
cRGD targeted and charge conversion-controlled release micelles for doxorubicin delivery
Xingang Guanab,
Xiuli Hua,
Zhihong Lic,
Hong Zhang*c and
Zhigang Xie*a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun 130022, P. R. China. E-mail: xiez@ciac.ac.cn; Fax: +86-431-85262775; Tel: +86-431-85262775
bLife Science Research Center, Beihua University, Jilin 132013, P. R. China
cDepartment of Thoracic Surgery, The First Hospital of Jilin University, Changchun 130021, P. R. China. E-mail: yveszhang@hotmail.com
First published on 16th February 2015
Abstract
Efficiently and selectively delivering chemotherapeutic agents to tumours still remains a challenge for the development of nanocarriers. In this study, polymer micelles based on cRGD targeting and pH-sensitive surface charge-switching were successfully prepared and used for doxorubicin (Dox) delivery. This nanoscale polymeric micelle indicated a high drug encapsulation efficiency of 90% and slightly negative charge. Drug release experiments showed a pH-enhanced release profile in PBS, which resulted from surface charge-switching by imidazole. Confocal laser scanning microscopy and flow cytometry experiments indicated that combining the capability of an RGD target and pH-sensitive charge-switching significantly enhanced the cellular uptake of B16F10 cells overexpressing αvβ3 integrins. An MTT assay also showed that our hybrid RGD-decorated micelles were much more cytotoxic to B16F10 cells than undecorated micelles. These results suggest the potential application of a cRGD target and pH-sensitive surface charge-switching polymeric micelles in the treatment of αvβ3 integrin-overexpressing cancers.
1 Introduction
Great progress has been made in understanding the mechanism of tumor origination and development; however, surgery, chemotherapy, and radiation are still the main methods for current cancer treatments.1,2 Although traditional chemotherapeutic agents can remarkably suppress tumor growth, their application is greatly hindered by the side effects to normal cells. Delivering the anti-cancer agents to tumor cells without affecting normal cells is the primary obstacle in the development of efficient anti-tumor carriers. Nanomedicines, which are drugs based on nanotechnology, hold great promise to improve the outcome of chemotherapy.3–9 Drug-delivery systems (DDSs) based on engineered nanoparticles possess many advantages such as reduced cytotoxicity, enhanced permeability and retention effect (EPR effect),10,11 responsiveness to the tumor microenvironment (e.g., pH, redox, enzymes, or others),12–15 and increased cellular uptake by equipping the target moiety (e.g., peptides, antibodies, and aptamers).16–18It is well-known that the pH value in the tumor extracellular environment is much lower (pH 5.8–6.5) than that of the blood (pH 7.4),19–21 and even much lower pH values (pH 5.0–5.5) have been detected in intracellular organelles (e.g., late endosomes and lysosomes).22 Accumulating reports have indicated that many types of pH-sensitive nanoparticles have been developed and will efficiently deliver their payload to the tumor environment.23–26 These pH-responsive nanocarriers can preserve the stable conformation or maintain the integrity of pH-sensitive linkages under physiological pH; however, when the nanocarriers were transported across the super-permeabilized vasculture to the tumor stroma (low pH) via the EPR effect, the breakage of pH-sensitive conformation or linkages resulted in the release of the drugs into the tumor extracellular environment. Recently, many reports have proven that charge conversion will improve the efficiency of drug delivery to tumor cells.27–29 The utilization of 1-(3-aminopropyl)imidazole (API) in mediating pH-responsive charge-switching via protonation leads to enhanced drug accumulation in tumor cells. However, in most studies, the drug accumulation in tumor cells mainly relied on the EPR effect, which is insufficient to significantly improve tumor cell uptake. Targeted delivery of anti-tumor agents will likely open a new era for chemotherapy treatment of cancers. By decoration of target ligands on the surface of nanocarriers, these drug nanocarriers can selectively bind with the corresponding receptors highly or uniquely expressed on tumor cells and then be internalized via endocytosis.30–32 DDSs based on active tumor targeting have been proven to significantly improve therapeutic efficacy. Among various target ligands, the Arg-Gly-Asp (RGD) peptide was preferentially used due to its low cost and high expression of its receptors (αvβ3 and αvβ5 integrins) in many solid tumors and tumor vasculture endothelial cells.33,34 Accumulating studies have indicated the key role of RGD peptides in targeted delivery of various nanoparticles to tumor cells overexpressing αvβ3 or αvβ5 integrins (Scheme 1).35–44
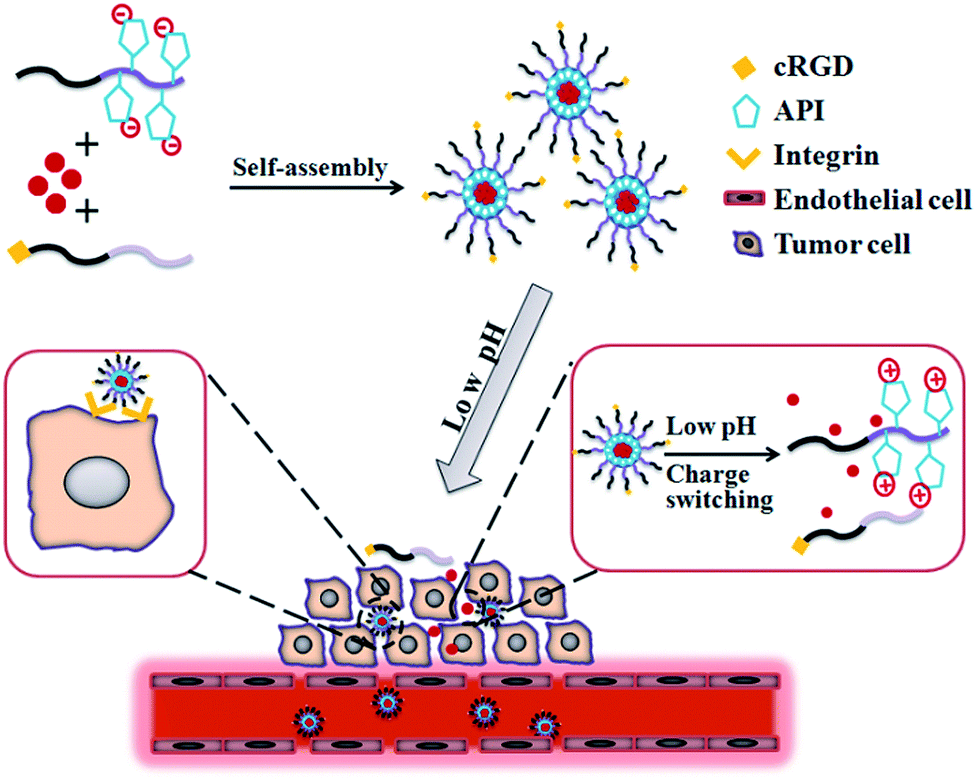 | ||
| Scheme 1 Schematic illustration of self-assembly and tumor cell uptake of the cRGD target that has been incorporated into pH-triggered surface charge-switching polymer micelles. | ||
In this study, in order to develop a DDS with excellent biocompatibility, polymer micelles based on poly(ethylene glycol)-block-poly(glutamic acid) were successfully synthesized. Poly(glutamic acid) was chosen as the backbone polymer not only for its excellent biocompatibility and biodegradability, but also for its abundant functional carboxyl groups, which are convenient for conjugation.45,46 By the conjugation of API on the side chains, a polymer micelle with a pH-responsive charge-switching property was prepared. By decoration with c(RGDfK) peptide, our charge conversion micelles were used to deliver doxorubicin (Dox) to mouse melanoma cells. The drug release profile in vitro, cellular uptake, and cytotoxicity analysis were evaluated in detail.
2 Material and methods
2.1 Materials
Monomethoxylpoly(ethylene glycol) (mPEG, average Mw = 5000), dicyclohexylcarbodiimide (DCC, 99%) and N-hydroxysuccinimide (NHS, 99%) were purchased from Aldrich (USA). N-(3-Aminopropyl)-imidazole (API) was ordered from Alfa Aesar. Doxrobicin (Dox) was purchased from Zhejiang Hisun Pharmaceutical Co. Ltd (Taizhou city, China). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Shanghai Yuanye Ltd (Shanghai, China). MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) and 9,10-phenan-threnequinone were purchased from Amresco (USA). Cyclic (RGDfK) peptide was customized from Shanghai China Peptides Ltd. (Shanghai, China). Ultrapure water was prepared with a Milli-Q system (Millipore, USA). All other chemicals were analytical grade or above.2.2 Synthesis of PEG–PGlu10
Poly(ethylene glycol)–poly(glutamic acid) (PEG–P(Glu)) block copolymers were synthesized using a previously reported synthetic method with slight modification.47 Ring-opening polymerization of the N-carboxyanhydride of β-benzyl-L-glutamate was carried out using mPEG–NH2 as a macroinitiator. Briefly, in a dried flask, 5.0 g of PEG–NH2 (1.0 mmol) and 2.63 g of BGL–NCA (10 mmol) were dissolved in dried chloroform (65 mL), and the solution was stirred for 72 h at 30 °C. The product mixture was precipitated with an excess of a mixture of acetic acid and methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) under vigorous stirring to give a white solid. PEG–PBGL was obtained under vacuum at 40 °C for 24 h.
:3, v/v) under vigorous stirring to give a white solid. PEG–PBGL was obtained under vacuum at 40 °C for 24 h.
In order to remove the protected benzyl group, mPEG-b-P(BLG10) (3.0 g) was dissolved in dichloroacetic acid (30 mL), and HBr/acetic acid (33 wt%, 12 mL) was added. After stirring for 1 h at 35 °C, the mixture was precipitated into excessive ice-cold diethyl ether. After drying under vacuum, the precipitate was dissolved in DMF and dialyzed against distilled water, and then freeze-dried to give the PEG–PGlu10 product. The 1H NMR spectra of the protected and unprotected PEG–PGlu10 are given in Fig. 1.
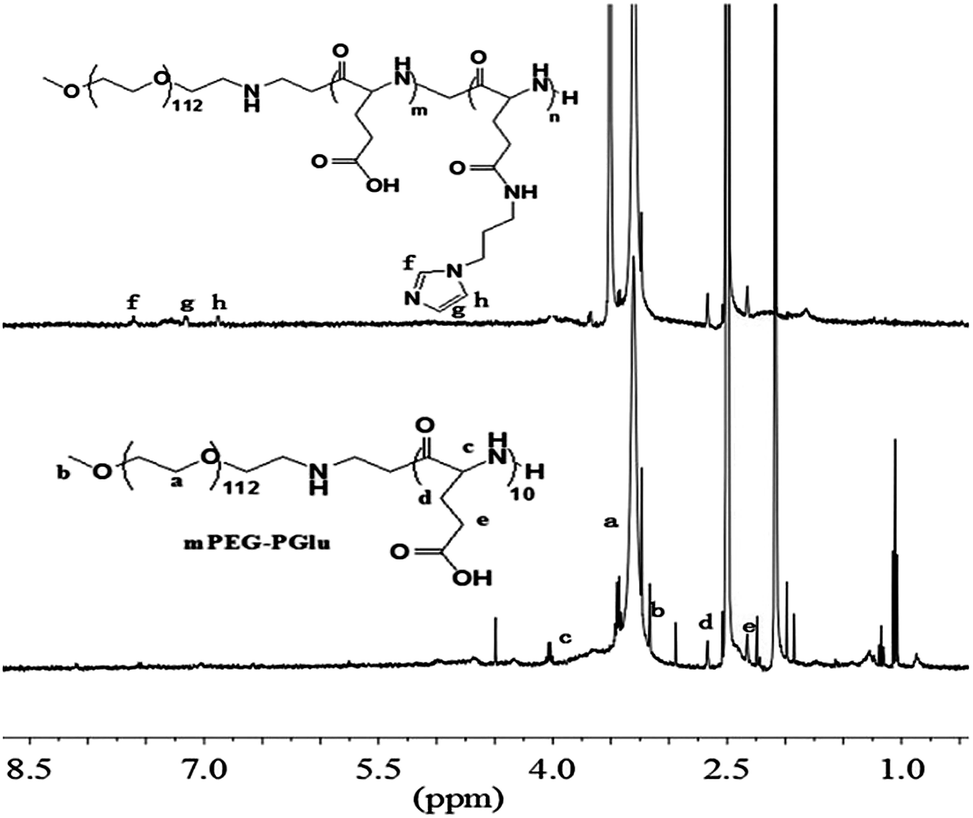 | ||
| Fig. 1 The 1H NMR spectra of PEG–PGA and PEG–PGA–API. | ||
2.3 Synthesis of PEG–PGA10–API
PEG–PGA10 (200 mg, 25 μmol) was dissolved in 5 mL DMSO, and then 7.74 mg dicyclohexylcarbodiimide (DCC) (37.5 μmol) and 4.32 mg N-hydroxysuccinimide (NHS) (37.5 μmol) were added. The reactions were conducted at room temperature for 2 hours, and then N-(3-aminopropyl)imidazole (API) (16 mg, 125 μmol) was added. After dialysis against distilled water for 12 hours (MWCO = 3500), the final suspension in the dialysis bag was freeze-dried.2.4 Synthesis of cRGD–PEG–PLA
cRGD–PEG–PLA was synthesized in a previous work.262.5 Preparation of Dox-loaded micelles M(Dox) and RGD–M(Dox)
Micelles (M) loaded with Dox were prepared as follows: 4 mg Dox–HCl, 36 mg PEG–PGA10–API, and 20 μL triethylamine (TEA) were dissolved in 2 mL DMSO, and the mixed solution was then added dropwise to 10 mL deionized water. Unincorporated Dox and DMSO were removed by dialysis (MWCO = 3500) against distilled water for 8 hours. The Dox-loaded micelles were stored at 4 °C and used within one month. When M(Dox) were used in target analysis compared with RGD–M(Dox), which contains 20% cRGD–PEG–PLA, M(Dox) were prepared by mixing 4 mg Dox–HCl, 28.8 mg PEG–PGA10–API, and 7.2 mg PEG–PLA, and using the same processes as described above.RGD–M(Dox) was prepared by mixing PEG–PGA10–API and cRGD–PEG–PLA with a weight ratio of 20/80, i.e., a mixture of 4 mg Dox–HCl, 28.8 mg PEG–PGA10–API, and 7.2 mg cRGD–PEG–PLA were used as the starting materials.
To determine the loading content of Dox, a standard calibration curve of free Dox solution in DMSO was constructed. Then, the freeze-dried Dox-loaded micelles were weighed and dissolved in DMSO, the UV absorbance of which was measured at 480 nm. The Dox content can thus be acquired according to the standard curve. The drug loading content (DLC) and drug loading efficiency (DLE) were calculated according to the following equations:
| DLC% = (weight of Dox in the micelle/weight of Dox + weight of all polymers) × 100 |
| DLE% = (weight of Dox in the micelle/weight of Dox added) × 100 |
2.6 Characterization of M(Dox) and RGD–M(Dox)
The morphology of polymer micelles was determined by transmission electron microscopy (TEM) (JEOL JEM-1011). The size distribution of micelles was measured by dynamic light scattering (DLS) with a vertically polarized He–Ne laser (DAWN EOS, Wyatt Technologies). The scattering angle was fixed at 90° for DLS measurement at 25 °C.2.7 Dox release in vitro
The release profile of Dox from M(Dox) was studied at 37 °C in phosphate-buffered saline (PBS) at different pH values (pH 5.8, pH 6.5, and pH 7.4). Briefly, a micelle dispersion containing 1 mg EPI was transferred into a dialysis bag (MWCO = 3000) followed by immersion in 20 mL PBS buffer (20 mM, pH 5.8, pH 6.5, and pH 7.4) with stirring at 37 °C. At certain time points, 1 mL of dialysis solution was taken out for UV-Vis measurement (480 nm) and replenished with 1 mL fresh PBS solution.2.8 Biocompatibility assay of polymers
In this study, cancer cells (B16F10 cells, originating from mouse skin melanoma) and non-cancer cells (L929 cells, mouse fibroblast cells) were used to examine polymer biocompatibility. After 5 passages, cells were seeded in 96-well plates at a density of 4 × 103 cells per well in 100 μL Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) for 12 h at 37 °C in 5% CO2. The medium was replaced with 200 μl DMEM containing different concentrations of PEG–PGA10 (ranging from 100 to 1000 μg mL−1) and PEG–PGA10–API (ranging from 50 to 500 μg mL−1) for 48 h. Next, 20 μl MTT (5 mg mL−1) was added to each well, the plates were incubated for 4 h, and then 150 μl DMSO was added to each well to dissolve the blue formazan formed in the live cells. The absorbance was measured on a microplate reader (BioTek, EXL808) at 490 nm. Experiments were repeated three times.2.9 Cytotoxicity analysis of M(Dox) and RGD–M(Dox)
The cytotoxicity of two types of Dox-loaded micelles was measured via MTT assay with free Dox as a control. Briefly, B16F10 melanoma cells were seeded in 96-well plates at 2 × 103 cells per well in 100 μL DMEM medium containing 10% fetal bovine serum (FBS) for 12 h of incubation at 37 °C in 5% CO2. The medium was replaced with 200 μl DMEM containing different concentrations of Dox or Dox-containing micelles (ranging from 0.01 to 10 μg mL−1) for 48 h. Next, 20 μl MTT (5 mg mL−1 in PBS buffer) was added to each well for 4 h of incubation, and then 150 μl DMSO was added to each well to dissolve the blue formazan formed in the live cells. The absorbance was measured on a microplate reader (BioTek, EXL808) at 490 nm. Experiments were repeated three times.2.10 Cellular uptake study
The cellular uptake of M(Dox) and RGD–M(Dox) was evaluated by confocal laser scanning microscopy (CLSM) and flow cytometry. For CLSM analysis, B16F10 cells were seeded in a six-well plate at a density of 4 × 105 cells per well for 12 h incubation. The medium was replaced with fresh DMEM medium containing Dox (20 μg mL−1) or Dox-loaded micelles (20 μg mL−1 Dox) for 1 h. After three washes with PBS, the cells were fixed with 4% (w/v) paraformaldehyde for 10 min at room temperature. The cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 15 min. CLSM images were captured via confocal microscope (Carl Zeiss LSM 780) under the same conditions. The mean fluorescence intensity of each cell was analyzed with Image Pro-Plus (IPP) software according to the inherent fluorescence of Dox. As for A549 carcinoma cell (a human cell line derived from respiratory epithelium) uptake, the protocol was the same as that followed for B16F10 cells.For flow cytometry, the cell seed and drug incubation process was the same as that used for the CLSM analysis. The cells were treated with 200 μl 0.25% trypsin for 2 min. After adding 1 mL DMEM medium, the cells were centrifuged at 300×g for 5 min. After removing the supernatant, the cells were resuspended in 0.5 mL PBS. Flow cytometry analysis was performed with a flow cytometer (Beckman, USA) that collected 1 × 104 gated events for each sample.
2.11 Statistics
All experiments were performed at least three times, and all results are expressed as the mean ± SD (standard deviation). Student's t-test was used to demonstrate statistical significance (P < 0.05).3 Results and discussion
3.1 Preparation and characterization of M(Dox) and RGD–M(Dox)
First, amphiphilic copolymer PEG–PGA–API was made through the amidation of mPEG–PGlu with API in the presence of EDC/NHS as shown in Scheme 2. The chemical structure was characterized by 1H NMR. As shown in Fig. 2, the presence of imidazole was confirmed by the characteristic signals at 6.9, 7.2, and 7.6 ppm. The API content in PEG–PGA–API calculated by the intensity of 1H NMR is approximately 5 wt%. cRGD was conjugated to PEG–PLA, and the results were confirmed according to previously published studies.26,48 The molar ratio of cRGD to PEG–PLA was determined to be 1:1.06 using a standard curve calculation, indicating that an average of 94% of polymer was labelled by cRGD.
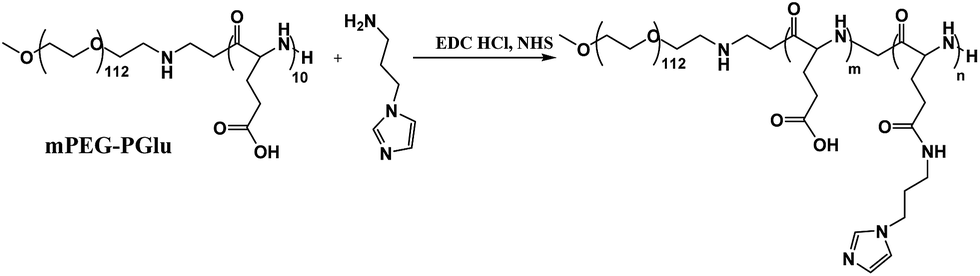 | ||
| Scheme 2 Synthetic routes for the preparation of PEG–PGA–API. | ||
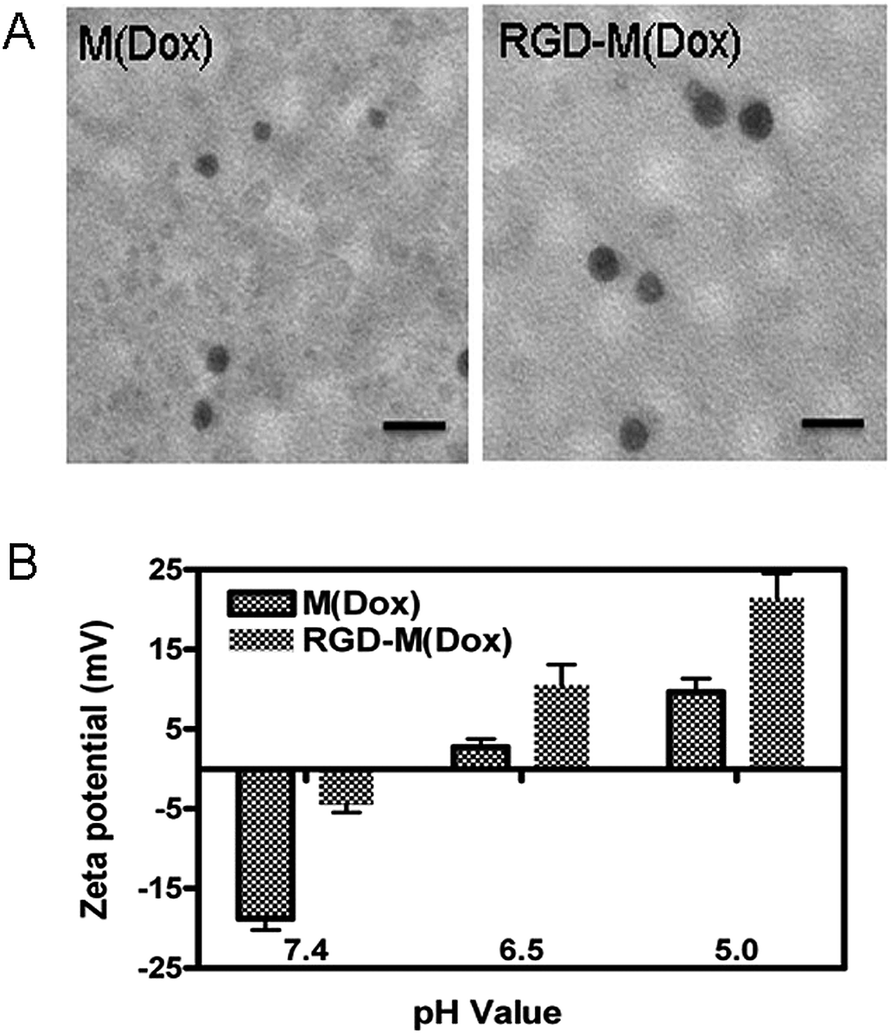 | ||
| Fig. 2 TEM analysis (A) and surface charge conversion of M(Dox) and RGD–M(Dox) at different pH values. The scale bar is equal to 50 nm. | ||
The amphiphilic block self-assembled into polymeric micelles in aqueous solution. Herein, the Dox-loaded micelles prepared with PEG–PGA–API were named M(Dox). The hybrid micelles containing cRGD decoration named RGD–M(Dox) were prepared using the co-assembly method. Considering the good target ability of 20% cRGD as described by Miura et al.,37 cRGD target micelles were prepared in this study using a mixture of cRGD–PEG-b-PLA and PEG–PGA–API in the ratio of 1:4. The characterization of the two micelles is shown in Table 1. The average diameters of M(Dox) and RGD–M(Dox) were 26 nm and 30 nm, respectively. The increase in diameter of RGD–M(Dox) could be attributed to the cRGD decoration on the surface of the micelles. The zeta potential of M(Dox) and RGD–M(Dox) was −18.8 and −4.4, respectively. TEM analysis indicated that both micelles displayed a nearly spherical nanosized structure, as shown in Fig. 2A. Two micelles exhibited nearly the same drug loading capability, (DLC 8.7%, DLE 86–88%), indicating that there was a negligible effect of cRGD decoration in loading the drugs. The charge conversion property of the two micelles conferred by imidazole was also analyzed in PBS buffer at different pH values. As shown in Fig. 2B, M(Dox) and RGD–M(Dox) micelles had a negative charge, and when the two micelles were immersed in an acidic environment, the micelle surface converted to a positive charge within 4 h, suggesting the important role of imidazole in surface conversion.
| Micelles | Size (nm) | PDI | Zeta potential (mV) | Drug loading content (DLC)% | Drug loading efficiency (DLE)% |
|---|---|---|---|---|---|
| M(Dox) | 26.42 ± 1.72 | 0.399 | −18.8 ± 1.41 | 8.73 | 87.25 |
| cRGD–M(Dox) | 30.12 ± 1.83 | 0.439 | −4.42 ± 1.07 | 8.72 | 86.13 |
The release of EPI from micelles was determined in PBS buffer (pH 5.8, pH 6.5, and pH 7.4). The results (Fig. 3) showed that both M(Dox) and RGD–M(Dox) exhibited a sustained-release in PBS at three different pH values. Compared with micelles, free Dox displayed a fast release at pH 7.4 and pH 5.0 PBS, with a cumulative release of approximately 76.8% after 4 h. After 36 h of release, the cumulative Dox release in pH 5.8, pH 6.5, and pH 7.4 PBS was 80.3%, 57.0%, and 45.4%, respectively; as for RGD-decorated micelles, the cumulative Dox release in the same PBS solutions was 43.4%, 56.1%, and 80.8%. The enhanced Dox release from M(Dox) and RGD–M(Dox) micelles suggested pH sensitivity. The result could be attributed to the pH-responsive imidazole groups in the micelles. No significant difference in cumulative Dox release (after 36 h) was detected between M(Dox) and RGD–M(Dox) micelles when the same PBS buffers at the same pH values were used.
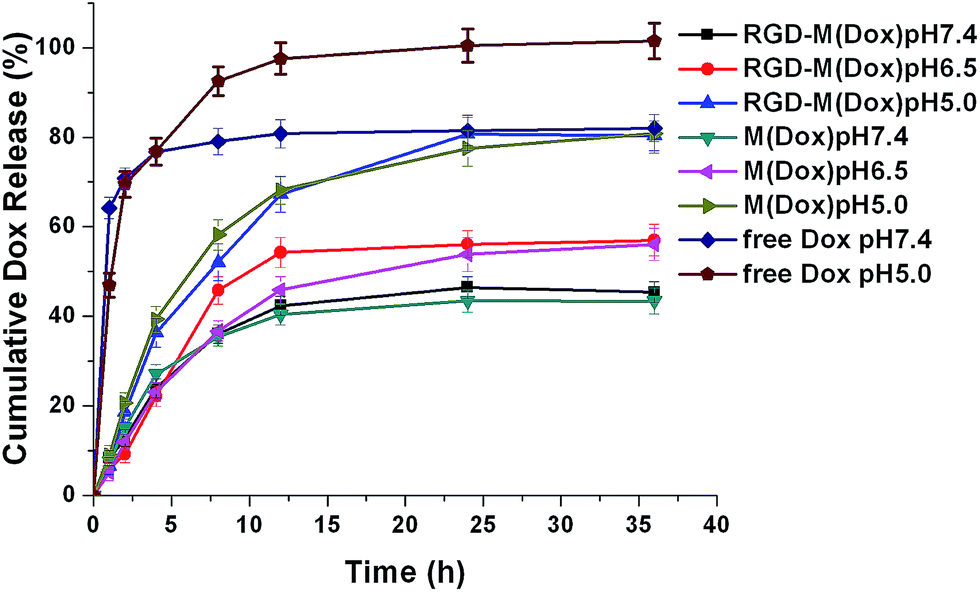 | ||
| Fig. 3 Cumulative release profiles of Dox from free Dox, M(Dox), and RGD–M(Dox) in vitro at different pH values (pH 5.8, 6.5, and 7.4) at 37 °C. | ||
3.2 Biocompatibility study of M(Dox) and RGD–M(Dox)
To evaluate if our micelles could be used for drug delivery, the biocompatibility of block copolymer PEG–PGA and PEG–PGA–API was investigated by MTT assay. PEG–PGA showed perfect biocompatibility at concentrations up to 1 mg mL−1 (>90%), and PEG–PGA–API also exhibited low cytotoxicity at concentrations up to 500 μg mL−1 (>80%) (shown in Fig. 4). The slightly reduced biocompatibility of PEG–PGA–API may be ascribed to the introduction of API.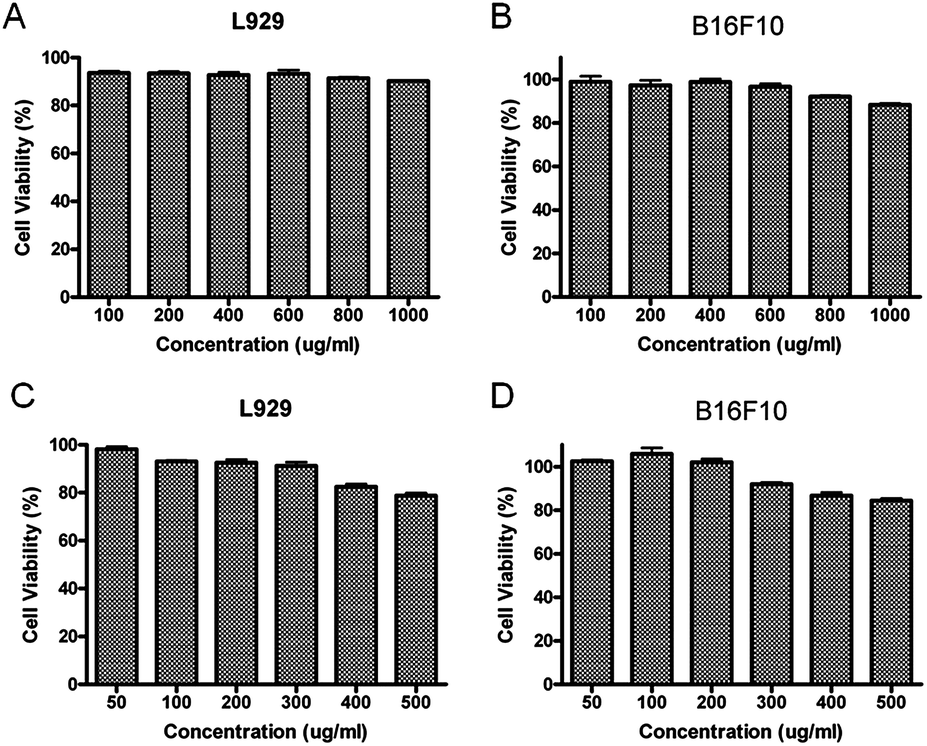 | ||
| Fig. 4 Cell viability of L929 and B16F10 cells treated with PEG–PGA (A and C) and PEG–PGA–API (B and D) at different concentrations after 48 h incubation at 37 °C. All the results were repeated three times and are presented as the mean ± SD. | ||
3.3 Cellular uptake
To evaluate the target ability of cRGD-decorated micelles, the cellular uptake of M(Dox) and RGD–M(Dox) by B16F10 and A549 cells was studied according to the inherent fluorescence of Dox. B16F10 cells express a high level of αvβ3 integrins, whereas nearly no such expression occurs in A549 cells, as previously described in many studies.26As shown in Fig. 5A, all cells displayed strong red fluorescence in the cell nucleus, and little fluorescence was distributed in the cytosol. Moreover, cRGD-decorated micelles showed much higher red fluorescence intensity in the nucleus than undecorated micelles in B16F10 cells, while no obvious change was observed in the fluorescence intensity of two micelles in A549 cells (Fig. 5A). The quantification analysis of the mean fluorescence intensity confirmed our conclusion (Fig. 5B). This result indicated that RGD decoration significantly improved the endocytosis of B16F10 cells that were overexpressing αvβ3 integrins. Due to the very low expression of αvβ3 integrins in A549 cells, the significantly enhanced endocytosis of RGD-decorated micelles was not observed. The increased cellular uptake of M(Dox) micelles by A549 cells as compared to B16F10 cells may be attributed to the different type of cell, i.e., the cellular uptake of micelles by respiratory epithelium cells (A549 cells) is apparently much easier than that in skin cells (B16F10 cells).
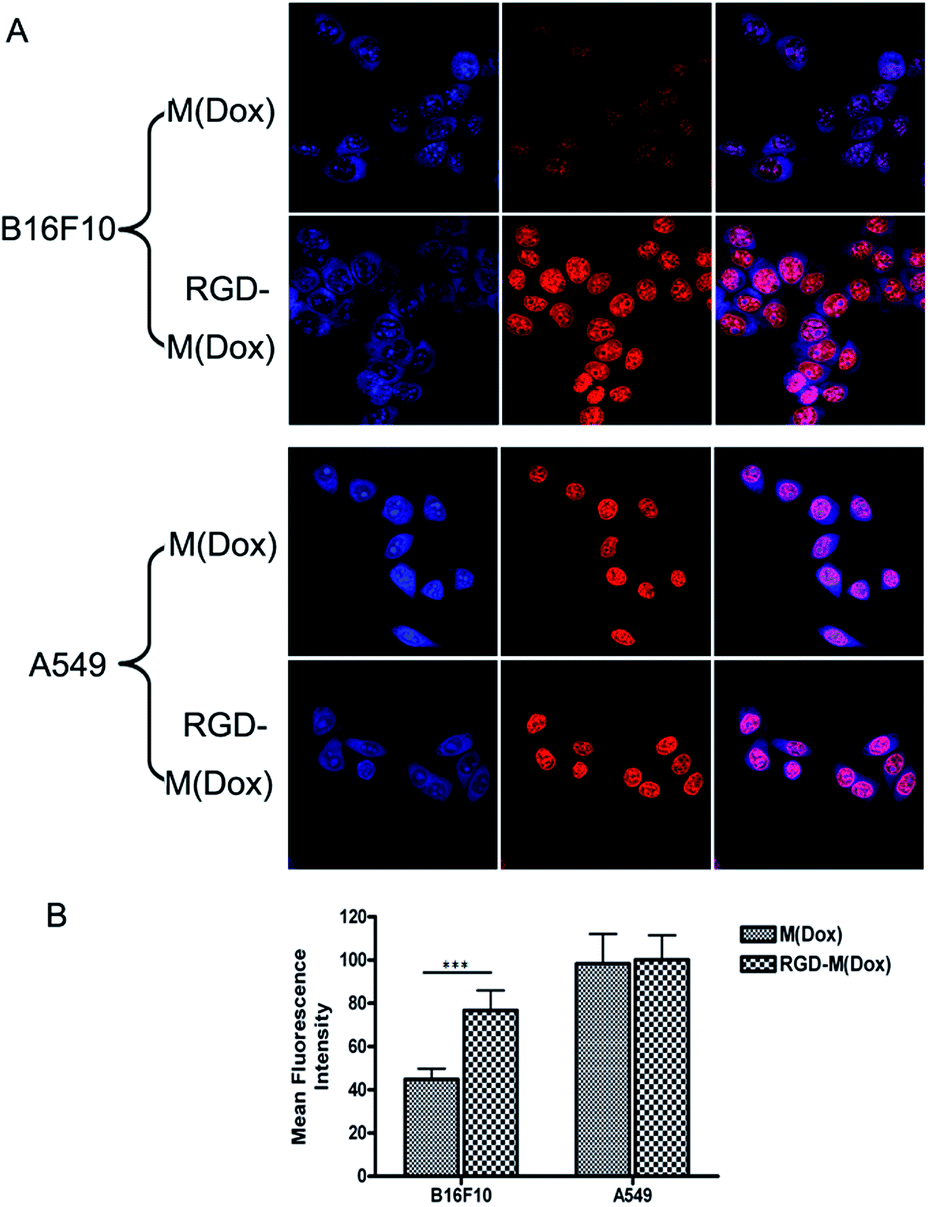 | ||
| Fig. 5 (A) CLSM images of B16F10 cells and A549 cells treated with M(Dox) and RGD–M(Dox) in PBS pH 7.4 after 1 h of incubation at 37 °C in vitro; (B) the quantification of the mean fluorescence intensity of each cell is also shown. | ||
Based on the target result, micelles combining cRGD decoration and a charge-switching imidazole group were developed with cRGD–PEG–PLA and PEG–PGA–API at the ratio of 1:4 (wt/wt). The RGD content in the copolymer of cRGD–PEG–PLA was 94%, and thus, the RGD content in RGD–M(Dox) micelles was calculated to be 18.8%. The cellular uptake of M(Dox) and RGD–M(Dox) in PBS (pH 7.4 and pH 5.8) was investigated in B16F10 cells. As shown in Fig. 6A and B, the RGD–M(Dox) micelles in pH 7.4 PBS (abbreviated as RGD-M/7.4) and M(Dox) in pH 5.8 PBS (abbreviated as M/5.8) showed much stronger fluorescence than M(Dox) in pH 7.4 PBS, which may be ascribed to the cRGD decoration and imidazole group. Moreover, the cells treated with RGD–M(Dox) micelles in pH 5.8 PBS (abbreviated as RGD-M/5.8) showed increased fluorescence as compared to the other three groups, indicating a synergetic effect of cRGD decoration and imidazole in improving cellular uptake. The cellular uptake was also examined by flow cytometric analysis, and the results of flow cytometric experiments supported the CLSM results (Fig. 7). In an acidic environment, the cellular uptake rate increased from 27.6% at pH 7.4 to 40.1% at pH 5.8, which could be interpreted as the occurrence of fast drug release caused by charge-switching of imidazole. After cRGD decoration, the cellular uptake rate increased to 55.7%, which may be attributed to the increased endocytosis by B16F10 cells mediated by cRGD decoration. When combining the cRGD target and an acidic environment, this group showed the highest cellular uptake rate in the four groups (77.2%), which was in accordance with the results of the CLSM experiments.
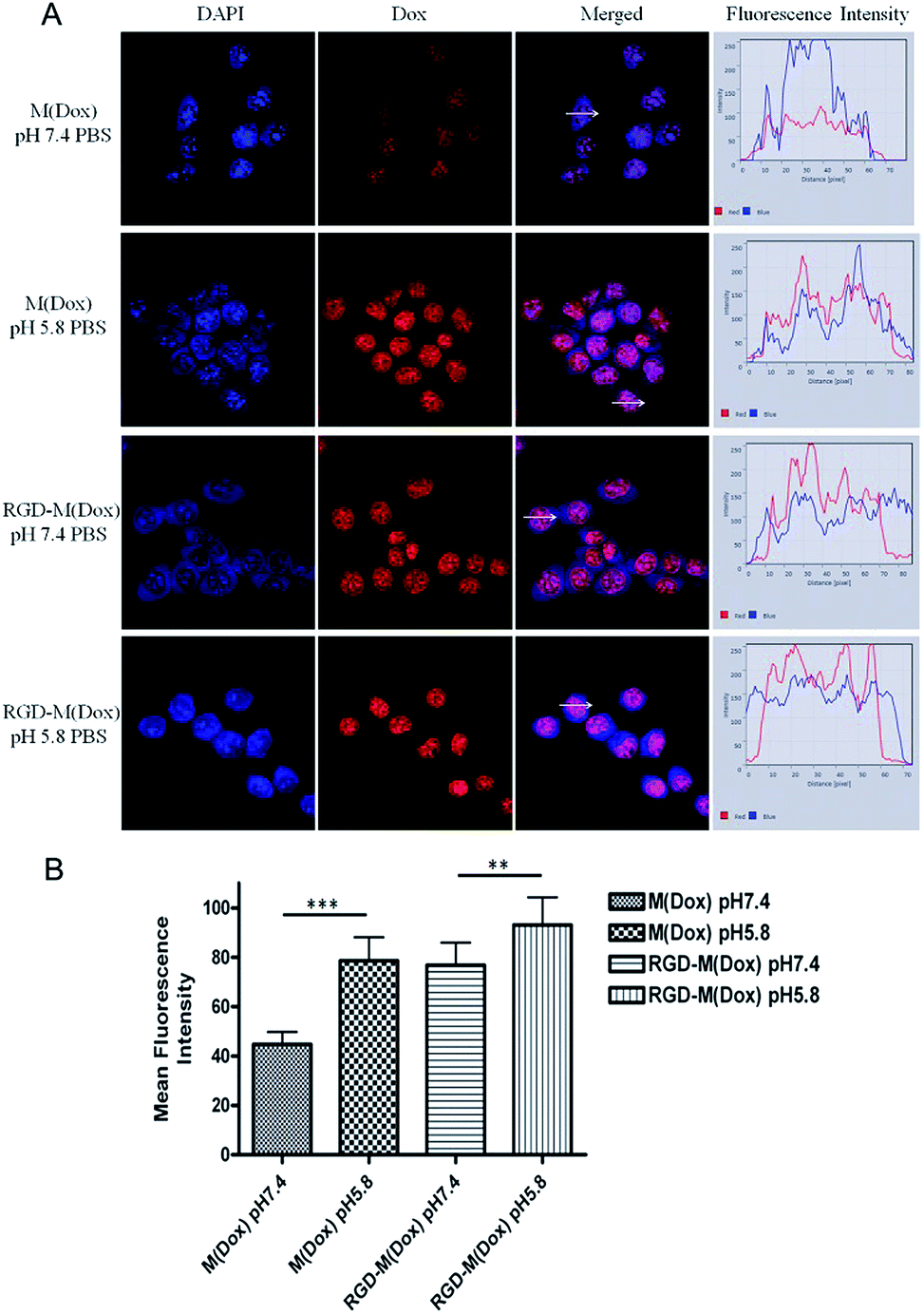 | ||
| Fig. 6 (A) CLSM images of B16F10 cells treated with M(Dox) in PBS at pH 7.4 and pH 5.8, and RGD–M(Dox) in PBS at pH 7.4 and pH 5.8 after 1 h of incubation at 37 °C in vitro; (B) the mean fluorescence intensity of Dox in each cell is also shown. | ||
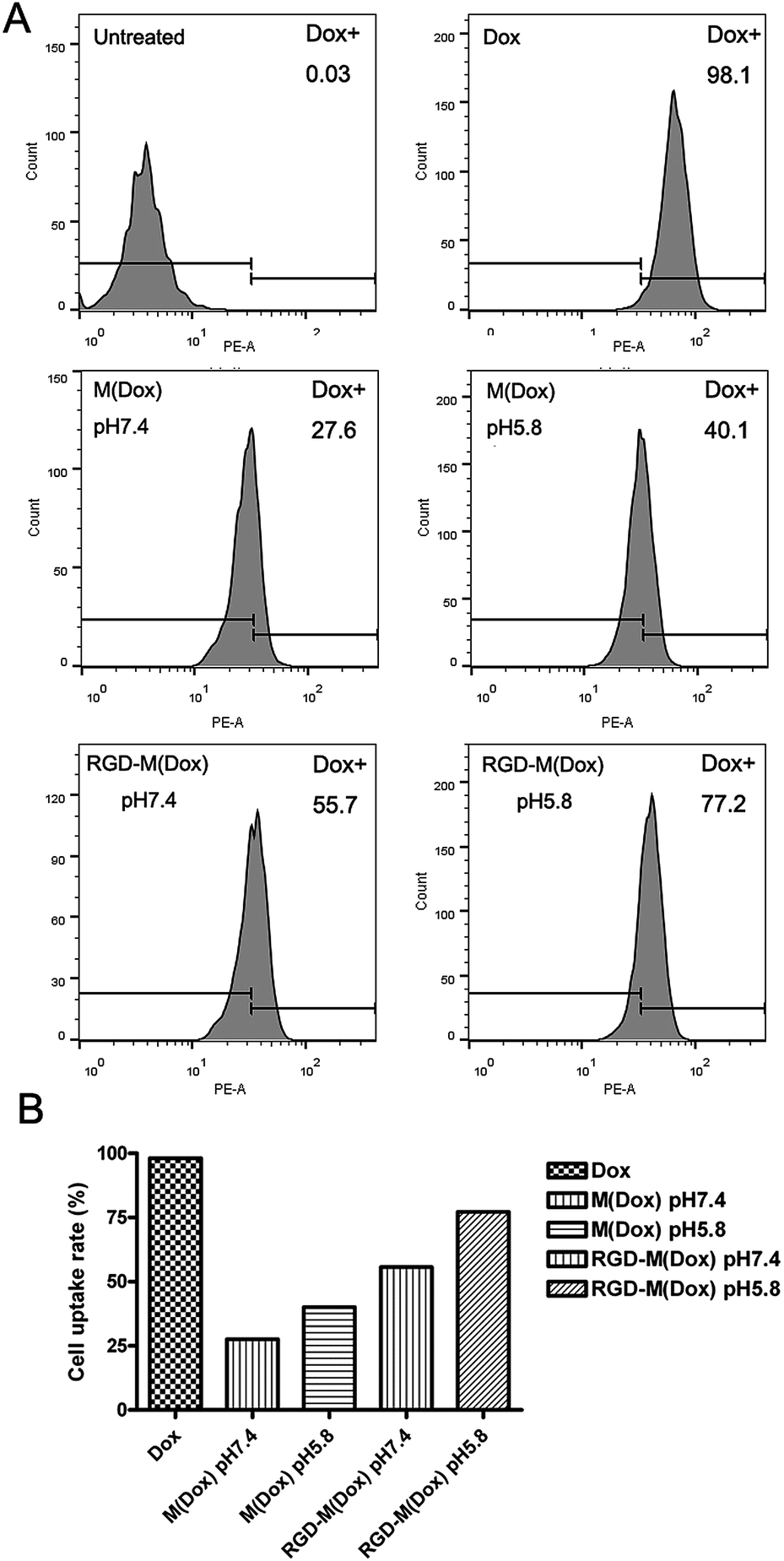 | ||
| Fig. 7 (A) Flow cytometry analysis of B16F10 cells treated with M(Dox) in PBS at pH 7.4 and pH 5.8 and RGD–M(Dox) in PBS at pH 7.4 and pH 5.8 after 1 h of incubation at 37 °C; Dox was used as a positive control; (B) the uptake rate of B16F10 cells is also shown. | ||
3.4 Cytotoxicity analysis
Based on the significantly improved cellular uptake results, we expected that our hybrid micelles could kill more cells than undecorated micelles. The cytotoxicity of Dox-loaded micelles was evaluated by MTT assay. The viability of B16F10 cells incubated with Dox, M(Dox), and RGD–M(Dox) for 48 h at the indicated Dox concentration is shown in Fig. 8, and a dose-dependent effect was observed for the three types of Dox formulations.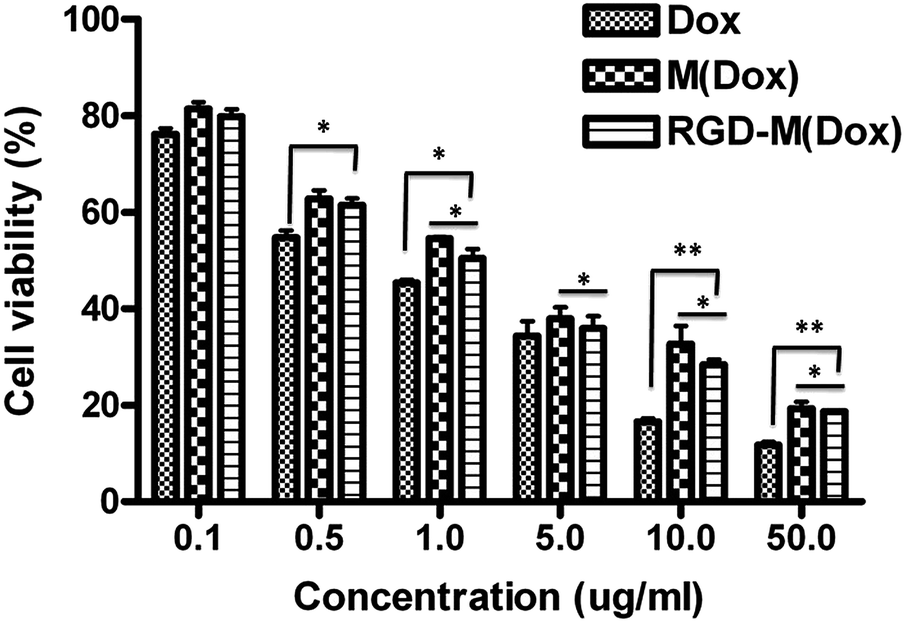 | ||
| Fig. 8 The cytotoxicity analysis of B16F10 cells treated with M(Dox) and RGD–M(Dox) after 48 h of incubation at 37 °C. All the results were repeated three times and are presented as the mean ± SD. | ||
Free Dox exhibited the most efficacious cytotoxicity against B16F10 cells among the three Dox formulations, which could be attributed to increased cellular uptake as compared to the two micelles (as shown in Fig. 6A). The cell viability at each concentration followed the order of Dox < RGD–M(Dox) < M(Dox), indicating that M(Dox) has decreased cytotoxicity as compared to RGD–M(Dox). Flow cytometry analysis revealed that RGD-decorated micelles displayed increased endocytosis by B16F10 cells as compared to undecorated micelles, which directly led to the more nuclear Dox accumulation indicated by CLSM. Therefore, RGD–M(Dox) micelles killed more cells than M(Dox) micelles, suggesting a higher cytotoxicity of RGD-decorated micelles.
4 Conclusion
In summary, we successfully developed Dox-loaded micelles combining cRGD targeting and charge conversion function. The micelles showed a hydrodynamic diameter of 30.1 nm, −4.42 mV of zeta potential, and approximately 90% of drug encapsulation efficiency. The Dox-loaded micelles exhibited a sustained, low pH-enhanced release profile in PBS. CLSM and flow cytometry analysis indicated significantly improved cellular uptake, mediated by selective binding of RGD to αvβ3 integrins and pH-triggered surface charge-switching. Our results in vitro demonstrated the key roles of combining RGD decoration and imidazoles for improving the delivery of anti-cancer agents to some tumours overexpressing αvβ3 integrins, which may be used to develop efficient drug delivery systems for cancer therapy in the future.Acknowledgements
The project was supported by the National Natural Science Foundation of China (Project no. 91227118 and 51373167).Notes and references
- S. D. Steichen, M. Caldorera-Moore and N. A. Peppas, Eur. J. Pharm. Sci., 2013, 48, 416–427 CrossRef CAS PubMed.
- F. Alexis, E. M. Pridgen, R. Langer and O. C. Farokhzad, in Drug Delivery, Springer, 2010, pp. 55–86 Search PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- P. Couvreur, Adv. Drug Delivery Rev., 2013, 65, 21–23 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2011, 47, 113–131 CrossRef.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nano, 2007, 2, 751–760 CrossRef CAS PubMed.
- X. Hu and X. Jing, Expert Opin. Drug Delivery, 2009, 6, 1079–1090 CrossRef CAS PubMed.
- X. Hu, J. Li, W. Lin, Y. Huang, X. Jing and Z. Xie, RSC Adv., 2014, 4, 38405–38411 RSC.
- X. Hu, S. Liu, Y. Huang, X. Chen and X. Jing, Biomacromolecules, 2010, 11, 2094–2102 CrossRef CAS PubMed.
- E. K.-H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv214 Search PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS.
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS PubMed.
- D. Ling, W. Park, S.-j. Park, Y. Lu, K. S. Kim, M. J. Hackett, B. H. Kim, H. Yim, Y. S. Jeon, K. Na and T. Hyeon, J. Am. Chem. Soc., 2014, 136, 5647–5655 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS PubMed.
- M. S. Muthu, C. V. Rajesh, A. Mishra and S. Singh, Nanomedicine, 2009, 4, 657–667 CrossRef CAS PubMed.
- R. Wang, X. Hu, S. Wu, H. Xiao, H. Cai, Z. Xie, Y. Huang and X. Jing, Mol. Pharmaceutics, 2012, 9, 3200–3208 CrossRef CAS PubMed.
- T. M. Allen, Nat. Rev. Cancer, 2002, 2, 750–763 CrossRef CAS PubMed.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2012, 64, 206–212 CrossRef PubMed.
- J. W. Nichols and Y. H. Bae, Nano Today, 2012, 7, 606–618 CrossRef CAS PubMed.
- O. Trédan, C. M. Galmarini, K. Patel and I. F. Tannock, JNCI, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef PubMed.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, D. E. Hu, J. H. Ardenkjaer-Larsen, R. Zandt, P. R. Jensen, M. Karlsson, K. Golman, M. H. Lerche and K. M. Brindle, Nature, 2008, 453, 940–943 CrossRef CAS PubMed.
- Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104–109 CrossRef CAS PubMed.
- W. She, N. Li, K. Luo, C. Guo, G. Wang, Y. Geng and Z. Gu, Biomaterials, 2013, 34, 2252–2264 CrossRef CAS PubMed.
- H. Wang, F. Xu, D. Li, X. Liu, Q. Jin and J. Ji, Polym. Chem., 2013, 4, 2004–2010 RSC.
- M. Curcio, I. Altimari, U. G. Spizzirri, G. Cirillo, O. Vittorio, F. Puoci, N. Picci and F. Iemma, J. Nanopart. Res., 2013, 15, 1–11 CrossRef.
- X. Guan, X. Hu, S. Liu, Y. Huang, X. Jing and Z. Xie, RSC Adv., 2014, 4, 55187–55194 RSC.
- X. Hu, X. Guan, J. Li, Q. Pei, M. Liu, Z. Xie and X. Jing, Chem. Commun., 2014, 50, 9188–9191 RSC.
- D. Ling, H. Xia, W. Park, M. J. Hackett, C. Song, K. Na, K. M. Hui and T. Hyeon, ACS Nano, 2014, 8, 8027–8039 CrossRef CAS PubMed.
- C.-S. Lee, W. Park, Y. U. Jo and K. Na, Chem. Commun., 2014, 50, 4354–4357 RSC.
- J. Yue, S. Liu, R. Wang, X. Hu, Z. Xie, Y. Huang and X. Jing, Mol. Pharmaceutics, 2012, 9, 1919–1931 CrossRef CAS PubMed.
- R. Wang, X. Hu, J. Yue, W. Zhang, L. Cai, Z. Xie, Y. Huang and X. Jing, J. Mater. Chem. B, 2013, 1, 293–301 RSC.
- X. Hu, R. Wang, J. Yue, S. Liu, Z. Xie and X. Jing, J. Mater. Chem., 2012, 22, 13303–13310 RSC.
- K. Temming, R. M. Schiffelers, G. Molema and R. J. Kok, Drug Resist. Updates, 2005, 8, 381–402 CrossRef CAS PubMed.
- N. Yonenaga, E. Kenjo, T. Asai, A. Tsuruta, K. Shimizu, T. Dewa, M. Nango and N. Oku, J. Controlled Release, 2012, 160, 177–181 CrossRef CAS PubMed.
- J. Yang, Y. Hou, G. Ji, Z. Song, Y. Liu, G. Dai, Y. Zhang and J. Chen, Eur. J. Pharm. Sci., 2014, 52, 180–190 CrossRef CAS PubMed.
- Z. Zhen, W. Tang, H. Chen, X. Lin, T. Todd, G. Wang, T. Cowger, X. Chen and J. Xie, ACS Nano, 2013, 7, 4830–4837 CrossRef CAS PubMed.
- Y. Miura, T. Takenaka, K. Toh, S. Wu, H. Nishihara, M. R. Kano, Y. Ino, T. Nomoto, Y. Matsumoto and H. Koyama, ACS Nano, 2013, 7, 8583–8592 CrossRef CAS PubMed.
- W. Song, Z. Tang, D. Zhang, Y. Zhang, H. Yu, M. Li, S. Lv, H. Sun, M. Deng and X. Chen, Biomaterials, 2014, 35, 3005–3014 CrossRef CAS PubMed.
- F. Danhier, B. Vroman, N. Lecouturier, N. Crokart, V. Pourcelle, H. Freichels, C. Jerome, J. Marchand-Brynaert, O. Feron and V. Preat, J. Controlled Release, 2009, 140, 166–173 CrossRef CAS PubMed.
- H. Tian, L. Lin, J. Chen, X. Chen, T. G. Park and A. Maruyama, J. Controlled Release, 2011, 155, 47–53 CrossRef CAS PubMed.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, Macromol. Biosci., 2011, 11, 219–233 CrossRef CAS PubMed.
- A. Shimoda, S. I. Sawada and K. Akiyoshi, Macromol. Biosci., 2011, 11, 882–888 CrossRef CAS PubMed.
- C. Zhan, B. Gu, C. Xie, J. Li, Y. Liu and W. Lu, J. Controlled Release, 2010, 143, 136–142 CrossRef CAS PubMed.
- H. A. Kim, K. Nam and S. W. Kim, Biomaterials, 2014, 35, 7543–7552 CrossRef CAS PubMed.
- W. Song, M. Li, Z. Tang, Q. Li, Y. Yang, H. Liu, T. Duan, H. Hong and X. Chen, Macromol. Biosci., 2012, 12, 1514–1523 CrossRef CAS PubMed.
- H. Uchino, Y. Matsumura, T. Negishi, F. Koizumi, T. Hayashi, T. Honda, N. Nishiyama, K. Kataoka, S. Naito and T. Kakizoe, Br. J. Cancer, 2005, 93, 678–687 CrossRef CAS PubMed.
- C. Deng, H. Y. Tian, P. B. Zhang, J. Sun, X. S. Chen and X. B. Jing, Biomacromolecules, 2006, 7, 590–596 CrossRef CAS PubMed.
- N. Graf, D. R. Bielenberg, N. Kolishetti, C. Muus, J. Banyard, O. C. Farokhzad and S. J. Lippard, ACS Nano, 2012, 6, 4530–4539 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |