
Pot-in-pot reactions: a simple and green approach to efficient organic synthesis
M. B. J. Atkinsond,
S. Oyola-Reynosoa,
R. E. Lunab,
D. K. Bwambokc and
M. M. Thuo*a
aDepartment of Materials Science and Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: mthuo@iastate.edu; Tel: +1-515-294-8581
bDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA
cWarner Babcock Institute for Green Chemistry, Wilmington, MA 01887, USA
dDepartment of Chemistry, University of Massachusetts Boston, MA 02125, USA
First published on 25th November 2014
Abstract
Incompatible organic reactions impede efficient green synthesis by making multi-component or cascade reactions a big challenge. This review highlights pot-in-pot reactions (multiple reactions carried out in one pot by separating key reactions with a thin polymeric membrane) as an efficient, green synthetic alternative to conventional synthesis. We discuss the advantages of homogeneous processes to develop new cascade reaction sequences by reviewing the use of polymeric thimbles as selective semi-permeable walls. These thimbles allow small organic molecules to diffuse through while retaining polar reagents, polar solvents, and/or organometallic catalysts. The dynamic and versatile nature of this technique is demonstrated by performing 2- and 3-step cascade reactions in one glass pot. A pot-in-pot reaction approach to synthesis circumvents the need to isolate intermediates, or handling of toxic/unpleasant by-products, therefore enabling synthesis of otherwise challenging molecules, improving the efficiency, or enabling greener approaches to modular synthesis.
1. Introduction
The ability to perform sequential/tandem reactions provides significant benefits to the chemical world.1–10 Nature is a constant source of inspiration in chemical synthesis due to its ability to perform extremely specific reactions with superb efficiency. For example, by isolating enzymes within individual organelles, the design of the cell allows for reactions to be compartmentalized, which eliminates potential catalyst poisoning, cross-reactivity, and/or any other types of inter-reaction interferences. While chemists do not have the requisite millions of years that it has taken nature to develop these efficient reactions, they use it as an inspiration to develop new methods for efficient and resource-sparing synthesis.11–31 Developments in this area are extremely belated considering the growth of the chemical industry – particularly in the synthesis of pharmaceutical compounds where 25–100 kg of waste is generated on average for every 1 kg of active pharmaceutical product.32 Waste management in the chemical industry is also a pressing issue. In 2005, for example, the U.S. Environmental Protection Agency reported that 25.1 billion pounds of production related chemical waste was generated in the United States and only 8.96 billion pounds (36%) of that waste was recycled.33 More recently, in 2012, a comparable proportion (35% of 23.5 billion pounds) was recycled.34 Development of new efficient and recycling technologies is vital to decreasing this waste and the negative impact of chemical synthesis on the environment. An alternative to waste management is the application of the 12 principles of green chemistry and/or engineer which would mitigate resource wastage, lower carbon footprint, while improving reaction efficiency.Current tools to achieve such a goal are limited by reagents and/or catalysts' compatibility. Recent advances in site-isolation of catalysts have provided few platforms geared towards this end.35–57 Notably, catalysts and reagents can be site-isolated from each other by microencapsulation, covalent attachment to a macromolecular ensemble or crystals, occlusion in an organic or inorganic matrix, entrapment in a coordination polymer or a single crystal assembly, and sometimes by heterogenization.6,58–78 Whereas each of these techniques offers a new arsenal to the organic chemist, most methods require chemical transformation of the catalyst. The materials used for site isolation may also be limited in their lifetime offering only temporal protection. Some techniques require a change in the reaction medium, thereby losing the benefits of already mature homogeneous catalyst and/or reagent.
In order to develop an efficient method for site isolation of catalysts and reagents, it is necessary to use a process that allows one-way selective flux – uni-direction transport of reaction products. Thermodynamic considerations, however, limit the existence of such a spontaneous system. A chemical potential driven molecular machine can, however, be conceptualized. This review summarizes such a system using thin polymeric membranes in the shape of thimbles that were used in “pot-in-pot” reaction sequences.
1.1 “Pot-in-pot reactions” as a method for site isolation
“Pot-in-pot reactions” is a term coined to describe a method of site-isolating catalysts and/or reagent(s) using a semi-permeable thimble(s) (pot 1) inside a glass flask or a second semi-permeable membrane (pot 2) as illustrated in Fig. 1. These pots need to be independently accessible without necessarily interfering with the progress of the reaction(s) to allow for real-time independent monitoring of reaction progress – otherwise, the system is no longer a ‘pot-in-pot’. In these reactions, one catalyst/reagent is kept dissolved inside a polymeric thimble using a non-permeating solvent(s) while small organic molecules flux across the polymeric walls and can be converted to another molecule(s) on the exterior of the thimbles. Several materials can be employed as semi-permeable membranes such as demonstrated here and in the “tea-bag” approach.79,80 Appropriate materials include ceramics, cellulose, and polymeric films.79,80 Pot-in-pot reactions make use of polydimethylsiloxane because it is easy to fabricate into various shapes and sizes without affecting its flux selectivity. Polydimethylsiloxane (PDMS), and similar polymers, exhibits high flux rates for small molecules and have previously been used in separating small organic molecules.25,81,82 By making the reaction vessel, i.e. the ‘pot’, from an active membrane, the diversity of solvents that can be used without interfering with downstream reactions in a cascade reaction sequence is greatly increased. | ||
| Fig. 1 Schematic illustrations of; (a) a generalized pot-in-pot reaction set-up, and, (b) rationalization of the application of thin polymer films as a active separation membranes based on flux of small molecules across them. Felicitous choice of solvents makes the flux uni-directional due to asymmetry in partition coefficients and differences in diffusion constant(s). | ||
Pot-in-pot reactions offer several advantages that make them appealing as a general reaction strategy. These advantages include; (i) enabling chemists to assess the progress of the reaction without interfering with, or stopping, the reaction for analysis – in multi-component and other cascade reactions, it is impossible to analyse a single reaction independently since involved steps are not truncated. (ii) Availability of ‘knobs’ (tuneable variables) for each reaction that are accessible through pot-in-pot, these include; running reactions under different reaction conditions (temperature, pressure, concentrations), use of different solvents, additives, and, reaction times per reaction. These flexibilities, albeit subtle in conventional reaction design, translates to increased capabilities in terms of yield, efficiency, and, adaptability. (iii) Pot-in-pot reactions allow for the addition of reagents to the reaction as well as removal of product or samples at any given time. (iv) Pot-in-pot reactions circumvent the need to isolate the product between each step, which reduces waste while increasing efficiency – making this a green alternative to conventional synthesis. (v) Manipulation of the permeability of the polymeric thimbles (inner pot) also allows for the flux of different sizes of molecules so that the products of the first reaction become the substrate (reagent) of the next reaction outside the thimble, but still within the same glass flask (outer pot). (vi) Since the catalyst is kept dissolved at all times, it remains active and can be re-used over prolonged periods of time. The use of a relatively larger amount of solvent on the exterior of the thimble ensures that the concentration of the target molecule is always lower on the exterior hence favouring one-way flux of the diffusant. A second catalyst or reagent can then be introduced on the outside of the thimble to allow for a second chemical transformation. This sequence can potentially continue Ad infinum.
In this review, we illustrate the usefulness of these reactions by showing site isolation of; (i) a reagent from a reagent, (ii) catalyst from a reagent, (iii) catalyst from a catalyst, and, (iv) recycling over many reaction cycles. We end the review by showing one-pot synthesis of molecules that have been documented to be otherwise challenging. For brevity, the last section illustrates the utility of pot-in-pot reactions by taking two known molecules and synthesizing them in better yield, less wastage, and, higher efficiency. This article concludes with an overview/discussion of why the reactions are successful and the rationale behind their success.
1.2 Polydimethylsiloxane (PDMS) as a suitable material for efficient site-isolation of reagents and catalysts
Polymers have found a variety of applications in site-isolation, providing avenues for both covalent and non-covalent attachment of catalysts or ligands to a support.65,66,83–98 While these polymer-based methods offer distinct advantages, none of them has previously site-isolated an entire reaction process, while granting access to the reaction as it progresses. Site-isolation using polymeric materials can thus be classified into two categories: (i) bulk dominated (diffusion coefficient, KD, is the selectivity determining factor): diffusion-controlled processes in which one or more components diffuse through the polymer more rapidly making the polymer an “active” membrane. This process is sensitive to size and functional groups on the molecules. (ii) Surface dominated (partition coefficient, KP, is the selectivity determining factor): partition controlled processes in which solubility of components, in the immediate solvent vs. in the polymer, is the critical step in site-isolation. In these types of processes, interface properties of the polymer are essential to site-isolation. Enhanced selectivity requires a set of molecules that are soluble in the polymer membrane and a second set which are not soluble. A balance between KD and KP dominated processes would offer the best control of selective flux.PDMS has been widely studied and its solid-state properties are well documented.99,100 PDMS has been used to fabricate micro-fluidic devices, which can be molded into various shapes and/or structures. It is cheap and readily available. PDMS is commercially available as a liquid, which can be readily cross-linked to give a rubbery solid that is firm and tough enough to withstand the rigor of organic reactions (e.g. stirring and heat).
The rate of diffusion of small molecules across thin PDMS membranes has been extensively studied.101–103 Most small, non-ionic molecules have diffusion rates within an order of magnitude of each other. This diffusion is directly proportional to temperature but inversely proportional to the molecular weight of the small molecule. Therefore the bigger the molecule, the slower it will diffuse through a PDMS membrane. The rate of diffusion, however, can be increased by raising the temperature at which the separation is performed making temperature a tuneable variable in selective flux. This is further supported by the classical relation between the rate of diffusion and energy summarized below (eqn (1)).
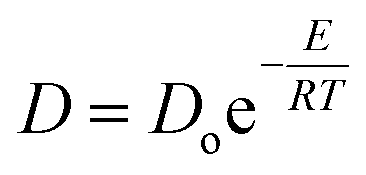 | (1) |
Although diffusion gives a general idea as to how this approach may work, it does not allow us to fully rationalize what might happen without understanding flux of various molecules in PDMS. A more complete picture of what might be anticipated would require an integration of Fick's laws, the Stokes–Einstein relation, and related mass transport laws, a discussion beyond the scope of this review. Understanding flux, moles of a substrate flowing through a defined area of material per unit time, would allow for prediction of how well the isolated catalyst may be slowed relative to a small molecule. The relationship between flux, rate of diffusion and solubility is complicated by the limited amount of data available on the diffusion properties of organometallic compounds in polymeric materials. This relationship is affected by molecular geometries, polarity and electronic effects.93,104–109 A simple way to relate these three vital properties is to generalize flux as a product of solubility and rate of diffusion (analogous to when the diffusant is a gas, P = D·S, where P = permeability coefficient, D = diffusion coefficient, S = solubility coefficient). This simple rationalization has been employed by others to separate organic molecules, like pyridine, from water using PDMS.110–113
Although small non-ionic molecules may have high solubility in PDMS, they may be challenging to use since they also swell the material.103 This can be a challenge in separation science, but careful use of this property may help reduce time needed for reactants to flux for the subsequent reaction down a cascade. Once the PDMS is swollen, the flux of small molecules through it is much higher.29,103
A procedure to fabricate cylindrical containers from PDMS (with even walls) in a quick and reproducible manner was desired. To achieve this, PDMS was cast from stainless steel rods (Fig. 2) by applying 2–3 coats of PDMS on a metal mold and allowing each coat to partially cure before applying the subsequent layer. Following this simple procedure, cylinders with wall thickness of 105 ± 22 μm were readily obtained.29
 | ||
| Fig. 2 PDMS thimbles next to a metal rod from which they were fabricated. A longitudinal and vertical view of two similar thimbles adjacent to a penny for comparison (left). A schematic of the thimbles shows the dimensions (right). | ||
2. Pot-in-pot reactions
In this section, we discuss the development of pot-in-pot reactions by examining different categories of site-isolation, viz.: (i) isolation of reagents from other incompatible reagents or solvents (Section 2.1), (ii) catalyst site-isolation from solvents (Section 2.2), (iii) isolation of catalysts from incompatible reagents (Section 2.3), (iv) isolation of catalysts from other incompatible catalysts (Section 2.4), and, (v) catalyst recycling (Section 2.5).2.1 Site-isolation of reagents from other reagents/solvents
When two immiscible solvents are placed in a container, a biphasic medium is formed (unless they are emulsified). In an analogous manner, when a polymeric membrane separates two immiscible solvents, one or both can diffuse into the polymer, but they cannot flux across the polymer film since the partition coefficient at the polymer membrane – solvent interface is too low for any significant flux to occur. On the other hand, if a solubilized compound in the first solvent is also soluble in the second solvent, this molecule would flux across the membrane. This is the underlying principle behind reagent–reagent or solvent–solvent isolation in a pot-in-pot setup. Polymeric materials and thimbles are ideal for isolating strongly ionic reagents from each other. Two forms of these reactions have been used as a general model to illustrate the versatility of pot-in-pot reactions in cases where the reagents and/or the solvents are incompatible.29 First, acid deprotection of a ketal, in an aqueous medium, is performed inside a thimble (Fig. 3). Upon completion of the deprotection, and subsequent flux to the exterior of the thimble, the generated ketone is readily reduced to the corresponding alcohol using LiAlH4. The success of this reaction sequence depends on the choice of solvent used, with the best solvents being hexanes or a mixture of hexanes/dichloromethane. The use of non-polar solvents in the deprotection step means that a phase transfer agent, in this case sodium dodecyl sulphate (SDS), must be used for the deprotection reaction.28 | ||
| Fig. 3 An illustration of a cascade reaction involving reagent–reagent site-isolation using a PDMS thimble. Deprotection of a ketal occurs in the interior of the thimble, followed by diffusion of the resultant ketone across the thimble wall to react with LiAlH4 in hexanes on the exterior. Copyright© 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.28 | ||
The site-isolation of nucleophilic reagents from acidic aqueous medium was achieved using a thin polymeric barrier and the two reactions gave >80% isolated yields over the two steps. These cascade reactions cannot be achieved otherwise as the reaction media are highly incompatible.28
2.2 Site-isolation of catalysts from solvents
Organometallic compounds, the basis of most homogenous catalysts, have low flux across polymeric membranes but flux well when the membranes are swollen with an appropriate solvent.29,114 To design efficient methods for isolation of organometallic catalyst, the effect of various solvents on catalyst leaching were investigated (using Grubbs' first generation catalyst).115 Whitesides and co-workers examined how PDMS swells when exposed to different solvents.103 Different solvent mixtures that would slightly swell PDMS, while fully dissolving Grubbs' metathesis catalysts, were investigated. Ionic liquids were deemed most ideal for this study, since they do not partition into PDMS but readily dissolve a large variety of compounds and their physico-chemical properties can be tuned as needed.Ionic liquids were chosen for site-isolation of the catalyst for four reasons: (1) they dissolve organometallic catalysts but they do not flux through PDMS;31,114,121,124–126 therefore, a mixture of ionic liquids and medium polarity solvents was most appealing. (2) Ionic liquids do not poison many catalysts.127,128 (3) They can mitigate partitioning of a catalyst into the polymer membrane but promote partition of non-polar or medium-polarity small molecules, i.e. products and reagents (due to their ionic nature). (4) They can be recycled with the catalyst.129–131
Having investigated the utility of ionic liquids for use as additional solvents for site-isolation, the following parameters were investigated: (i) the degree of swelling, which can be controlled via careful choice of solvent(s) without affecting the reactivity of the catalyst. (ii) Effect of the thickness of walls of the thimble which would affect flux. (iii) The reaction conditions including the effect of different solvent mixtures, catalyst concentration, and temperature. Cylindrical PDMS rods were used to investigate the swelling. Changes in the length and diameter were measured (Table 1). As expected, the amount of swelling is proportional to the amount, and identity, of non-polar solvent added. A swelling ratio of <20% was deemed to be sufficient to significantly increase the flux of small organic molecules for use in pot-in-pot reactions.115
| Entry | Solvent | Swellinga, S, (D/Do) | |||
|---|---|---|---|---|---|
| Length | Diameter | Average | Reportedb S | ||
| a The Whitesides swelling ratio.b Lee, J. N. et al. 2003.c Methoxy ethyl ether. | |||||
| 1 | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2:[BMIM][PF6] :1 CH2Cl2:[BMIM][PF6] |
1.15 | 1.14 | 1.14 | — |
| 2 | 1:1 CH2Cl2:[BMIM][PF6] |
1.08 | 1.03 | 1.05 | — |
| 3 | 3:1 THF:[BMIM][PF6] |
1.24 | 1.21 | 1.23 | — |
| 4 | CH2Cl2 | 1.18 | 1.21 | 1.20 | 1.22 |
| 5 | Pentane | 1.35 | 1.28 | 1.32 | 1.44 |
| 6 | [BMIM][PF6] | 1.00 | 1.00 | 1.00 | — |
| 7 | MEEc | 1.04 | 1.05 | 1.04 | — |
| 8 | MeOH | 1.00 | 1.02 | 1.01 | 1.02 |
| 9 | 1:1 MeOH:H2O |
1.00 | 1.02 | 1.00 | — |
Based on the swelling ratios of various solvents (Table 1), olefin metathesis reaction was performed under different solvent mixtures, using the same solvent both on the interior and exterior of the thimble.115 The substrate was placed on the outside of the thimble and allowed to diffuse into the thimble, react and the product diffuses out (Fig. 4). The conversion to the product was monitored only on the outside. At ambient temperature, no solvent was found to give satisfactory conversions of diethyl diallyl malonate to the ring closed product. Elevating the temperature to 45 °C gave slightly higher conversions but with the substrate being added on the exterior, 100% conversion was not achieved even after 18 h reaction times with the best solvent (1:1 CH2Cl2:[BMIM][PF6]). This observation can be understood from mass balance across the interface where the concentration of the reactant asymptotically decreases on the exterior of the thimble. Similarly, the concentration of the product follows a similar flux profile as its concentration increases on the inside.
 | ||
| Fig. 4 Schematic summary of a pot-in-pot reaction involving site-isolated Grubbs catalyst (G2) in the interior of the thimble. Solvent 1 and 2 may be the same or different solvents. The substrate, diethyl diallylmalonate, is added on the exterior, and reacts with the Grubbs catalyst after diffusing to the interior. The product subsequently diffuses to the exterior. Copyright© 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.29 | ||
When the substrate was added to the interior and the product allowed to flux out, 100% conversion to the product was achieved in a short reaction time. Despite slight loss of the catalyst to the exterior, the catalyst could be recycled over five cycles illustrating that a simple polymer membrane could drastically reduce waste and cost while improving efficiency in synthesis. Over short reaction times (<4 h), <5% of the catalyst had been lost when 1:1 CH2Cl2:[BMIM][PF6] was used in both the interior and exterior. For longer reaction times (>8 h), a significant amount (≥10%) of catalyst had leached as determined by ICP-MS.115 This study demonstrates that relying on partition coefficients across the polymer membrane alone might be useful for fast reactions, but may not be ideal for cases where two catalysts or reagents are highly incompatible or for slow reactions.
To limit catalyst leaching, and achieve complete site-isolation, the solvent on the exterior of the thimble was selected based on the insolubility of the catalyst in it. This criteria meant that the partition coefficient of catalyst to the exterior of the thimble was drastically reduced and therefore the leaching could be minimized. To test the strategy, ring closing olefin metathesis was performed as previously described and the amount of ruthenium metal leaching to the exterior of the thimble was monitored by ICP-MS (Fig. 5). When a mixture of water and MeOH (1:1) was adopted on the exterior of the thimble, >99% of the catalyst was retained within the interior of the thimble.
 | ||
| Fig. 5 Summary of ICP-MS data showing over 99.5% retention of ruthenium using different solvents and our thimbles (triangles) but gradual leaching is observed (diamonds) when the same solvent is used on the interior and exterior of the thimble. | ||
Having developed a system in which the catalyst was fully contained inside the thimble, a series of olefin metathesis reactions were performed to test the new platform (Fig. 6a). The substrate was added on the exterior of the thimble and allowed to diffuse into the interior to react with the catalyst, followed by product leaching back to the exterior. To improve solubility into the polar solvent, while also improving the rate of the reactions, the reactions were heated to 45 °C. Using the two solvents in pot-in-pot reactions, both olefin ring closing and cross metathesis reactions were performed with good yields albeit sometimes over extended reaction times – since it is a diffusion limited process (Fig. 6b). What was most encouraging about this reaction set up was the almost perfect catalyst site-isolation. Fig. 6c shows a picture of the reaction set up with the site-isolated catalyst after 16 h. Although this solvent system showed high catalyst containment, there was no direct method to ascertain that the ruthenium metal identified by ICP-MS was the active catalyst or not.
 | ||
| Fig. 6 Site-isolated metathesis reactions. (a) Small molecules were added to the exterior of the thimbles and diffused into the thimbles to react with Grubbs' catalyst (G2). (b) Sample reactions and their corresponding yields. (c) A picture of a PDMS thimble containing the Grubbs' catalyst (coloured solution), sitting on top of a white stir bar, immersed in an incompatible solvent – 1:1 MeOH:H2O. | ||
Using a highly active metathesis substrate, diethyl diallyl malonate, a ring closing metathesis reaction was first performed to completion. A second equivalent of the substrate was added on the exterior and after thorough mixing; the ratio of the substrate to the product was checked. Then, half the solvent on the exterior was transferred into a clean flask and subjected to the same conditions as the remaining mixture in the pot-in-pot reaction (with the thimble). The newly split reactions were allowed to proceed independently for an additional 17 h after the separation. These control experiments showed that the pot-in-pot reaction still contained an active catalyst as indicated by an increase in the amount of ring closed product. On the other hand, no increase in the product was observed for the portion of the solution that was transferred into a new clean flask – without the thimble.
2.3 Site-isolation of catalysts from reagents
Pot-in-pot was then extended to include a cascade reaction with the Grubbs' second generation catalyst, on the interior, and m-chloroperoxybenzoic acid (MCPBA), on the exterior, (Fig. 7).29 These reagents react vigorously with each other such that both are rapidly poisoned. As a control experiment, diethyl diallyl malonate was treated with Grubbs' second generation catalyst followed by MCPBA all in the same reaction vessel. This reaction failed to give the desired product due to decomposition of MCBPA by ruthenium despite a Ru:MCPBA ratio of 1:3000.29 Thus, even a small amount of the Grubbs' catalyst will significantly poison MCPBA.
 | ||
| Fig. 7 Cascade reactions with encapsulated Grubbs' catalyst and MCPBA. (a) Reagents were added to the interior of the PDMS thimbles to react by olefin metathesis, upon 100% conversion, MeOH and MCPBA were added to the exterior of the thimble. Grubbs' catalysts remained encapsulated but the intermediates diffused from the interior of the thimbles. (b) A summary of some overall reactions completed via this scheme, their yields and reaction times are shown. Copyright© 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.29 | ||
A series of cascade reactions were then carried out with the CH2Cl2:[BMIM][PF6] on the interior, and MeOH:H2O on the exterior of the thimble (Fig. 7). The product of the metathesis reaction diffused from the thimble and reacted with MCPBA in good yields (Fig. 7). It is critical to note that these reactions would have failed without a PDMS thimble to site-isolate the Grubbs' catalyst since we had a Ru to MCBPA ratio of 1 to 125.
Besides the examples given above, the pot-in-pot method has proven effective at site-isolating PdCl2 as well (Fig. 8).27 Because of its ionic nature, PdCl2 is not soluble in PDMS and will therefore not flux across the polymer membrane. The catalyst was shown to be >99% site-isolated. The Pd catalyst was also incorporated in cascade reactions involving a Wacker–Tsuji oxidation of a substrate on the interior of the thimble followed by the addition of a Grignard reagent to the exterior. Not only was it necessary to site-isolate the catalyst and reagent from each other, but the Wacker–Tsuji oxidation takes place in the presence of water, which would spontaneously quench the Grignard reagent with concomitant expedition of a large amount of energy. The Pd catalyst was recycled for the Wacker–Tsuji oxidation of styrene, and retained its activity through five cycles. The ability to efficiently site-isolate Pd catalysts has major consequences in materials synthesis where even trace amounts of the metal can lead to large losses of organic electronic materials.
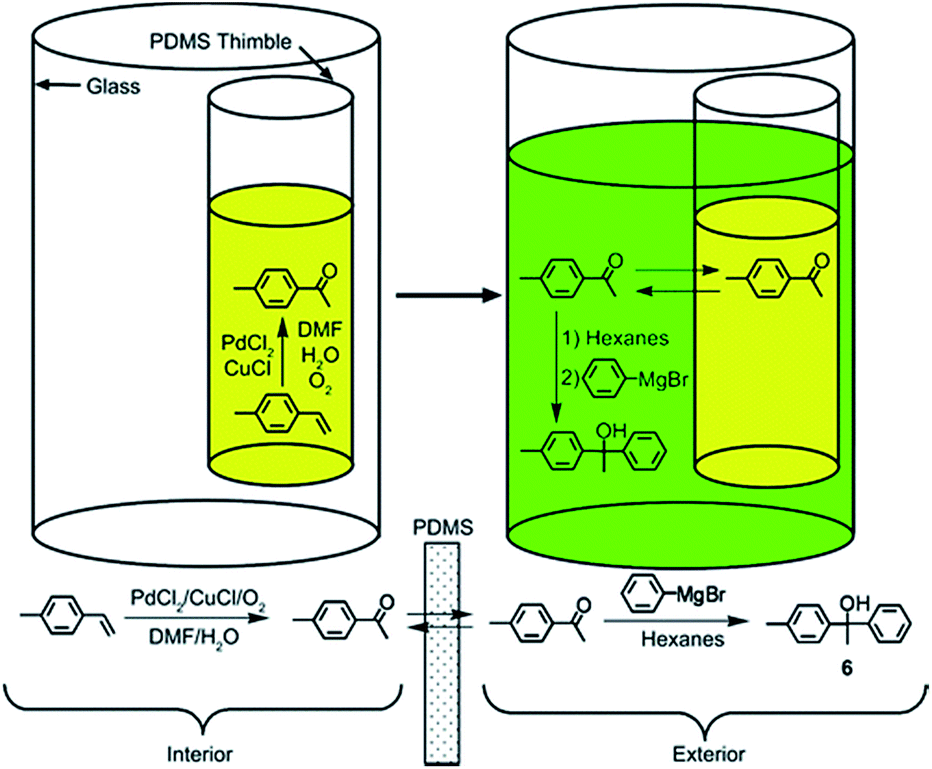 | ||
| Fig. 8 Schematic reaction showing Pd-catalyzed oxidation of alkene to ketone in the interior of the thimble. Subsequent addition of hexanes followed by the addition of a Grignard reagent to the exterior upon completion of oxidation reaction affords the tertiary alcohol product. Reprinted with permission from (J. Org. Chem., 2009, 74, 4834–4840). Copyright© 2009 American Chemical Society.27 | ||
2.4 Catalyst–catalyst site-isolation
 | ||
| Fig. 9 a) Site-isolation of two incompatible catalysts, – Grubbs' catalyst and the Sharpless dihydroxylation catalyst using a PDMS membrane. (b) Metathesis–dihydroxylation cascade reactions with catalysts dissolved in 1:1 (v/v) CH2Cl2:[BMIM][PF6] on the interior (metathesis), and a solvent mixture of acetone/water/ionic liquid on the exterior (dihydroxylation) of the thimble. | ||
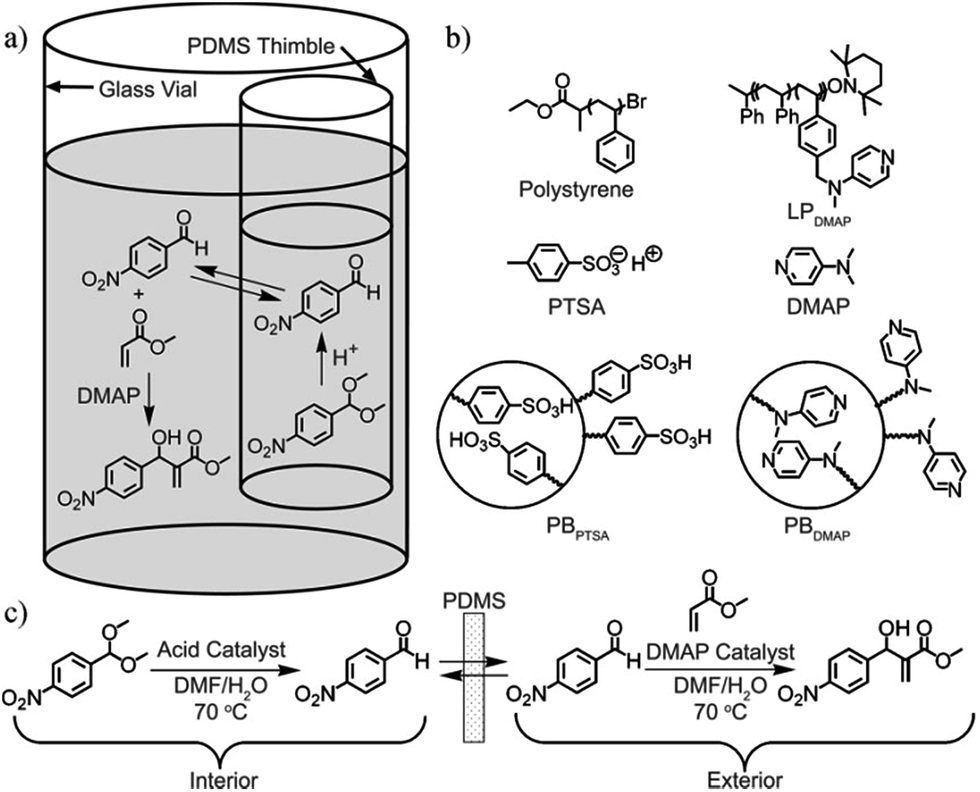 | ||
| Fig. 10 a) Experimental scheme showing acid (p-toluenesulfonic acid (PTSA)) and base (4-dimethyaminopyridine (DMAP)) catalyst. (b) Molecules and polymer derivatives site-isolated between the PDMS membrane. (b) Detailed reaction scheme showing the product of acid catalyzed reaction permeate between the PDMS membrane to the exterior where it becomes a reactant and undergoes base-catalyzed reaction. Copyright© 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.82 | ||
The use of a PDMS thimble to separate polymer-bound organic catalysts allows for simultaneous reactivity of the acid and base catalysts, thus eliminating the need to incorporate changes in the structure of the catalyst. Catalysts immobilized on polymeric beads are large and cannot partition into the PDMS even with swelling. Fig. 10 shows an example of the reactivity of an aldehyde using bound and unbound pTSA and DMAP. It is important to note that while pTSA had insignificant flux through PDMS (owing to the ionic structure). DMAP and the aldehyde had considerable flux (0.057 mmol h−1 cm−2 and 0.013 mmol h−1 cm−2, respectively). The measurable flux of DMAP through PDMS prompted the use of polymer-bound DMAP, which had no measureable flux across the membrane.
2.5 Recycling of catalysts from site-isolation
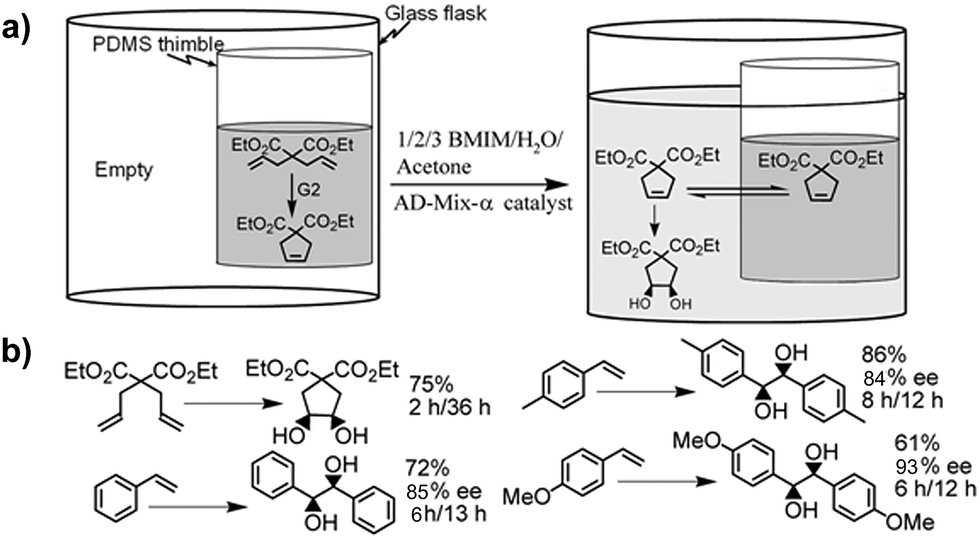
A key aspect to site-isolation is the possibility of recycling the catalyst (Fig. 11).29 A pot-in-pot approach allows for the ease of recycling of a catalyst by merely removing the solvent on the exterior after the reaction is complete. The metathesis–dihydroxylation sequence is used to illustrate that, even under challenging conditions, the catalysts can be re-used (Fig. 11). The conversions were quantitative and the yields were high over seven recycling steps. Measurement of the leakage of Ru to the exterior was found to be insignificant (<1%) indicating that this approach could lead to significant reuse of the catalysts at very low cost. To further demonstrate the importance of this method, we recycled the Grubbs' catalyst even as it was integrated into a cascade sequence with MCPBA. It is particularly noteworthy that high yields were obtained for these reactions even after several recycling steps. In all the cases discussed above, the catalysts were recycled in an analogous manner.26,28,29,31,115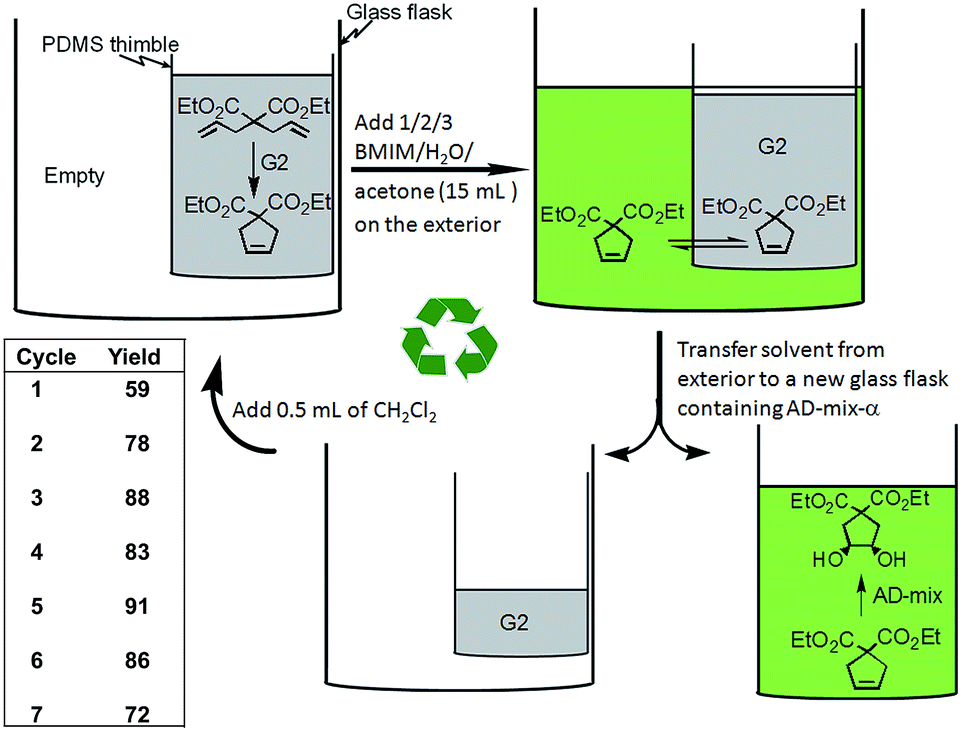 | ||
| Fig. 11 a) Pot-in-pot approach to site-isolate two catalytic processes using a PDMS membrane for otherwise impossible cascade reactions. Recycling of Grubbs' catalyst followed by dihydroxylation using AD-mix-α. The metathesis reaction is allowed to go to completion. The product leached to the exterior of the thimble, then placed in a new flask and dihydroxylated. The Grubbs catalyst (G2) is recycled. | ||
2.6 Pot-in-pot reactions as an alternative to challenging reactions
Besides the ability to site-isolate solvents, reagents, or catalysts, pot-in-pot reactions can be used to circumvent tedious, inefficient or difficult to handle reaction schemes. To illustrate this point, we discuss a few examples viz.; (i) pot-in-pot conversion of alcohols to amides as alternative to the elegant, but expensive to make, Milstein's catalysts, (ii) pot-in-pot synthesis of cyclic sulfoxide (thiophene dioxide) as an alternative to a tedious, multi-step and low yield synthesis, (iii) pot-in-pot metathesis with amines, substrate that readily poison the Grubbs' catalysts unless first converted to quaternary amines (Fig. 12). | ||
| Fig. 12 Application of pot-in-pot reactions as an alternative to tedious, inefficient, or challenging synthesis. (a) Dihydroxy thiophene dioxide is produced by ring closing metathesis of diallyl sulfane followed by oxidation. The intermediate 2,5-dihydrothiophene is volatile, odorous, and difficult to purify. (b) Conversion of an alcohol to an amide in a pot-in-pot reaction involving a Mo'Hansen oxidation and acid amidation. (c) Protonation of a diallyl amine allows its use in olefin metathesis, a subsequent deprotonation allowed the produced 2,3-dihydropyrolle to flux across PDMS. Dihydroxylation gives a chiral cyclic dihydroxypyrrolidine. | ||
Fig. 12a shows a scheme illustrating the ring-closing metathesis of dially mercaptan to give a volatile, pungent, and difficult to handle product. Purification of this product and subsequent isolation presents a major challenge in the synthesis of dihydroxy thiophene dioxide. Alternatives to this purification led to at least a 13 step synthesis with only 23% yield.133,134 Under pot-in-pot reactions, the same chiral compound was synthesized in a single day with >75% yield.26 To circumvent development of expensive, and often complex to synthesize catalysts – like the Milstein's catalyst, a felicitous choice of reaction conditions under a pot-in-pot arrangement facilitates the realization of the same products as would be obtained with such catalysts. To illustrate this idea, a Mo'Hansen oxidation followed by coupling of an amine to an acid leads to the synthesis of an amide from an alcohol (Fig. 12b).80 An even simpler case is the synthesis of a dihydropyrollidine, via olefin metathesis of diallyl amine, by first protonation of an amine to mitigate chelation to a metathesis catalyst followed by oxidation of resultant olefin. Amines (1° and 2°) are incompatible with many organometallic catalysts due to their ability to chelate at the metal centre. Upon protonation, amine containing molecules can be subjected to olefin metathesis using Grubbs catalysts. Protonation, however, would limit their flux across polymers but this limitation can be overcome by in situ deprotonation. This idea was demonstrated with good yields in a pot-in-pot reaction sequence where diallyl amine is protonated, cyclized via a Grubbs catalysts followed by deprotonation and subsequent flux to the exterior of the thimble where the 2,5-dihydropyrolle is asymmetrically dihydroxylated using AD-mix (Fig. 12c).28
3. Conclusions
This paper reviews progress in pot-in-pot reactions, a method to site-isolate homogeneous catalysts without altering their structures. This site-isolation allowed for the development of new cascade reaction sequences that are not possible with free catalysts in homogeneous settings. This method is efficient and convenient because it does not require change to the catalysts structure nor does it require development of new reaction media. Desirable properties of known catalysts are maintained while adding many of the benefits of using multi-phasic systems with concomitant increase in overall reaction efficiency. Since the pot-in-pot approach is a general method that uses concepts of diffusion through polymer membranes and solubility of catalysts rather than chemical transformations to modify and accommodate the catalyst structure, we believe that this approach will be applicable in diverse synthesis schemes and make an impact in how molecules are synthesized. Pot-in-pot approach is an efficient, low cost, and environmentally benign approach to chemical synthesis qualifying it as a green approach to organic synthesis. This method possesses advantages for green chemistry particularly the ability to recycle catalysts/reagents/solvents, efficiency in procedural and personnel resources, and, minimizes wastage of time and resources. Pot-in-pot reactions possess the potential to be expanded to include three, four, or more catalysts in one reaction vessel to carry out longer, multistep cascade sequences.Acknowledgements
This work was supported by Iowa State University through start-up funds.Notes and references
- C. Aubert, L. Fensterbank, V. Gandon and M. Malacria, Top. Organomet. Chem., 2006, 19, 259–294 CrossRef CAS.
- G. Balme, D. Bouyssi and N. Monteiro, Top. Organomet. Chem., 2006, 19, 115–148 CrossRef CAS.
- J. M. Bowness, Science, 1966, 152, 1370–1371 CrossRef CAS.
- C. Bruneau, S. Derien and P. H. Dixneuf, Top. Organomet. Chem., 2006, 19, 295–326 CrossRef CAS.
- Y. Chi, S. T. Scroggins and J. M. J. Frechet, J. Am. Chem. Soc., 2008, 130, 6322–6323 CrossRef CAS PubMed.
- A. Corma, Catal. Rev.: Sci. Eng., 2004, 46, 369–417 CAS.
- M. Malacria, Chem. Rev., 1996, 96, 289–306 CrossRef CAS PubMed.
- S. F. Mayer, W. Kroutil and K. Faber, Chem. Soc. Rev., 2001, 30, 332–339 RSC.
- T. J. J. Mueller, Top. Organomet. Chem., 2006, 19, 149–205 CrossRef CAS.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed.
- E. Zhang, T. Xu, W. Wei, T. Huang, M. Yuan, W. Zeng and Y. Zou, Synthesis, 2014, 46, 1167–1176 CrossRef PubMed , 1110 pp.
- W. You and M. K. Brown, J. Am. Chem. Soc., 2014, 136, 14730–14733 CrossRef CAS PubMed.
- J. Song, J. Moss, D.-C. Yang, Z. Guan and Y.-H. He, RSC Adv., 2014, 4, 54032–54038 RSC.
- K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2014, 53, 11960–11964 CrossRef CAS PubMed.
- J.-J. Liang, J.-Y. Pan, D.-C. Xu and J.-W. Xie, Tetrahedron Lett., 2014, 55, 6335–6338 CrossRef CAS PubMed.
- S. Jia, S. Su, C. Li, X. Jia and J. Li, Org. Lett., 2014, 16, 5604–5607 CrossRef CAS PubMed.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Catal., 2014, 4, 4548–4559 CrossRef.
- Y. He, X. Zhang and X. Fan, Chem. Commun., 2014, 50, 14968–14970 RSC.
- B. Han, X. Xie, W. Huang, X. Li, L. Yang and C. Peng, Adv. Synth. Catal., 2014, 356, 3676–3682 CrossRef CAS.
- S. Goswami, A. Manna, M. Mondal and D. Sarkar, RSC Adv., 2014, 4, 62639–62643 RSC.
- C.-N. Feng, Y.-C. Hsieh and Y.-T. Wu, Chem. Rec., 2014 DOI:10.1002/tcr.201402066 , ahead of print.
- C.-W. Fan, G.-C. Xu, B.-D. Ma, Y.-P. Bai, J. Zhang and J.-H. Xu, J. Biotechnol., 2014 DOI:10.1016/j.jbiotec.2014.10.026 , ahead of print.
- E. Brenna, M. Crotti, F. G. Gatti, D. Monti, F. Parmeggiani, A. Pugliese and S. Santangelo, J. Mol. Catal. B: Enzym., 2014 DOI:10.1016/j.molcatb.2014.10.006 , ahead of print.
- M. Ayaz, G. Martinez-Ariza and C. Hulme, Synlett, 2014, 25, 1680–1684 CrossRef PubMed , 1685 pp.
- A. Gupta, T. R. Long, D. G. Rethwisch and N. B. Bowden, Chem. Commun., 2011, 47, 10236–10238 RSC.
- M. T. Mwangi, M. D. Schulz and N. B. Bowden, Org. Lett., 2009, 11, 33–36 CrossRef CAS PubMed.
- A. L. Miller II and N. B. Bowden, J. Org. Chem., 2009, 74, 4834–4840 CrossRef PubMed.
- M. B. Runge, M. T. Mwangi, A. L. Miller II, M. Perring and N. B. Bowden, Angew. Chem., Int. Ed., 2008, 47, 935–939 CrossRef CAS PubMed.
- M. T. Mwangi, M. B. Runge, K. M. Hoak, M. D. Schulz and N. B. Bowden, Chem.–Eur. J., 2008, 14, 6780–6788 CrossRef CAS PubMed.
- A. L. Miller II and N. B. Bowden, Adv. Mater., 2008, 20, 4195–4199 Search PubMed.
- M. T. Mwangi, M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2006, 128, 14434–14435 CrossRef CAS PubMed.
- K. Kümmerer, Annu. Rev. Environ. Resour., 2010, 35, 57–75 CrossRef.
- US EPA, 2005 Toxics Release Inventory (TRI) Public Data Release Report, United State Environmental Protetion Agency, Washington DC, USA, 2007, http://www.epa.gov/tri Search PubMed.
- US EPA, 2012 Toxic Release Inventory National Analysis Overview, United State Environmental Protection Agency, Washington DC, USA, 2014, pp. 1–45 Search PubMed.
- A. Hamel, M. Sacco, N. Mnasri, F. Lamaty, J. Martinez, F. De Angelis, E. Colacino and C. Charnay, ACS Sustainable Chem. Eng., 2014, 2, 1353–1358 CrossRef CAS.
- M. G. Kulkarni, Y. B. Shaikh, A. S. Borhade, S. W. Chavhan, A. P. Dhondge, D. D. Gaikwad, M. P. Desai, D. R. Birhade and N. R. Dhatrak, Tetrahedron Lett., 2013, 54, 2293–2295 CrossRef CAS PubMed.
- A. J. Yap, A. F. Masters and T. Maschmeyer, Handbook of Green Chemistry, 2012, vol. 8, pp. 137–163 Search PubMed.
- Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, Angew. Chem., Int. Ed., 2012, 51, 9164–9168 CrossRef CAS PubMed.
- H. Hagiwara, Synlett, 2012, 23, 837–850 CrossRef CAS PubMed.
- Y. Wei, S. Soh, M. M. Apodaca, J. Kim and B. A. Grzybowski, Small, 2010, 6, 857–863 CrossRef CAS PubMed.
- D. Schoeps, K. Buhr, M. Dijkstra, K. Ebert and H. Plenio, Chem.–Eur. J., 2009, 15, 2960–2965 CrossRef CAS PubMed.
- Y. Motoyama, K. Kamo and H. Nagashima, Org. Lett., 2009, 11, 1345–1348 CrossRef CAS PubMed.
- T. Brendgen, T. Fahlbusch, M. Frank, D. T. Schuehle, M. Sessler and J. Schatz, Adv. Synth. Catal., 2009, 351, 303–307 CrossRef CAS.
- E. Mamedov, Appl. Catal., A, 2014, 474, 34–39 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, H. Yang, F. Cheng and Z. Du, ChemSusChem, 2014, 7, 1888–1900 CrossRef CAS PubMed.
- M. Asadi, S. Bonke, A. Polyzos and D. W. Lupton, ACS Catal., 2014, 4, 2070–2074 CrossRef CAS.
- B. R. Goldsmith, E. D. Sanderson, D. Bean and B. Peters, J. Chem. Phys., 2013, 138, 204105/204101–204105/204111 CrossRef PubMed.
- X. Fan, C. Rodriguez-Escrich, S. Sayalero and M. A. Pericas, Chem.–Eur. J., 2013, 19, 10814–10817 CrossRef CAS PubMed.
- K. K. Sharma, A. Anan, R. P. Buckley, W. Ouellette and T. Asefa, J. Am. Chem. Soc., 2008, 130, 218–228 CrossRef CAS PubMed.
- G. Centi and S. Perathoner, Top. Catal., 2001, 15, 145–152 CrossRef CAS.
- D. A. Annis and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 4147–4154 CrossRef CAS.
- D. Zhang, J. Xu, Q. Zhao, T. Cheng and G. Liu, ChemCatChem, 2014, 6, 2998–3003 CAS.
- M. Tada and S. Muratsugu, Heterogeneous Catalysts for Clean Technology, Wiley-VCH, 2014, ch. 7, pp. 173–191 Search PubMed.
- M. E. Potter, M. E. Cholerton, J. Kezina, R. Bounds, M. Carravetta, M. Manzoli, E. Gianotti, M. Lefenfeld and R. Raja, ACS Catal., 2014, 4, 4161–4169 CrossRef CAS.
- X. Fan, C. Rodriguez-Escrich, S. Wang, S. Sayalero and M. A. Pericas, Chem.–Eur. J., 2014, 20, 13089–13093 CrossRef CAS PubMed.
- B. Hu, N. M. Schweitzer, M. P. Lanci, J. T. Miller and A. S. Hock, Prepr. - Am. Chem. Soc., Div. Energy Fuels, 2013, 58, 183–184 CAS.
- C. S. Chen, Y. T. Lai, T. W. Lai, J. H. Wu, C. H. Chen, J. F. Lee and H. M. Kao, ACS Catal., 2013, 3, 667–677 CrossRef CAS.
- P. Biggs, L. Jones II and G. Lewis, PMSE Prepr., 2007, 96, 498 CAS.
- J. O. Brubaker, R. T. Patil, T. J. Speaker and P. A. Offit, J. Immunol. Methods, 2000, 237, 85–93 CrossRef CAS.
- Y. Jiang, L. Zhang, D. Yang, L. Li, Y. Zhang, J. Li and Z. Jiang, Ind. Eng. Chem. Res., 2008, 47, 2495–2501 CrossRef CAS.
- S. Kobayashi and R. Akiyama, Chem. Commun., 2003, 449–460 RSC.
- E. Kokufuta, T. Sodeyama and T. Katano, Chem. Commun., 1986, 641–642 RSC.
- S. V. Ley, C. Ramarao, A.-L. Lee, N. Ostergaard, S. C. Smith and I. M. Shirley, Org. Lett., 2003, 5, 185–187 CrossRef CAS PubMed.
- K. Okamoto, R. Akiyama and S. Kobayashi, Org. Lett., 2004, 6, 1987–1990 CrossRef CAS PubMed.
- S. L. Poe, M. Kobaslija and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 15586–15587 CrossRef CAS PubMed.
- K. E. Price, B. P. Mason, A. R. Bogdan, S. J. Broadwater, J. L. Steinbacher and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 10376–10377 CrossRef CAS PubMed.
- J. D. Rule, N. R. Sottos and S. R. White, Polymer, 2007, 48, 3520–3529 CrossRef CAS PubMed.
- M. S. Vishwanath and S. E. Gibson, Adv. Synth. Catal., 2002, 344, 619–621 CrossRef.
- Y. H. Tseng, M. H. Fang, P. S. Tsai and Y. M. Yang, J. Microencapsulation, 2005, 22, 37–46 CrossRef CAS PubMed.
- C. K. Yeom, S. B. Oh, J. W. Rhim and J. M. Lee, J. Appl. Polym. Sci., 2000, 78, 1645–1655 CrossRef CAS.
- L. Zhuo and S. Chen, Int. J. Polym. Mater., 2004, 53, 385–393 CrossRef CAS.
- A. G. M. Barrett, B. T. Hopkins and J. Koebberling, Chem. Rev., 2002, 102, 3301–3323 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS PubMed.
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615–3640 CrossRef CAS PubMed.
- D. E. De Vos and P. A. Jacobs, Stud. Surf. Sci. Catal., 2001, 137, 957–985 CrossRef CAS.
- Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3466 CrossRef CAS PubMed.
- A. K. Kakkar, Chem. Rev., 2002, 102, 3579–3587 CrossRef CAS PubMed.
- I. Vankelecom, Chem. Rev., 2002, 102, 3779–3810 CrossRef CAS PubMed.
- A. V. Gaikwad, V. Boffa, J. E. ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2008, 47, 5407–5410 CrossRef CAS PubMed.
- M. Gaab, S. Bellemin-Laponnaz and L. H. Gade, Chem.–Eur. J., 2009, 15, 5450–5462 CrossRef CAS PubMed.
- J. M. Watson and P. A. Payne, J. Membr. Sci., 1990, 49, 171–205 CrossRef CAS.
- T. R. Long, A. Gupta, A. L. Miller II, D. G. Rethwisch and N. B. Bowden, J. Mater. Chem., 2011, 21, 14265–14276 RSC.
- T. C. O. Mac Leod, V. P. Barros, A. L. Faria, M. A. Schiavon, I. V. P. Yoshida, M. E. C. Queiroz and M. D. Assis, J. Mol. Catal. A: Chem., 2007, 273, 259–264 CrossRef CAS PubMed.
- A. R. Bogdan, B. P. Mason, K. T. Sylvester and D. T. McQuade, Angew. Chem., Int. Ed., 2007, 46, 1698–1701 CrossRef CAS PubMed.
- S. L. Poe, M. Kobaslija and D. T. McQuade, J. Am. Chem. Soc., 2007, 129, 9216–9221 CrossRef CAS PubMed.
- J.-I. Park, S. L. Roth, K. E. Price and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3271–3278 CrossRef CAS.
- D. T. McQuade, Polym. Prepr., 2006, 47, 879 CAS.
- J. L. Steinbacher and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6505–6533 CrossRef CAS.
- K. E. Price and D. T. McQuade, Chem. Commun., 2005, 1714–1716 RSC.
- S. J. Broadwater, S. L. Roth, K. E. Price, M. Kobaslija and D. T. McQuade, Org. Biomol. Chem., 2005, 3, 2899–2906 CAS.
- H. M. Jung, K. E. Price and D. T. McQuade, J. Am. Chem. Soc., 2003, 125, 5351–5355 CrossRef CAS PubMed.
- J. W. Yang, H. Han, E. J. Roh, S. Lee and C. E. Song, Org. Lett., 2002, 4, 4685–4688 CrossRef CAS PubMed.
- K. E. Price, S. J. Broadwater, A. R. Bogdan, I. Keresztes, J. L. Steinbacher and D. T. McQuade, Macromolecules, 2006, 39, 7681–7685 CrossRef CAS.
- M. Kobaslija and D. T. McQuade, Macromolecules, 2006, 39, 6371–6375 CrossRef CAS.
- B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS PubMed.
- A. Sarkar, P. Ilankumaran, P. Kisanga and J. G. Verkade, Adv. Synth. Catal., 2004, 346, 1093–1096 CrossRef CAS.
- M. Kobaslija, A. R. Bogdan, S. L. Poe, F. Escobedo and D. T. McQuade, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2309–2315 CrossRef CAS.
- B. P. Mason, A. R. Bogdan, A. Goswami and D. T. McQuade, Org. Lett., 2007, 9, 3449–3451 CrossRef CAS PubMed.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- E. Baltussen, P. Sandra, F. David, H.-G. Janssen and C. Cramers, Anal. Chem., 1999, 71, 5213–5216 CrossRef CAS.
- P. Mayer, W. H. J. Vaes and J. L. M. Hermens, Anal. Chem., 2000, 72, 459–464 CrossRef CAS.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
- J. Du Pleiss, W. J. Pugh, A. Judefeind and J. Hadgraft, Eur. J. Pharm. Sci., 2002, 15, 63–69 CrossRef.
- H. Kaya, A. Erzan and F. Kadirgan, J. Chem. Phys., 1993, 98, 9030–9033 CrossRef CAS PubMed.
- V. Sarveiya, J. F. Templeton and H. A. E. Benson, J. Pharm. Pharmacol., 2004, 56, 717–724 CrossRef CAS PubMed.
- H. Takagi and K. Kaneko, Int. J. Bifurcation Chaos Appl. Sci. Eng., 2002, 12, 2579–2598 CrossRef CAS.
- S. Weinketz, J. Chem. Phys., 1994, 101, 1632–1637 CrossRef CAS PubMed.
- R. Woesler, P. Schuetz, M. Bode, M. Or-Guil and H.-G. Purwins, Phys. D, 1996, 91, 376–405 CrossRef CAS.
- A. Hasanoglu, Y. Salt, S. Keleser, S. Ozkan and S. Dincer, Chem. Eng. Process., 2007, 46, 300–306 CrossRef CAS PubMed.
- Y. K. Hong and W. H. hong, J. Membr. Sci., 1999, 159, 29–39 CrossRef.
- Y. Luo, S. Tan, H. Wang, F. Wu, X. Liu, L. Li and Z. Zhang, Chem. Eng. J., 2008, 137, 496–502 CrossRef CAS PubMed.
- X. K. Zhang, Y. Poojari, L. E. Drechsler, C. M. Kuo, J. R. Fried and S. J. Clarson, J. Inorg. Organomet. Polym. Mater., 2008, 18, 246–252 CrossRef CAS PubMed.
- M. B. Runge, M. T. Mwangi and N. B. Bowden, J. Organomet. Chem., 2006, 691, 5278–5288 CrossRef CAS PubMed.
- M. Thuo, PhD thesis, University of Iowa, 2008.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238–2264 CrossRef CAS PubMed.
- J. G. Huddleston and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- T. D. Ho, C. Zhang, L. W. Hantao and J. L. Anderson, Anal. Chem., 2013, 86, 262–285 CrossRef PubMed.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- G. Cevasco and C. Chiappe, Green Chem., 2014, 16, 2375–2385 RSC.
- D. K. Bwambok, M. M. Thuo, M. B. J. Atkinson, K. A. Mirica, N. D. Shapiro and G. M. Whitesides, Anal. Chem., 2013, 85, 8442–8447 CrossRef CAS PubMed.
- T. Schäfer, R. E. D. Paolo, R. Franco and J. G. Crespo, Chem. Commun., 2005, 2594–2596, 10.1039/b501507c.
- A. I. Horowitz and M. J. Panzer, Angew. Chem., Int. Ed., 2014, 53, 9780–9783 CrossRef CAS PubMed.
- A. Figoli, T. Marino, S. Simone, E. Di Nicolo, X. M. Li, T. He, S. Tornaghi and E. Drioli, Green Chem., 2014, 16, 4034–4059 RSC.
- X. Ding, X. Lv, B. Hui, Z. Chen, M. Xiao, B. Guo and W. Tang, Tetrahedron Lett., 2006, 47, 2921–2924 CrossRef CAS PubMed.
- S. M. Rountree, M. C. Lagunas, C. Hardacre and P. N. Davey, Appl. Catal., A, 2011, 408, 54–62 CrossRef CAS PubMed.
- N. Audic, H. Clavier, M. Mauduit and J.-C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248–9249 CrossRef CAS PubMed.
- R. C. Buijsman, E. van Vuuren and J. G. Sterrenburg, Org. Lett., 2001, 3, 3785–3787 CrossRef CAS PubMed.
- P. Sledz, M. Mauduit and K. Grela, Chem. Soc. Rev., 2008, 37, 2433–2442 RSC.
- M. T. Mwangi, M. D. Schulz and N. B. Bowden, Org. Lett., 2008, 11, 33–36 CrossRef PubMed.
- T. Naka, N. Nishizono, N. Minakawa and A. Matsuda, Tetrahedron Lett., 1999, 40, 6297–6300 CrossRef CAS.
- N. Nishino, R. Baba, C. Nakamura, K. Oda and M. Machida, Org. Biomol. Chem., 2003, 1, 3692–3697 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |