
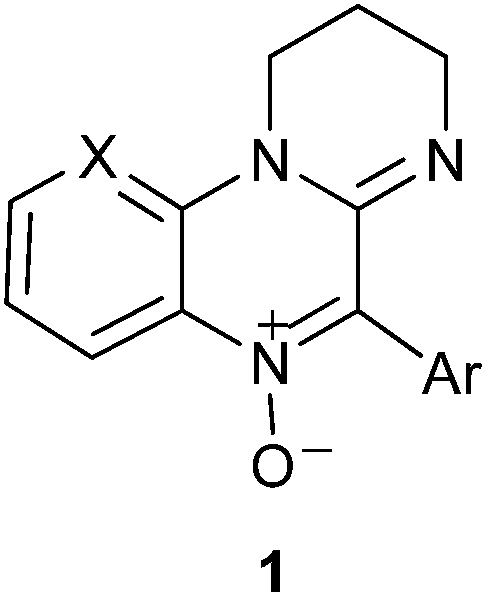
Amidinoquinoxaline N-oxides as novel spin traps†
Nadia Gruberabc,
Lidia L. Piehlb,
Emilio Rubin de Celisb,
Jimena E. Díaza,
María B. Garcíaa,
Pierluigi Stipa*c and
Liliana R. Orelli*a
aDepartamento de Química Orgánica, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, CONICET, Junín 956, (1113), Buenos Aires, Argentina. E-mail: lorelli@ffyb.uba.ar; Fax: +54 11 49648252; Tel: +54 11 49648252
bCátedra de Física, Departamento de Fisicomatemática, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Junín 956 (1113), Buenos Aires, Argentina
cS.I.M.A.U. Department Chemistry Division, Università Politecnica delle Marche, via Brecce Bianche 12, I-60131 Ancona, Italy. E-mail: pstipa@univpm.it
First published on 1st December 2014
Abstract
A novel type of spin traps 1 derived from the pyrimidoquinoxaline N-oxide heterocyclic core is reported. EPR technique was used to evaluate their ability to trap methyl radicals generated in a Fenton reaction in the presence of DMSO. All the synthesized nitrones showed spin trapping properties and the corresponding nitroxides 2 were characterized by EPR. The novel spin traps showed remarkably persistent signals, as confirmed in a competition experiment with DMPO. The addition rate constants leading to the spin adducts (kadd) were determined, and very good correlations were found with steric and electronic parameters of the parent nitrones. The spin adducts decomposition rate constants (kdec) and the corresponding half-life times (t1/2) were also determined. DFT and MP2 calculations were used in order to rationalize the adducts hfcc and the structural factors influencing their addition and decomposition rates.
Introduction
Electron paramagnetic resonance (EPR) spin trapping represents one of the most specific and reliable techniques for detecting and identifying transient free radicals, such as those produced in chemical and biological processes, whose lifetime is too short in the EPR spectroscopic time scale. This technique, widely used since its introduction about 40 years ago,1 is based upon the fast reaction between a suitable diamagnetic molecule (a spin trap) and short-lived free radicals with formation of relatively long lived radicals (spin adducts), whose EPR signals are persistent enough to be recorded and analyzed. Spectral parameters such as hyperfine coupling constants (hfcc) and g-factors are generally characteristic of the type of radical initially trapped. This technique has been successfully used in biological systems, with many applications in a series of human diseases like ischemia-reperfusion syndrome, Friedreich's ataxia, atherosclerosis,2 neurodegenerative diseases3 and cellular aging.4 Nitrones (N-oxides) are very efficient spin traps,5 being able to undergo fast radical additions by C- and O-centered radicals, to yield aminoxyls (nitroxides) as spin adducts, these species being among the most persistent organic free radicals in liquid solution. Among the commercially available nitrones, N-tert-butylbenzylideneamine N-oxide (PBN) and 5,5-dimethyl-3,4-dihydro-2H-pyrrole N-oxide (DMPO)6 are the most popular, but their use is not without limitations. For example, PBN and its analogues give spin adducts with similar EPR spectra generally consisting of a triplet of doublets with a relatively small variation in the doublet splitting depending on the radical trapped, and this may be a source of misinterpretations in spin trapping experiments.7 On the other hand, the use of DMPO is limited by its sensitivity to nucleophilic attack by water and the relatively low stability of its superoxide spin adduct, which decomposes rapidly.8 A continuous effort has been devoted to the synthesis of PBN and DMPO analogues9 and of other nitrones10 to be used in spin trapping experiments without the above mentioned drawbacks. To this end, we synthesized a series of 5-aryl-2,3-dihydro-1H-pyrimido[1,2-a]quinoxaline 6-oxides 1a–h and a fused pyridyl analogue 1i and evaluated their spin trapping ability by EPR spectroscopy. 2,3-Dihydro-1H-pyrimido[1,2-a]quinoxaline N-oxides represent a heterocyclic core of interest due to its pharmacological properties. Some suitably substituted derivatives possess antineoplastic activity,11 especially against hypoxic tumours. Other pyrimidoquinoxaline 6-oxides have been employed as antiamoebic12 and antianaerobic agents.13 As part of our research on nitrogen heterocycles, we reported a novel methodology for their synthesis,14 and investigated some of their chemical,15 spectroscopic16 and pharmacological17 properties.Two structural features of these compounds which could both have a stabilizing effect on the corresponding spin adducts prompted us to test these nitrones as potential spin traps: the absence of hydrogens α- to the N-oxide function and the presence of a conjugated amidine moiety. In fact, it is known that aminoxyls bearing α-hydrogens are unstable and may disproportionate to the corresponding nitrone and a hydroxylamine,18 while it has been observed that electron-withdrawing substituents close to the N–O function could stabilize spin adducts.9e,19
Taking advantage of recent developments in the field,20 in the present paper we also present DFT21 and MP2 calculations which were able to describe the EPR features of the novel spin adducts as well as to rationalise some trends concerning their formation and decomposition rates.
Results and discussion
The compounds under study are shown in Table 1, and were chosen so as to include 5-aryl groups with variable stereoelectronic features or the presence of a strongly electron withdrawing group in the fused ring, which might in turn stabilize the resulting spin adduct.
|
||
|---|---|---|
| Compound 1 | Ar | X |
| a | C6H5 | CH |
| b | 4-OCH3C6H4 | CH |
| c | 4-ClC6H4 | CH |
| d | 4-NO2C6H4 | CH |
| e | 2-OCH3C6H4 | CH |
| f | 2-ClC6H4 | CH |
| g | 2-IC6H4 | CH |
| h | 3-C4H3S | CH |
| i | C6H5 | N |
We had previously described compounds 1a–d, f.14,16 The remaining compounds were prepared by a modification of our previously reported method.14
All compounds showed spin trap properties in a Fenton reaction system in the presence of DMSO, where methyl radicals are formed,22 in agreement with Scheme 1. This system, involving the presence of oxygen and aqueous medium, was chosen as a first approximation to biocompatible conditions.
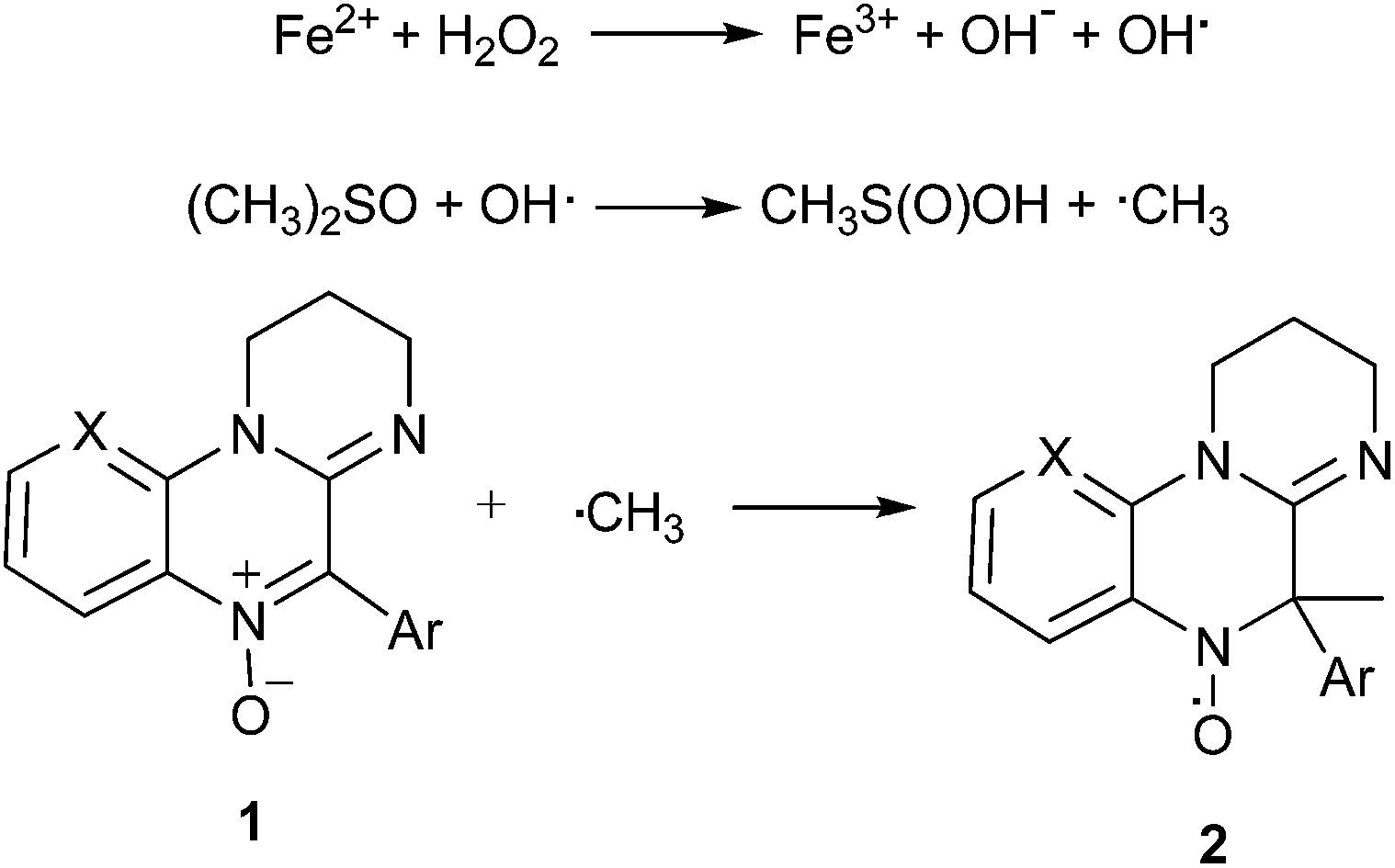 | ||
| Scheme 1 Spin-trapping reaction. | ||
For the sake of comparison, DMPO adducts were also obtained with the same trapping Fenton reaction system (Fig. 1a). The simulated spectrum of DMPO adduct (Fig. 1b) was compatible with the DMPO/CH3 adduct (aN: 16.1 Gauss, aH: 22.8 Gauss, percentage > 95).
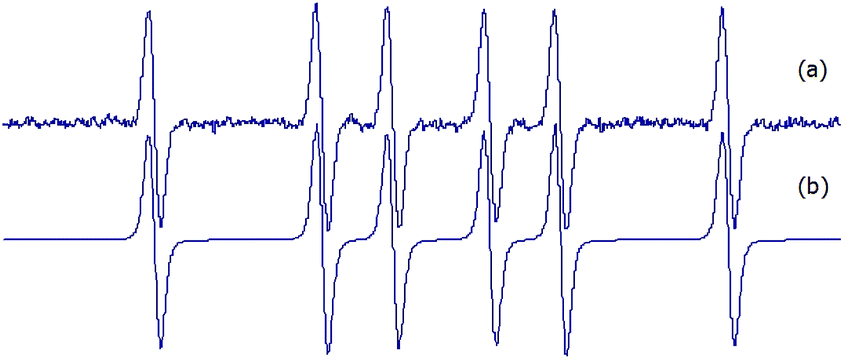 | ||
| Fig. 1 (a) Experimental and (b) simulated EPR spectrum of DMPO (30 mM final concentration) adduct obtained by addition of the spin trap to methyl radicals produced by a Fenton reaction (1.33 mM FeSO4 plus 3.33 mM H2O2, final concentrations) in PBS pH 7.6 containing 33% DMSO. | ||
Spin adducts 2 displayed similar EPR spectra and were characterized by simulation, showing a typical EPR trace with 9 resolved peaks. Fig. 2 shows the EPR spectrum of 2a as a representative example.
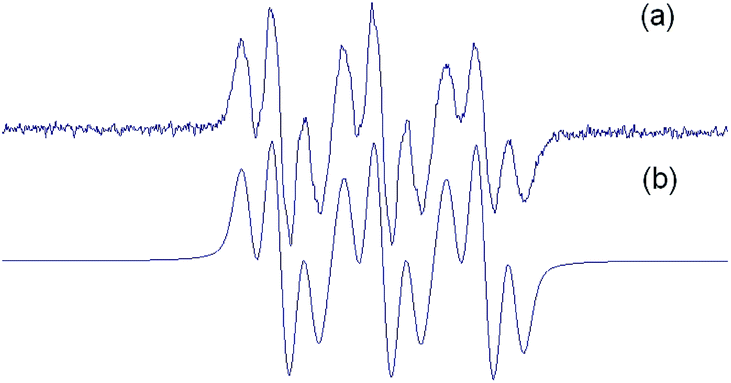 | ||
| Fig. 2 (a) Experimental and (b) simulated EPR spectrum of the spin adduct of 1a (30 mM final concentration) obtained by addition of methyl radicals produced by a Fenton reaction (1.33 mM FeSO4 plus 3.33 mM H2O2, final concentrations) in PBS pH 7.6 containing 33% DMSO. | ||
In general, the spin density in aminoxyls is mainly located on the N–O group, with typical N-hfcc values of 14–15 G for alkyl aminoxyls23 and of 10–11 G for aryl aminoxyls.24 Consistently, the N-hfcc for the nitroxides under study also display values of 10–11 G, which can be attributed to the extensive electron delocalization in this system, mainly in the fused aromatic ring. A plot of spin density in nitroxide 2a (Fig. 3) illustrates this feature.
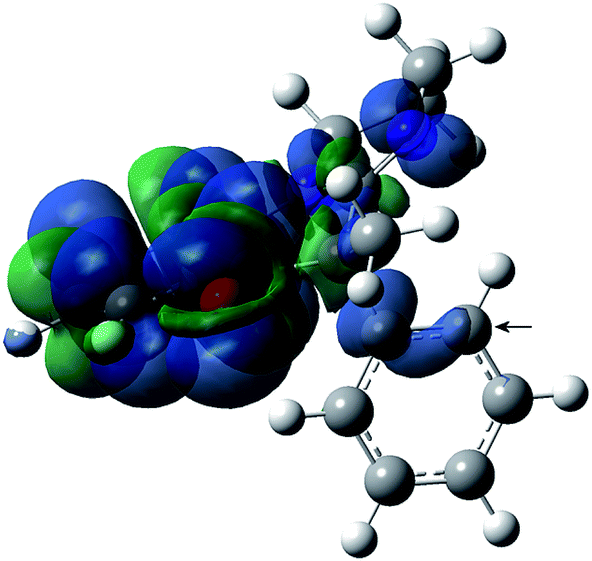 | ||
| Fig. 3 Spin density distribution (α–β) of nitroxide 2a computed at the B3LYP/EPR-III level (positive values in blue and negative in green). C-24 has been arrowed (see text). | ||
This is in accordance with previous findings in other heteroaromatic analogues, such as indolinonic25 and benzoxazinic nitroxides.26 In line with this, the EPR spectra of nitroxides 2 were interpreted on the basis of hfcc typical of this kind of radicals, in which the nitrogen three lines are mainly split by two different couples of aromatic hydrogens of the heteroaromatic ring moiety (Fig. 4), H(13) and H(15) with a larger hfcc (ca. 3 Gauss) and H(12) and H(14) with a smaller one (ca. 1 Gauss). In addition, the splitting of the amidine ring nitrogen N-21 was considered. The assignments, confirmed by means of appropriate DFT calculations previously described,25 are reported in Table 2. However, because the experimental conditions followed in our study (aqueous medium in the presence of molecular oxygen) did not allow us to achieve a better spectral resolution than that shown in Fig. 2, the hfcc values given in the last two columns of Table 2 are tentative and mainly based on DFT computational predictions.
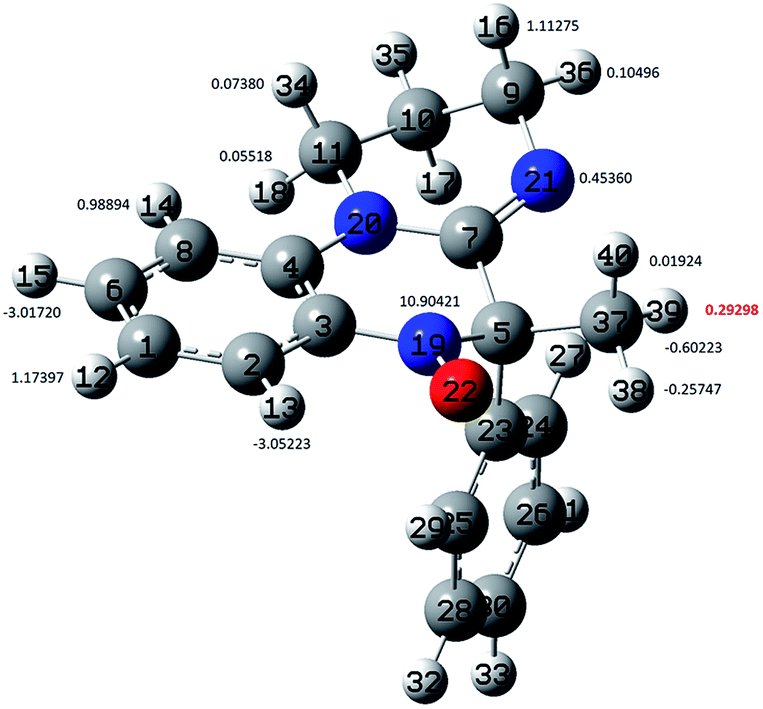 | ||
| Fig. 4 Ball and stick representation of the optimized geometry of nitroxide 2a showing the arbitrary atom numbering and the corresponding hfcc computed at the B3LYP/EPR-III level. | ||
| Nitroxide 2 | N-19 | H-13, 15 | H-12, 14 | N-21 |
|---|---|---|---|---|
| a | 10.77 | 3.13 | 0.88 | 0.29 |
| b | 10.84 | 3.12 | 0.89 | 0.23 |
| c | 10.76 | 3.08 | 0.88 | 0.31 |
| d | 10.64 | 3.07 | 0.91 | 0.41 |
| e | 10.62 | 3.09 | 0.98 | 0.57 |
| f | 10.53 | 3.08 | 0.87 | 0.47 |
| g | 10.55 | 3.04 | 0.83 | 0.67 |
| h | 10.90 | 3.15 | 0.80 | 0.24 |
| i | 10.45 | 3.48 | 0.89; 0.82 (N) | 0.34 |
The isotropic hfcc (aX) arises from the Fermi contact interaction between the unpaired electron and the nucleus (X), and is correlated to the corresponding spin density (ρX) by:
| aX = 2/3μ0geμBgXρX |
In order to evaluate the stability of the novel spin adducts, their EPR signals were followed by recording the corresponding spectra at different times until disappearance. Fig. 5 shows the time-dependent EPR signal intensity of one of the new spin traps (compound 1d) adduct. Comparatively, the DMPO/CH3 adduct was less stable as a function of time, as can be seen in Fig. 6.
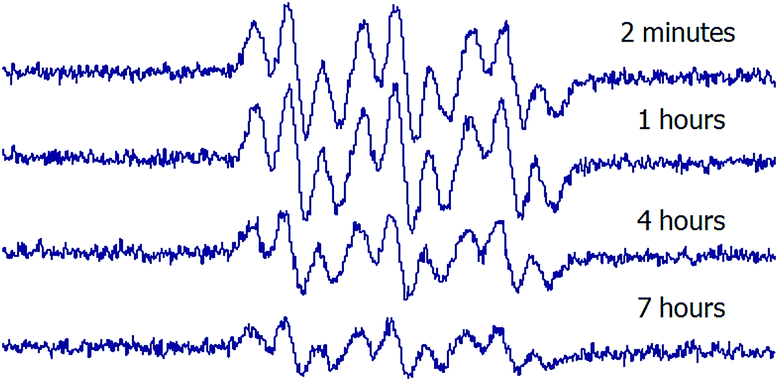 | ||
| Fig. 5 Time-dependent EPR spin adduct spectra of 1d (10 mM final concentration) upon reaction with methyl radicals produced by a Fenton reaction (1.33 mM FeSO4 plus 3.33 mM H2O2, final concentrations) in PBS pH 7.6 containing 33% DMSO. | ||
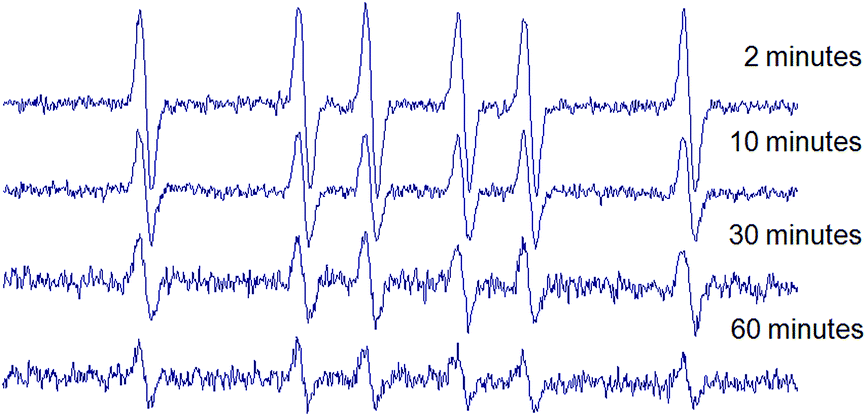 | ||
| Fig. 6 Time-dependent EPR spectra of DMPO/CH3 spin adduct. DMPO (10 mM final concentration) was tested in a Fenton reaction (1.33 mM FeSO4 plus 3.33 mM H2O2, final concentrations) in PBS pH 7.6 containing 33% DMSO. | ||
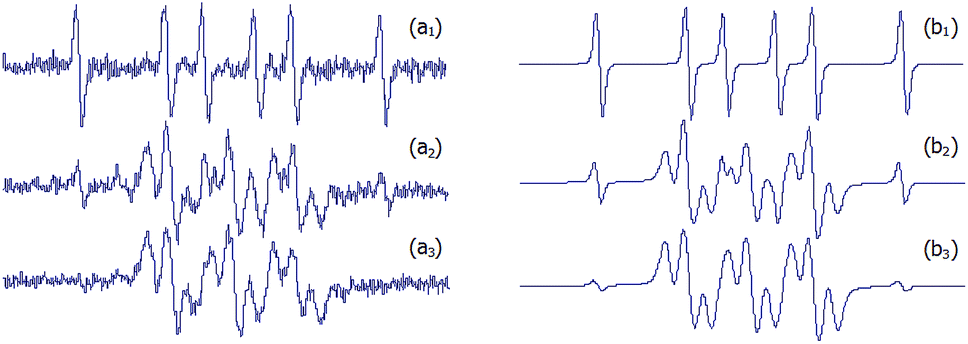 | ||
| Fig. 7 (a) Experimental and (b) simulated EPR spectra of the competition assay between DMPO and 1a adducts. FeSO4 (1.33 mM, final concentration) and H2O2 (3.33 mM, final concentration) were added to a solution containing both DMPO and compound 1a in 33% DMSO–PBS pH 7.6. Spectra a1, a2 and a3 are the experimental EPR signals obtained at 1, 15 and 30 minutes and b1, b2 and b3 are their respective simulated EPR spectra. | ||
To further investigate this difference, a competition experiment between DMPO and 1a was performed, and EPR spectra are shown in Fig. 7. This experiment reveals the persistence of the signal of the novel adducts after DMPO adduct signal disappearance. Hence, even if the newly synthesized nitrones react more slowly with methyl radicals than DMPO, the corresponding adducts resulted much more persistent. The enhanced stability of adducts 2 would result from the absence of a hydrogen α- to the N–O·group, which is the main issue concerning the stability of DMPO spin adducts.18
The kinetics of formation and decomposition of the spin adducts generated by methyl radicals addition to the new nitrones 1 were then studied. The experimental conditions were modified to prevent precipitation of some derivatives and to enhance signal intensity, using a higher proportion of DMSO and H2O2 (see Experimental). Fig. 8 shows the spin adduct formation and decomposition kinetics for amidinoquinoxaline N-oxide 1a in the trapping-Fenton reaction system.
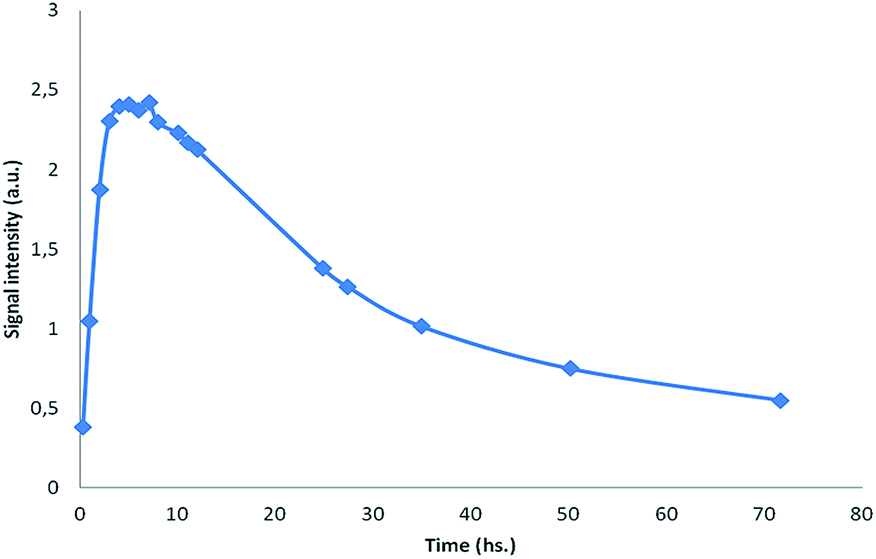 | ||
| Fig. 8 Spin adduct formation and decay kinetics of the spin trap 1a in the Fenton reaction system. | ||
Since the EPR signal recorded during the kinetic experiments was due exclusively to the aminoxyls under investigation, it was possible to use high modulation conditions to merge together all the small signal splittings; in such conditions the EPR signal was reduced to a broad triplet of triplets, which could be easily handled for kinetic measurements. The experimental addition rate constants are listed in Table 3.
| Compound 1 | kadd × 104 | Dihedral angle (Fig. 9) (°) |
|---|---|---|
| a | 2.559 | −45.29 |
| b | 3.655 | −41.33 |
| c | 3.947 | −43.66 |
| d | 3.319 | −44.86 |
| e | 0.486 | −60.26 |
| f | 0.380 | −66.31 |
| g | 0.311 | −75.55 |
| h | 1.034 | −43.38 |
| i | 0.521 | −45.70 |
The rate of the addition is low, a fact that may be attributed to steric hindrance of the 5-aryl group, which is twisted with respect to the heterocyclic core in the ground state (Fig. 9 and Table 3).
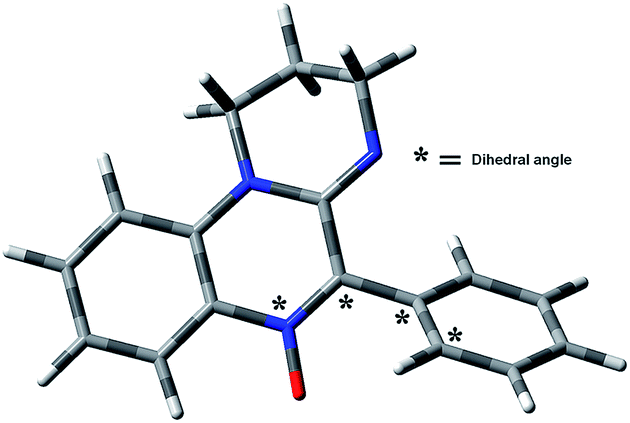 | ||
| Fig. 9 MPW1K/6-31G(d) ground state geometry of nitrone 1a. | ||
The steric hindrance is more important for the ortho substituted derivatives 1e–g, which have restricted rotation around the C5–Ar bond.16 The rate constants for this subgroup follow the order OCH3 > Cl > I, which parallels the increasing Van der Waals radii of the ortho substituents. A plot of −ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kadd vs. steric parameters (Sternhell scale28) shows an excellent linear correlation (Fig. 10). The steric volume of the ortho substituent determines the dihedral angle between the Ar group and the heterocyclic core in the ground state (Table 3), which in turn influences the steric hindrance exerted by the 5-substituent.
kadd vs. steric parameters (Sternhell scale28) shows an excellent linear correlation (Fig. 10). The steric volume of the ortho substituent determines the dihedral angle between the Ar group and the heterocyclic core in the ground state (Table 3), which in turn influences the steric hindrance exerted by the 5-substituent.
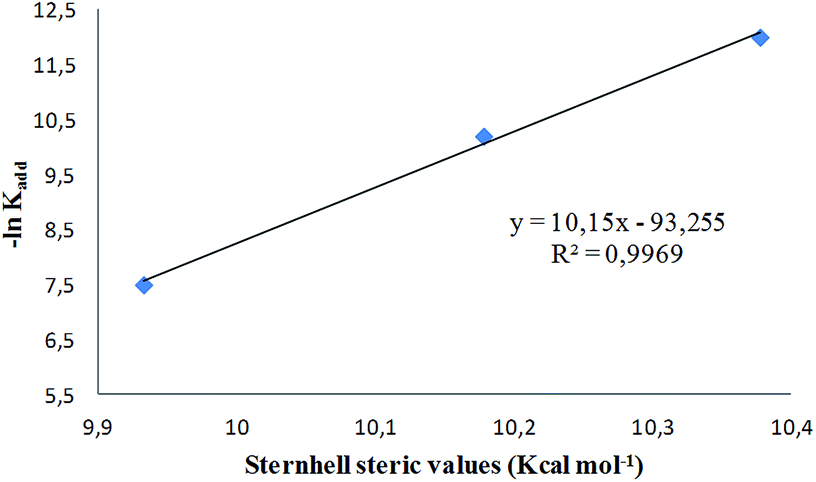 | ||
| Fig. 10 −lnkadd vs. Sternhell steric parameters. | ||
On the other hand, para substitution (compounds 1a–d) does not induce significant changes in kadd, while modifications in the fused ring (compound 1i) or in the nature of the 5-substituent (compound 1h) bring about noticeable effects on the addition rates. In order to assess the importance of electronic effects on the addition rate constants, a NBA analysis was carried out at the MP2/6-31G(d) level to compute the corresponding atomic charges at the nitrone α-carbon (Table 4). The plot of −lnkadd vs. MP2 charges, reported in Fig. 11, shows a clear dependence of the rate constant from atomic charge at the nitrone α-carbon, according to literature reports.29
| Compound 1 | C5 MP2 charge |
|---|---|
| a | 0.13710 |
| b | 0.13673 |
| c | 0.13554 |
| d | 0.13582 |
| h | 0.14087 |
| i | 0.14543 |
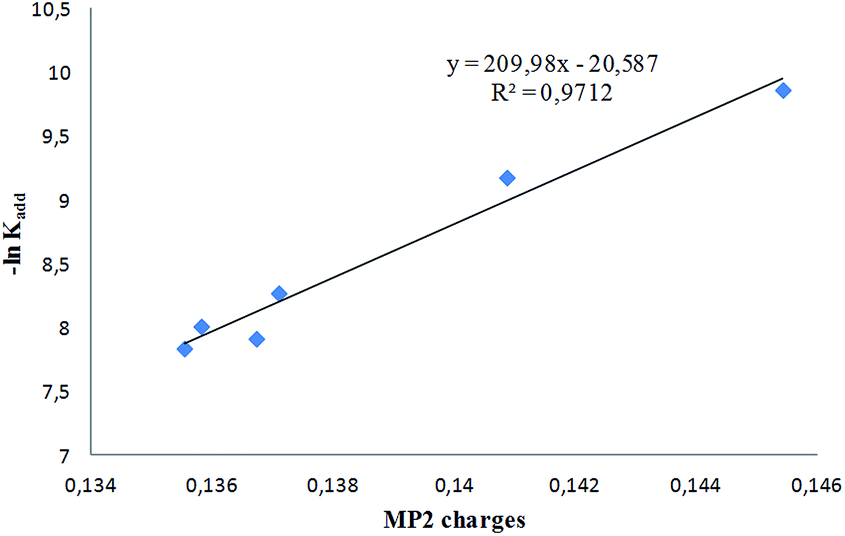 | ||
| Fig. 11 −lnkadd as a function of MP2 charge densities. | ||
Spin adducts decomposition of all spin traps followed a pseudo-first order kinetics. Their respective kdec and half-life times (t1/2) are informed in Table 5.
| Spin adduct 2 | kdec × 105 (s−1) | t1/2 × 10−3 (s) |
|---|---|---|
| a | 0.792 | 87.51 |
| b | 1.070 | 64.80 |
| c | 1.321 | 52.46 |
| d | 1.511 | 45.87 |
| e | 0.265 | 261.32 |
| f | 0.487 | 142.21 |
| g | 0.397 | 174.42 |
| h | 1.730 | 40.07 |
| i | 1.157 | 59.91 |
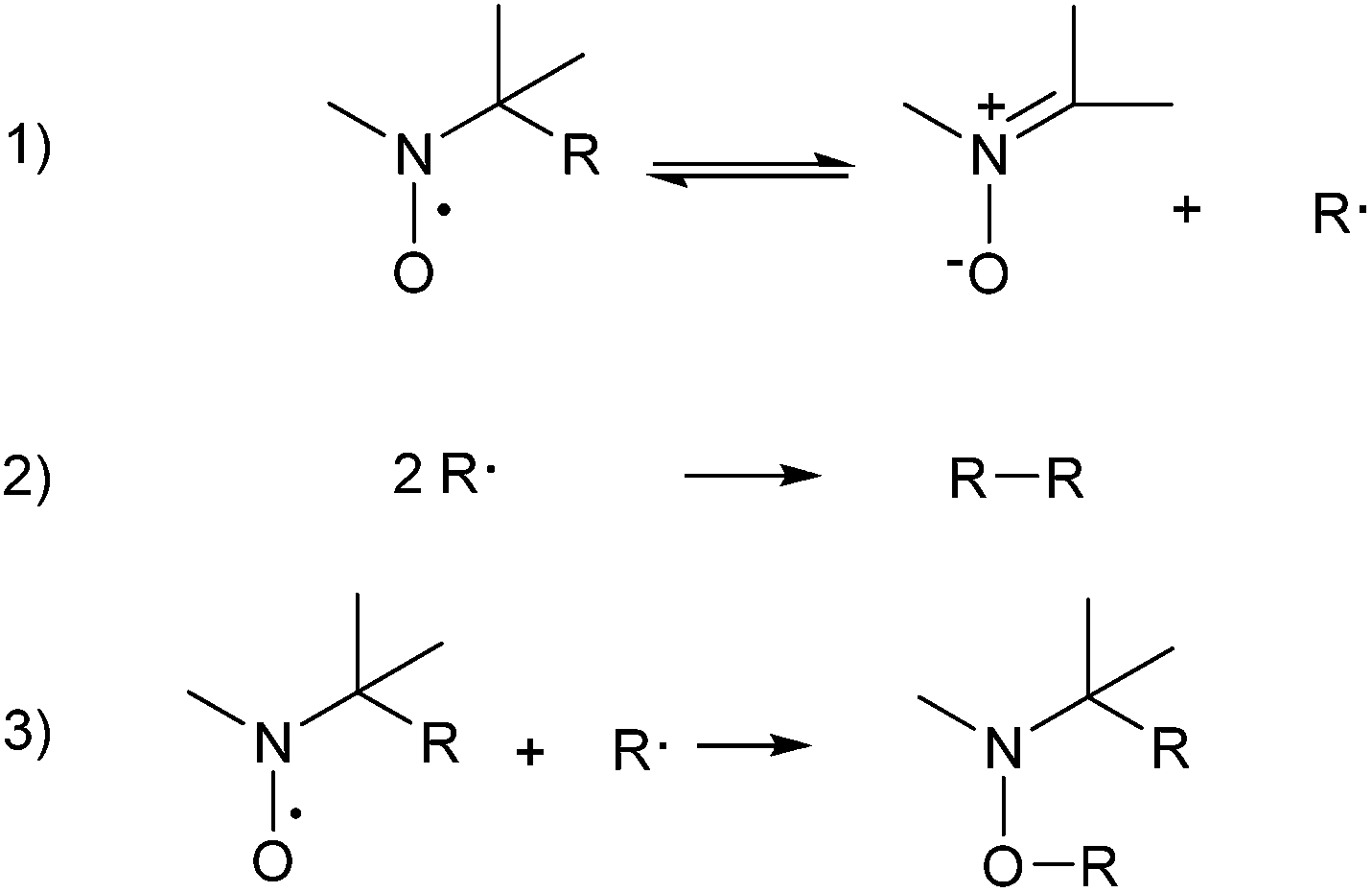 | ||
| Scheme 2 Reactions involved in the spin adduct decomposition. | ||
The overall spin adducts decomposition could be simplified as in Scheme 2. Assuming the reversibility of the first reaction, the C-centered radicals formed in this step can undergo self-coupling and/or react with the nitroxide in excess present in the reaction medium. Both these two last processes are extremely fast and largely shift the equilibrium of the first step to the right, also allowing to approximate the observed EPR signal decay to a pseudo first order process. The t1/2 values reported in Table 5 have been determined following this assumption.
In spite of the previous considerations, other chemical reactions cannot be ruled out in the oxidative aqueous reaction medium. Therefore, no clear relationship between structure and decomposition rates of the spin adducts comprising all the compounds under study could be established. In spite of this, analysis of the results reported in Table 5 clearly show that ortho substitution stabilizes the spin adducts, as can be seen on comparing the decomposition rate constants of compounds 2e and 2f with the corresponding para isomers (2b and 2c, respectively). An attempt to correlate the nitroxide decomposition rate constant with the corresponding dihedral angle has been reported in Fig. 12.
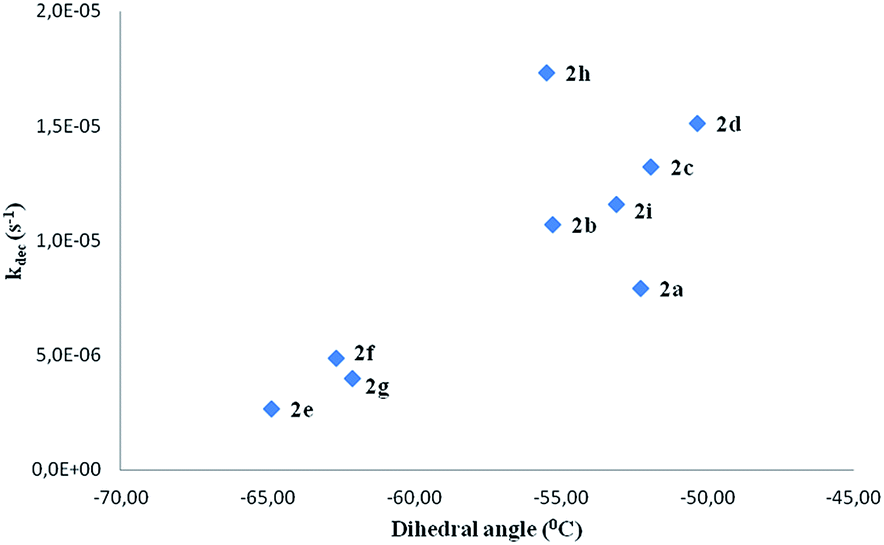 | ||
| Fig. 12 Decomposition rate constants (kdec in s−1) of nitroxides 2 vs. the corresponding dihedral angle N19–C5–C23–C25 (in degrees) (see Fig. 4). | ||
Upon this basis, it could be hypothesized that the more “twisted” the nitrone, the more persistent is the corresponding spin adduct. This could be likely due to the fact that in nitroxides 2e–g it becomes more difficult to reach the planarity necessary for the formation of the nitrone N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond required by the adduct decomposition process as shown in Fig. 13, where the computed geometry of the transition state for nitroxide 2a decomposition has been reported as an example.
C double bond required by the adduct decomposition process as shown in Fig. 13, where the computed geometry of the transition state for nitroxide 2a decomposition has been reported as an example.
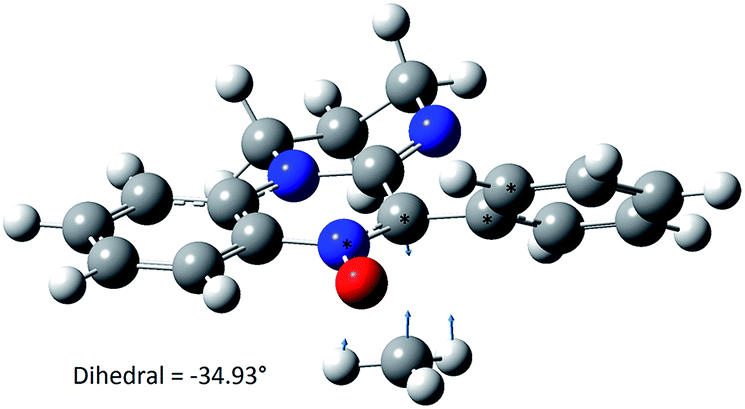 | ||
| Fig. 13 Decomposition transition state geometry of aminoxyl 2a computed at the MPW1K/6-31G(d) level. | ||
Conclusions
We present in this work a novel type of spin traps derived from the pyrimidoquinoxaline N-oxide heterocyclic core. The structural variations comprise the steric and electronic features of the aryl substituent at the nitrone α-carbon and the nature of the fused aryl ring. EPR spin trapping technique showed that all the derivatives were able to trap methyl radicals generated in a Fenton reaction system in the presence of DMSO. The corresponding nitroxides were characterized by EPR and their hfcc were assigned on the basis of related heterocyclic systems and confirmed by DFT calculations. The values of the N-hfcc were indicative of extensive electron delocalization in the adducts due to the high level of coplanarity of the condensed rings within the amidinoquinoxaline system. As a consequence, the novel spin traps showed remarkably persistent signals, as evidenced in a competition experiment between derivative 1a and DMPO.The kinetics of formation and decomposition of the spin adducts were studied. In both processes, a clear dependence on ortho substitution at the 5-aryl group was identified as the main general trend. In fact, both rates of formation and decomposition are significantly lower in ortho substituted derivatives. For the decomposition rate constants, a clear dependence on structural features could not be established. On the other hand, very good correlations were found between steric and electronic parameters of the nitrones and their CH3 addition rates. Taken together, these results represent an insight into the relevant structural features that influence the spin trapping efficiency of this novel type of nitrones, to be taken into account for the future design of new derivatives based on this heterocyclic scaffold.
Experimental
Synthesis
Reagents, solvents, and starting materials were purchased from standard sources and purified according to literature procedures.
Synthesis of 5-aryl-2,3-dihydro-1H-pyrimido[1,2-a]quinoxaline 6-oxides 1
:methanol 9:1). The solvent was then removed in vacuo and the crude product was purified by column chromatography (silica gel, chloroform:methanol 10:0–9:1). Compounds 1a–d,14 f16 were described in the literature. Yields and analytical data of compounds 1e, g–i are as follows.
5-(2-Methoxyphenyl)-2,3-dihydro-1H-pyrimido[1,2-a]quinoxa-line 6-oxide (1e). This compound was obtained as a yellow solid, (0.141 g, 46%), mp 191–193 °C (from hexane/ethyl acetate). 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 8.38 (1H, dd, J = 8.2, 1.4), 7.51–7.54 (1H, m), 7.43 (1H, ddd, J = 8.5, 7.5, 1.7), 7.30 (1H, dd, J = 7.5, 1.7), 7.19 (1H, ddd, J = 8.2, 7.3, 1.1), 7.12–7.14 (1H, m), 7.08 (1H, td, J = 7.5, 0.9), 7.03 (1H, dd, J = 8.5, 0.9), 3.92 (2H, t, J = 6.2), 3.82 (3H, s), 3.56–3.69 (2H, m, bs), 3.41 (2H, s), 2.02–2.09 (2H, m). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 157.2, 145.4, 139.2, 135.0, 131.6, 131.0, 130.6, 130.4, 121.9, 121.2, 120.8, 119.1, 111.5, 111.2, 55.9, 44.0, 43.8, 19.5. Anal. calcd for C18H17N3O2: C, 70.3; H, 5.6; N, 13.7. Found: C, 70.1; H, 5.8; N, 13.6%.
5-(2-Iodophenyl)-2,3-dihydro-1H-pyrimido[1,2-a]quinoxaline 6-oxide (1g). This compound was obtained as a yellow solid, (0.206 g, 51%), mp 153–155 °C (from hexane/ethyl acetate). 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 8.40 (1H, dd, J = 8.2, 1.4), 7.95 (1H, dd, J = 8.0, 0.9), 7.56 (1H, dd, J = 8.6, 7.1), 7.50–7.53 (1H, m), 7.32 (1H, dd, J = 7.8, 1.4), 7.20 (1H, ddd, J = 8.2, 7.1, 1.0), 7.16–7.18 (1H, m), 7.13 (1H, dd. J = 8.6, 1.0), 3.93–3.98 (1H, m), 3.86–3.91 (1H, m), 3.62–3.68 (1H, m), 3.52–3.57 (1H, m), 2.05–2.11 (2H, m). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 144.43, 142.61, 139.01, 136.48, 135.36, 131.91, 130.53, 130.33, 130.01, 128.41, 121.69, 121.28, 110.99, 97.44, 44.49, 43.64, 19.58. Anal. calcd for C17H14IN3O: C, 50.6; H, 3.5; N, 10.4. Found: C, 50.6; H, 3.7; N, 10.3%.
5-(3-Thienyl)-2,3-dihydro-1H-pyrimido[1,2-a]quinoxaline 6-oxide (1h). This compound was obtained as a yellow solid (0.153 g, 54%), mp 160–161 °C (from hexane/chloroform). 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 8.61 (1H, dd, J = 3.1, 1.0), 8.40 (1H, dd, J = 8.4, 1.3), 7.89 (1H, dd, J = 5.1, 1.0), 7.45–7.49 (1H, m), 7.30 (1H, dd, J = 5.1, 3.1), 7.15–7.18 (1H, m), 7.03 (1H, d, J = 8.5), 3.87 (2H, t, J = 6.2), 3.66 (2H, t, J = 5.5), 2.08 (2H, m). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 144.5, 136.2, 134.4, 132.6, 131.2, 130.2, 130.1, 128.6, 122.4, 121.7, 121.1, 110.5, 44.4, 43.9, 19.9. Anal. calcd for C15H13N3OS: C, 63.6; H, 4.6; N, 14.8. Found: C, 63.4; H, 4.7; N, 14.7%.
8H-Pyrido[3′,2′:5,6]pyrazino[1,2-a]pyrimidine-9,10-dihydro-6-phenyl-5-oxide (1i). This compound was obtained as a yellow solid (0.142 g, 51%), mp 215–217 °C (from hexane/chloroform). 1H NMR (500 MHz, CDCl3, 25 °C, TMS): δ = 8.55 (1H, dd, J = 8.0, 1.7), 8.42 (1H, dd, J = 4.8, 1.7), 7.59–7.62 (2H, m), 7.47–7.51 (2H, m), 7.42–7.46 (1H, m), 7.10 (1H, dd, J = 8.0, 4.8), 4.22 (2H, t, J = 6.2), 3.66 (2H, t, J = 5.6), 1.99–2.03 (2H, m). 13C NMR (125 MHz, CDCl3, 25 °C): δ = 149.9, 145.3, 144.8, 141.2, 130.1, 129.6, 129.3, 129.1, 128.0, 126.2, 117.3, 45.7, 41.3, 19.6. Anal. calcd for C16H14N4O: C, 69.05; H, 5.1; N, 20.1. Found: C, 70.1; H, 5.2; N, 20.0%.
Spin adducts characterization and competition experiments
EPR spectra were recorded at 20 °C using an X-band EPR Spectrometer Bruker ECS 106. General EPR spectrometer settings: center field 3480 Gauss; sweep width 80 Gauss; time constant 2.56 ms; conversion time 2.56 ms; modulation amplitude 0.1 Gauss; modulation frequency 50 kHz; receiver gain 2 × 104; microwave power 10 mW. EPR spectra simulations were carried out by means of the Winsim program, freely available from NIEHS.30Kinetic experiments
Each nitrone (5 mM final concentration) was tested in a Fenton reaction (0.72 mM FeSO4 plus 147 mM H2O2, final concentrations) in DMSO with 33% PBS (phosphate buffered saline) pH 7.6. Nitroxide radical signal was measured as the peak-to-peak intensity variation. Kinetic experiments were performed at least three times and rate constants were determined as averaged values from the independent runs.EPR spectra were recorded at 20 °C using an X-band EPR Spectrometer Bruker EMX Plus. General EPR spectrometer settings: center field 3512 Gauss; sweep width: 100 Gauss; time constant 5.12 ms; conversion time 5.12 ms; modulation amplitude: 0.75 G; modulation frequency 50 kHz, receiver gain 1 × 105; microwave power 10 mW. The spectra are the result of the accumulation of a variable number of scans in order to improve signal-to-noise ratio.
Computational details
Density functional theory calculations21 were carried out using the GAUSSIAN 09 suite of programs31 on an EURORA EUROTECH Cluster at Cineca Supercomputing Center.32 All aminoxyls geometries were optimized at the B3-LYP/6-31G(d) level of theory and were carried out with the unrestricted formalism, giving 〈S2〉 = 0.7501 ± 0.0003 for spin contamination (after annihilation). Aminoxyls conformations were systematically screened by means of appropriate relaxed (i.e., with optimization at each point) potential energy surface scans to ensure that species were global minimum energy structures. In addition, in frequency calculations, imaginary (negative) values were never found, confirming that the computed geometries were always referred to a minimum. EPR parameters calculations were performed following the multistep procedure previously described.25 Transition state optimizations were performed employing the MPW1K functional33 in conjunction with the 6-31G(d) basis set for optimizations and 6-31+G(d,p) for frequency calculations; in these last runs, all optimized stationary points were found to have the appropriate number of imaginary frequencies, and the imaginary modes (negative sign) corresponded to the correct reaction coordinates, also confirmed by their visualization with appropriate programs.Acknowledgements
This work was supported by the University of Buenos Aires (20020100100935) and by CONICET (PIP 286). MIUR (Ministero dell'Università e della Ricerca Scientifica e Tecnologica) is kindly acknowledged for financial support (2010PFLRJR – project PROxi) (P.S.) and Cineca Supercomputing Center for computational resource allocation (ISCRA grant ALKOXY-B, code: HP10CYGED5).Notes and references
- (a) A. Mackor, Th. A. J. W. Wajer and Th. J. de Boer, Tetrahedron Lett., 1966, 7, 2115 CrossRef; (b) M. Iwamura and N. Inamoto, Bull. Chem. Soc. Jpn., 1967, 40, 702 CrossRef CAS; (c) G. R. Chalfont, M. J. Perkins and A. Horsfield, J. Am. Chem. Soc., 1968, 90, 7141 CrossRef CAS; (d) E. G. Janzen and B. J. Blackburn, J. Am. Chem. Soc., 1968, 90, 5909 CrossRef CAS; (e) C. Lagerscrantz and S. Forshult, Nature, 1968, 218, 1247 CrossRef.
- J. A. Berliner and J. W. Heinecke, Free Radical Biol. Med., 1996, 20, 707 CrossRef CAS.
- (a) R. T. Dean, S. Fu, R. Stocker and M. J. Davies, Biochem. J., 1997, 324, 1 CAS; (b) S. Fu, M. J. Davies, R. Stocker and R. T. Dean, Biochem. J., 1998, 333, 519 CAS.
- (a) J. Moskovitz, M. B. Yim and P. B. Chock, Arch. Biochem. Biophys., 2002, 397, 354 CrossRef CAS PubMed; (b) R. A. Floyd and K. Hensley, Neurobiol. Aging, 2002, 23, 795 CrossRef CAS.
- (a) E. G. Janzen, Acc. Chem. Res., 1971, 4, 31 CrossRef CAS; (b) C. A. Evans, Aldrichimica Acta, 1979, 12, 23 CAS; (c) C. Mottley and R. P. Mason, in Biological Magnetic Resonance 8, ed. L. J. Berliner and J. Reuben, Plenum Publishers, New York, 1989, p. 489 Search PubMed; (d) P. Tordo, Electron Paramagn. Reson., 1998, 16, 116 CAS; (e) G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259 CrossRef CAS; (f) E. G. Janzen and J. I.-P. Liu, J. Magn. Reson., 1973, 9, 510 CAS; (g) E. G. Janzen, C. A. Evans and J. I.-P. Liu, J. Magn. Reson., 1973, 9, 513 CAS; (h) E. G. Janzen, in Free Radicals in Biology, ed. W. A. Prior, Academic Press, New York, 1980, p. 115 Search PubMed.
- C. Fréjaville, H. Karoui, B. Tuccio, F. Le Moigne, M. Culcasi, S. Pietri, R. Lauricella and P. Tordo, J. Med. Chem., 1995, 38, 258 CrossRef.
- Y. Kotake and E. G. Janzen, J. Am. Chem. Soc., 1991, 113, 9503 CrossRef CAS.
- (a) K. Makino, T. Hagiwara, H. Imaishi, M. Nishi, S. Fuji, H. Ohya and A. Murakami, Free Radical Res. Commun., 1990, 9, 233 CrossRef CAS; (b) E. Finkelstein, G. M. Rosen and E. J. Rauckman, Mol. Pharmacol., 1982, 21, 262 CAS.
- (a) R. D. Hinton and E. G. Janzen, J. Org. Chem., 1992, 57, 2646 CrossRef CAS; (b) A. Zeghdaoui, B. Tuccio, J.-P. Finet, V. Cerri and P. Tordo, J. Chem. Soc., Perkin Trans. 1, 1995, 2087 RSC; (c) E. G. Janzen, Y.-K. Zhang and D. L. Haire, Magn. Reson. Chem., 1994, 32, 711 CrossRef CAS; (d) C. Fréjaville, H. Karoui, B. Tuccio, F. Le Moigne, M. Culcasi, S. Pietri, R. Lauricella and P. Tordo, J. Chem. Soc., Chem. Commun., 1994, 1793 RSC; (e) C. Fréjaville, H. Karoui, B. Tuccio, F. Le Moigne, M. Culcasi, S. Pietri, R. Lauricella and P. Tordo, J. Med. Chem., 1995, 38, 258 CrossRef; (f) K. Stolze, N. Udilova and H. Nohl, Biol. Chem., 2002, 383, 813 CrossRef CAS PubMed; (g) K. Stolze, N. Udilova, T. Rosenau, A. Hofinger and H. Nohl, Biol. Chem., 2003, 384, 493 CrossRef CAS PubMed; (h) H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599 CrossRef CAS; (i) G. Olive, A. Mercier, F. Le Moigne, A. Rockenbauer and P. Tordo, Free Radical Biol. Med., 2000, 28, 403 CrossRef CAS.
- (a) P. Tsai, S. Pou, R. Straus and G. M. Rosen, J. Chem. Soc., Perkin Trans. 2, 1999, 1759 RSC; (b) G. M. Rosen, P. Tsai, E. D. Barth, G. Dorey, P. Casara, M. Spedding and H. J. Halpern, J. Org. Chem., 2000, 65, 4460 CrossRef CAS; (c) P. Astolfi, M. Marini and P. Stipa, J. Org. Chem., 2007, 72, 8677 CrossRef CAS PubMed.
- G. E. Adams, E. M. Fielden, M. A. Naylor and I. J. Stratford, UK Pat. Appl. GB 2257360, 1993.
- P. C. Parthasarathy, B. S. Joshi, M. R. Chaphekar, D. H. Gawad, L. Anandan, M. A. Likhate, M. Hendi, S. Mudaliar, S. Iyer, D. K. Ray and V. B. Srivastava, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1983, 22, 1250 Search PubMed.
- G. J. Ellames, K. R. Lawson, A. A. Jaxa-Chamiec and R. M. Upton, EP 0256545, 1988.
- M. B. García, L. R. Orelli, M. L. Magri and I. A. Perillo, Synthesis, 2002, 2687 Search PubMed.
- M. B. García, L. R. Orelli and I. A. Perillo, J. Heterocycl. Chem., 2006, 43, 1703 CrossRef.
- J. E. Díaz, M. B. García and L. R. Orelli, J. Mol. Struct., 2010, 982, 50 CrossRef PubMed.
- M. L. Lavaggi, G. Aguirre, L. Boiani, L. Orelli, B. García, H. Cerecetto and M. González, Eur. J. Med. Chem., 2008, 43, 1737 CrossRef CAS PubMed.
- (a) R.-M. Dupeyre and A. Rassat, J. Am. Chem. Soc., 1966, 88, 3180 CrossRef CAS; (b) K. Adamic, D. F. Bowman, T. Gillan and K. U. Ingold, J. Am. Chem. Soc., 1971, 93, 902 CrossRef.
- (a) H. Karoui, C. Nsanzumuhire, F. Le Moigne and P. Tordo, J. Org. Chem., 1999, 64, 1471 CrossRef CAS PubMed; (b) A. Allouch, V. Roubaud, R. Lauricella, J.-C. Bouteiller and B. Tuccio, Org. Biomol. Chem., 2005, 3, 2458 RSC.
- For a review see R. Improta and V. Barone, Chem. Rev., 2004, 104, 1231 CrossRef CAS PubMed.
- (a) R. G. Parr and W. Yang, in Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, NY, 1998 Search PubMed; (b) W. Koch and M. C. A. Holthausen, in Chemist's Guide to Density Functional Theory, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- M. K. Eberhardt and R. Colina, J. Org. Chem., 1988, 53, 1071 CrossRef CAS.
- (a) A. Di Matteo, C. Adamo, M. Cossi, V. Barone and P. Rey, Chem. Phys. Lett., 1999, 310, 159 CrossRef CAS; (b) A. Di Matteo, C. Adamo, V. Barone and P. Rey, J. Phys. Chem., 1999, 103, 3481 CrossRef; (c) A. Di Matteo, A. Bencini, M. Cossi, V. Barone, M. Mattesini and F. Totti, J. Am. Chem. Soc., 1998, 120, 7069 CrossRef; (d) V. Barone, A. Grand, D. Luneau, P. Rey, C. Minichino and R. Subra, New J. Chem., 1993, 17, 545 CAS; (e) J. Cirujeda, J. Vidal-Gancedo, O. Jürgens, F. Mota, J. J. Novoa, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2000, 122, 11393 CrossRef CAS; (f) A. Zheludev, V. Barone, M. Bonnet, B. Delley, A. Grand, E. Ressouche, P. Rey, R. Subra and J. Schweizer, J. Am. Chem. Soc., 1994, 116, 2019 CrossRef CAS; (g) S. M. Mattar and A. L. Stephens, Chem. Phys. Lett., 2000, 319, 601 CrossRef CAS.
- A. Alberti, in Nitroxide Radicals and Nitroxide Based High-Spin Systems, Landolt-Börnstein-Group II Molecules and Radicals, 2005, vol. 26D, pp. 1–6 Search PubMed.
- P. Stipa, Chem. Phys., 2006, 323, 501 CrossRef CAS PubMed.
- P. Astolfi and P. Stipa, J. Org. Chem., 2011, 76, 9253 CrossRef CAS PubMed.
- W. Weltner, in Magnetic Atoms and Molecules, Dover, New York, 1989 Search PubMed.
- G. Bott, L. D. Field and S. Sternhell, J. Am. Chem. Soc., 1980, 102, 5618 CrossRef CAS.
- F. A. Villamena, C. M. Hadad and J. L. Zweier, J. Am. Chem. Soc., 2004, 126, 1816 CrossRef CAS PubMed.
- D. Duling, PEST Winsim, version 0.96, National Institute of Environmental Health Sciences, Triangle Park, NC, 1996 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01 Search PubMed.
- Cineca Supercomputing Center, via Magnanelli 6/3, I-40033 Casalecchio di Reno, Bologna, Italy, http://www.cineca.it/HPSystems.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 481 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data for all the new compounds. See DOI: 10.1039/c4ra14335c |
| This journal is © The Royal Society of Chemistry 2015 |