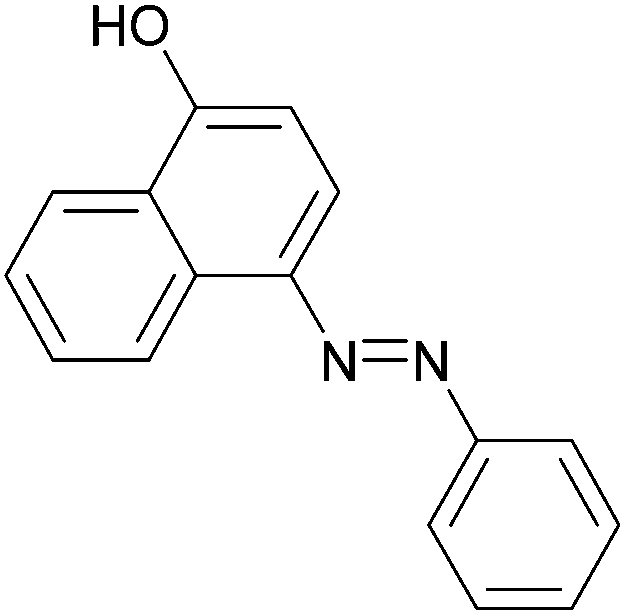
| Entry | Amine | 2-Arylazo-1-naphthol (a′) | 4-Arylazo-1-naphthol (b′) | Yielda (%) | MPfound (Lit.) in a′/found (Lit.) in b′ (°C) |
| a Total isolated yield of 2-arylazo-1-naphthols and 4-arylazo-1-naphthols after chromatography. | |||||
| 1 | 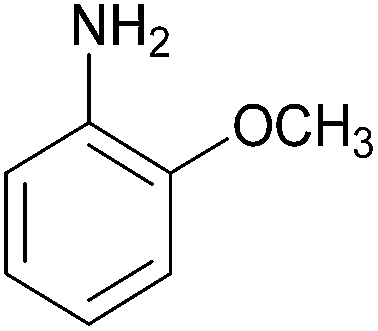 |
 |
 |
92 | 181–183 (180.5–181)19/176–178 (177–178)20 |
| 2 | 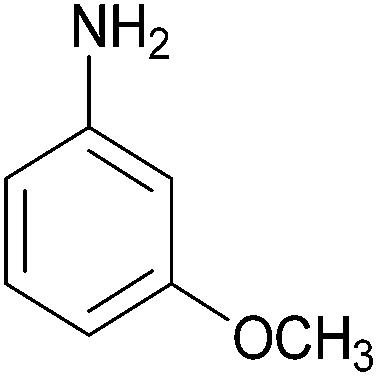 |
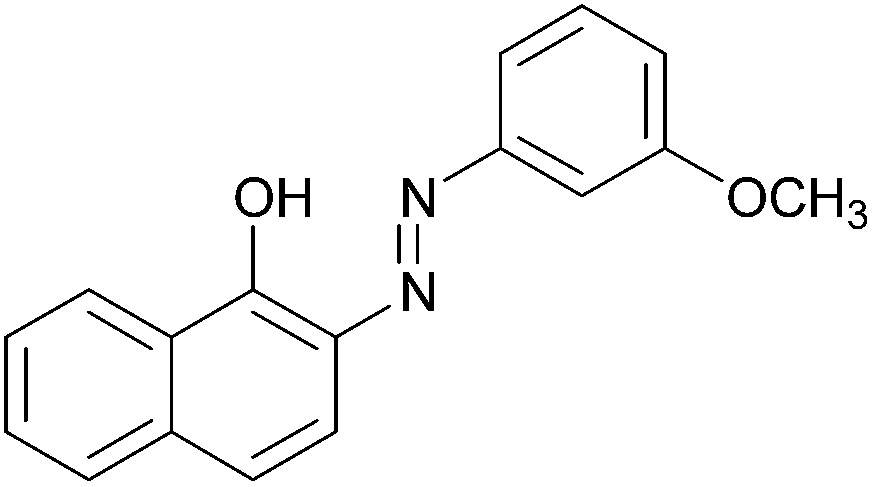 |
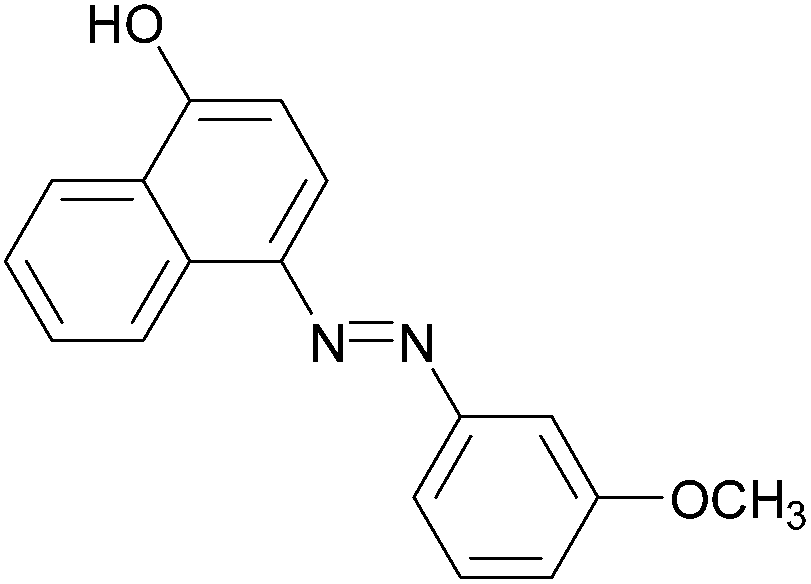 |
89 | 116–118/157–159 (159)20 |
| 3 |  |
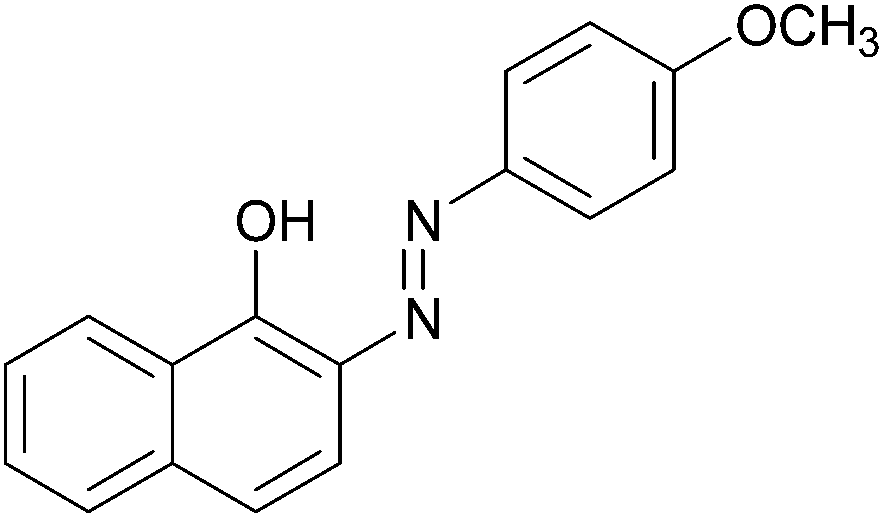 |
 |
93 | 127–129 (127.5–128)19/173–175 (173)20 |
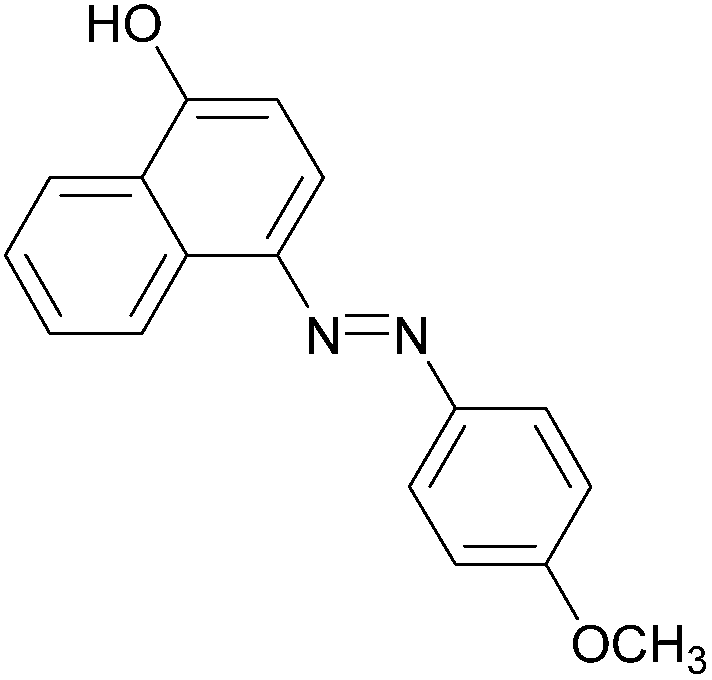
| 4 | 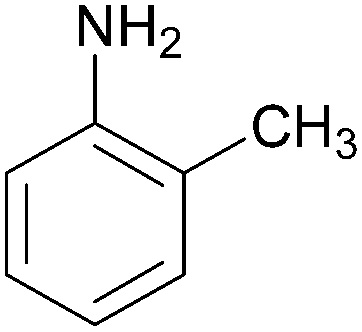 |
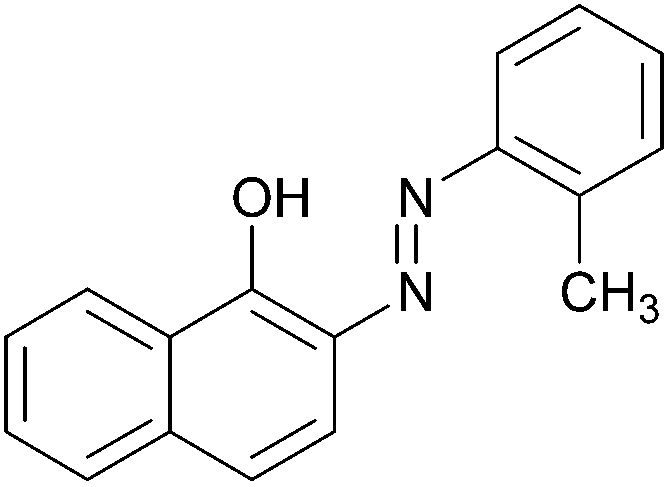 |
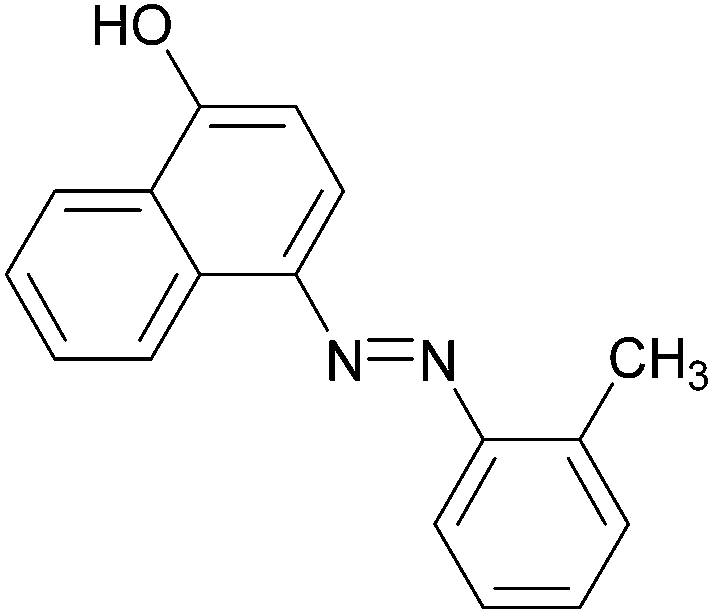 |
91 | 157–159 (156)19/160–162 (162–163)20 |
| 5 | 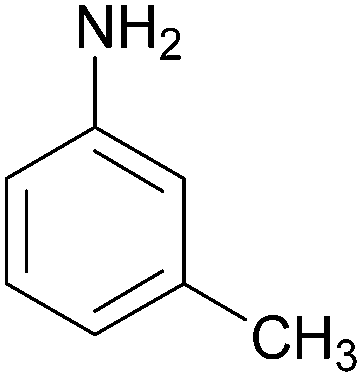 |
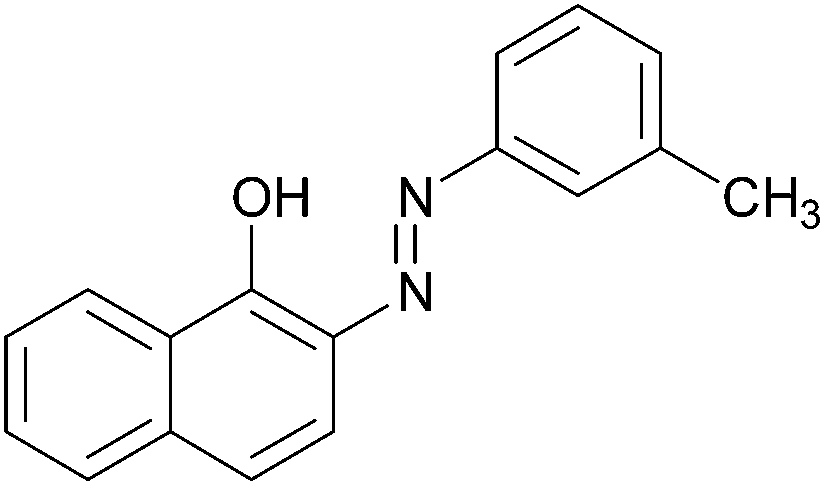 |
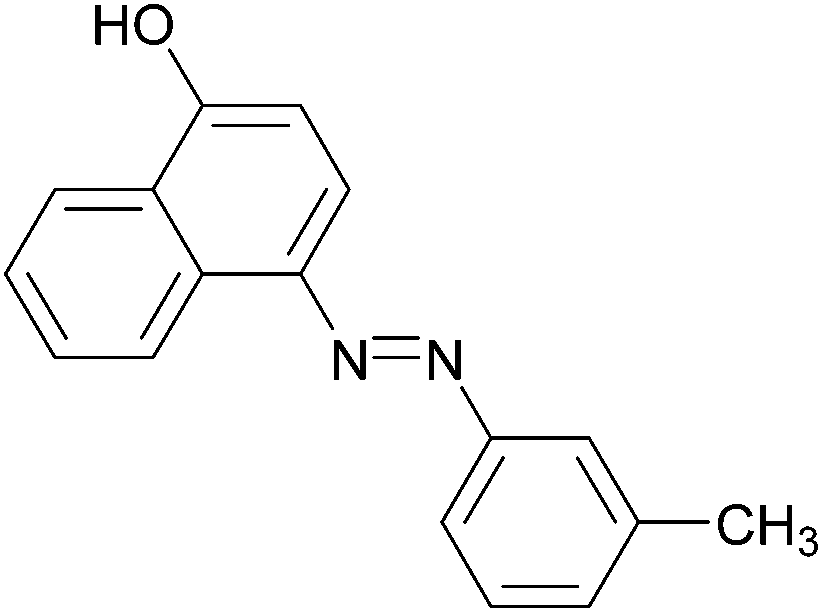 |
90 | 116–118 (117–118)21/201–203 (200)20 |
| 6 | 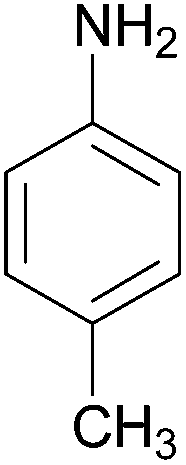 |
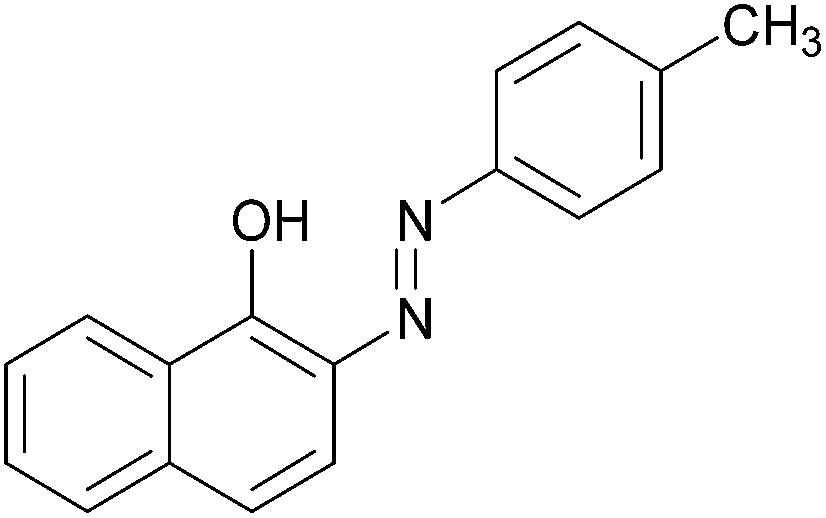 |
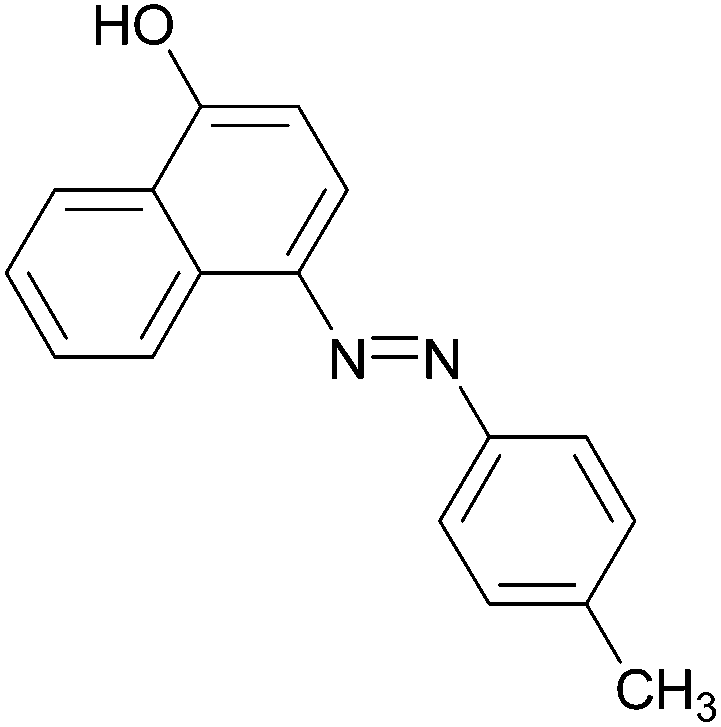 |
96 | 148–150 (151)19/211–213 (211.5)20 |
| 7 | 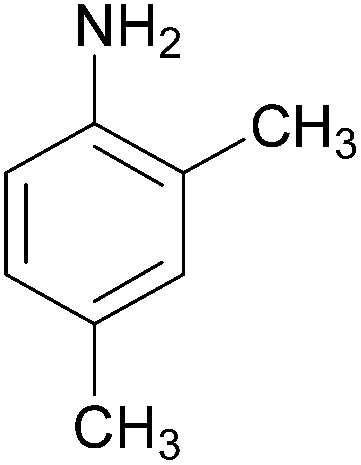 |
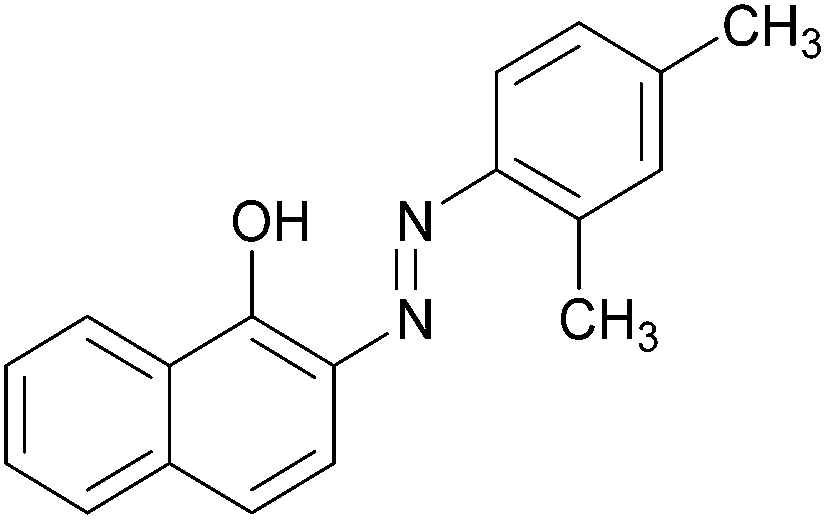 |
 |
87 | 184–186 (186)21/138–140 |
| 8 | 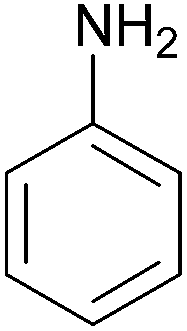 |
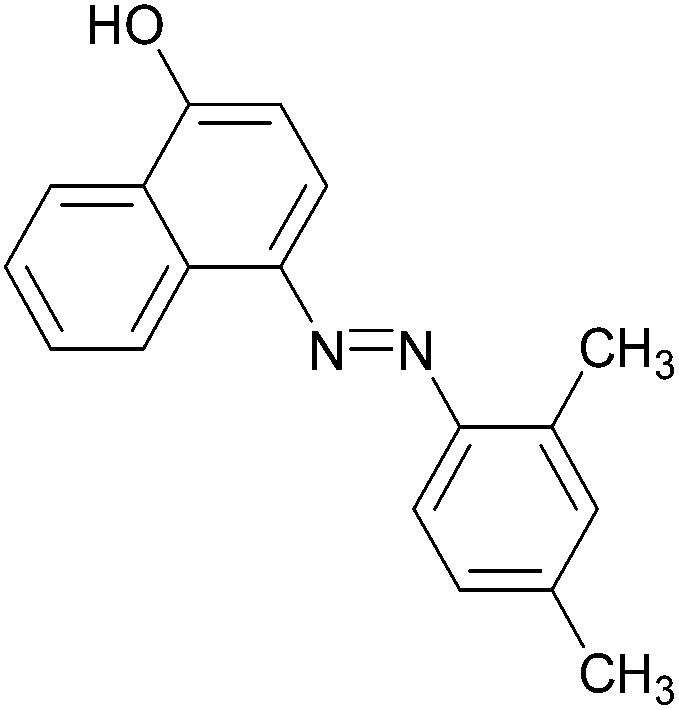
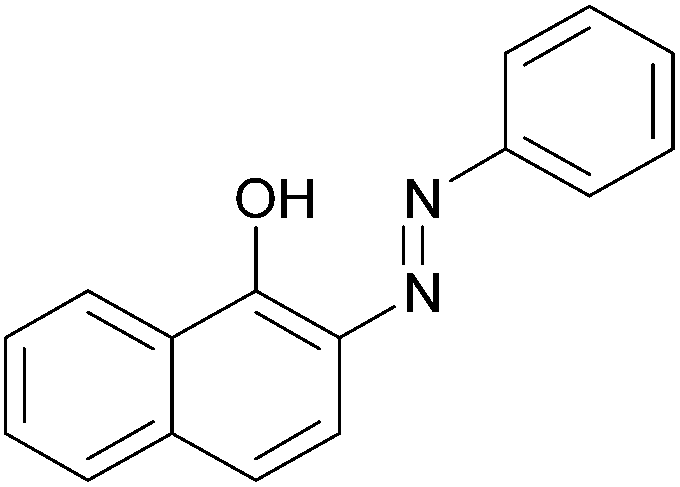 |
 |
89 | 137–139 (138)22/207–209 (208)20 |
| 9 | 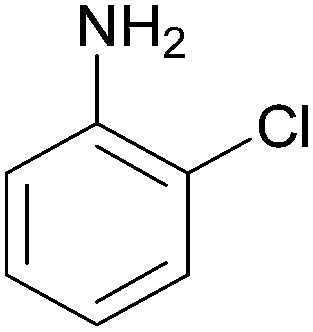 |
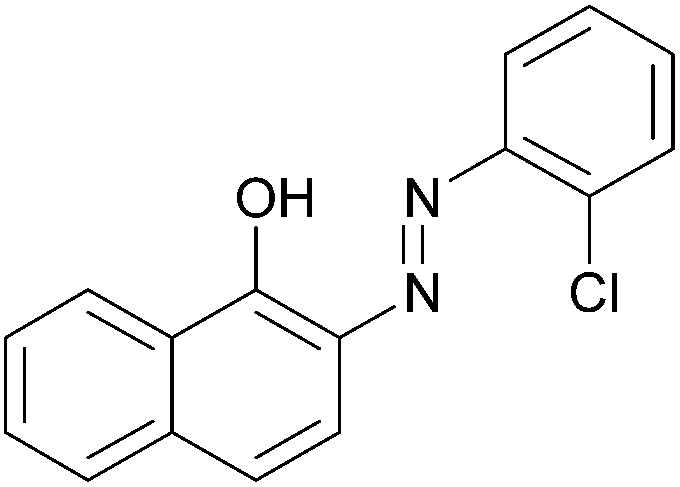 |
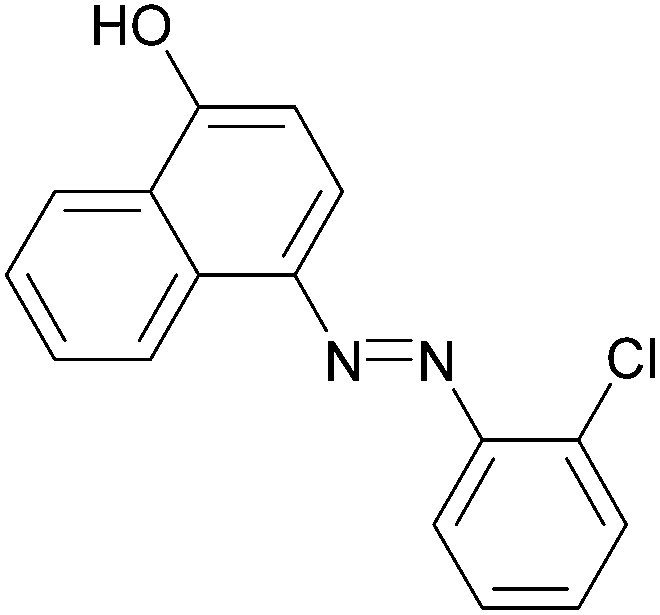 |
92 | 172–174/188–190 (189.5–190.5)20 |
| 10 | 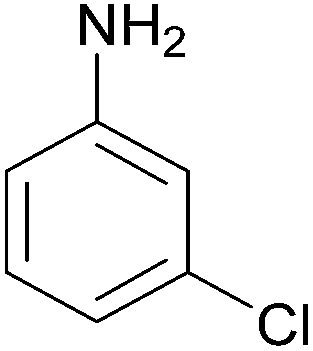 |
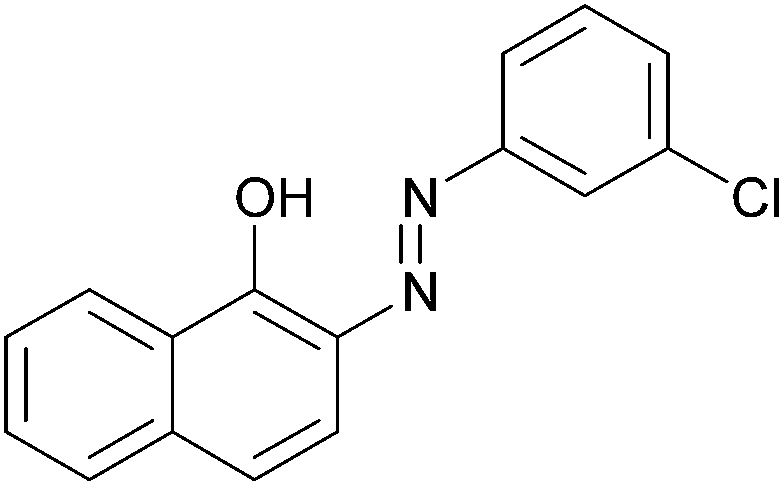 |
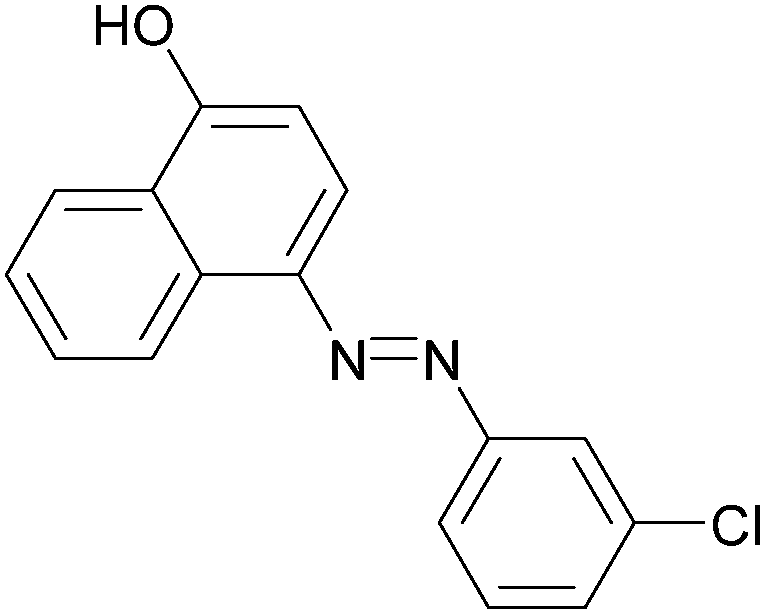 |
90 | 152–154/228–230 (227.5–228)20 |
| 11 | 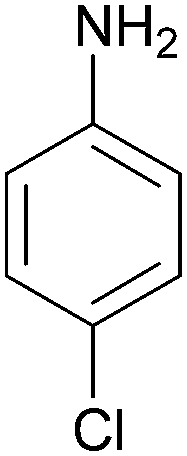 |
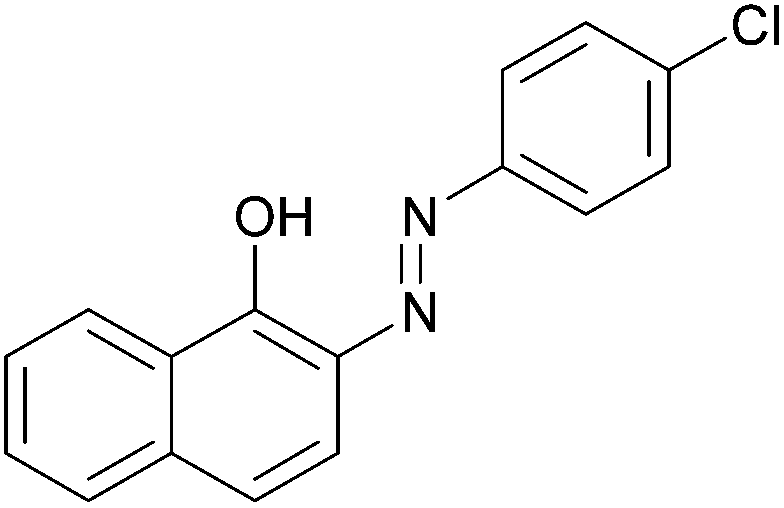 |
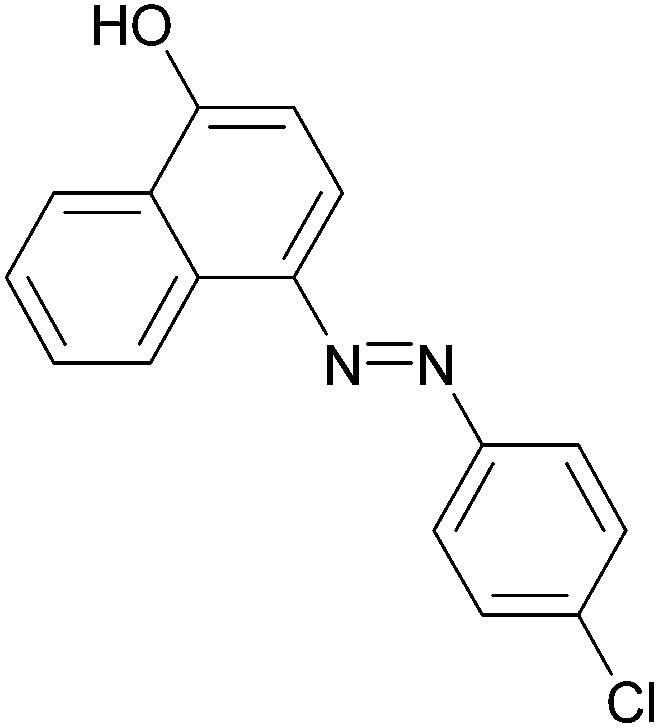 |
95 | 187–189 (187)21/229–231 (229.5)20 |
| 12 | 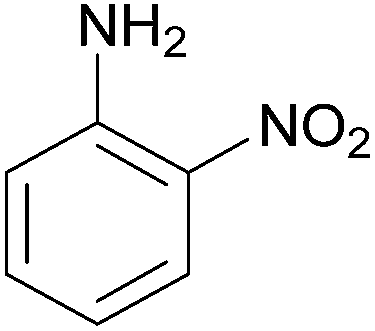 |
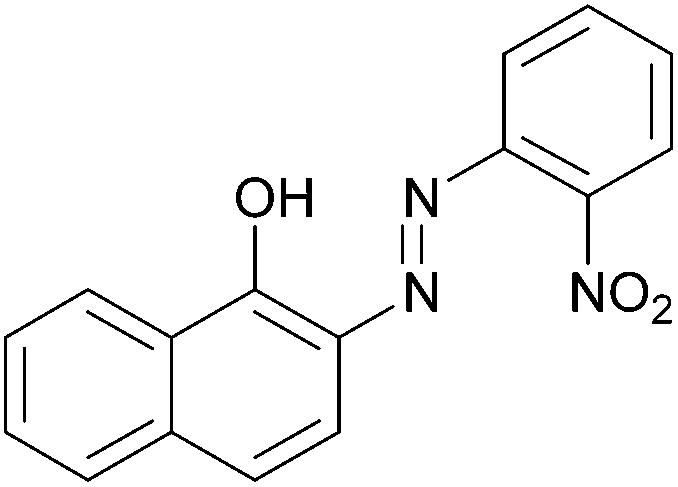 |
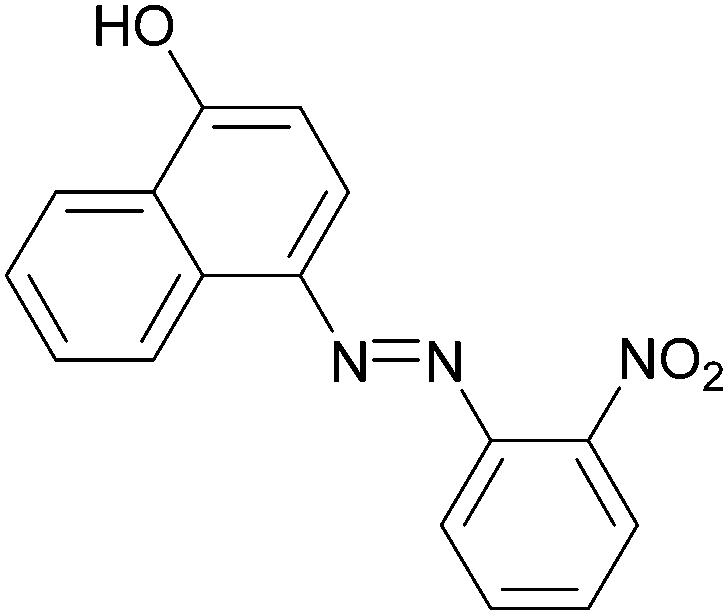 |
85 | 216–218 (216)23/258–260 (256–257)20 |
It is also important to note that the electronic effects of aryl diazonium salt substituents do not play significant role in the rate of diazo coupling step. However, the coupling reaction occurred rapidly regardless of the substituent effects.
Altogether, in this research, we investigated the application of Fe3O4@SiO2–BF3 MNPs as a strong and useful solid acid reagent for the synthesis of azo dyes based on 1-naphthol via a green procedure.
Experimental
Materials and apparatus
Chemicals and solvents were purchased from Merck and Sigma-Aldrich Companies. Melting points were obtained with a micro melting point apparatus (Electrothermal, Mk3) and are uncorrected. 1H-NMR spectra were recorded on a Bruker DRX-400 Avance spectrometer. Tetramethyl silane (TMS) was used as an internal reference, and CDCl3 and DMSO-d6 were used as solvents. FT-IR spectra were obtained on a Nicolet Magna 550 spectrometer. The ultrasonic equipment used for the synthesis of MNPs (Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2–BF3) was a Sonica 2200ETH S3 SOLTEC ultrasonic bath (Italy) with a working frequency of 40 KHz. XRD patterns were acquired using a Philips Xpert MPD diffractometer equipped with a Cu Kα anode (λ = 1.54 Å) in the 2θ range from 10° to 80°. The magnetization of the samples was recorded as a function of the applied magnetic field sweeping between ±10 kOe at room temperature. All the measurements were performed on a vibrating sample magnetometer device (Meghnatis Daghigh Kavir Co.; Kashan Kavir; Iran). The morphology of the synthesized samples was studied by a Mira II LMU Tescan FE-SEM made in the Czech Republic. The elemental composition of the three above mentioned MNPs was investigated by EDS spectroscopy (SAMX, France). The average size of Fe3O4 and Fe3O4@SiO2–BF3 MNPs was analyzed by TEM using a Philips CM120 with a LaB6 cathode at an accelerating voltage of 120 kV.Synthesis of Fe3O4@SiO2–BF3 MNPs
The synthesis of Fe3O4 nanoparticles was carried out according to a known procedure using a chemical co-precipitation method with a little modification of the methodology already reported in the literature.24 Briefly, FeCl3·6H2O (8.0 g, 0.029 mol) and FeCl2·4H2O (3.5 g, 0.017 mol) with a molar ratio of approximately 2:1 were dissolved in 38 mL of deoxygenated 0.4 M HCl solution. Then, 375 mL of deoxygenated 0.7 M ammonia solution was quickly added into the reaction mixture with sonication and under nitrogen atmosphere. This resulted in the immediate formation of a black precipitate of Fe3O4 (magnetite). The sonication of magnetite dispersion was continued for 30 min. Finally, the precipitates were collected using an external magnetic field and washed for several times with distilled water and ethanol. The synthesized Fe3O4 MNPs were suspended in 50 mL of distilled water to be used in the next steps.
A modified Stöber sol–gel process25 was used for coating magnetite nanoparticles with a silica shell. Typically, 50 mL of magnetite suspended in water was added to 250 mL ethanol and sonicated at room temperature for 20 min under nitrogen flow. Then, 11.85 mL PEG 200, 50 mL distilled water, and 25 mL NH3 (28%) were added, after 15 min, 5 mL of TEOS was introduced into the suspension and the mixture was again sonicated for 6 h. Fe3O4@SiO2 nanoparticles were centrifuged at 3000 rpm for 10 min. The solvent was discarded and nanoparticles were washed three times with water and ethanol and then dried in vacuum at room temperature.
In the final stage, BF3·Et2O (0.45 mL) was added drop-wise to a slurry containing Fe3O4@SiO2 core–shell nanoparticles (4.5 g) and ethanol (15 mL). The mixture was sonicated for 1 h at room temperature. The resulting suspension was filtered and dried at room temperature to obtain a brown solid named nano Fe3O4@SiO2–BF3 (10 wt%).
Typical procedure for the synthesis of arylazo-1-naphthol dyes
For the synthesis of arylazo-1-naphthol derivatives, we mixed aromatic amines (2 mmol) with sodium nitrite (3 mmol) and Fe3O4@SiO2–BF3 nanoparticles (0.3 g) in a mortar with a pestle by rapid grinding. The progress of reaction was monitored by TLC (ethyl acetate/n-hexane). Moreover, for the preparation of 1-naphtoxide salt, 2 mmol of 1-naphthol and 10 mmol NaOH were ground in another mortar. Then, aryl diazonium salt was added to 1-naphthoxide salt and mixing and grinding were performed for a short time (about 3 min). After the completion of the reaction and formation of two main products (2-arylazo-1-naphthol and 4-arylazo-1-naphthol), the mixture was washed with distilled water (3 × 10 mL) and acetone (4 × 5 mL). Two final products were separated by flash column chromatography with silica mesh of 230–400 (40–63 μm).Conclusion
Fe3O4@SiO2–BF3 MNPs as a novel heterogeneous solid acid reagent was prepared by the support of BF3·Et2O on the surface of Fe3O4@SiO2 core–shell nanoparticles and characterized by various methods. Then, some brilliant arylazo-1-naphthol derivatives were successfully synthesized by the diazotization of aniline derivatives in the presence of Fe3O4@SiO2–BF3 MNPs and their diazo coupling with 1-naphthol at room temperature. The use of Fe3O4@SiO2–BF3 and a solvent-free procedure simplified the synthesis; in addition the procedure did not require special conditions, such as liquid acids and low temperature, and the method was compatible with environment. The procedure gave efficient yields using short reaction times, making it attractive to synthesize a variety of important dyes. The structure and stability of the aryl diazonium salt supported on Fe3O4@SiO2–BF3 MNPs were also studied.Acknowledgements
The authors are grateful to the University of Kashan for supporting this work by Grant no. 159189/41.Notes and references
- (a) T. Kaicharla, S. R. Yetra, T. Roy and A. T. Biju, Green Chem., 2013, 15, 1608–1614 RSC; (b) M. V. Reddy and Y. T. Jeong, Tetrahedron Lett., 2013, 54, 3546–3549 CrossRef PubMed; (c) M. Özil and M. Canpolat, Polyhedron, 2013, 51, 82–89 CrossRef PubMed; (d) Z. N. Siddiqui and T. Khan, RSC Adv., 2014, 4, 2526–2537 RSC; (e) J. Kamalraja and P. T. Perumal, Tetrahedron Lett., 2014, 55, 3561–3564 CrossRef CAS PubMed; (f) D. Lanari, O. Rosati and M. Curini, Tetrahedron Lett., 2014, 55, 1752–1755 CrossRef CAS PubMed.
- H. S. Freeman and A. T. Peters, Colorants for Non-Textile Applications, Elsevier Science B. V., Amsterdam, 2000 Search PubMed.
- P. Gregory, High Technology Applications of Organic Colorants, Plenum, New York, 1991, ch. 9 Search PubMed.
- Y. Kubo, S. Maeda, S. Tokita and M. Kubo, Nature, 1996, 382, 522–524 CrossRef CAS.
- R. Steinsträsser and L. Pohl, Angew. Chem., Int. Ed., 1973, 12, 617–630 CrossRef.
- Y. He, X. Gu, M. Guo and X. Wang, Opt. Mater., 2008, 31, 18–27 CrossRef CAS PubMed.
- S. Pieraccini, S. Masiero, G. P. Spada and G. Gottarelli, Chem. Commun., 2003, 598–599 RSC.
- M. Tatsuta and T. Kitao, Reagent for Detecting and Diagnosing Cancer, publication no. JP 01-207247 A, 1989 Search PubMed.
- A. Aszalos, J. L. Weaver and P. S. Pine, Methods of Using Azo Dyes and Their Derivatives, US Pat. no. 5,468,469, 1995 Search PubMed.
- R. M. Christie, Colour Chemistry, Royal Society of Chemistry, Cambridge, 2001, ch. 3 Search PubMed.
- (a) A. B. Atar and Y. T. Jeong, Tetrahedron Lett., 2013, 54, 1302–1306 CrossRef CAS PubMed; (b) F. Jing, B. Katryniok, E. Bordes-Richard and S. Paul, Catal. Today, 2013, 203, 32–39 CrossRef CAS PubMed; (c) Y. B. Huang and Y. Fu, Green Chem., 2013, 15, 1095–1111 RSC; (d) M. Billamboz, F. Mangin, N. Drillaud, C. Chevrin-Villette, E. Banaszak-Léonard and C. Len, J. Org. Chem., 2014, 79, 493–500 CrossRef CAS PubMed; (e) C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060–1063 CrossRef CAS PubMed; (f) K. M. Hello, H. R. Hasan, M. H. Sauodi and P. Morgen, Appl. Catal., A, 2014, 475, 226–234 CrossRef CAS PubMed; (g) J. G. Hernández-Cortez, M. Manríquez, L. Lartundo-Rojas and E. López-Salinas, Catal. Today, 2014, 220–222, 32–38 CrossRef PubMed.
- (a) J. H. Clark, Catalysis of Organic Reactions by Supported Inorganic Reagents, VCH, New York, 1994 Search PubMed; (b) T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef CAS PubMed.
- (a) A. K. Shah, K. J. Prathap, M. Kumar, S. H. R. Abdi, R. I. Kureshy, N. H. Khan and H. C. Bajaj, Appl. Catal., A, 2014, 469, 442–450 CrossRef CAS PubMed; (b) M. Korzec, P. Bartczak, A. Niemczyk, J. Szade, M. Kapkowski, P. Zenderowska, K. Balin, J. Lelatko and J. Polanski, J. Catal., 2014, 313, 1–8 CrossRef CAS PubMed; (c) Z. Dong, X. Le, X. Li, W. Zhang, C. Dong and J. Ma, Appl. Catal., B, 2014, 158–159, 129–135 CrossRef CAS PubMed; (d) X. Le, Z. Dong, W. Zhang, X. Li and J. Ma, J. Mol. Catal. A: Chem., 2014, 395, 58–65 CrossRef CAS PubMed; (e) T. R. Mandlimath, B. Umamahesh and K. I. Sathiyanarayanan, J. Mol. Catal. A: Chem., 2014, 391, 198–207 CrossRef CAS PubMed.
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- (a) Y. H. Liu, J. Deng, J. W. Gao and Z. H. Zhang, Adv. Synth. Catal., 2012, 354, 441–447 CrossRef CAS; (b) X. Zhang, X. He, L. Chen and Y. Zhang, J. Mater. Chem., 2012, 22, 16520–16526 RSC; (c) X. Jin, K. Zhang, J. Sun, J. Wang, Z. Dong and R. Li, Catal. Commun., 2012, 26, 199–203 CrossRef CAS PubMed; (d) H. J. Xu, X. Wan, Y. Y. Shen, S. Xu and Y. S. Feng, Org. Lett., 2012, 14, 1210–1213 CrossRef CAS PubMed; (e) G. M. Ucoski, F. S. Nunes, G. F. Silva, Y. M. Idemori and S. Nakagaki, Appl. Catal., A, 2013, 459, 121–130 CrossRef CAS PubMed; (f) F. Nemati and R. Saeedirad, Chin. Chem. Lett., 2013, 24, 370–372 CrossRef CAS PubMed; (g) S. Wang, Z. Zhang, B. Liu and J. Li, Catal. Sci. Technol., 2013, 3, 2104–2112 RSC; (h) M. R. Nabid, Y. Bide and M. Niknezhad, ChemCatChem, 2014, 6, 538–546 CrossRef CAS; (i) F. Zhang, M. Chen, X. Wu, W. Wang and H. Li, J. Mater. Chem. A, 2014, 2, 484–491 RSC; (j) R. K. Sharma, Y. Monga and A. Puri, J. Mol. Catal. A: Chem., 2014, 393, 84–95 CrossRef CAS PubMed; (k) E. Kolvari, N. Koukabi and O. Armandpour, Tetrahedron, 2014, 70, 1383–1386 CrossRef CAS PubMed.
- (a) B. F. Mirjalili, A. Bamoniri and A. Akbari, Tetrahedron Lett., 2008, 49, 6454–6456 CrossRef CAS PubMed; (b) B. F. Mirjalili, A. Bamoniri, M. A. Karimi Zarchi and H. Emtiazi, J. Iran. Chem. Soc., 2010, 7, 95–99 CrossRef CAS; (c) B. F. Mirjalili, A. Bamoniri and A. Akbari, J. Iran. Chem. Soc., 2011, 8, 135–140 CrossRef; (d) A. Bamoniri, A. Ghorbani-Choghamarani and B. F. Mirjalili, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 381–388 CrossRef CAS; (e) A. Bamoniri, B. F. Mirjalili, A. A. Jafari and F. Abasaltian, Iran. J. Catal., 2012, 2, 73–76 Search PubMed; (f) B. F. Mirjalili, A. Bamoniri and M. A. Mirhoseini, Chem. Heterocycl. Compd., 2012, 48, 856–860 CrossRef CAS PubMed; (g) B. F. Mirjalili, A. Bamoniri and L. Zamani, Sci. Iran., 2012, 19, 565–568 CrossRef CAS PubMed; (h) A. Bamoniri, B. F. Mirjalili and S. Nazemian, Curr. Chem. Lett., 2013, 2, 27–34 CrossRef CAS; (i) A. Bamoniri, B. F. Mirjalili and S. Nazemian, J. Iran. Chem. Soc., 2014, 11, 653–658 CrossRef CAS.
- K. Wilson and J. H. Clark, Chem. Commun., 1998, 2135–2136 RSC.
- (a) F. Toda and K. Tanaka, Chem. Rev., 2000, 100, 1025–1074 CrossRef PubMed; (b) K. Tanaka, Solvent-Free Organic Synthesis, John Wiley & Sons, New York, 2003 Search PubMed.
- L. N. Ogoleva and B. I. Stepanov, Ž. Org. Chim., 1965, 1, 2083–2086 CAS.
- K. J. Morgan, J. Chem. Soc., 1961, 2151–2159 RSC.
- G. Charrier and G. Ferreri, Gazz. Chim. Ital., 1914, 44, 234 Search PubMed.
- W. R. Brode and L. E. Herdle, J. Org. Chem., 1941, 6, 713–721 CrossRef CAS.
- E. Bamberger, Chem. Ber., 1897, 30, 513–516 CrossRef CAS.
- M. Emadi, E. Shams and M. K. Amini, J. Chem., 2013, 787682, DOI:10.1155/2013/787682.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |