
Surface chemistry modulated introduction of multifunctionality within Co3O4 nanocubes†
Monalisa Pal*,
Ashutosh Kumar Singh,
Rupali Rakshit and
Kalyan Mandal
Department of Condensed Matter Physics and Material Sciences, S. N. Bose National Centre for Basic Sciences, Block JD, Sector III, Salt Lake, Kolkata 700 098, India. E-mail: monalisa.pal6@gmail.com; monalisa12@bose.res.in
First published on 19th January 2015
Abstract
Co3O4 as a multifunctional nanomaterial, possessing simultaneously, unique optical, magnetic, and catalytic properties is sparse in the existing literature. We have activated intrinsic multicolor fluorescence covering the entire visible region by the functionalization of Co3O4 nanocubes (NCs) with Na-tartrate. The functionalized Co3O4 NCs show excellent catalytic efficiency in the degradation of both biologically and environmentally harmful dyes, which opens up a way for the possible application of the NCs toward therapeutic and wastewater treatment. Systematic investigations using UV-visible absorption, steady state, and time-resolved photoluminescence studies reveal the mechanistic origin behind the generation of multicolor fluorescence from the ligand-functionalized Co3O4 NCs. Moreover, from the magnetic study we have found that the room temperature antiferromagnetic nature of Co3O4 NCs turns to ferromagnetic after ligand functionalization due to the surface modification of the NCs. We believe that the developed multifunctional Co3O4 NCs could open up a new horizon toward their diverse applications in the present era of multitasking.
Introduction
The aggressive scaling down of transition metal oxides to the nanometer size scale is the cornerstone for creating integrated systems with imposed multifunctionality. Although still in its infancy, the field of multifunctional nanoparticles has shown great promise to solve a number of major global issues.The vast medical potential of nanoscale magnetic materials has revolutionized the diagnosis of ailing tissues by magnetic resonance, ultrasound,1 and optical imaging, along with targeted drug delivery,2 separation and purification of cells,3 tissue repairing,4 hyperthermia for cancer treatment,5 etc. Fluorescent magnetic nanoparticles are special candidates for many applications as they can be concurrently utilized in diagnostic and therapeutic applications. To date, fluorescent magnetic nanoparticles have been prepared either by molecular functionalization with dyes or by attachment with quantum dots (QDs). In spite of their remarkable efficiency in the field of biomedical applications, due to some serious drawbacks like photo-bleaching and the instability of organic dyes and inherent toxicity of QDs, nanoprobes with intrinsic fluorescence are highly desirable, but unfortunately, are sparsely found in the existing literature.
Advanced oxidation processes (AOPs), such as sonolysis, radiolysis, and photocatalysis, have assisted in environmental remediation by mineralizing organic compounds in aqueous media, namely by producing hydroxyl radicals as the primary oxidant.6,7 Nanocatalysis is a versatile, low-cost, and environmentally benign technology for the treatment of a host of pollutants. The ability of AOPs to destroy low levels of persistent organic pollutants, as well as microorganisms in water, has been widely demonstrated and, progressively, the technology is now being commercialized in many areas of the world, including in developing nations. Nowadays, research has been focused in the development of nontoxic, cheaper, smarter catalysts with enhanced performance and reusability.8–11
As an important transition metal oxide, Co3O4 has attracted immense attention owing to its applications in many fields, such as, catalysis,12 toxic gas sensing,12,13 in lithium ion batteries,14 and in supercapacitors.15–18 The development of nanostructured Co3O4 with desirable size and morphologies, like cubes, rods, wires, tubes, dendrites, and sheets, have been widely explored for their superior performance in the traditional arena and also to give other unique properties.19,20 Recently, Co3O4 was found to be an active agent for the photocatalytic degradation of organic pollution under light irradiation, which is a novel finding that paves the way for the exploitation and application of Co3O4 in such oxidative ventures.21 Nowadays, Co3O4-based nano-materials are also being promoted for biomedical applications by protein-assisted synthesis or surface modification or by designing suitable nanoarchitechtures.22,23 However, to the best of our knowledge, the development of intrinsic fluorescence upon utilizing the surface chemistry through facile functionalization and incorporation of multifuntionality within a single Co3O4 nanostructure with intrinsic fluorescence, ferromagnetism, and catalytic properties, simultaneously, has not yet been reported.
Herein, we have synthesized Co3O4 NCs by a solvothermal route following a previous report by Zhang et al., with some modification,24 where solutions of cobalt acetate in ethanol were treated with aqueous ammonium hydroxide and heated at 150 °C in an teflon-lined stainless steel autoclave for 3 h. Detailed characterization through XRD (X-ray diffraction) and TEM (Transmission electron microscopy) confirmed the formation of uniform Co3O4 NCs of small size (∼8 nm) with a narrow size distribution. We generated intrinsic multicolor fluorescence covering the whole visible region, ranging from blue, cyan, and green to red by functionalization of Co3O4 NCs with Na-tartrate. Systematic investigations through UV-visible absorption, steady state and time-resolved photoluminescence studies revealed that the ligand-to-metal charge transfer (LMCT) transition from tartrate ligand to the lowest unoccupied energy level of Co2+/3+ of the NCs and d–d transitions centered over Co2+/3+ ions in the NCs play the important role in the generation of multiple fluorescence from the ligand-functionalized Co3O4 NCs. The FTIR (Fourier transform infrared spectroscopy) study inferred the attachment of the ligand to the Co3O4 NCs' surface. Interestingly, we found that the magnetic behavior of the NCs can be tuned from room temperature antiferromagnetic to ferromagnetic, by surface modification of NCs with the ligands. Then, we tested the functionalized Co3O4 NCs in catalysis. The functionalized Co3O4 NCs showed very important catalytic efficiency (in the absence of any photoactivation) toward the degradation of bilirubin (BR, the pigment responsible for yellow coloration of the skin in jaundice), along with photocatalytic activity in the degradation of methylene blue (MB, a model water pollutant), which opens up possible applications toward therapeutic uses, as well as in wastewater treatment. So, by combining the manifold beneficial activities within a single entity at the nanoscale, we have developed multifunctional Co3O4 NCs with great potential toward diverse applications, ranging from bioimaging, drug delivery, and therapeutics, to wastewater treatment.
Results and discussion
A TEM study was carried out to evaluate the morphology and monodispersity of the solvothermally synthesized Co3O4 NCs. As shown in Fig. 1a, as-prepared Co3O4 NCs have a nearly homogeneous size distribution (6–11 nm), with an average size of 8.65 ± 0.22 nm (Fig. 1b). The corresponding SAED (selected area electron diffraction) pattern in Fig. 1c and the HRTEM (high resolution transmission electron microscopy) image in Fig. 1d confirm the crystallinity of the NCs. The calculated interplanar distance between the lattice fringes is about 0.243 nm, which corresponds to the distance between the (311) planes of Co3O4 crystal lattice. The EDX (energy dispersive X-ray) spectroscopic analysis of Co3O4 NCs (Fig. 1e) confirms the elemental composition of only cobalt, and oxygen. Fig. 1f shows the XRD pattern of the as-prepared Co3O4 NCs, where all the diffraction peaks in the figure perfectly match with the cubic spinel structure (JCPDS 42-1467) of Co3O4 NCs from the literature.24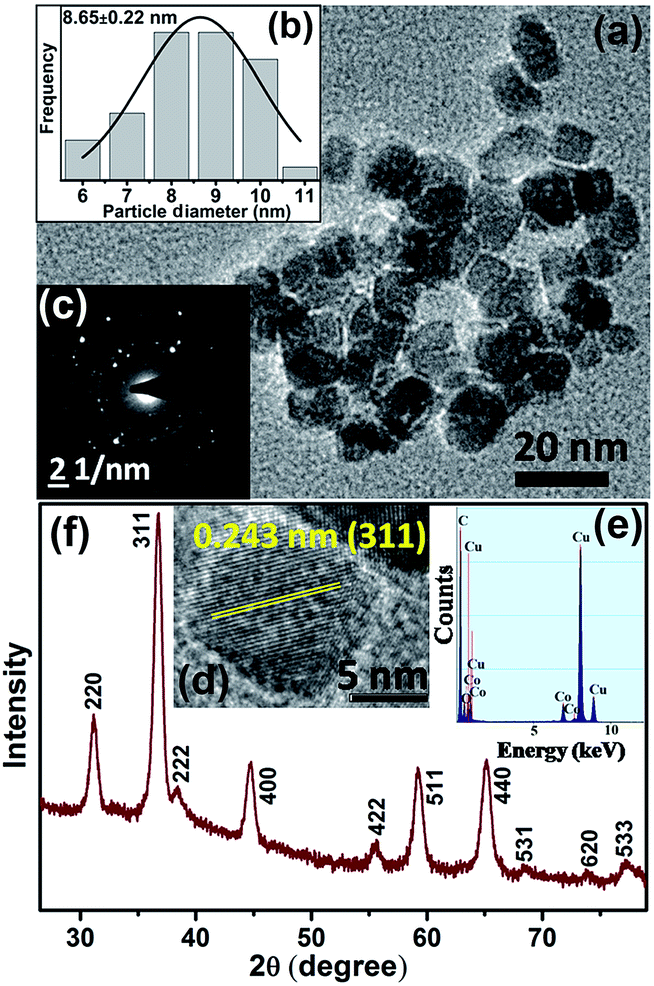 | ||
| Fig. 1 (a) TEM image of the as-prepared bare Co3O4 NCs. (b) Size distribution of the as-prepared Co3O4 NCs. (c) SAED pattern of the as-prepared bare Co3O4 NCs (d) corresponding HRTEM image indicating the high crystallinity and showing the lattice fringes. (e) EDX spectrum of the NCs indicating the presence of only Co and O. (f) XRD pattern of as-prepared Co3O4 NCs. All the diffraction peaks in the figure are perfectly indexed in the literature to the cubic spinel structure of Co3O4 crystal lattice. | ||
In order to solubilize Co3O4 NCs, we functionalized the NCs with tartrate ligands. Fig. 2a shows the TEM image of functionalized Co3O4 NCs with an average size of ∼7.4 nm (Fig. 2b). From the SAED pattern (lower inset of Fig. 2a) and the HRTEM image (upper inset of Fig. 2a) of functionalized Co3O4 NCs, the highly crystalline nature of the NCs is clearly evident. The calculated interplanar distance between the fringes was found to be 0.243 nm, corresponding to the (311) plane of the crystal lattice.
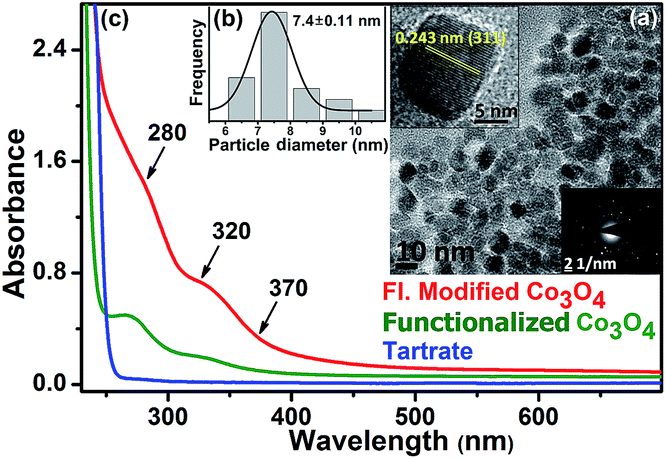 | ||
| Fig. 2 (a) TEM image of highly dispersible surface-modified Co3O4 NCs, in aqueous medium. In the upper inset, lattice fringes in the corresponding HRTEM image indicate the highly crystalline nature of the NCs even after functionalization. In the lower inset, SAED pattern of the functionalized Co3O4 NCs. (b) Size distribution of Co3O4 NCs after solubilization in water. (c) UV-visible absorption spectrum of the functionalized Co3O4 NCs before and after heat and pH treatment along with only Na-tartrate. | ||
As shown in Fig. 2c, the functionalized Co3O4 NCs (at pH ∼7) exhibit a distinct absorption pattern in the UV-visible region, which indicates a significant variation in the surface electronic structure of the NCs upon functionalization with the tartrate ligand. The observed peak at around 280 nm and 320 nm has a relatively higher optical density compared to another peak at 370 nm. Interestingly, upon exciting the sample at both 280 nm and 322 nm, we observed a photoluminescence peak with the maxima at 418 nm. While exciting at 355 nm, we obtained a photoluminescence peak with a maxima at 460 nm, although with a low intensity. To increase the photoluminescence intensity, we carried out further surface modification of the solubilized NCs by heating after first increasing the pH, which gave rise to the generation of two more photoluminescence peaks with multiple fold increases in the overall intensity upon excitation at proper wavelengths. The reason for the enhancement of the photoluminescence intensity, as well as for the generation of multiple optical bands upon surface modification, could be due to the increased coordination between the ligand functional groups (carboxylate and hydroxyl moieties) and the Co2+/3+ centers at the NC surface, as evident from Fig. S2 in the ESI.†
Fig. 3a shows the normalized steady-state photoluminescence emission spectra obtained from surface-modified Co3O4 NCs. Upon excitation at wavelengths of 322, 355, 410, and 520 nm, the NCs solution gave rise to intense photoluminescence peaks at 418, 460, 503, and 562 nm, respectively. Photographs of the aqueous solution of surface-modified Co3O4 NCs under visible and UV light are presented in the inset of Fig. 3a. The amazing fluorescence micrographs, as shown in Fig. 3c, demonstrate that the black powder of functionalized Co3O4 NCs under a bright field generates fluorescent colors like cyan, green, and red upon excitation at 365, 436 and 546 nm, respectively, by using proper filters. Fig. S1† shows that the fluorescence micrographs of the as-prepared bare Co3O4 NCs under identical conditions have no such fluorescence. Fluorescence quantum yields (QY) of the functionalized Co3O4 NCs were calculated by following the relative method of Williams et al.,25 involving the use of well-characterized standard fluorescent compounds with known QY values. The fluorescence QYs of 12% (for the 418 nm band), 1.3% (for the 460 nm band), 5.38% (for the 503 nm band), and 0.23% (for the 562 nm band) were obtained relative to the standard fluorescent compounds such as 2-aminopurine (2AP), 4′,6-diamidino-2-phenylindole (DAPI), Hoechst 33258 and Rhodamine B (RhB), respectively. Thus, the emergence of multicolor fluorescence in Co3O4 NCs was induced by tartrate ligand functionalization, the mystery of which can be solved by ligand field theory. The observed absorption peaks at around 285 nm and 320 nm, for functionalized Co3O4 NCs, which give rise to photoluminescence at 418 nm can be attributed to the LMCT involving HOMO (highest occupied molecular orbital) of the tartrate ligand and LUMO (lowest unoccupied molecular orbital) centered over the metal ion Co2+/3+ on the NCs' surface.26,27 Additionally, three photoluminescence peaks with maxima at 460, 503, and 562 nm, against excitation at 355, 410, and 520 nm, can arise due to the possible d–d transitions involving Co2+/3+ ions in the functionalized Co3O4 NCs' surface (the presence of both Co2+ and Co3+ before and after functionalization of Co3O4 NCs is confirmed by the XPS, i.e., X-ray photoelectron spectroscopy, study as shown in Fig. S3 in the ESI†). We can explain the generation of the three excitation bands (375, 410, and 510 nm as shown in the excitation spectra in Fig. 3b), in terms of spectroscopic term symbols, due to transitions of 3T1g(F) → 3A2g(F), 3T1g(F) → 3T1g(P), and 3T1g(F) → 3T2g(F), respectively, where these energy levels are obtained from the Tanabe Sugano diagram of Co2+.26 Additional contributions to the emission peaks with maxima at 460 nm and 503 nm may also come from the d–d transitions (1A1g → 1T2g and 1A1g → 1T1g, respectively) involving the energy levels of Co3+, as obtained from Tanabe Sugano diagram.26 Among the d–d transitions, except for the peak at around 370 nm, other excitation bands at around 410 nm and 520 nm were not observed in the absorption spectrum (Fig. 2c), presumably due to masking by the more intense LMCT bands.
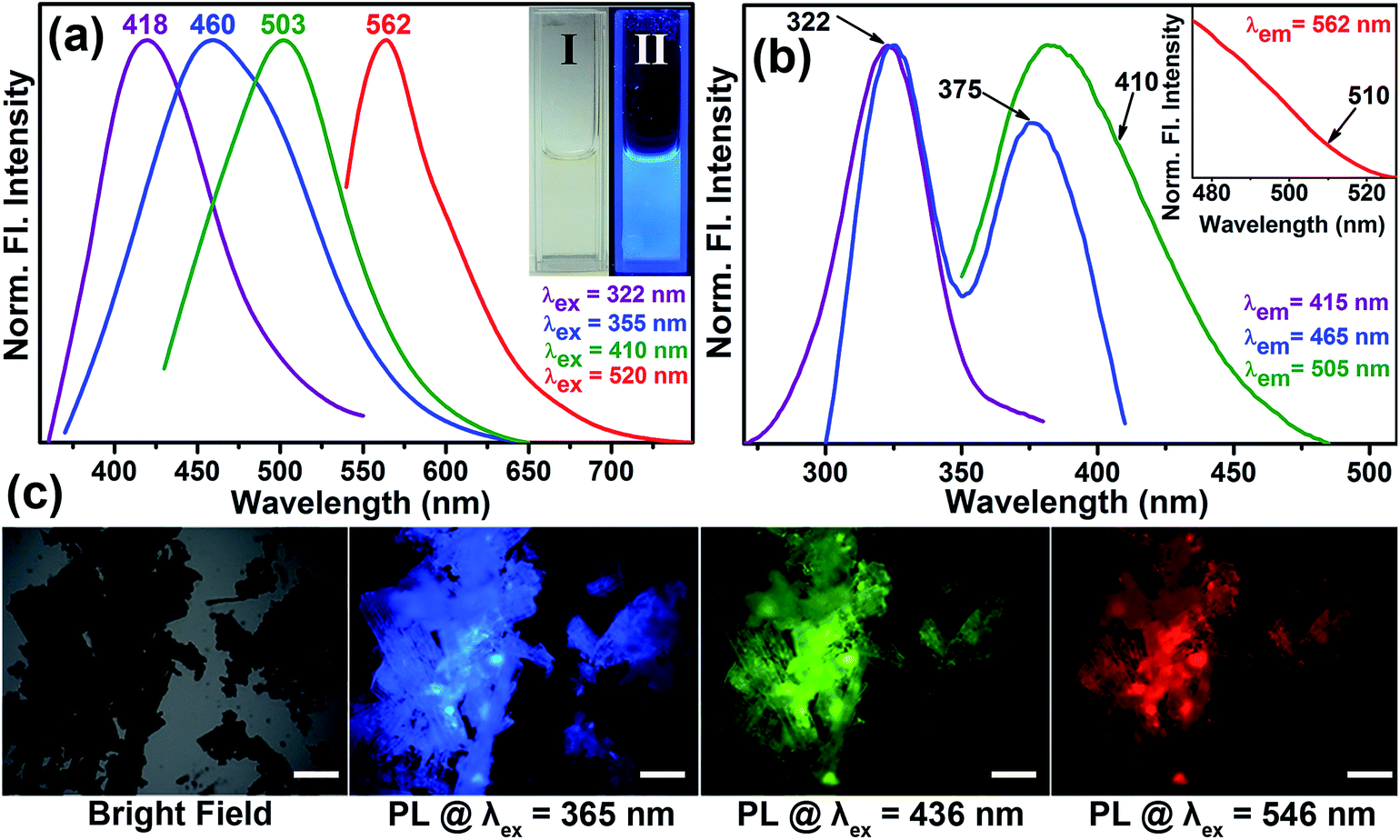 | ||
| Fig. 3 (a) Normalized steady-state fluorescence emission spectra collected from the surface-modified Co3O4 NCs with four different excitation wavelengths of 322, 355, 410, and 520 nm. Image I and II in the inset show the digital image of the aqueous surface-modified Co3O4 NCs solution under visible and UV light, respectively. (b) Fluorescence excitation spectra of surface-modified Co3O4 NCs at different emission maximum of 415, 465, 505, and 562 (inset) nm. (c) Fluorescence micrographs of surface-modified Co3O4 NCs powder under bright field, UV (365 nm), blue (436 nm) and green (546 nm) light irradiation. The scale bars in all the images are 500 μm. | ||
To obtain further reliable evidence for our proposed mechanism for the appearance of multiple photoluminescence, we performed picosecond-resolved fluorescence decay transient measurements of the functionalized Co3O4 NCs employing the TCSPC (time-correlated single-photon counting) technique. Fig. 4a represents the time-resolved fluorescence decay transients of functionalized Co3O4 NCs at two different fluorescence maxima of 410 nm and 460 nm using two different pulsed diode laser excitation sources of 294 nm and 377 nm wavelengths, respectively. As evident from the figure, a significantly larger average excited-state lifetime (τav) of the functionalized Co3O4 NCs is observed for the 410 nm fluorescence band (2.81 ns) compared to that for 460 nm fluorescence band (0.79 ns), strongly suggesting a mechanistic difference in the origin of these two fluorescence peaks. The lifetime components and their corresponding weight percentages are given in Table 1. With this photoluminescence lifetime study, proposition with regard to the origin of this multiple fluorescence made from the steady state experiments are verified, and we can thus rationally conclude that the LMCT excited state is responsible for the 410 nm photoluminescence, while the 460 nm photoluminescence is attributed to d–d transitions. Moreover, we can safely state that the other two lower energy photoluminescence at 503 nm and 562 nm also correspond to other plausible d–d transitions.
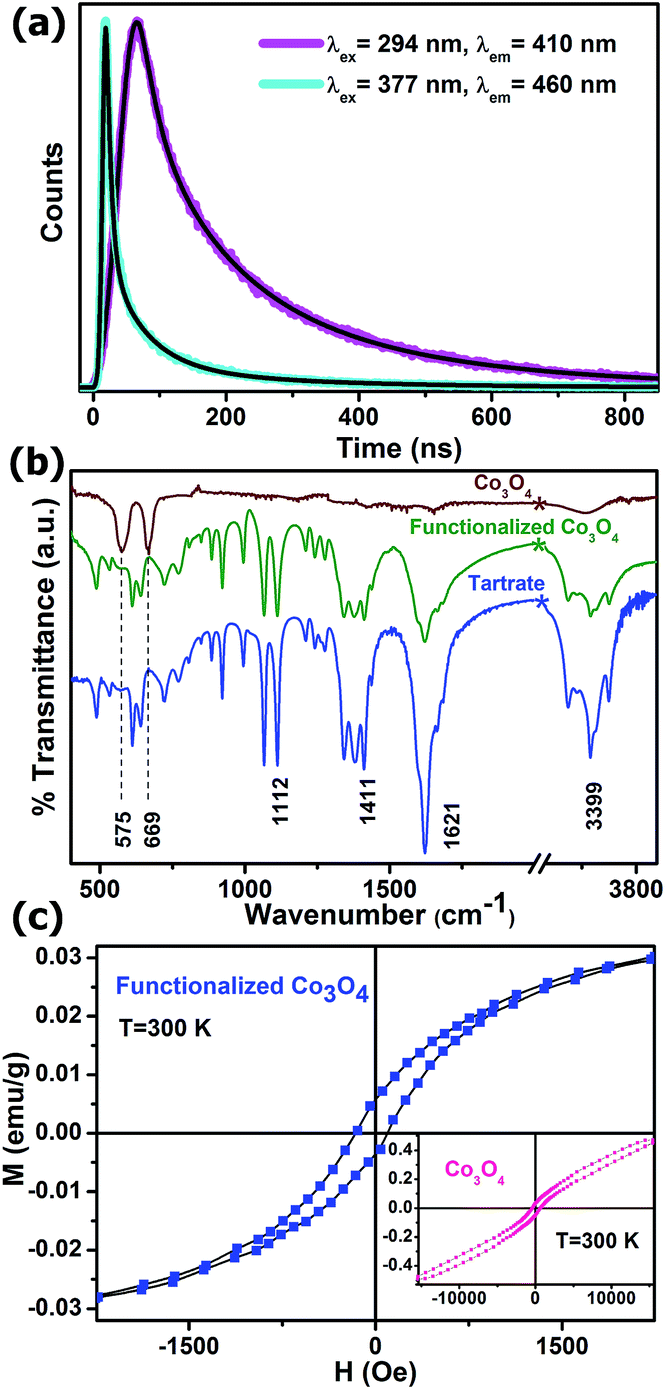 | ||
| Fig. 4 (a) Picosecond-resolved fluorescence transients of functionalized Co3O4 NCs studied at emission wavelengths of 410 nm and 460 nm upon excitation with laser sources of 294 nm and 377 nm wavelengths, respectively. (b) FTIR spectra of as-prepared Co3O4 NCs and functionalized Co3O4 NCs along with only Na-tartrate. (c) Plot of magnetization versus applied magnetic field (M–H) for functionalized Co3O4 NCs at 300 K. Inset shows an M–H plot for the as-prepared Co3O4 NCs at 300 K. | ||
| System | Excitation wavelength, λex (nm) | Fluorescence peak, λem (nm) | τ1 (ns) | τ2 (ns) | τ3 (ns) | τav (ns) |
|---|---|---|---|---|---|---|
| Functionalized Co3O4 NCs | 294 | 410 | 3.27 (29.1) | 8.55 (18.4) | 0.55 (52.5) | 2.81 |
| 377 | 460 | 1.73 (19.04) | 6.9 (3.76) | 0.26 (77.2) | 0.79 |
In order to affirm the attachment of tartrate ligands to the NCs' surface, a comparative FTIR spectroscopic study was carried out on bare and tartrate-functionalized Co3O4 NCs, along with Na-tartrate alone (Fig. 4b). Co3O4 NCs show two strong bands at 575 cm−1 (stretching vibration of (Co3+)–O–(Co3+)3) and 669 cm−1 (stretching vibration of (Co2+)–(Co3+)–O3),28 which disappear after functionalization. In the case of tartrate, the two sharp peaks arising at 1066 cm−1 and 1112 cm−1 are due to the C–OH stretching modes,29 and peaks at 1412 cm−1 and 1622 cm−1 are associated with the symmetric and asymmetric stretching modes of the carboxylate groups (COO−) of tartrate, respectively.30 Upon interaction with the nanoparticle surface, i.e., in the case of functionalized Co3O4 NCs, all these different bands of tartrate are perturbed notably, accompanied with the band at 3400 cm−1, which corresponds to the stretching vibrational modes of hydroxyl group (O–H),29 clearly proving the involvement of both –COO− and –OH groups in the functionalization process.
Fig. 4c shows the magnetic study of both the as-prepared and functionalized Co3O4 NCs at room temperature. The inset shows the plot of magnetization versus the applied magnetic field for the as-prepared Co3O4 NCs, which is approximately linear with a fine hysteresis loop with a coercivity of 532 Oe and a remanent magnetization of 0.04 emu g−1; however, the maximum field applied (15 kOe) cannot saturate the magnetization, and the magnetization goes up to 0.47 emu g−1. So, it is evident that the as-prepared Co3O4 NCs show antiferromagnetic behavior with very weak ferromagnetic nature. In spite of the antiferromagnetic behavior arising due to super exchange among each Co2+ ion in the tetrahedral-site and its four neighboring Co2+ ions of opposite spins in case of bulk Co3O4,31 the very weak ferromagnetic behavior of the as-prepared NCs can be attributed to the finite size effects leading to uncompensated surface spins.32 Moreover, the ferromagnetic nature becomes more prominent after functionalization with Na-tartrate, which can be ascribed to the ligand-mediated strong pinning of the uncompensated spins of the surface Co2+ ions. However, the coercivity decreases significantly to 130 Oe, along with the decrease in the saturation magnetization to 0.03 emu g−1. This phenomenon can be explained by ligand field theory. The tartrate ligand containing both the σ-donor (–OH) and π-donor (–COO−) functional groups,33 favors quenching of the magnetic moments of Co2+ ions on the surface of functionalized Co3O4 NCs, leading to a decrease in the saturation magnetization.34 Again, quenching of the magnetic moments reduces its spin–orbit coupling, resulting in a diminution of the magnetocrystalline anisotropy, which causes a reduction in the coercivity35 in the case of functionalized Co3O4 NCs as compared to the as-prepared NCs.
Considering recent significant progress in nanocatalysis through rational engineering and the manipulation of various materials at the nanoscale to accelerate the rate of different beneficial reactions with direct biological and environmental significance, we aimed to utilize the functionalized Co3O4 NCs in the degradation of the biologically relevant organic dye, bilirubin (BR), the pigment responsible for the yellow coloration of the skin in jaundice. Fig. 5a shows the degradation of BR over time, in the presence of functionalized Co3O4 NCs at pH ∼7, which was found to follow the first order rate equation with a kinetics rate constant (k) of 2.2 × 10−2 min−1. We also verified the reusability of the catalyst, in the degradation of BR, by adding the same amount (which was added 1st time to the reaction mixture, i.e., 7.8 μM) of BR to the reaction mixture every 100 min for up to five doses, keeping the catalyst concentration fixed (adding only once at the 1st cycle), and measured the BR decomposition rate of the different cycles by monitoring the decrease of BR absorbance at 440 nm using UV-visible spectroscopy. As shown in Fig. 5b, the plot of the relative concentration of BR versus time, up to five consecutive cycles, confirms the reusability of the functionalized Co3O4 NCs catalyst, with an almost consistent degradation rate. Notably, as shown in Fig. 5a, an isosbestic point was obtained at ∼370 nm due to the increase in the absorbance at 319 nm with a simultaneous decrease in the absorbance at 440 nm over time. According to the reported literature on BR, the peak at 319 nm arises due to the absorption of methylvinylmaleimide (MVM), a photo-oxidation product of BR in aqueous medium.36 When we carried out the reaction in the presence of a radical quencher, EtOH, (as shown in Fig. 5c) the rate of the reaction decreased significantly, with a rate constant (k) of 7.86 × 10−3 min−1, which indicates that the reaction follows a radical pathway. Performing the reaction in the presence of the oxidizing agent H2O2, (as shown in Fig. 5c), we also observed a decrease in the rate of reaction (k = 1.93 × 10−2 min−1), which can be explained by combining the redox and radical pathway. The production of the oxidized product, MVM, reveals that a reduction procedure, i.e., conversion of Co3+ to Co2+ must have happened during the reaction. However, in the presence of the strong oxidizing agent H2O2, the reduction process (acceptance of a single electron from BR by Co3+ for the conversion to Co2+) via a radical pathway was overcompensated by the oxidation of Co2+ to Co3+, leading to an overall slowing down of the reaction. This reveals that functionalized Co3O4 NCs-assisted BR degradation takes place via a radical pathway involving the reduction of Co3+ to Co2+.
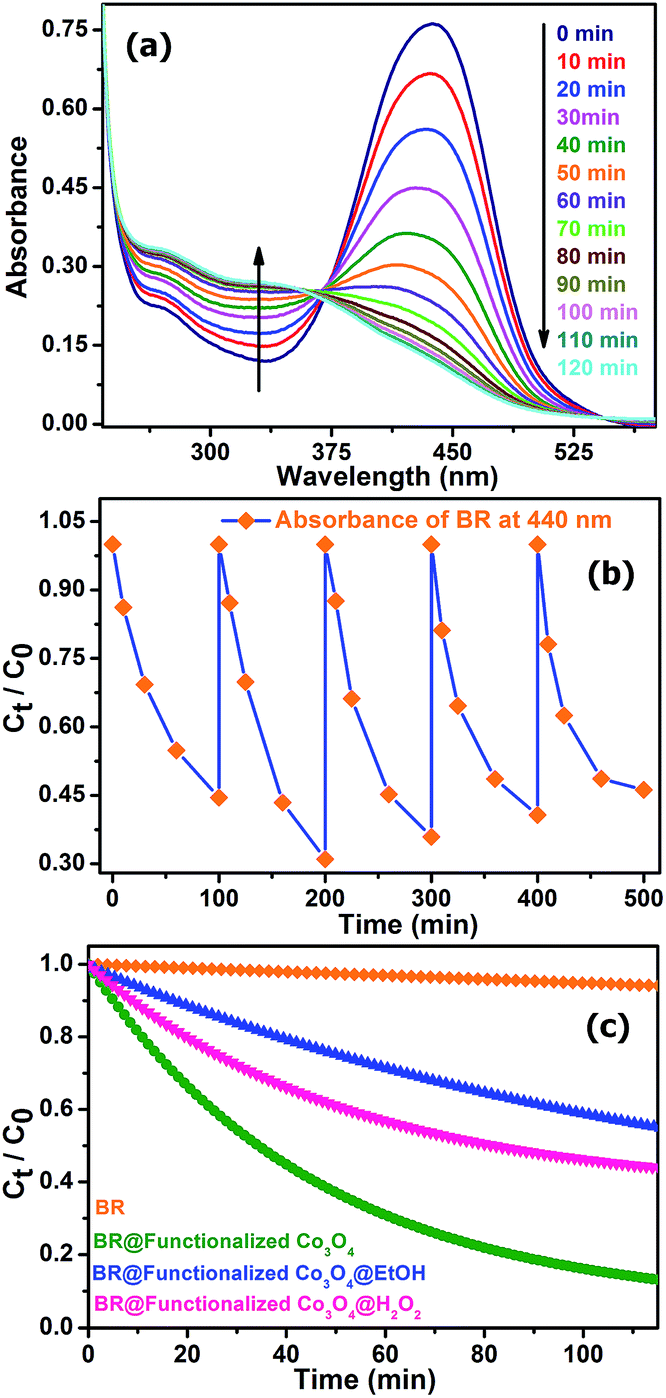 | ||
| Fig. 5 (a) UV-visible spectral changes of an aqueous solution of bilirubin (BR) in the presence of functionalized Co3O4 NCs (at pH ∼7) over time. (b) Plot of the relative concentration of BR monitored at 440 nm versus time for consecutive five cycles, showing the reusability of the functionalized Co3O4 NCs in BR degradation. (c) The comparative rate of degradation of BR (monitored at 440 nm) alone and in the presence of functionalized Co3O4 NCs, functionalized Co3O4 NCs@EtOH, and functionalized Co3O4 NCs@H2O2. | ||
Furthermore, inspired by the significant efficiency of functionalized Co3O4 NCs in the degradation of biologically harmful pigments, we attempted to utilize the strong excitation of the functionalized Co3O4 NCs in the UV region in photocatalysis for wastewater treatment. Functionalized Co3O4 NCs showed excellent photocatalytic property (as shown in Fig. 6a) toward the degradation of methylene blue, a model water-contaminant and commonly used dye in textile industries, upon UV light irradiation. We found that the functionalized Co3O4 NCs catalyzed photodegradation of MB takes place exponentially with time, following a first-order rate equation with a kinetic rate constant (k) of 2.23 × 10−2 min−1. To check the reusability of the catalyst, we added the same dose of MB (5 μM) into the reaction mixture for up to five doses, keeping the catalyst concentration fixed (i.e., without the addition of extra catalyst after the 1st cycle). The MB decomposition rates of different cycles were monitored by measuring the decrease of MB absorbance at 660 nm with UV-visible spectroscopy. Fig. 6b demonstrates the plots of the relative concentration of MB versus time, for up to five consecutive cycles, indicating the reusability of the functionalized Co3O4 NCs catalyst with a consistent degradation rate. Having evidence from several other photochemical reactions, we propose that the photodegradation process may follow a reactive oxygen species (ROS)-mediated radical pathway.
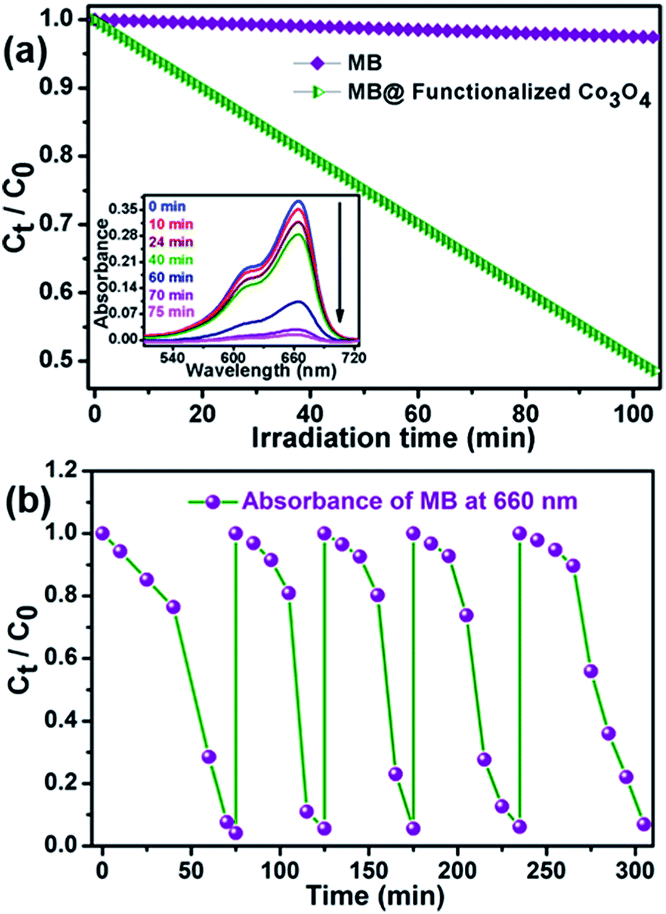 | ||
| Fig. 6 (a) The plot of the relative concentration of MB monitored at 660 nm versus time with and without functionalized Co3O4 NCs and inset shows the UV-visible spectral changes of aqueous solution of methylene blue (MB) in the presence of functionalized Co3O4 NCs over time under UV irradiation. (b) The plot of the relative concentration of MB monitored at 660 nm versus time for consecutive five cycles showing the reusability of the functionalized Co3O4 NCs in MB degradation under UV light. | ||
Conclusions
In summary, we have demonstrated an easy fabrication procedure for multifunctional Co3O4 NCs. By facile functionalization of as-prepared NCs with Na-tartrate ligand, we have been able to develop intrinsic multicolor fluorescence covering almost the entire visible region. Systematic investigations through UV-visible absorption, steady state, and time-resolved photoluminescence studies reveal that the LMCT transition from the tartrate ligand to the lowest unoccupied energy level of Co2+/3+ of the NCs and d–d transitions centered over Co2+/3+ ions in the NCs play the important role in the generation of multiple fluorescence from the ligand-functionalized Co3O4 NCs. A magnetic study exhibited that the antiferromagnetic nature of Co3O4 NCs turns to ferromagnetic after surface modification. The newly developed magnetofluorescent Co3O4 NCs can open up a new horizon in the field of bioimaging and drug delivery. The functionalized Co3O4 NCs also show good catalytic efficiency for the degradation of BR, a biologically harmful pigment, which reveals their developing therapeutic application in severe hyperbilirubinemia. Moreover, their good photocatalytic efficiency in the degradation of MB, a model water contaminant, may give rise to their potential application in wastewater treatment.Acknowledgements
We thank DST for financial grants. Authors would like to thank Mr Mahesh Agarwal and Dr Deepak Kumar Sinha from IACS, Kolkata for providing fluorescence microscopy facility used in this study. Authors would like to thank Mr Sukanta Barman and Dr Krishnakumar S. R. Menon from SINP, Kolkata for providing XPS facility used in this study. We thank Mr Samik Roy Moulik for performing TEM measurements.References
- F. Yang, Y. Li, Z. Chen, Y. Zhang, J. Wu and N. Gu, Superparamagnetic Iron Oxide Nanoparticle-Embedded Encapsulated Microbubbles as Dual Contrast Agents of Magnetic Resonance and Ultrasound Imaging, Biomaterials, 2009, 30(23–24), 3882–3890 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Design and Fabrication of Magnetic Nanoparticles for Targeted Drug Delivery and Imaging, Adv. Drug Delivery Rev., 2010, 62(3), 284–304 CrossRef CAS PubMed.
- M. Tamura, F. Yanagawa, S. Sugiura, T. Takagi, K. Sumaru, H. Matsui and T. Kanamori, Optical Cell Separation from Three-Dimensional Environment in Photodegradable Hydrogels for Pure Culture Techniques, Sci. Rep., 2014, 4, 4793 Search PubMed.
- N. Tran and T. J. Webster, Nanotechnology for Bone Materials, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1(3), 336–351 CrossRef CAS PubMed.
- M. Johannsen, U. Gneveckow, L. Eckelt, A. Feussner, N. Waldofner, R. Scholz, S. Deger, P. Wust, S. A. Loening and A. Jordan, Clinical Hyperthermia of Prostate Cancer Using Magnetic Nanoparticles: Presentation of a New Interstitial Technique, Int. J. Hyperthermia, 2005, 21(7), 637–647 CrossRef CAS.
- N. L. Stock, J. Peller, K. Vinodgopal and P. V. Kamat, Combinative Sonolysis and Photocatalysis for Textile Dye Degradation, Environ. Sci. Technol., 2000, 34(9), 1747–1750 CrossRef CAS.
- A. Giri, N. Goswami, M. Pal, M. T. Zar Myint, S. Al-Harthi, A. Singha, B. Ghosh, J. Dutta and S. K. Pal, Rational surface modification of Mn3O4 nanoparticles to induce multiple photoluminescence and room temperature ferromagnetism, J. Mater. Chem. C, 2013, 1(9), 1885–1895 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Nano-Magnetite (Fe3O4) as a Support for Recyclable Catalysts in the Development of Sustainable Methodologies, Chem. Soc. Rev., 2013, 42(8), 3371–3393 RSC.
- M. B. Gawande, V. D. B. Bonifacio, R. S. Varma, I. D. Nogueira, N. Bundaleski, C. A. A. Ghumman, O. M. N. D. Teodoro and P. S. Branco, Magnetically Recyclable Magnetite-Ceria (Nanocat-Fe-Ce) Nanocatalyst-Applications in Multicomponent Reactions Under Benign Conditions, Green Chem., 2013, 15(5), 1226–1231 RSC.
- M. Pal, R. Rakshit and K. Mandal, Surface Modification of MnFe2O4 Nanoparticles to Impart Intrinsic Multiple Fluorescence and Novel Photocatalytic Properties, ACS Appl. Mater. Interfaces, 2014, 6(7), 4903–4910 CAS.
- A. Giri, N. Goswami, C. Sasmal, N. Polley, D. Majumdar, S. Sarkar, S. N. Bandyopadhyay, A. Singha and S. K. Pal, Unprecedented catalytic activity of Mn3O4 nanoparticles: potential lead of a sustainable therapeutic agent for hyperbilirubinemia, RSC Adv., 2014, 4(10), 5075–5079 RSC.
- J. Mu, Y. Wang, M. Zhao and L. Zhang, Intrinsic peroxidase-like activity and catalase-like activity of Co3O4 nanoparticles, Chem. Commun., 2012, 48(19), 2540–2542 RSC.
- Z. Wen, L. Zhu, W. Mei, Y. Li, L. Hu, L. Sun, W. Wan and Z. Ye, A Facile Fluorine-Mediated Hydrothermal Route to Controlled Synthesis of Rhombus-Shaped Co3O4 Nanorod Arrays and their Application in Gas Sensing, J. Mater. Chem. A, 2013, 1(25), 7511–7518 CAS.
- L. Hu, N. Yan, Q. Chen, P. Zhang, H. Zhong, X. Zheng, Y. Li and X. Hu, Fabrication Based on the Kirkendall Effect of Co3O4 Porous Nanocages with Extraordinarily High Capacity for Lithium Storage, Chem.–Eur. J., 2012, 18(29), 8971–8977 CrossRef CAS PubMed.
- H.-W. Shim, A.-H. Lim, J.-C. Kim, E. Jang, S.-D. Seo, G.-H. Lee, T. D. Kim and D.-W. Kim, Scalable one-pot bacteria-templating synthesis route toward hierarchical, porous-Co3O4 superstructures for supercapacitor electrodes, Sci. Rep., 2013, 3, 2325 Search PubMed.
- S. Xiong, C. Yuan, X. Zhang, B. Xi and Y. Qian, Controllable Synthesis of Mesoporous Co3O4 Nanostructures with Tunable Morphology for Application in Supercapacitors, Chem.–Eur. J., 2009, 15(21), 5320–5326 CrossRef CAS PubMed.
- Y. Xiao, S. Liu, F. Li, A. Zhang, J. Zhao, S. Fang and D. Jia, hierarchical nanoarchitectures: 3D hierarchical Co3O4 twin-spheres with an urchin-like structure: large-scale synthesis, multistep-splitting growth, and electrochemical pseudocapacitors, Adv. Funct. Mater., 2012, 22(19), 4051 CrossRef.
- G. Wang, X. Shen, J. Horvat, B. Wang, H. Liu, D. Wexler and J. Yao, Hydrothermal Synthesis and Optical, Magnetic, and Supercapacitance Properties of Nanoporous Cobalt Oxide Nanorods, J. Phys. Chem. C, 2009, 113(11), 4357–4361 CAS.
- H. Pang, F. Gao, Q. Chen, R. Liu and Q. Lu, Dendrite-like Co3O4 nanostructure and its applications in sensors, supercapacitors and catalysis, Dalton Trans., 2012, 41(19), 5862–5868 RSC.
- J. Mu, L. Zhang, M. Zhao and Y. Wang, Catalase Mimic Property of Co3O4 Nanomaterials with Different Morphology and its Application as a Calcium Sensor, ACS Appl. Mater. Interfaces, 2014, 6(10), 7090–7098 CAS.
- J. Kou, C. Bennett-Stamper and R. S. Varma, Hierarchically Triangular Prism Structured Co3O4: Self-Supported Fabrication and Photocatalytic Property, Nanoscale, 2011, 3(12), 4958–4961 RSC.
- E. Papis, F. Rossi, M. Raspanti, I. Dalle-Donne, G. Colombo, A. Milzani, G. Bernardini and R. Gornati, Engineered Cobalt Oxide Nanoparticles Readily Enter Cells, Toxicol. Lett., 2009, 189(3), 253–259 CrossRef CAS PubMed.
- J. Zhao, Z. Huang, J. Zeng, M. Deng, G. Yin, X. Liao and J. Gu, Histidine-Assisted Synthesis and Cellular Compatibility of Magnetic Cobalt Oxide Nanoparticles at Room Temperature, J. Inorg. Organomet. Polym. Mater., 2012, 22(2), 492–499 CrossRef CAS.
- Y. Dong, K. He, L. Yin and A. Zhang, A Facile Route to Controlled Synthesis of Co3O4 Nanoparticles and their Environmental Catalytic Properties, Nanotechnology, 2007, 18(43), 435602 CrossRef.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Relative fluorescence quantum yields using a computer-controlled luminescence spectrometer, Analyst, 1983, 108(1290), 1067–1071 RSC.
- J. E. Huheey, E. A. Keiter, R. L. Keiter and O. K. Medhi, Inorganic Chemistry: Principles of Structure and Reactivity, Pearson Education, 2006 Search PubMed.
- A. Giri, N. Goswami, M. S. Bootharaju, P. L. Xavier, R. John, N. T. K. Thanh, T. Pradeep, B. Ghosh, A. K. Raychaudhuri and S. K. Pal, Emergence of Multicolor Photoluminescence in La0.67Sr0.33MnO3 Nanoparticles, J. Phys. Chem. C, 2012, 116(48), 25623–25629 CAS.
- A. Agiral, H. S. Soo and H. Frei, Visible Light Induced Hole Transport from Sensitizer to Co3O4 Water Oxidation Catalyst across Nanoscale Silica Barrier with Embedded Molecular Wires, Chem. Mater., 2013, 25(11), 2264–2273 CrossRef CAS.
- N. Kaneko, M. Kaneko and H. Takahashi, Infrared and Raman Spectra and Vibrational Assignment of Some Metal Tartrates, Spectrochim. Acta, Part A, 1984, 40(1), 33–42 CrossRef.
- V. Ramakrishnan and J. M. T. Maroor, IR and Raman Studies of Gel Grown Manganese Tartrate, Infrared Phys., 1988, 28(4), 201–204 CrossRef CAS.
- P. Dutta, M. S. Seehra, S. Thota and J. Kumar, A Comparative Study of the Magnetic Properties of Bulk and Nanocrystalline Co3O4, J. Phys.: Condens. Matter, 2008, 20(1), 015218 CrossRef.
- T. Ozkaya, A. Baykal, M. S. Toprak, Y. Koseoğlu and Z. Durmuş, Reflux Synthesis of Co3O4 Nanoparticles and its Magnetic Characterization, J. Magn. Magn. Mater., 2009, 321(14), 2145–2149 CrossRef CAS PubMed.
- M. Pal, R. Rakshit and K. Mandal, Facile functionalization of Fe2O3 nanoparticles to induce inherent photoluminescence and excellent photocatalytic activity, Appl. Phys. Lett., 2014, 104(23), 233110 CrossRef PubMed.
- R. Rakshit, M. Mandal, M. Pal and K. Mandal, Tuning of Magnetic Properties of CoFe2O4 Nanoparticles through Charge Transfer Effect, Appl. Phys. Lett., 2014, 104(9), 092412 CrossRef PubMed.
- C. R. Vestal and Z. J. Zhang, Effects of Surface Coordination Chemistry on the Magnetic Properties of MnFe2O4 Spinel Ferrite Nanoparticles, J. Am. Chem. Soc., 2003, 125(32), 9828–9833 CrossRef CAS PubMed.
- W. E. Kurtin, Spectroscopy and Photochemistry of Bilirubin Photoproducts. I. Methylvinylmaleimide*, Photochem. Photobiol., 1978, 27(5), 503–509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section containing material used, detailed synthesis procedure and functionalization of Co3O4 NCs, characterization techniques, and fluorescence micrographs of bare Co3O4 NCs under bright field, UV, blue, and green light irradiation. See DOI: 10.1039/c4ra12901f |
| This journal is © The Royal Society of Chemistry 2015 |