
Ni cluster nucleation and growth on the anatase TiO2(101) surface: a density functional theory study†
Yanxin Wanga,
Yan Sub,
Mingyuan Zhua and
Lihua Kang*a
aCollege of Chemistry and Chemical Engineering, Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, Shihezi University, Shihezi, Xinjiang 832000, China. E-mail: kanglihua@shzu.edu.cn; Fax: +86-0993-2057270; Tel: +86-0993-2057213
bSchool of Physics and Optoelectronic Technology and College of Advanced Science and Technology, Dalian University of Technology, Dalian 116024, China
First published on 30th January 2015
Abstract
Density functional theory (DFT) calculations are carried out to study the nucleation and growth rule of Ni clusters on both a perfect and defective anatase TiO2(101) surface using supported Nin (n = 1–6) cluster models. Our results show that a single Ni atom prefers to adsorb at the bridge site formed by two-coordinated oxygen (2cO) atoms on the perfect TiO2(101) surface and at the 3cO-bridge site on the defective TiO2(101) surface. The active site for Ni cluster growth on the perfect TiO2(101) surface shifts from the bridge site of two 2cO atoms or the 2cO–6cTi–3cO bridge site for Ni1, Ni2, and Ni3 clusters to the 2cO–5cTi bridge site for Ni4, Ni5, and Ni6. The Ni cluster cohesive energy remains constant with cluster size variation on both the perfect and defective surface. The Ni–TiO2 interaction is the main driving force of the initial Ni nucleation stage, and the Ni–Ni interaction begins to control the Nin cluster growth process with increased cluster size.
1. Introduction
Transition metal clusters supported on metal-oxide substrates are attracting considerable attention because of their importance in heterogeneous catalysis.1 A number of studies have indicated that the interaction between metal and support significantly affects the activity and selectivity of catalytic reactions.2–5 Therefore, understanding metal cluster nucleation and growth on oxide support mechanisms is important to further explore the metal–support interaction and improve the catalytic activity of the systems.Among the metal-oxide supports, TiO2 is the most widely used in many processes such as hydrogenation,6–8 CO oxidization,9–11 and water splitting.12–14 The two most common TiO2 polymorphs are rutile and anatase. Rutile TiO2 is the most widely studied form both experimentally and theoretically, whereas, anatase TiO2 has been found to be dominant in the nanocrystalline phase.15 Extensive experimental and theoretical investigations have been performed on metal cluster nucleation and growth patterns, including Au,16–22 Pt,23–27 and Pd,28–33 on both rutile and anatase TiO2 surfaces.
Recently, some experiment and theoretical studies concerning Ni cluster deposition on rutile TiO2(110) surfaces have been reported. Chen et al.34 studied Ni and Cu growth on TiO2(110) using scanning tunneling microscope (STM). They reported that Ni islands grew three dimensionally at room temperature, and although the Ni–TiO2 interfacial energy should be greater than that of Cu–TiO2, the Ni islands did not grow flatter on the surface compared to Cu, indicating that Ni islands were located preferably at step edges with high step edge density. This was further confirmed by the Tanner group's STM study.35 Asakura and co-workers36 investigated Ni cluster growth mode and morphology on a TiO2(110) surface with a wide terrace using low coverage STM. They observed that many flat and small Ni islands were located on the terrace, not at step edges with low step density. The initial stage bonding structure and binding character of Ni thin-film growth on a rutile (110) surface using first-principle density functional theory were studied by Ellis et al.37 Their results showed that Ni atoms in the first monolayer were preferentially adsorbed on top of bridging oxygen atoms and upon secondary surface oxygen. The bond strength between Ni adatom and substrate was much stronger than that between Ni adatoms. However, for Ni deposition on anatase TiO2(101), almost no report was found until J. Chen and co-workers38 found that nickel catalysts supported on ZrO2 (monoclinic phase), SiO2, TiO2 (anatase phase), and γ-Al2O3, Ni/TiO2 (anatase phase) showed the best catalytic hydrogenation performance of o-chloronitrobenzene to o-chloroaniline. The good Ni/TiO2 performance was attributed to the strong N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band polarization induced by TiOx oxygen vacancies which was produced by high temperature reduction. Therefore, to explore Ni cluster deposition nucleation and growth mechanism deposition on anatase TiO2(101) surface is essential. Meanwhile, the role of oxygen vacancies on the TiO2 surface in dispersing and nucleating Ni clusters should be understood.
O band polarization induced by TiOx oxygen vacancies which was produced by high temperature reduction. Therefore, to explore Ni cluster deposition nucleation and growth mechanism deposition on anatase TiO2(101) surface is essential. Meanwhile, the role of oxygen vacancies on the TiO2 surface in dispersing and nucleating Ni clusters should be understood.
In the present work, we use anatase TiO2(101) surface as the oxide support for depositing Nin (n = 1–6) clusters. DFT calculations on Ni cluster nucleation and growth mechanism on perfect and defective support surfaces were carried out. This work provides a fundamental understanding to further disclose the metal–support interaction and gives helpful design and synthesis instructions of supported Ni-based catalysts in heterogeneous catalysis.
2. Model catalysts and computational details
The calculated bulk anatase TiO2 structure lattice constants: a = 3.776 Å, c = 9.486 Å were comparable with experimental data (ICSD #24276). The (101) surface was modeled with 11.33 × 10.21 × 17.84 Å dimensions, which included 1 × 3 sized supercell slab with 12 atomic layers. The vacuum region separating the slabs in the direction perpendicular to the surface was set to 12 Å. The atomic positions of the six uppermost layers were always fully relaxed. In addition, the oxygen vacancy was obtained by removing one surface 2cO atom (marked with green circle in Fig. 1b) from the 1 × 3 surface unit cell, corresponding to 1/6 oxygen vacancy density. The properties of isolated Nin clusters were calculated using a 15 × 15 × 15 Å cubic unit cell. Side (left panel) and top (right panel) view of the anatase TiO2(101) surface are shown in Fig. 1.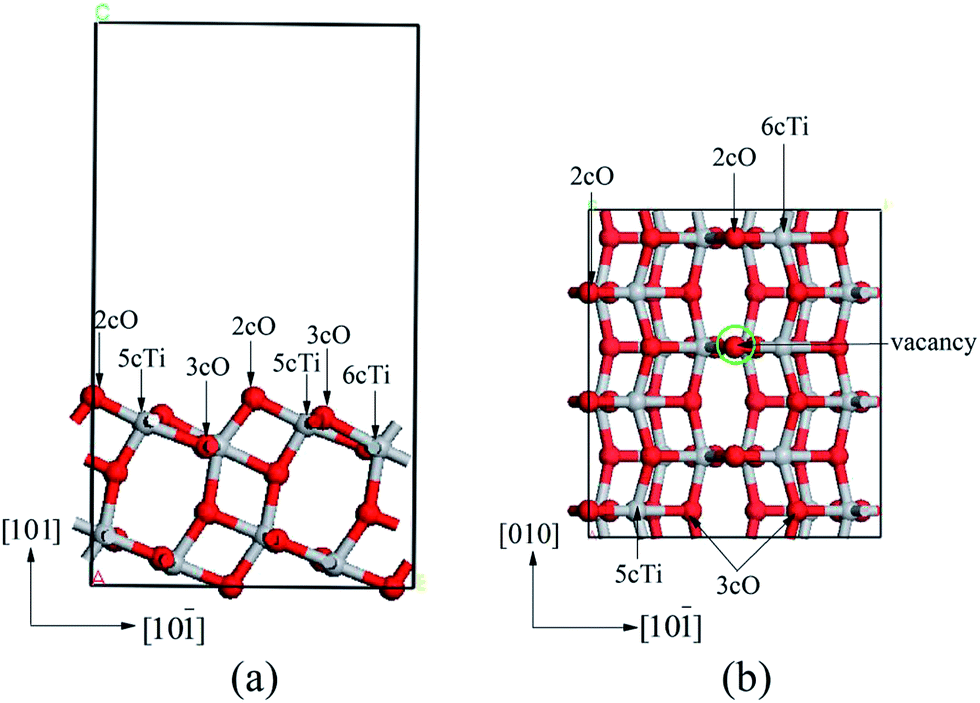 | ||
| Fig. 1 Side (a) and top (b) views of anatase TiO2(101) supercell. The red is an O atom and the argent is an Ti atom. | ||
The fully periodic plane-wave DFT calculations implemented in the Vienna Ab initio Simulation Program (VASP)39,40 were employed. DFT calculations were performed with the generalized gradient approximation PW91 function41 implemented with projector augmented wave function (PAW)42,43 for representing the non-valence core electrons. For better calculation accuracy, a 2 × 2 × 1 k-point grid determined by the Monkhorst–Pack method was used. The plane-wave cutoff energy was optimized at 400 eV. The convergence criteria for electronic self-consistent iteration were set to 1.0 × 10−4 eV, and the atomic positions were optimized by means of a conjugate-gradient algorithm until atomic forces were smaller than 0.02 eV Å−1. Spin-polarized calculations were performed to account for the magnetic properties of nickel. Charge distributions were estimated using Bader's atoms in molecules (AIM) theory of using the algorithm developed by Henkelman.44,45
To describe the interaction between Ni cluster and titania surface, adsorption energy is defined as follows:33
| Eads = −[E(Nin/TiO2) − E(TiO2) − E(Nin)] | (1) |
3. Results and discussion
3.1 Perfect and defective TiO2(101) surface
Anatase TiO2(101) surface presents a stepped structure, as shown in Fig. 1. 3-Fold-coordinational oxygen atoms (3cO) and 6-fold-coordinational titanium atoms (6cTi) were fully saturated whereas the 2cO and 5cTi atoms were coordinately unsaturated (these atoms are marked with black arrows in the side and top views of the surface in Fig. 1). After optimization, atomic positions of the six uppermost layers changed. The 3cO and 6cTi atoms rose by 0.41 and 0.21 Å, respectively, and the 2cO and 5cTi by 0.12 and 0.03 Å, respectively, relative to their bulk position. However, the 2cO–6cTi and 2cO–5cTi bond lengths shortened by 0.05 and 0.13 Å, respectively.The oxygen vacancy was obtained by removing a bridging 2cO atom from a (1 × 3) surface supercell (vacancy coverage = 1/6 ML). Oxygen vacancy produced gave rise to significant structure relaxations and important changes of the electronic properties (changes of the electronic properties will be shown in 3.2.3 section). The original 6cTi and 5cTi atoms bound to the 2cO atom at the vacancy site become five- and four-coordinational, denoted as Tiv-5c and Tiv-4c, respectively. The 3cO atom under the oxygen vacancy relaxed upward by 0.29 Å. Tiv-5c and Tiv-4c relaxed inward by 0.06 and 0.09 Å, respectively. Both relaxed away from each other, which led to Tiv-5c–3cO–Tiv-4c angle change from 101.92° to 126.51° on the perfect surface.
The formation energy of an oxygen vacancy was defined with respect to the energy of an oxygen molecule and calculated according to: E = −[Eperfect − Edefective − 1/2EO2]. Our calculated oxygen vacancy formation energy was 4.80 eV. The slab was kept neutral after 2cO was removed, causing some unpaired electrons. Bader charge analysis showed that the excess electrons were mainly localized at the Tiv-5c and Tiv-4c atoms.
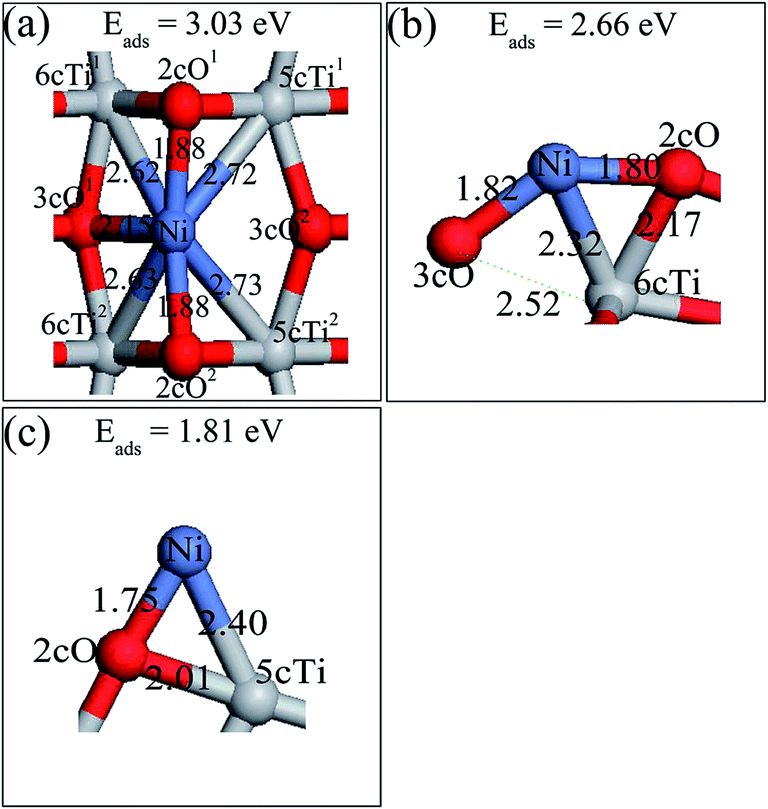 | ||
| Fig. 2 Optimized structures of a single Ni adatom and their corresponding adsorption energy on a perfect anatase TiO2(101) surface: (a) Ni1,a; (b) Ni1,b; (c) Ni1,c. Their corresponding adsorption energy on the surface are also shown in the figure. | ||
Six possible single Ni adatom adsorption sites on the defective TiO2(101) surface have been searched. The relaxed structures (Ni1,A and Ni1,B) and their adsorption energies are shown in Fig. 3. The description of Ni1,B is shown in ESI.† Completely different from Au, Pt and Pd,22,26,33 the preferable adsorption site for the Ni adatom on the defective surface was not the oxygen vacancy, but the bridge site of two 3cO atoms, with the Ni–3cO bond lengths of 1.87 (left) and 1.89 Å (right), respectively. Ni insertion in the middle of two 3cO atoms pulled these two 3cO atoms close to each other by 0.13 Å, and the right 3cO moved to left by 0.14 Å compared to the clean surface. The 3cO atom movement weakened the 3cO bond strength with its left and right side Ti atoms (Tia and Tib, respectively), and especially the interaction force of 3cO–Tib was nearly zero. The interaction between the Ni adatom and the remaining 2cO atom was much weaker than that in the perfect surface with the Ni–2cO bond length of 2.02 Å. The adsorption energy of Ni1A was 3.10 eV, which is a little higher than the 3.03 eV of Ni1a in the perfect surface.
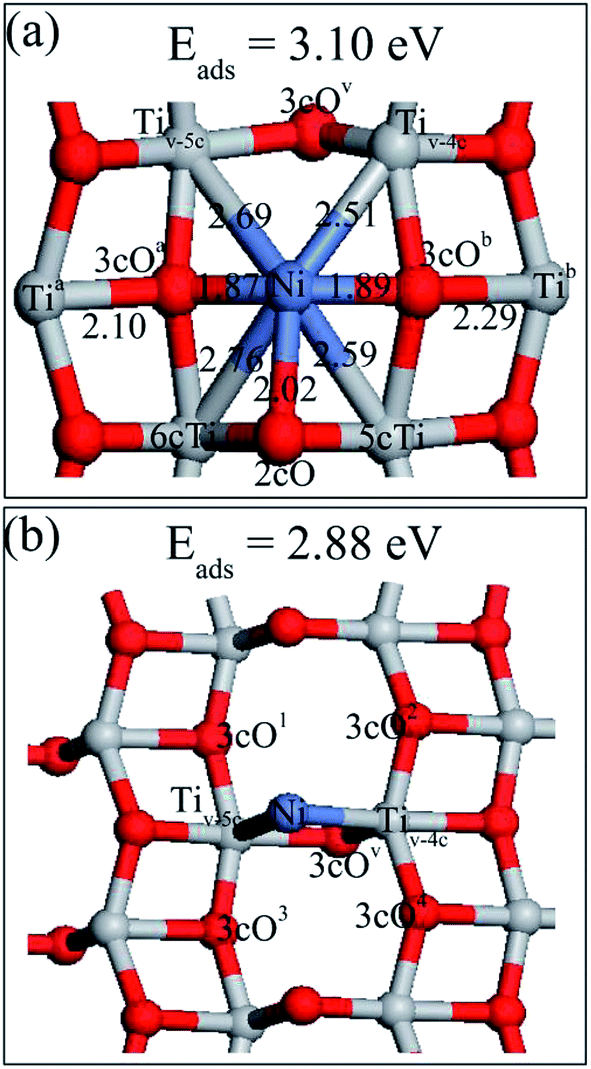 | ||
| Fig. 3 Optimized structures of a single Ni adatom and their corresponding adsorption energy on a defective anatase TiO2(101) surface: (a) Ni1,A; (b) Ni1,B. Their corresponding adsorption energy on the surface are also shown in the figure. | ||
 | ||
| Fig. 4 Density of state for the (a) perfect TiO2(101) surface (top) and Ni1,a (bottom) and (b) defective TiO2(101) surface (top) and Ni1,A (bottom). The local DOS of Ni, which contributes to the total DOS in Ni1,a and Ni1,A, is represented in red color. | ||
To gain further insight of the bonding nature between the supported Ni adatom and the TiO2 support, Bader charge analysis was conducted. The Bader charges of Ni1,a and the surface atoms which are directly associated with the Ni adatom on the perfect anatase TiO2(101) surface, are shown in Table 1S (in the ESI†). The Ni atom was positively charged with a charge of 0.55e, suggesting charge transfer occurrence from the Ni atom to the TiO2 surface. After Ni atom adsorption on the TiO2 surface, 2cO1, 2cO2, 3cO1 and 3cO2 atoms that directly bonded to Ni atom became more negative owing to receiving a charge of 0.11e, 0.11e, 0.03e and 0.02e, respectively, while 5cTi and 6cTi atoms became less positive. This indicated further that all Ti and O atom types which directly interact with Ni atoms, are all electron receptors. Furthermore, the amounts of charge order transferred from Ni to 2cO, 3cO, and Ti atoms were consistent to their bond strengths. The Ni–2cO bond length was the shortest compared to Ni–Ti and Ni–3cO, which means the strongest bond strength, hence, 2cO1 and 2cO2 gained the most charge amount (both 0.11e) from Ni atom. The results indicate that the interaction between Ni atom and the surface TiO2 atoms was formed via the electron transported among them.
The Ni1,A and Ni1,B atom net charge analysis and of their adjacent atoms with defective surface, is shown in Tables 2S and 3S, respectively (in the ESI†). With the same situation as Table 1S,† Ni1,A was also an electron donor of the defective surface and the net charges were not equally distributed among all atoms bonded with Ni1,A. Surprisingly, although Ni–3cOa and Ni–3cOb bond strengths are the strongest, the charge amount 3cOa obtained was less than that of 2cO, Tiv-5c, and Tiv-4c atom gains, while the charge density of 3cOb was almost constant. From the previous analysis, we know that Ni insertion pulls 3cOa and 3cOb atoms close together resulting to 3cOa–Tia and 3cOb–Tib bond strength weakening. Therefore, in addition to charge amount from Ni atom, 3cOa and 3cOb atoms can only accept less charge amounts from Tia and Tib, respectively, after Ni1,A disposition on the defective TiO2(101) surface. In spite of higher adsorption energy of Ni1,A than Ni1,a on the TiO2(101) surface, Ni1,A atom transfers less charge amount (0.35e) compared to Ni1,a. No direct correlation between the stability of the configuration and the amount of charge that is transferred was apparent. When the Ni1,B occupied the oxygen vacancy site, it was negatively charged with a charge of −0.29e, indicating that some charges were transferred from the TiO2(101) surface to Ni atom. Moreover, the Ni atom deposition on the TiO2 surface resulted in charge density increase on the Tiv-5c and Tiv-4c atoms and charge density decrease on the 3cO atom (marked with 3cOv in Fig. 3b) under the oxygen vacancy and 3cO atoms (marked with 3cO1, 3cO2, 3cO3 and 3cO4 in Fig. 3b) directly bound to Tiv-5c and Tiv-4c atoms. Therefore, Ni atom interaction with the defective surface was similar to a back-donation interaction47 where the Ni metal promotes a charge transfer from the surface oxygen to the Tiv-5c and Tiv-4c atoms.
3.2 Adsorption of Ni2 on the perfect and defective surface
After searching different possible sites for Ni dimer adsorption on perfect anatase TiO2(101) surface, four stable structures denoted as Ni2,a, Ni2,b, Ni2,c and Ni2,d as well as their adsorption energies were obtained and they are shown in Fig. 5. Ni2,b, Ni2,c and Ni2,d are described in ESI.† The Ni–Ni bonds are parallel to the TiO2(101) surface in all four structures. Ni2,a was built by adding the second Ni atom to the most stable single Ni adsorption (Ni1,a) and allowing the structures to relax again. It presents the most stable structure with a Ni atom located at the 2cO bridge site, another Ni adatom adsorbed at the 2cO–6cTi–3cO bridge site. The Ni1–Ni2 distance was 2.56 Å, which is significantly longer than the 2.09 Å of the calculated gas-phase Ni dimer. Combined with various single Ni atom adsorption energies on different surface sites, it can be concluded that the decreasing sequence for Ni adsorption active site is 2cO bridge site, 2cO–6cTi–3cO bridge site, and 2cO–5cTi bridge site. | ||
| Fig. 5 Adsorption structures and energies of Ni dimer on the perfect anatase TiO2(101) surface. (a) Ni2,a; (b) Ni2,b; (c) Ni2,c; and (d) Ni2,d. | ||
Three stable adsorbed structures of Ni dimer on defective TiO2 surface as well as their adsorption energies were obtained, namely, Ni2,A, Ni2,B, Ni2,C and they are shown in Fig. 6. The description of Ni2,B and Ni2,C are presented in ESI.† The preferable Ni dimer adsorption sites on defective TiO2(101) surface are shown in Fig. 5a, wherein Ni1 adatom is located at the oxygen vacation, and Ni2 sites are in between Ni1 and one nearby 2cO site. Moreover, Ni2 atom preferred a slight displacement to the left of Ni1 atom thus Ni2 atom has no interaction with its neighboring right 3cO atom. The Ni1–Ni2 bond length was 2.27 Å, which is much shorter than that of Ni2 on a perfect surface, but is longer than the Ni–Ni bond (2.09 Å) in the gas-phase. This indicates stronger interaction of Ni1 and Ni2 in defective surface than in the perfect surface. The adsorption energy of Ni2,A was 3.81 eV, which is higher by about 0.80 eV than that of the Ni dimer on a perfect surface. However, this most stable Ni dimer structure adsorbed on the defective TiO2(101) surface is different from the adsorbed Au dimer and Pd dimer,22,33 but similar with the Pt dimer.27
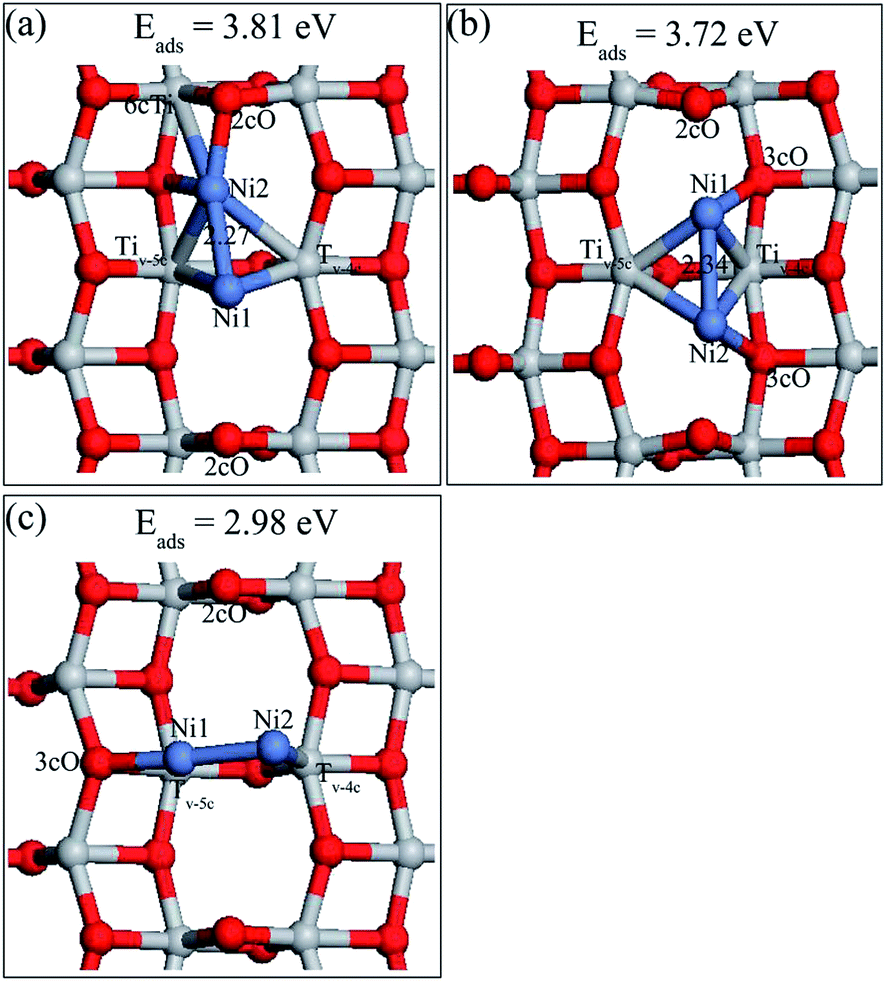 | ||
| Fig. 6 Adsorption structures and energies of Ni dimer on the defective anatase TiO2(101) surface. (a) Ni2,A; (b) Ni2,B; (c) Ni2,C. | ||
3.3 Ni3 adsorption on the perfect and defective surface
Two kinds of Ni trimer structures at the gas phase were considered (i.e., linear and triangular configurations) as the initial states absorbed on the perfect surface. The configurations of Ni3,a, Ni3,b, Ni3,c, Ni3,d as well as their adsorption energies were determined in Fig. 7. The description of Ni3,b, Ni3,c and Ni3,d are shown in ESI.† Ni3,a, the most stable adsorption configurations, was obtained by adding a third Ni adatom to the Ni2,b structure, in which two Ni atoms symmetrically occupied 2cO–6cTi–3cO bridge sites. The third Ni atom was bound at the 2cO-bridge site and the plane formed by these three Ni atoms was almost parallel with the TiO2 terrace. The Ni1–Ni2, Ni1–Ni3 and Ni2–Ni3 bond lengths were 2.50, 2.45 and 2.46 Å, respectively. The Ni1–Ni2 bond parallel to the 2cO–2cO bond was slightly longer than the other two Ni–Ni bonds, but shorter compared to the dimer Ni2,d, indicating Ni3 adatom strengthens the interaction between Ni1 and Ni2 atoms. The stability order of these Ni3 trimers on the perfect anatase TiO2(101) surface further demonstrated that the 2cO–6cTi–3cO bridge site was much more active than the 2cO–5cTi bridge site for Ni cluster adsorption. | ||
| Fig. 7 Top (left) and side views (right) of the stable structures and energies for Ni trimer adsorption on the perfect anatase TiO2(101) surface: (a) Ni3,a; (b) Ni3,b; (c) Ni3,c; (d) Ni3,d. | ||
Various possible Ni3 adsorptions (triangular and linear) on the defective anatase TiO2(101) surface were investigated. Four Ni3 stable adsorption structures (Ni3,A, Ni3,B, Ni3,C and Ni3,D) as well as their adsorption energies were determined and they are shown in Fig. 8. The description of Ni3,C and Ni3,D are shown in ESI.† Ni3,A and Ni3,B were the most stable structures with similar adsorption energies (4.79 and 4.74 eV, respectively). Ni3,A resulted from relaxing initial structures constructed from adding a third Ni atom over the Tiv-5c atom of Ni2,B. In this structure, the Ni3 atom rose above the surface with Ni3–Tiv-4c distance of 2.35 Å, which made the triangle plane almost perpendicular to the terrace of TiO2(101) surface. The Ni1–Ni2, Ni1–Ni3, and Ni2–Ni3 bond lengths were 2.46, 2.28 and 2.26 Å, respectively. Ni3,B was obtained based on Ni2,B by adding a third atom on the 3cO and Tiv-5c bridge site. The Ni3 triangle was almost parallel to the TiO2(101) terrace and the Ni1–Ni2, Ni1–Ni3, and Ni2–Ni3 lengths were 2.46, 2.29 and 2.29 Å, respectively. In contrast to Ni2,A dimer, the Ni1–Ni2 length was elongated by 0.19 Å with the Ni3 atom in both Ni3,A and Ni3,B structure.
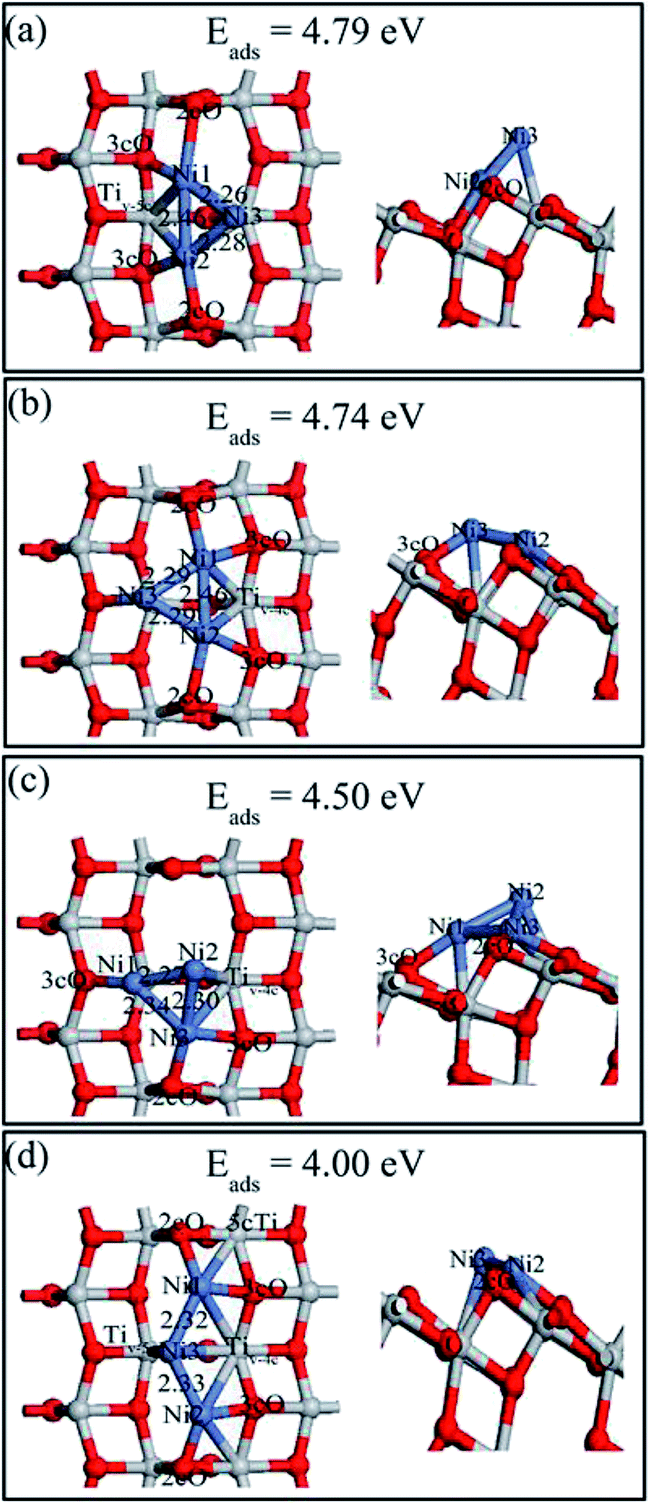 | ||
| Fig. 8 Top (left) and side views (right) of the stable structures and energies for Ni trimer adsorption on the defective anatase TiO2(101) surface: (a) Ni3,A; (b) Ni3,B; (c) Ni3,C and (d) Ni3,D. | ||
3.4 Ni4, Ni5, and Ni6 adsorptions on the perfect and defective surface
Both 2D and 3D structures of Nin (n = 4, 5, and 6) and their different possible adsorption sites on the perfect anatase TiO2(101) surface have been searched. Three stable Ni4 configurations and the most stable Ni5 and Ni6 structures, and their adsorption energies are shown in Fig. 9. The description of Ni4,b and Ni4,c are shown in ESI.† Ni4 cluster most preferred a tetrahedral structure (Fig. 8a) by adding the fourth Ni atom on top of the triangle Ni plane based on the configuration of Ni3,c. Interestingly, the Ni trimer active sites grown to Ni tetrahedron on the perfect anatase TiO2(101) surface was shifted. Ni3 would likely grow based on the 2cO–6cTi–3cO bridge site while Ni4 prefered nucleation at the 2cO–5cTi bridge site. The Ni5 and Ni6 clusters both preferred 3D geometries (Ni5,a and Ni6,a) than 2D structures and their last atom was introduced based on the Nin−1 geometry.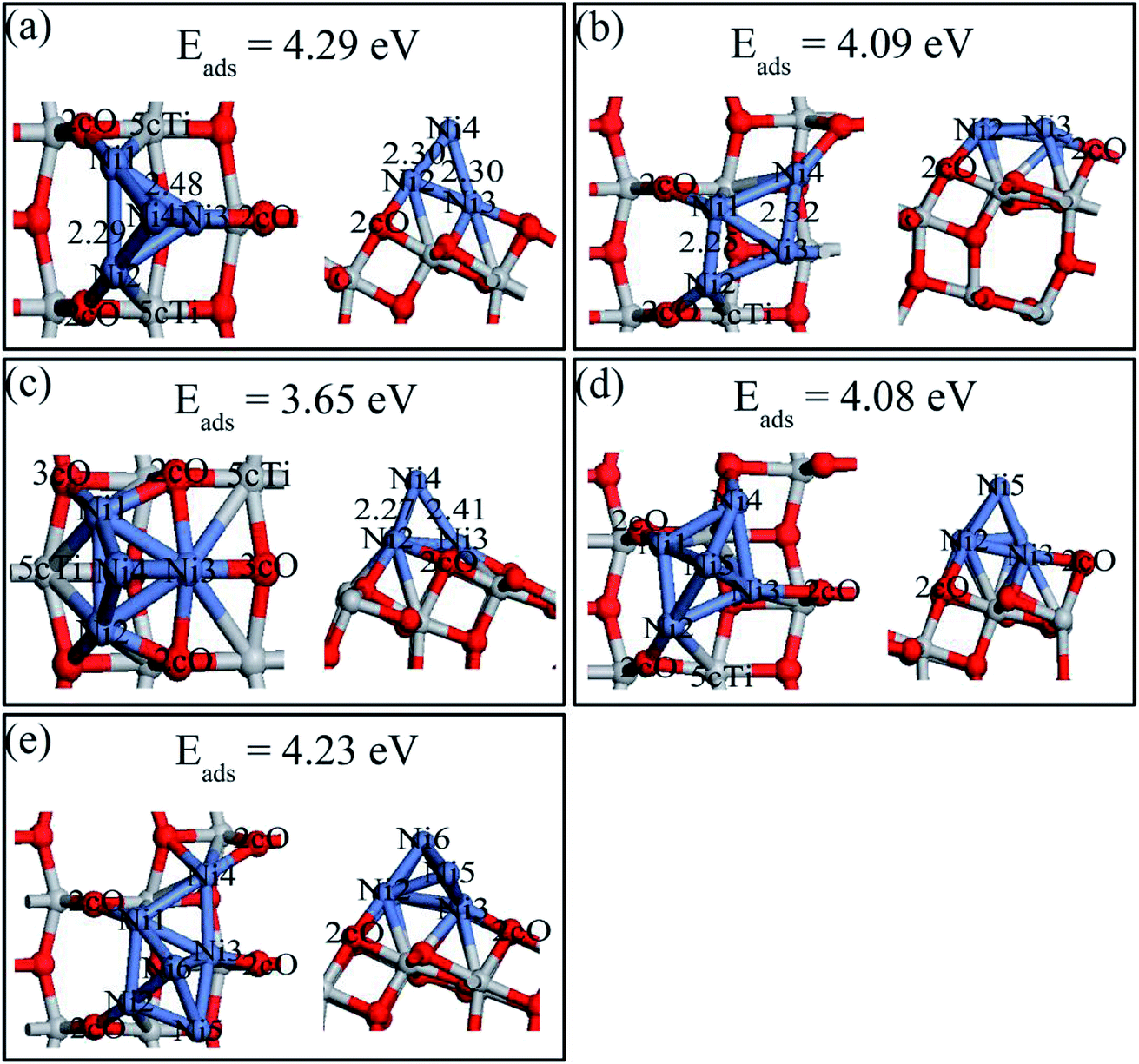 | ||
| Fig. 9 Top (left) and side views (right) of the stable structures and energies for Ni4, Ni5 and Ni6 adsorption on the perfect anatase TiO2(101) surface: (a) Ni4,a; (b) Ni4,b; (c) Ni4,c; (d) Ni5,a; (e) Ni6,a. | ||
Various possible 2D and 3D Nin (n = 4, 5, and 6) cluster structures adsorbed on the defective anatase TiO2(101) surface, were considered and only the most stable geometries with their adsorption energies are shown in Fig. 10. For Ni4,A, the geometry can be treated as a combination of Ni3,A and Ni3,B configurations in which Ni1 and Ni2 atoms almost symmetrically distribute two sides of the oxygen vacancy site, Ni3 atom bridges the 3cO and Tiv-5c and Ni4 atom binds with Tiv-4c. In Ni5,A configuration, the fifth Ni atom was added right above the Ni1, Ni3, and Ni4 triangular plane. In Ni6,A, Ni4 and Ni5 atoms symmetrically occupied two sides of Tiv-4c while other atoms were nearly distributed at the same sites as those of Ni4,A. Nin (n = 4, 5, and 6) clusters on the defective surface also tended to present 3D geometries. As atom number increased, the Nin cluster transformed from 2D (n < 4) to 3D geometry (n = 4, 5, and 6) on both perfect and defective anatase TiO2(101) surfaces, which has similar trend to that of Pd and Ru clusters on the TiO2(101) surface.33,46
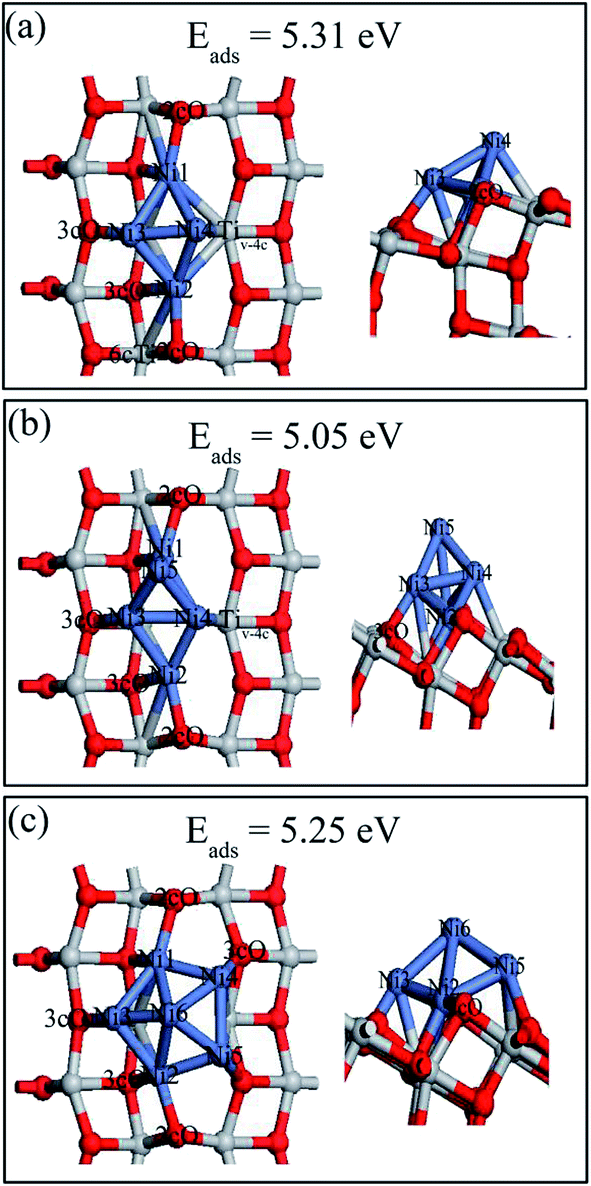 | ||
| Fig. 10 Top (left) and side views (right) of the stable structures and energies for Ni4, Ni5 and Ni6 adsorption on the defective anatase TiO2(101) surface: (a) Ni4,A; (b) Ni5,A; (c) Ni6,A. | ||
3.5 Ni cluster nucleation and growth rule on anatase TiO2(101) surface
According to the various geometry of Nin (n = 1–6) stable adsorption on the perfect and defective TiO2(101) surfaces, we made some conclusions: a single Ni atom prefers to adsorb at the 2cO-bridge site on the perfect TiO2(101) surface, and 3cO-bridge site on the defective TiO2(101) surface, whereas the oxygen vacancy is the most active site for Au, Pt and Pd deposition on the defective surface.22,27,33 Furthermore, a single Ni atom prefers 2cO-bridge site rather than 2cO–5cTi bridge site, and the Ni atoms of Ni2 and Ni3 clusters both prefer to occupy the 2cO-bridge site or 2cO–6cTi–3cO bridge site on the perfect TiO2(101) surface. However, when n was greater than four, the Nin growth was based on the 2cO–5cTi bridge site. For Nin (n = 1–6) clusters on the defective TiO2(101) surface, the nucleation site was constant with Ni atoms bridging two 3cO atoms. Like Pd and Ru clusters,33,46 the adsorbed Ni3 clusters prefer to form planar triangles and the adsorbed Ni4, Ni5 and Ni6 clusters tend to three-dimensional structures on both perfect and defective TiO2 surfaces.To further understand the rule and mechanism of Nin clusters nucleation on the TiO2(101) surface, the cohesive energy (Ecoh) is defined in this paper in addition to Eads. The definition of the cohesive energy of adsorbed Ni clusters is as follows:46
| Ecoh = −[E(Nin/TiO2) − E(TiO2) − nE(Ni)]/n | (2) |
 | ||
| Fig. 11 Cohesive energy of Ni, Au and Pt on the perfect and defective anatase TiO2(101) surfaces at different cluster sizes. | ||
What then is the driving force for the growth of Ni clusters? Exploring the definition essence of the cohesive energy, Ni–Ni and Ni–TiO2 interaction jointly dominated the nucleation behavior of Nin clusters. Therefore, we can divide the cohesive energy (Ecoh) of adsorbed Ni cluster at the perfect and defective TiO2(101) surface into two parts: one is ENi–TiO2 and the other one is ENi–Ni. In addition, the average binding energy of isolated Nin (n = 2–6) clusters, Ebind (Nin), was introduced. The definition of ENi–TiO2, ENi–Ni (ref. 33) and Ebind (Nin)48 are as follows:
| E(Ni–TiO2) = −[E(Nin/TiO2) − E(TiO2) − E(Nin*)]/n | (3) |
| E(Ni–Ni) = Ecoh − ENi–TiO2 | (4) |
| Ebind(Nin) = −[E(Nin) − n × E(Ni)]/n | (5) |
The variation of ENi–TiO2 and ENi–Ni of bare and adsorbed Nin clusters with the cluster sizes are shown in Fig. 12. The interaction between a single Ni atom and both the perfect and defective surface was the strongest and then upon Ni adatom addition, the interactions both gradually weakened. However, the Ni–Ni interactions began to increase from zero to one with Ni atoms in both the perfect and defective surfaces. For Ni1 and Ni2 clusters absorbed on the perfect and defective surface, the Ni–TiO2 interaction was much larger than the Ni–Ni interaction, indicating that the Ni–TiO2 interaction was the main driving force at the initial stage of Ni nucleation. With regard to Ni4, Ni5 and Ni6 clusters, the Ni–Ni interactions have greatly exceeded the Ni–TiO2 interaction, showing that the Ni–Ni interaction begins to control the growth process of Nin clusters as the cluster size gets larger. This is why Ni1, Ni2 and Ni3 clusters preferably occupy the active sites like 2cO-bridge site or 2cO–6cTi–3cO bridge site, whereas Ni4, Ni5 and Ni6 tend to aggregate at the inactive 2cO–5cTi bridge site to reduce the Ni–TiO2 interaction. The Ni–Ni interaction for Ni2 and Ni3 clusters on the perfect surface was stronger than on the defective surface, while that for Ni4, Ni5 and Ni6 clusters were nearly equal on these two kinds of surfaces. Compared to bare clusters, the perfect surface weakens the strength of Ni–Ni interaction to a certain degree, whereas the strength would not be affected by the defective surface.
 | ||
| Fig. 12 Variation of ENi–TiO2 and ENi–Ni with the cluster sizes of Ni on the perfect and defective anatase TiO2(101) surface as well as ENi–Ni for bare Ni. | ||
4. Conclusion
First principles DFT calculations were carried out to investigate the energetically stable adsorption structures of Nin (n = 1–6) clusters at both perfect and defective anatase TiO2(101) surface and further disclose the nucleation and growth rule of Ni clusters on these two kinds of surfaces. Our results show that a single Ni atom prefers to adsorb at the 2cO-bridge site on the perfect TiO2(101) surface, and 3cO-bridge site on the defective TiO2(101) surface (not the oxygen vacancy). The active site for Ni cluster growth on the perfect TiO2(101) surface shifted from the bridge site of two 2cO atoms or 2cO–6cTi–3cO bridge site for Ni1, Ni2 and Ni3 clusters to the 2cO–5cTi bridge site for Ni4, Ni5 and Ni6. The adsorbed Ni3 clusters prefer to form planar triangles and the adsorbed Ni4, Ni5 and Ni6 clusters tend to 3D structures on both perfect and defective TiO2 surfaces.The cohesive energy of Ni cluster was almost kept constant with variation of the cluster sizes on both the perfect and defective surface. Moreover, the clustering energy on the defective surface was larger than that on the perfect surface. The Ni–TiO2 interaction was the main driving force at the initial stage of Ni nucleation and the Ni–Ni interaction began to control the growth process of Nin clusters as the cluster size gets larger.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (NSFC, Grant no. 11304208), the National Natural Science Funds of China (NSFC, Grant no. U1203293) and the Ph.D. Programs Foundation of the Xinjiang Production and Construction Corps (no. 2013BB010).References
- V. E. Henrich and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University Press, Cambridge, UK, 1994 Search PubMed.
- P. Castillo-Villalon and J. Ramirez, Spectroscopic Study of the Electronic Interactions in Ru/TiO2 HDS Catalysts, J. Catal., 2009, 268, 39–48 CrossRef CAS PubMed.
- J. C. Kang, S. L. Zhang, Q. H. Zhang and Y. Wang, Ruthenium Nanoparticles Supported on Carbon Nanotubes as Efficient Catalysts for Selective Conversion of Synthesis Gas to Diesel Fuel, Angew. Chem., Int. Ed., 2009, 48, 2565–2568 CrossRef CAS PubMed.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skala, A. Bruix, F. Illas and K. C. Prince, et al., Support Nanostructure Boosts Oxygen Transfer to Catalytically Active Platinum Nanoparticles, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- J. Sa, A. Goguet, S. F. R. Taylor, R. Tiruvalam, C. J. Kiely, M. Nachtegaal, G. J. Hutchings and C. Hardacre, Influence of Methyl Halide Treatment on Gold Nanoparticles Supported on Activated Carbon, Angew. Chem., Int. Ed., 2011, 50, 8912–8916 CrossRef CAS PubMed.
- F. Wang, S. T. Zhang, C. M. Li, J. Liu, S. He, Y. F. Zhao, H. Yan, M. Wei, D. G. Evans and X. Duan, Catalytic behavior of supported Ru nanoparticles on the (101) and (001) facets of anatase TiO2, RSC Adv., 2014, 4, 10834–10840 RSC.
- C. Torres, C. Campos, J. L. G. Fierro, M. Oportus and P. Reyes, Nitrobenzene Hydrogenation on Au/TiO2 and Au/SiO2 Catalyst: Synthesis, Characterization and Catalytic Activity, Catal. Lett., 2013, 143, 763–771 CrossRef CAS PubMed.
- K. Sun, M. Kohya, S. Tanaka and S. Takeda, A Study on the Mechanism for H2 Dissociation on Au/TiO2 Catalysts, J. Phys. Chem. C, 2014, 118, 1611–1617 CAS.
- Y. G. Wang, Y. Yoon, V. A. Glezakou, J. Li and R. Rousseau, The Role of Reducible Oxide–Metal Cluster Charge Transfer in Catalytic Processes: New Insights on the Catalytic Mechanism of CO Oxidation on Au/TiO2 from Ab Initio Molecular Dynamics, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS PubMed.
- I. X. Green, W. J. Tang, M. Neurock and J. T. Yates, Insights into Catalytic Oxidation at the Au/TiO2 Dual Perimeter Sites, Acc. Chem. Res., 2014, 47, 805–815 CrossRef CAS PubMed.
- H. Einaga, N. Urahama, A. Tou and Y. Teraoka, CO Oxidation Over TiO2-Supported Pt–Fe Catalysts Prepared by Coimpregnation Methods, Catal. Lett., 2014, 144, 1653–1660 CrossRef CAS PubMed.
- J. G. Yu, L. F. Qi and M. Jaroniec, Hydrogen Production by Photocatalytic Water Splitting over Pt/TiO2 Nanosheets with Exposed (001) Facets, J. Phys. Chem. C, 2010, 114, 13118–13125 CAS.
- P. A. DeSario, J. J. Pietron, D. E. DeVantier, T. H. Brintlinger, R. M. Stroudb and D. R. Rolison, Plasmonic Enhancement of Visible-Light Water Splitting with Au–TiO2 Composite Aerogels, Nanoscale, 2013, 5, 8073–8083 RSC.
- F. Wu, X. Y. Hu, J. Fan, E. Z. Liu, T. Sun, L. M. Kang, W. Q. Hou, C. J. Zhu and H. C. Liu, Photocatalytic Activity of Ag/TiO2 Nanotube Arrays Enhanced by Surface Plasmon Resonance and Application in Hydrogen Evolution by Water Splitting, Plasmonics, 2013, 8, 501–508 CrossRef CAS.
- H. Z. Cheng and A. Selloni, Surface and Subsurface Oxygen Vacancies in Anatase TiO2 and Differences with Rutile, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 092101 CrossRef.
- C. H. Sun and S. C. Smith, Strong Interaction Between Gold and Anatase TiO2(001) Predicted by First Principle Studies, J. Phys. Chem. C, 2012, 116, 3524–3531 CAS.
- E. Lira, J. Hansen, L. R. Merte, P. T. Sprunger, Z. S. Li, F. Besenbacher and S. Wendt, Growth of Ag and Au Nanoparticles on Reduced and Oxidized Rutile TiO2(110) Surfaces, Top. Catal., 2013, 56, 1460–1476 CrossRef CAS.
- L. Zhang, F. Cosandey, R. Persaud and T. E. Madey, Initial Growth and Morphology of Thin Au Films on TiO2(110), Surf. Sci., 1999, 439, 73–85 CrossRef CAS.
- R. P. Galhenage, H. Yan, S. A. Tenney, N. Park, G. Henkelman, P. Albrecht, D. R. Mullins and D. A. Chen, Understanding the Nucleation and Growth of Metals on TiO2: Co Compared to Au, Ni, and Pt, J. Phys. Chem. C, 2013, 117, 7191–7201 CAS.
- R. P. Galhenage, S. C. Ammal, H. Yan, A. S. Duke, S. A. Tenney, A. Heyden and D. A. Chen, Nucleation, Growth, and Adsorbate-Induced Changes in Composition for Co–Au Bimetallic Clusters on TiO2, J. Phys. Chem. C, 2012, 116, 24616–24629 CAS.
- Y. J. Chen, G. H. Tian, K. Pan, C. G. Tian, J. Zhou, W. Zhou, Z. Y. Ren and H. G. Fu, In Situ Controlled Growth of Well-Dispersed Gold Nanoparticles in TiO2 Nanotube Arrays as Recyclable Substrates for Surface-Enhanced Raman Scattering, Dalton Trans., 2012, 41, 1020–1026 RSC.
- A. Vittadini and A. Selloni, Small Gold Clusters on Stoichiometric and Defected TiO2 Anatase (101) and their Interaction with CO: A Density Functional Study, J. Chem. Phys., 2002, 117, 353 CrossRef CAS PubMed.
- X. Y. Pan and Y. J. Xu, Defect-Mediated Growth of Noble-Metal (Ag, Pt, and Pd) Nanoparticles on TiO2 with Oxygen Vacancies for Photocatalytic Redox Reactions under Visible Light, J. Phys. Chem. C, 2013, 117, 17996–18005 CAS.
- X. Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, Small Au and Pt Clusters at the Anatase TiO2(101) Surface: Behavior at Terraces, Steps, and Surface Oxygen Vacancies, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS PubMed.
- Y. Zhou, C. L. Muhich, B. T. Neltner, A. W. Weimer and C. B. Musgrave, Growth of Pt Particles on the Anatase TiO2(101) Surface, J. Phys. Chem. C, 2012, 116, 12114–12123 CAS.
- Y. Han, C. J. Liu and Q. F. Ge, Interaction of Pt Clusters with the Anatase TiO2(101) Surface: A First Principles Study, J. Phys. Chem. B, 2006, 110, 7463–7472 CrossRef CAS PubMed.
- Y. Han, C. J. Liu and Q. F. Ge, Effect of Surface Oxygen Vacancy on Pt Cluster Adsorption and Growth on the Defective Anatase TiO2(101) Surface, J. Phys. Chem. B, 2007, 111, 16397–16404 CAS.
- C. Xu, X. Lai, G. W. Zajac and D. W. Goodman, Scanning Tunneling Microscopy Studies of the TiO2(110) Surface: Structure and the Nucleation Growth of Pd, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13464–13482 CrossRef CAS.
- M. D. Negra, N. M. Nicolaisen, Z. Li and P. J. Moller, Study of the Interactions between the Overlayer and the Substrate in the Early Stages of Palladium Growth on TiO2(110), Surf. Sci., 2003, 540, 117–128 CrossRef.
- W. N. Zhao, H. X. Lin, Y. Li, Y. F. Zhang, X. Huang and W. K. Chen, Growth mechanism of palladium clusters on rutile TiO2(110) surface, J. Nat. Gas Chem., 2012, 21, 544–555 CrossRef CAS.
- S. V. Ong and S. N. Khanna, Theoretical Studies of the Stability and Oxidation of Pdn (n = 1–7) Clusters on Rutile TiO2(110): Adsorption on the Stoichiometric Surface, J. Phys. Chem. C, 2012, 116, 3105–3111 CAS.
- J. F. Sanz and A. Marquez, Adsorption of Pd Atoms and Dimers on the TiO2 (110) Surface: A First Principles Study, J. Phys. Chem. C, 2007, 111, 3949–3955 CAS.
- J. L. Zhang, M. Zhang, Y. Han, W. Li, X. K. Meng and B. N. Zong, Nucleation and Growth of Palladium Clusters on Anatase TiO2(101) Surface: A First Principle Study, J. Phys. Chem. C, 2008, 112, 19506–19515 CAS.
- J. Zhou, Y. C. Kang and D. A. Chen, Controlling Island Size Distributions: A Comparison of Nickel and Copper Growth on TiO2(110), Surf. Sci. Lett., 2003, 537, L429–L434 CrossRef CAS.
- R. E. Tanner, K. Goldfarb, M. R. Castell and G. A. D. Briggs, The Evolution of Ni Nanoislands on the Rutile TiO2(110) Surface with Coverage, Heating and Oxygen Treatment, Surf. Sci., 2001, 486, 167–184 CrossRef CAS.
- K. Fujikawa, S. Suzuki, Y. Koike, W. J. Chun and K. Asakura, Self-Regulated Ni Cluster Formation on the TiO2(110) Terrace Studied Using Scanning Tunneling Microscopy, Surf. Sci., 2006, 600, L117–L121 CrossRef CAS PubMed.
- P. L. Cao, D. E. Ellis and V. P. Dravid, First-principles study of initial stage of Ni thin-film growth on a TiO2(110) surface, J. Mater. Res., 1999, 14, 3684–3689 CrossRef CAS.
- J. Xiong, J. X. Chen and J. Y. Zhang, Liquid-Phase Hydrogenation of o-Chloronitrobenzene over Supported Nickel Catalysts, Catal. Commun., 2007, 8, 345–350 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculation Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficiency of Ab Initio Total Energy Calculation for Metals and Semi conductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, Molecules, Solids, and Surfaces: Applications of the Generalized Gradient Approximation for Exchange and Correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved Grid-Based Algorithm for Bader Charge Allocation, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. A. Jonsson, Fast and Robust Algorithm for Bader Decomposition of Charge Density, Comput. Mater. Sci., 2006, 36, 354–460 CrossRef PubMed.
- S. T. Zhang, C. M. Li, H. Yan, M. Wei, D. G. Evans and X. Duan, Density Functional Theory Study on the Metal–Support Interaction between Ru Cluster and Anatase TiO2(101) Surface, J. Phys. Chem. C, 2014, 118, 3514–3522 CAS.
- L. G. V. Briquet, C. A. R. Catlow and S. A. French, Comparison of the Adsorption of Ni, Pd, and Pt on the (0001) Surface of α-Alumina, J. Phys. Chem. C, 2008, 112, 18948–18954 CAS.
- S. Nigam and C. Majumder, Growth Pattern of Agn (n = 1–8) Clusters on the α-Al2O3(0001) Surface: A First Principles Study, Langmuir, 2010, 26, 18776–18787 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13975e |
| This journal is © The Royal Society of Chemistry 2015 |