
Bioluminescent tools for the analysis of G-protein-coupled receptor and arrestin interactions
Mitsuru Hattori and
Takeaki Ozawa*
Department of Chemistry, School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: ozawa@chem.s.u-tokyo.ac.jp; Fax: +81-3-5802-2989; Tel: +81-3-5841-4351
First published on 15th January 2015
Abstract
G protein-coupled receptors (GPCRs) play crucial roles in numerous physiological and disorder-related processes. Because GPCRs are regarded as a target of many therapeutics, various methods for their analyses have been developed and applied for high-throughput screening of large chemical libraries. As a complement to the traditional analysis of second messengers and gene expression, direct monitoring of GPCR behavior is now indispensable for the identification and accurate analysis of novel chemicals. This review presents new protein-based bioluminescent probes for monitoring GPCR interaction with β-arrestin, a cytoplasmic protein that binds to GPCRs. The principle is based mainly on bioluminescence resonance energy transfer (BRET) and protein fragment complementation (PCA) techniques, which can advance GPCR drug discovery technologies.
1. Introduction
Seven transmembrane spanning receptors, G protein coupled receptors (GPCRs), constitute the largest protein family in the human genome, with over 800 members.1 Actually, GPCRs are activated with extracellular stimuli of many kinds, including neurotransmission, amino acids, peptides, hormones, ions, and photons.2,3 The GPCRs mainly function to facilitate the conversion of extracellular stimuli into intracellular signals, which subsequently regulate physiological functions such as hormone and enzyme release, proliferation, differentiation, chemotaxis, and inflammation.4–6 Many GPCRs have been implicated in human diseases, as evidenced by the fact that GPCRs are now in the spotlight of drug discovery in the pharmaceutical industry. In fact, more than 40% of current therapeutic drugs target GPCRs.7Binding of ligands produces conformational changes in GPCRs, which initiate GPCR-related signal transduction.8 The conformational change induces the release of a GDP from G protein alpha subunit (Gα) from GPCRs and exchanges its GDP to GTP. The activated G proteins catalyze the synthesis of second messenger such as adenosine 3′,5′-cyclic monophosphate (cAMP), diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3). Together with development of a measuring system, a series of G protein activations and signal transduction with second messengers have become markers of GPCR activation.9–11 However, these downstream pathways often tightly link to other signal cascades. When we apply the measuring methods for high-throughput screening of GPCR ligands, identification of many false positive compounds presents a serious problem.
Another strategy to measure GPCR activation is to examine the spatial dynamics of GPCRs. Through ligand-induced signal transduction, GPCRs interact temporally with several signaling factors. GPCR homo–hetero dimerization is also involved in the signaling events.12 Furthermore, activated GPCRs are internalized through clathrin-dependent endocytosis for degradation or recycling them to the plasma membrane.13 One trigger of such GPCR dynamics is an intracellular protein, arrestin, which belongs to a small cytoplasmic protein family. It is a popular target protein used to monitor GPCR activation. Ligand-bound GPCRs are suppressed by G protein catalysis upon signal termination. G protein-coupled receptor kinases (GRKs) are recruited to GPCR and phosphorylate the C-terminal region, which are the binding site of arrestins, to interrupt the association of G protein.14 In short, arrestin binding can constitute specific evidence that GPCR activation occurs. Because the GPCR–arrestin interaction is maintained until sequestration, the amounts of its complex become a good index of how GPCR activates upon ligand binding. The arrestins are categorized into three types: visual arrestins, non-visual arrestins, and core arrestin.15 Non-visual arrestins, β-arrestin1 and β-arrestin2 are mainly used for GPCR analysis.
To elucidate the potential of GPCRs as a drug targets, methods to measure GPCR activation and inhibition should be quantitative under natural cellular conditions. Bioluminescence tools are established as a general approach for monitoring the actual state of living cells. Monitoring protein expression and functions using bioluminescence is important to elucidate the mechanisms and processes of biological phenomena.16 Because a luciferase emits photons with an enzyme substrate as a chemical reaction, excitation light is not necessary, in contrast to fluorescence-imaging techniques. Light irradiation readily generates background noise from cell tissues and interferes with correct measurements. Moreover, some chemicals have fluorescent properties, leading to a misinterpretation of their effects when fluorescence techniques are used. For these reasons, applications of luciferase have been widely developed with highly quantitative properties.
This review specifically describes bioluminescence methods for monitoring the status of GPCR–arrestin interactions using genetically encoded probes. Ligand-activated GPCRs are measured quantitatively through the GPCR–arrestin interaction. Techniques based on bioluminescence resonance energy transfer (BRET) and protein fragment complementation (PCA) are particular topics that are applicable for discovery of drugs targeted to GPCR.
2. Bioluminescence resonance energy transfer (BRET) for monitoring GPCR activity through arrestin behavior
In single-cell imaging, Förster (or Fluorescence) Resonance Energy Transfer (FRET) is a useful technique for the sensing of protein conformational changes and protein–protein interactions with a fluorescence signal.17 Bioluminescence Resonance Energy Transfer (BRET) is an advanced technique of FRET, which replaces the donor fluorophore with luciferase protein. When the donor luciferase is placed close to a fluorescent acceptor with close proximity, transfer of energy occurs with luciferase-substrate oxidation.18 By genetic fusion of each donor or acceptor molecule to proteins of interest, we can detect protein–protein interaction through measurement of the resultant photons of the acceptor relative to the donor bioluminescence photon counts. Whereas BRET signals are much weaker than FRET signals because of lower photon numbers, BRET methods are widely applied for quantitative evaluation in cell populations and living tissues.192.1 BRET-based assays for monitoring the interaction of GPCR with β-arrestin
Renilla luciferase (RLuc; λmax = 480 nm) has been used exclusively in BRET strategy because of the ideal wavelength as a donor protein. For monitoring the GPCR–β-arrestin interaction by BRET, RLuc is a major donor protein. Angers et al. prepared two cDNA constructs encoding C-terminal end of β2-adrenergic receptor (β2AR) fused to RLuc, whereas N-terminal end of enhanced red-shifted GFP (YFP; λem = 527 nm) fused to β-arrestin2 (ref. 20) (Fig. 1A). HEK293 cells coexpressing the β2AR–RLuc and β-arrestin2–YFP were measured. Their BRET ratio was calculated (acceptor emission to donor emission) from bioluminescence photon counts. The BRET ratio showed an increase in acceptor fluorescence in a dose-dependent manner upon addition of agonist of β2AR, isoproterenol (Fig. 1B). The selection of the fluorescence molecule is crucial for improvement of the BRET signal intensity with higher efficiency. Hamdan et al. used a Venus protein, which is a modified YFP increasing in the rate of chromophore maturation,21 as an acceptor fused with each of different GPCRs.22 The pair of probes, GPCR–Venus and β-arrestin2–RLuc, showed BRET with higher signal intensity. Venus was also applied as BRET probes for fusions with the N-terminal end of arrestins.23–26 In another example, a BRET pair of Renilla green fluorescent protein (RGFP) and RLuc was demonstrated for application to GPCRs–β-arrestin interaction because of the highly efficient resonance energy transfer.27 Usefulness of the RGFP based BRET probes has been demonstrated by monitoring the activities of GPCRs: β2AR, δ-opioid receptor (DOP), and vasopressin-2 receptor (V2R).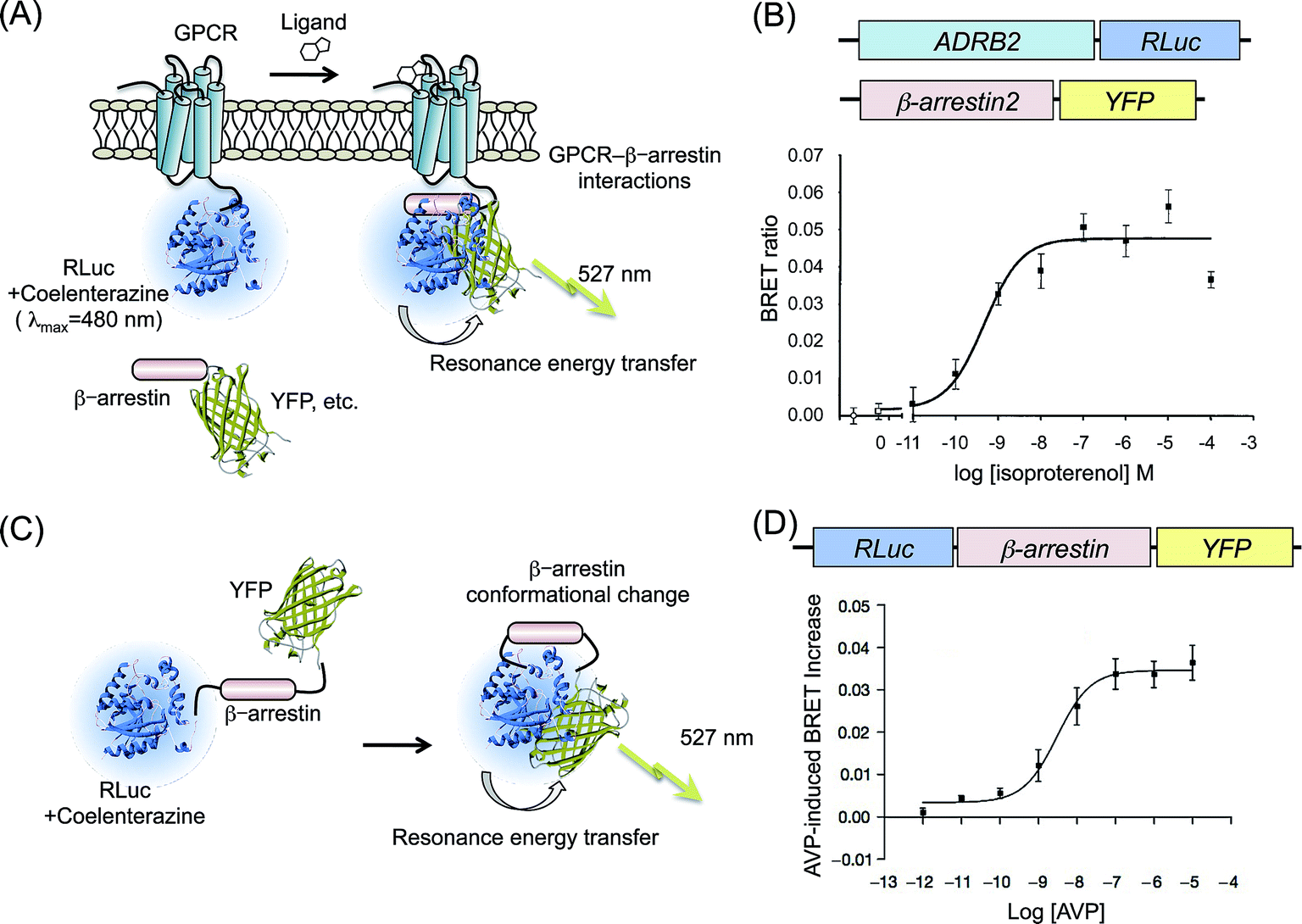 | ||
| Fig. 1 BRET assays based on GPCR–β-arrestin interactions. (A) Schematic diagram showing the BRET probes used to detect a GPCR–β-arrestin interaction. The GPCR and fluorescence protein (YFP, Venus, etc.) are attached, respectively, to RLuc and β-arrestin. Binding of a ligand to GPCR induces recruitment of β-arrestin to GPCR. The interaction brings RLuc and fluorescence protein into proximity. Then resonance energy transfer occurs. (B) Agonist dependence on β2AR–β-arrestin2 interactions evaluated by BRET. Upper figures present schematic structures of cDNA constructs transformed into cells. The lower graph shows BRET ratios in the presence of increasing concentrations of β2AR-agonist, isoproterenol. The graph was modified from an earlier report.20 (C) Schematic showing the principle of intramolecular BRET for β-arrestin. RLuc and YFP were fused with the N-terminal and C-terminal of β-arrestin. When it undergoes a conformational change, RLuc becomes sufficiently close to YFP, resulting in resonance energy transfer. (D) A conformational change of β-arrestin depending on the concentration of V2R agonist. The upper figure shows a schematic structure of the cDNA construct. The lower graph shows eBRET kinetics with V2R-agonist AVP dosing. The graph was modified from an earlier report.33 | ||
The bioluminescence reaction of RLuc occurs normally with RLuc substrates coelenterazine or DeepBlueC™. However, one shortcoming of the use of RLuc is that the substrates are relatively unstable in aqueous solutions. To overcome this shortcoming, Pfleger et al. developed a modified BRET technique for real-time experiments in living cells: extended BRET (eBRET).28 eBRET uses a protected form of coelenterazine h, EnduRen™. Enhanced GFP (EGFP; λex = 488 nm) was selected as an acceptor of eBRET because RLuc with EnduRen generated a shorter wavelength of bioluminescence than with coelenterazine h. With application of eBRET, time-lapse monitoring of β-arrestin1 binding to GPCRs such as thyrotropin-releasing hormone receptor 1 (TRHR1), orexin receptor 2 (OxR2), and angiotensin II receptor type 1a (AT1aR) was demonstrated. The data clearly showed temporal changes in ligand induced GPCR activation. Because the BRET signal ratio is stable against environmental changes such as temperature and substrate concentrations, it is widely applied in various cell types. Moreover, the character of reversible and rapid reactions is useful for monitoring temporal GPCR activity in living cells.
2.2 Intramolecular BRET assays for monitoring conformational changes of β-arrestin
Mutagenesis studies of β-arrestin structures suggest a conformational rearrangement of the molecule upon GPCR binding.29–32 Therefore, detecting the intramolecular distance of amino-terminal and carboxy-terminal domains of β-arrestin is noticeable as another strategy for indirect monitoring of the translocation of β-arrestin to C-terminal of GPCRs.Charest et al. designed an intramolecular BRET probe: RLuc was fused to the N-terminal end of β-arrestin, whereas YFP was fused to the C-terminal end33 (Fig. 1C). The BRET reaction of the probe was detected in cells that overexpressed GPCRs V2R. Under dosing its specific ligand arginine vasopressin (AVP), concentration-dependent increases in the BRET ratio were clearly confirmed, from which the EC50 was calculated (Fig. 1D). Shukla et al. measured the conformational changes of β-arrestin upon stimulation of GPCRs: β2AR, AT1aR, and parathyroid hormone peptide receptor (PTH1R).34 Monitoring the variation of the BRET ratio, they confirmed the inhibition of agonist-activated G protein signaling by an antagonist. Furthermore, results demonstrated that a conformational change of β-arrestin was dependent on the phosphorylation of the GPCR, not a result of binding of β-arrestin to the GPCR. Intramolecular BRET probes based on β-arrestin are not applicable for chemical library screening of GPCRs because the structural changes in the β-arrestin are not specific for a particular GPCR but a common event for many GPCRs. However, the general applicability for different GPCRs is a strong advantage for the analysis of β-arrestin signaling.
BRET techniques for monitoring GPCR–β-arrestin interaction present the benefit of higher luminescence counts than those of other bioluminescence methods. Even if there is no information related to the spatial arrangement of donor and acceptor molecules in a probe designing process, BRET techniques will be applicable to measuring GPCR signaling pathways, especially in the reaction with rapid alteration.
3. Split-reporter complementation for monitoring the interaction of GPCR with arrestin
Another increasingly popular technique for detecting protein–protein interactions in living cells is protein fragment complementation assays (PCAs). In principle, a reporter protein is dissected into two fragments that consequently have less enzymatic activity. These fragments are fused, respectively, to two proteins of interest. Upon interaction of these two proteins, fragments of the reporter protein are brought into proximity, leading the fragments to refold spontaneously and to reconstitute their activity. As the reporter protein, enzymes, and fluorescent and bioluminescent proteins is often used. Their best dissection sites have already been identified. The restored enzymatic activity or emission light can be used to detect the timing of protein–protein interactions. Because PCA has the benefit of a low signal-to-background-noise ratio, the method provides quantitative information with wider dynamic ranges in many cell events.3.1 Complementation assays with split luciferase fragments
PCAs based on split luciferase fragments convert the protein–protein interaction directly to bioluminescence signals. In contrast to the irreversible complementation of fluorescent protein fragments, complementation of the luciferase fragments is known to be reversible. Because of this, we are able to monitor the dissociation of protein–protein interactions.35–37 Therefore, PCA methods using luciferase fragments provide data of temporal variation of the interactions. Luciferases are categorized by the type of substrate: coelenterazine or D-luciferin. The dissection sites of RLuc and Gaussia Luciferase (GLuc), using coelenterazine, have been determined.38–41 However, these luciferases are unstable in terms of the bioluminescence reaction in living cells, although the intensities are high. The type of luciferases using D-luciferin, originated from fireflies, click beetles, and railroad worms, have a property for sustaining the luminescence intensity. Therefore, in PCAs, The latter luciferases are appropriate for temporal measurements of biological phenomena in living cells and animals. Several patterns of split fragments of each luciferase have been applied to biological studies, which are useful as probes to sense intermolecular and intramolecular protein interactions.36,37,42,43 GPCR–β-arrestin interactions are also targets of bioluminescence analysis based on luciferase complementation.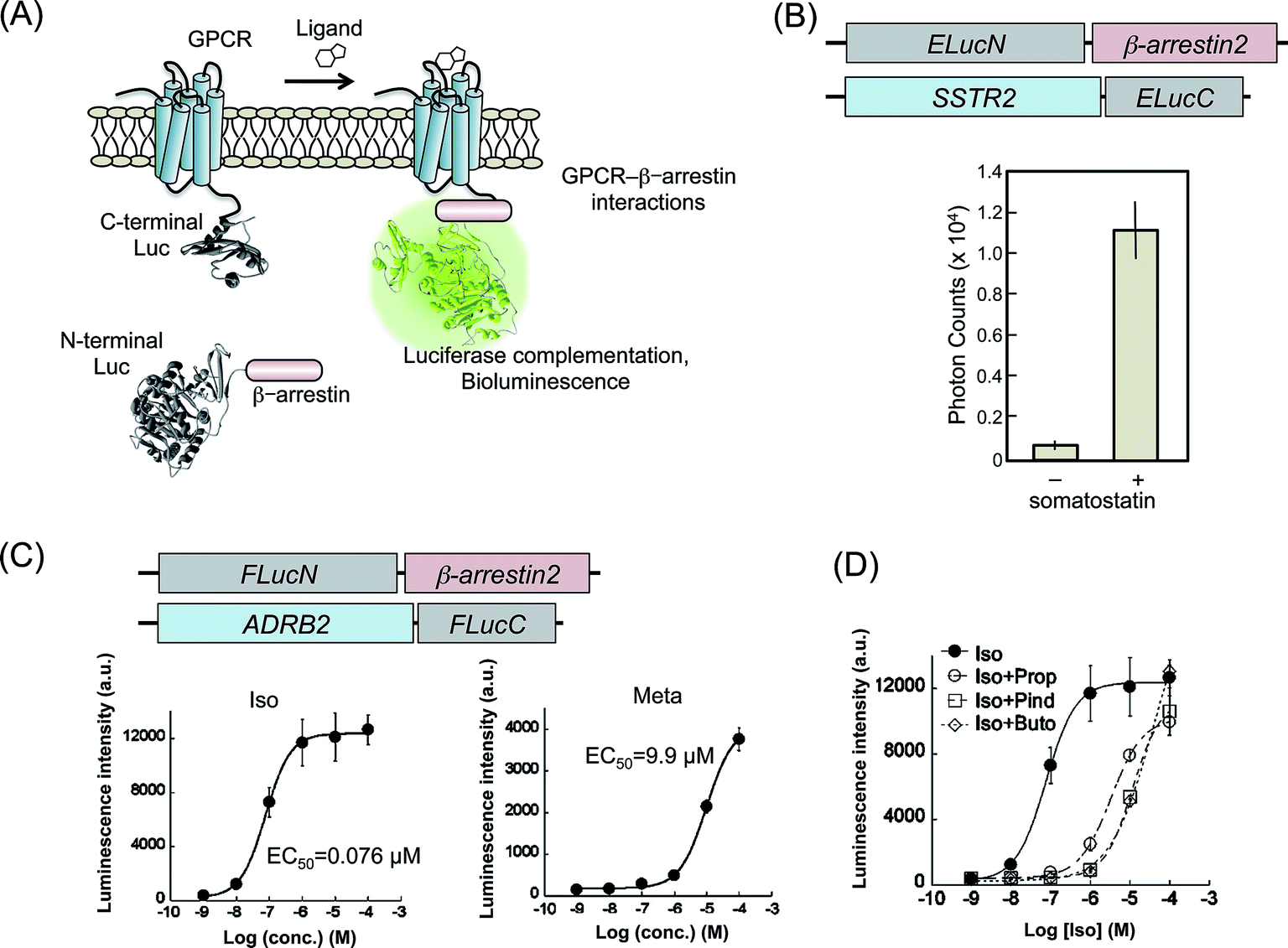 | ||
| Fig. 2 Luciferase fragment complementation assays for the detection of GPCR–β-arrestin interaction. (A) A schematic diagram of the measurement of GPCR–β-arrestin interactions by luciferase fragment complementation. The N-terminal and C-terminal fragments of luciferase are fused, respectively, to GPCR and β-arrestin. Ligand-induced interaction of GPCR with β-arrestin brings two luciferase fragments into proximity, in subsequent reconstitution. The diagram was modified from an earlier report.51 (B) Agonist dependence of SSTR2–β-arrestin2 interactions evaluated using luciferase-based PCAs. Upper figures present schematic structures of cDNA constructs transformed into cells. The lower graph shows measured photon counts with or without a SSTR2 agonist: somatostatin. The graph was modified from an earlier report.44 (C) Dose–response curves for agonists of β2AR based on β2AR–β-arrestin2 interactions evaluated using luciferase-based PCAs. Upper figures show schematic structures of cDNA constructs. The lower graph shows dose–response curves using agonists of β2AR, isoproterenol (Iso) and metaproterenol (Meta). Calculated EC50 values are also shown. Graphs were modified from an earlier report.45 (D) Dose–response curves for Iso in the presence of competitive inhibitors, propranolol (Prop), pindolol (Pind), and butoxamine (Buto). The graph was modified from an earlier report.45 | ||
SSTR2 was demonstrated for trial experiments of luciferase-based PCAs.44 Emerald luciferase (ELuc) has a highest luminescence intensity (λmax = 537 nm) in luciferases using D-luciferin. The C-terminal fragment of ELuc (ELucC) was fused to the C-terminal end of SSTR2, whereas the N-terminal fragment of ELuc (ELucN) was fused to N-terminal end of β-arrestin2 (Fig. 2B). These two constructs were transfected into HEK293 cells to establish cell lines that stably express the fusion proteins. The cell lines were cultured on a 96-well plate and were exposed to different concentrations of a specific ligand, somatostatin. The luminescence photon counts increased concomitantly with increasing somatostatin concentration. The results demonstrated that the interaction of β-arrestin2 to GPCR induced increased bioluminescence.
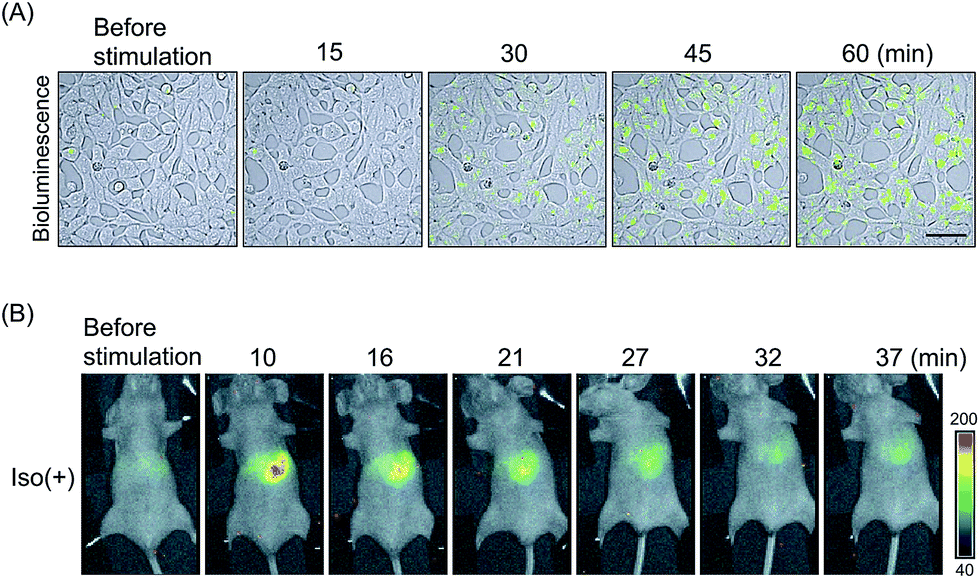 | ||
| Fig. 3 Real-time imaging of the ligand-induced GPCR–β-arrestin interaction with luciferase-based PCAs. (A) Time course analysis of SSTR2–β-arrestin2 interactions. ELucN–β-arrestin2 and SSTR2–ELucC probes were expressed in the cells. Then the luminescence was measured after somatostatin administration. Bioluminescence images were merged on DIC images. Bar = 50 μm. (B) Bioluminescence in vivo imaging of β2AR–β-arrestin interactions. FLucN–β-arrestin2 and β2AR-FLucC were expressed in the liver of mice. Variation of luminescence was monitored in the mice after injection of isoproterenol. The images were modified from an earlier report.45 | ||
Bioluminescence measurements including luciferase-based PCAs show highly quantitative data enabling the correct examination of GPCR ligands. However, enzymatic activity of luciferases is influenced by concentrations of their substrate and ATP. In addition, activities of some GPCRs were well investigated under inflammation conditions in tissues. In such conditions, fluctuation of the cell number and condition easily affected the precise measurements. For that reason, it is practically useful to normalize the probe's signal against for such an environmental factor. Luciferases have multi-color variations that are mutually separable with individual wavelength. Red luciferase from railroad worms (Phrixothrix hirtus; RWLuc, λmax = 623 nm) was applied for the internal control of PCA with ELuc.46 Full-length of RWLuc was coexpressed into cells with split ELuc probes targeting GPCRs. The photon counts of bioluminescence from ELuc were increased concomitantly with increasing concentrations of agonists. However, standard deviations of the data indicated strongly that the photon counts fluctuated considerably. To correct the unreliable counts, the bioluminescence intensities from ELuc were normalized against the counts of RWLuc. The improvement of standard deviations was confirmed by comparing the coefficient of variation (CV). Using dual color bioluminescence analysis, quantitative monitoring for GPCR activity is further refined.
From the reason that the class of GPCRs has been determined by the localization profiles in the cell, the classes of so many GPCRs remain unclear. No information is available related to the classes of endothelin receptor type B (EDNRB), endothelial differentiation G-protein/coupled receptor 3 (EDG3), or angiotensin receptor-like 1/aperin receptor (AGTRL1). These GPCR probes fused to ELucC were cotransfected with ELucN-β-arrestin1 or ELucN-β-arrestin2. The EDNRB probes transiently reached the maximum of bioluminescence intensity. Then the counts decreased rapidly close to the initial level. For EDG3 and AGTRL1 probes, the temporal patterns of photon counts were similar to those of AGTR1 probes, which stably hold the increasing bioluminescence intensities. These results suggest that EDNRB is categorized into Class-A, and that EDG3 and AGTRL1 are categorized into Class-B. Using luciferase-based PCAs, the class of GPCR can be determined by temporal monitoring of the interaction of GPCRs with β-arrestin.
In the same way as other in vivo bioluminescence analysis of drug sensitivity, the mouse is also a popular model for GPCR activation analysis. For in vivo imaging, luciferases that have an emission light with longer wavelength are effective for tissue penetration. Based on the principle of FLuc fragments complementation, Luker et al. developed a system of detecting chemokine receptor CXCR4 activation by CXCL12 binding.56 Intraperitoneal HEK293T cells expressing the FLuc probes fused with CXCR4 and β-arrestin2 were reacted to CXCL12 administration as an increase in bioluminescence counts. We also generated FLuc based probes with ADRB2 and β-arrestin2.45 HEK293 cells stably expressing the probes were implanted under mice skin. After intraperitoneal injection of isoproterenol, time-course increases in the bioluminescence intensity were monitored. This imaging method demonstrated the effects of competitive inhibitor of GPCR in living mice.
GPCRs are expressed in many animal organs. Therefore, probe localization in assays is indispensable for drug evaluation. Hydrodynamic tail vein (HTV) method can express the β2AR probes in mice liver cells. Living mice were treated with HTV using plasmids encoding FLuc-based probes.45 After 16–24 h of injection, isoproterenol-induced bioluminescence was validated in the mice liver (Fig. 3B). The bioluminescence increase was abolished upon β2AR inhibitor dosing. These results indicated that in vivo imaging of GPCR reaction by luciferase-based PCAs contributes to the development of new GPCR targeting drugs as a pharmacokinetic model.
An issue of the luciferase-based PCAs at the present stage is that the luminescence photon counts fundamentally lower than those in other assays with a full-length luciferase such as BRET. Nevertheless, the techniques are strongly advantageous for high-throughput screening of GPCR ligands because luciferase-based PCAs show high sensitivity and low false positive rate for ligands.
3.2 Complementation assays based on β-galactosidase
A product of a bacterial lacZ gene, β-galactosidase (β-gal) is a famous reporter enzyme in the study of transcriptional machinery. It is traditionally used for measuring transcriptional activity of promoters. Because PCAs are adequate to control the reporter enzyme activity in living cells, they have been applied for β-gal in prokaryotic and eukaryotic cells.57,58 β-gal-based PCAs need a pair of mutants with impaired enzyme activity. Each mutant is named Δα, which lacks 11–41 amino acids of β-gal, and Δω, which has an α donor. When these mutants are fused to target proteins respectively, the protein interaction drives complementation of the β-gal enzyme activity. Mostly, the activity is measured by the chemiluminescent substrate of β-gal or luciferase reporter system.Yan et al. developed a system that detected the interaction of GPCR–β-arrestin by β-gal-based PCAs.59 They connected the Δα with C-terminal end of ADRB2; whereas Δω was connected with the C-terminal end of β-arrestin (Fig. 4A). Activities of β-gal with the agonists were measured using chemiluminescence, which showed dose–responses curves. Additionally, the β-gal probes demonstrated the inhibition of ADRB2 activity by antagonists. Based on the system, chemical screening of β2AR agonists and antagonists was demonstrated with lower false positive than by previous methods. Because of the high enzymatic activity of reconstituted β-gal, β-gal-based PCAs were also demonstrated in living mice.60 Cells expressing the β-gal probes were generated and injected under the mouse skin. The mice showed ligand-dependent complementation of β-gal visualized by the FLuc reporter.
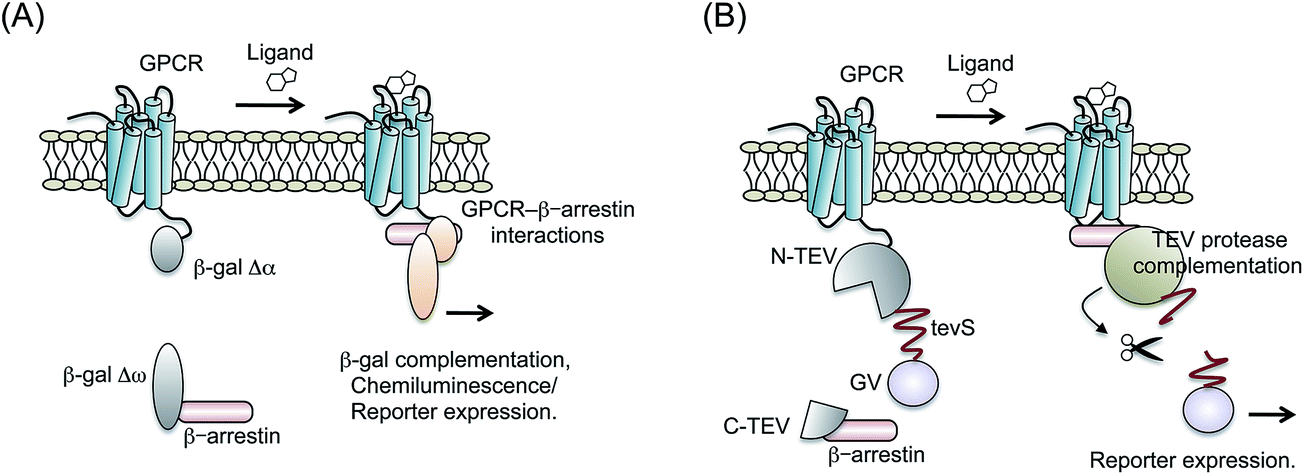 | ||
| Fig. 4 PCA based β-galactosidase (β-gal) and tobacco etch virus (TEV) protease. (A) Schematic diagram showing the split β-gal fragment probes used to detect the interaction of GPCR with β-arrestin. The fragments of β-gal (Δα and Δω) are combined, respectively, with GPCR and β-arrestin. Ligand-induced interaction of GPCR with β-arrestin brings reconstitution of β-gal. (B) Schematic figure showing the principle of split TEV-protease fragment probes. N-terminal fragments of TEV-protease (N-TEV) are connected with GPCR, tevS sequence and GV. A C-terminal fragment of TEV-protease (C-TEV) is fused to β-arrestin. The GPCR–β-arrestin interaction induces recovery of the enzyme activity of TEV-protease subsequent in cleavage of tevS. | ||
Internalization of GPCRs by clathrin-dependent endocytosis follows the β-arrestin binding to GPCR. Hammer et al. simultaneously analyzed GPCRs–β-arrestin interactions and GPCR internalization using β-gal-based PCAs and fluorescence imaging.61 The results revealed that the frequency of internalization was not always correspondent to the GPCR activity. Furthermore, the relation was dependent on the concentration of ligands and temperature.
3.3 Complementation assay based TEV-protease
Tobacco etch virus (TEV) protease is widely used for cleavage of a specific amino acid sequence.62,63 If two arbitrary proteins are mutually connected through the TEV-protease cleavage site (tevS), then they can be detached when TEV-protease access to tevS.64 Wehr et al. determined the most efficient site of TEV-protease-based PCAs by monitoring several membrane proteins' behavior.65 The C-terminal fragment of TEV-protease (C-TEV) was fused to a target protein-A, whereas N-terminal fragment of TEV-protease (N-TEV) was fused to a target protein-B. In addition, a GV domain (yeast Gal4 DNA-binding domain and the herpes simplex VP16 transactivation domain) was connected with N-TEV via tevS. Upon the protein-A interaction with the protein-B, the complemented TEV-protease cleaves tevS. Subsequently, the GV domain translocates into the nucleus and activates reporter gene expression.Djannatian et al. applied the system for some GPCRs and β-arrestin2 (ref. 66) (Fig. 4B). To measure complementation of TEV-protease, luciferase reporter system to GV domain was co-transfected with the probes. When specific ligands for GPCRs were administered, the cells harboring TEV-protease-based probes showed increased bioluminescence. After comparing the reaction property of TEV-protease-based PCAs with different cultured-cell types, they presented the possibility that GPCR phosphorylation and sequestration are regulated in a cell-type dependent manner.
In the TEV-protease-based complementation assays, the activity of GPCR is detected as a signal of reporter gene expression. Therefore, it is impossible to obtain the information about temporal activity of GPCRs. On the other hand, different reporters can be applied for the assay systems, which is an advantage of massive chemical library screening.
4. Conclusions and perspectives
In the present review, we described bioluminescence methods for measuring GPCR–arrestin interactions based on BRET and PCA. Because there are many well-established strategies to detect GPCR activities, researchers need to consider which method is suitable for the purpose of each study. For example, screening of unknown chemicals requires higher sensitivity and signal-to-background ratio, which influence on accurate identification of agonists and antagonist with their high affinity. From this point of view, luciferase-based PCAs are strong candidates and appropriate for chemical library screening. In contrast, BRET assays show higher temporal resolution and strong luminescence intensities than other luminescence methods. Therefore, it is often used for analyzing mechanism of GPCR signaling. In any methods shown in this review, we need to consider some artifacts originated from the fusion of the luminescent probes and their overexpression in target cells.Interactions of arrestin with GPCRs are regarded as signal suppression of GPCR after G protein signal transduction. The extent of the complex formation is reflected directly by phosphorylation of GPCRs. Therefore, the interaction is an excellent index of GPCR activation with a specific ligand. Practically, the roles of arrestin are not only signal suppression but controls of more intricate intracellular signals. Sequestrated GPCRs determine the fate of degradation or recycling to plasma membranes. Thereby the integral control of GPCR is necessary for the signal duration. Arrestins are categorized into several types, each of which has an individual property for binding to GPCRs. For example, some GPCRs are selectively bound to β-arrestin2 rather than β-arrestin1.48 Our experiments also showed that distinctive amino acid sites of a GPCR are necessary for the selection of β-arrestin types.51 Therefore, the binding properties of arrestins are expected to be important factors for the regulation of such a complicated GPCR deactivation and relocation mechanism. We need to consider another interactions of β-arrestin with G-protein beta (β) and gamma (γ) subunits, which could associate with the bias of GPCR recycling.67 Furthermore, it was reported that internalization of particular GPCRs occurs through alternative pathways without β-arrestin.68 Arrestins have a distinct signaling pathway from G-protein mediated signaling. β-arrestins facilitate ERK1/2 signaling with Raf, MEK1 and c-Src as a scaffold and signal transducer.69–71 Strategies to monitor arrestin behavior based on BRET and PCA techniques can elucidate various arrestin functional manner.
Most GPCR ligand screening systems use an end point of G protein activation. Additionally, monitoring of GPCR–arrestin interactions becomes an indirect method of markers of the GPCR activity. In recent years, oligomerization of GPCRs on the plasma membrane has been emphasized as an important signal event. Numerous reports have described experiments demonstrating that at least 30 kinds of GPCRs form homodimers, and that 20 pairs of GPCRs form heterodimers.72 Some reports describe trials to detect the formation of GPCR oligomerization using BRET technique.20,73 In the case of the expanded analysis, i.e., combination of luciferase-based PCA and BRET technologies, the dimerization-dependent GPCR association to G protein was monitored using luminescence signals.74 However, although mechanisms of GPCR oligomerization and GPCR–arrestin interaction have been investigated independently, crosstalk of these two events remains unclear. BRET and PCA methods can monitor multiple protein–protein interactions simultaneously.75 Analysis of the combination of GPCR dimerization and arrestin interaction can serve as an important new approach for ligand screening of GPCR.
The BRET and PCA techniques introduced in this review present the important benefit that an event can be detected at the place where interactions natively occur in living cells. Therefore, they dynamically represent the actual circumstances related to GPCR–arrestin interactions. Regarding the arrestin function as a main target for bioluminescence-based studies, we can gain further understanding of GPCR signaling processes.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS), Japan Science and Technology (JST), and MEXT, Japan.References
- R. Grisshammer, Expert Rev. Proteomics, 2013, 10, 1–3 CrossRef CAS PubMed.
- A. Musnier, B. Blanchot, E. Reiter and P. Crepieux, Cell. Signalling, 2010, 22, 707–716 CrossRef CAS PubMed.
- R. C. Stevens, V. Cherezov, V. Katritch, R. Abagyan, P. Kuhn, H. Rosen and K. Wuthrich, Nat. Rev. Drug Discovery, 2013, 12, 25–34 CrossRef CAS PubMed.
- P. J. Conn, A. Christopoulos and C. W. Lindsley, Nat. Rev. Drug Discovery, 2009, 8, 41–54 CrossRef CAS PubMed.
- J. Yan, V. Mihaylov, X. Xu, J. A. Brzostowski, H. Li, L. Liu, T. D. Veenstra, C. A. Parent and T. Jin, Dev. Cell, 2012, 22, 92–103 CrossRef CAS PubMed.
- F. X. Yu, Y. Zhang, H. W. Park, J. L. Jewell, Q. Chen, Y. Deng, D. Pan, S. S. Taylor, Z. C. Lai and K. L. Guan, Genes Dev., 2013, 27, 1223–1232 CrossRef CAS PubMed.
- S. M. Foord, T. I. Bonner, R. R. Neubig, E. M. Rosser, J. P. Pin, A. P. Davenport, M. Spedding and A. J. Harmar, Pharmacol. Rev., 2005, 57, 279–288 CrossRef CAS PubMed.
- H. Unal and S. S. Karnik, Trends Pharmacol. Sci., 2012, 33, 79–88 CrossRef CAS PubMed.
- K. Liu, S. Titus, N. Southall, P. Zhu, J. Inglese, C. P. Austin and W. Zheng, Curr. Chem. Genomics, 2008, 1, 70–78 CrossRef CAS PubMed.
- A. Koval, D. Kopein, V. Purvanov and V. L. Katanaev, Anal. Biochem., 2010, 397, 202–207 CrossRef CAS PubMed.
- S. A. Martins, J. R. Trabuco, G. A. Monteiro, V. Chu, J. P. Conde and D. M. Prazeres, Trends Biotechnol., 2012, 30, 566–574 CrossRef CAS PubMed.
- J. Gonzalez-Maeso, Mol. Brain, 2011, 4, 20–26 CrossRef CAS PubMed.
- F. Jean-Alphonse and A. C. Hanyaloglu, Mol. Cell. Endocrinol., 2011, 331, 205–214 CrossRef CAS PubMed.
- E. V. Gurevich, J. J. Tesmer, A. Mushegian and V. V. Gurevich, Pharmacol. Ther., 2012, 133, 40–69 CrossRef CAS PubMed.
- J. G. Krupnick and J. L. Benovic, Annu. Rev. Pharmacol. Toxicol., 1998, 38, 289–319 CrossRef CAS PubMed.
- D. Scott, E. Dikici, M. Ensor and S. Daunert, Annu. Rev. Anal. Chem., 2011, 4, 297–319 CrossRef CAS PubMed.
- A. Miyawaki, Annu. Rev. Biochem., 2011, 80, 357–373 CrossRef CAS PubMed.
- Z. Xia and J. Rao, Curr. Opin. Biotechnol., 2009, 20, 37–44 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, C. T. Chan, A. De, T. F. Massoud and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12060–12065 CrossRef CAS PubMed.
- S. Angers, A. Salahpour, E. Joly, S. Hilairet, D. Chelsky, M. Dennis and M. Bouvier, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3684–3689 CAS.
- A. Rekas, J. R. Alattia, T. Nagai, A. Miyawaki and M. Ikura, J. Biol. Chem., 2002, 277, 50573–50578 CrossRef CAS PubMed.
- F. F. Hamdan, M. Audet, P. Garneau, J. Pelletier and M. Bouvier, J. Biomol. Screening, 2005, 10, 463–475 CrossRef CAS PubMed.
- N. Audet, I. Charfi, O. Mnie-Filali, M. Amraei, A. J. Chabot-Dore, M. Millecamps, L. S. Stone and G. Pineyro, J. Neurosci., 2012, 32, 4827–4840 CrossRef CAS PubMed.
- L. E. Gimenez, S. Kook, S. A. Vishnivetskiy, M. R. Ahmed, E. V. Gurevich and V. V. Gurevich, J. Biol. Chem., 2012, 287, 9028–9040 CrossRef CAS PubMed.
- M. L. Rives, M. Rossillo, L. Y. Liu-Chen and J. A. Javitch, J. Biol. Chem., 2012, 287, 27050–27054 CrossRef CAS PubMed.
- L. E. Gimenez, S. A. Vishnivetskiy, F. Baameur and V. V. Gurevich, J. Biol. Chem., 2012, 287, 29495–29505 CrossRef CAS PubMed.
- P. Molinari, I. Casella and T. Costa, Biochem. J., 2008, 409, 251–261 CrossRef CAS PubMed.
- K. D. Pfleger, J. R. Dromey, M. B. Dalrymple, E. M. Lim, W. G. Thomas and K. A. Eidne, Cell. Signalling, 2006, 18, 1664–1670 CrossRef CAS PubMed.
- M. Han, V. V. Gurevich, S. A. Vishnivetskiy, P. B. Sigler and C. Schubert, Structure, 2001, 9, 869–880 CrossRef CAS.
- S. A. Vishnivetskiy, J. A. Hirsch, M. G. Velez, Y. V. Gurevich and V. V. Gurevich, J. Biol. Chem., 2002, 277, 43961–43967 CrossRef CAS PubMed.
- T. Zhuang, S. A. Vishnivetskiy, V. V. Gurevich and C. R. Sanders, Biochemistry, 2010, 49, 10473–10485 CrossRef CAS PubMed.
- M. Kim, S. A. Vishnivetskiy, N. Van Eps, N. S. Alexander, W. M. Cleghorn, X. Zhan, S. M. Hanson, T. Morizumi, O. P. Ernst, J. Meiler, V. V. Gurevich and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18407–18412 CrossRef CAS PubMed.
- P. G. Charest, S. Terrillon and M. Bouvier, EMBO Rep., 2005, 6, 334–340 CrossRef CAS PubMed.
- A. K. Shukla, J. D. Violin, E. J. Whalen, D. Gesty-Palmer, S. K. Shenoy and R. J. Lefkowitz, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9988–9993 CrossRef CAS PubMed.
- V. Villalobos, S. Naik, M. Bruinsma, R. S. Dothager, M. H. Pan, M. Samrakandi, B. Moss, A. Elhammali and D. Piwnica-Worms, Chem. Biol., 2010, 17, 1018–1029 CrossRef CAS PubMed.
- N. Hida, M. Awais, M. Takeuchi, N. Ueno, M. Tashiro, C. Takagi, T. Singh, M. Hayashi, Y. Ohmiya and T. Ozawa, PLoS One, 2009, 4, e5868 Search PubMed.
- M. Hattori, S. Haga, H. Takakura, M. Ozaki and T. Ozawa, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9332–9337 CrossRef CAS PubMed.
- S. B. Kim, M. Sato and H. Tao, Anal. Chem., 2009, 81, 67–74 CrossRef CAS PubMed.
- S. B. Kim, T. Ozawa, S. Watanabe and Y. Umezawa, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11542–11547 CrossRef CAS PubMed.
- R. Paulmurugan and S. S. Gambhir, Anal. Chem., 2003, 75, 1584–1589 CrossRef CAS.
- M. Hirohama, A. R. Voet, T. Ozawa, H. Saitoh, Y. Nakao, K. Y. Zhang, A. Ito and M. Yoshida, Anal. Biochem., 2013, 448, 92–94 CrossRef PubMed.
- L. Yang, Y. Nasu, M. Hattori, H. Yoshimura, A. Kanno and T. Ozawa, Anal. Chem., 2013, 85, 11352–11359 CrossRef CAS PubMed.
- M. Takeuchi, Y. Nagaoka, T. Yamada, H. Takakura and T. Ozawa, Anal. Chem., 2010, 82, 9306–9313 CrossRef CAS PubMed.
- N. Misawa, A. K. M. Kafi, M. Hattori, K. Miura and T. Ozawa, Anal. Chem., 2010, 82, 2552–2560 CrossRef CAS PubMed.
- H. Takakura, M. Hattori, M. Takeuchi and T. Ozawa, ACS Chem. Biol., 2012, 7, 901–910 CrossRef CAS PubMed.
- A. K. M. Kafi, M. Hattori, N. Misawa and T. Ozawa, Pharmaceuticals, 2011, 4, 457–469 CrossRef CAS PubMed.
- R. T. Kendall and L. M. Luttrell, Cell. Mol. Life Sci., 2009, 66, 2953–2973 CrossRef CAS PubMed.
- R. H. Oakley, S. A. Laporte, J. A. Holt, M. G. Caron and L. S. Barak, J. Biol. Chem., 2000, 275, 17201–17210 CrossRef CAS PubMed.
- R. H. Oakley, S. A. Laporte, J. A. Holt, L. S. Barak and M. G. Caron, J. Biol. Chem., 1999, 274, 32248–32257 CrossRef CAS PubMed.
- R. Irannejad, J. C. Tomshine, J. R. Tomshine, M. Chevalier, J. P. Mahoney, J. Steyaert, S. G. Rasmussen, R. K. Sunahara, H. El-Samad, B. Huang and M. von Zastrow, Nature, 2013, 495, 534–538 CrossRef CAS PubMed.
- M. Hattori, M. Tanaka, H. Takakura, K. Aoki, K. Miura, T. Anzai and T. Ozawa, Mol. BioSyst., 2013, 9, 957–964 RSC.
- A. Emami-Nemini, T. Roux, M. Leblay, E. Bourrier, L. Lamarque, E. Trinquet and M. J. Lohse, Nat. Protoc., 2013, 8, 1307–1320 CrossRef PubMed.
- J. Bubnell, P. Pfister, M. L. Sapar, M. E. Rogers and P. Feinstein, PLoS One, 2013, 8, e74941 CAS.
- J. A. Prescher and C. H. Contag, Curr. Opin. Chem. Biol., 2010, 14, 80–89 CrossRef CAS PubMed.
- R. S. Dothager, K. Flentie, B. Moss, M. H. Pan, A. Kesarwala and D. Piwnica-Worms, Curr. Opin. Biotechnol., 2009, 20, 45–53 CrossRef CAS PubMed.
- K. E. Luker, M. Gupta and G. D. Luker, Anal. Chem., 2008, 80, 5565–5573 CrossRef CAS PubMed.
- B. T. Blakely, F. M. V. Rossi, B. Tillotson, M. Palmer, A. Estelles and H. M. Blau, Nat. Biotechnol., 2000, 18, 218–222 CrossRef CAS PubMed.
- F. Verkaar, W. M. Blankesteijn, J. F. Smits and G. J. Zaman, FASEB J., 2010, 24, 1205–1217 CrossRef CAS PubMed.
- Y. X. Yan, D. M. Boldt-Houle, B. P. Tillotson, M. A. Gee, B. J. D'Eon, X. J. Chang, C. E. Olesen and M. A. Palmer, J. Biomol. Screening, 2002, 7, 451–459 CrossRef CAS PubMed.
- G. von Degenfeld, T. S. Wehrman, M. M. Hammer and H. M. Blau, FASEB J., 2007, 21, 3819–3826 CrossRef CAS PubMed.
- M. M. Hammer, T. S. Wehrman and H. M. Blau, FASEB J., 2007, 21, 3827–3834 CrossRef CAS PubMed.
- T. Henrichs, N. Mikhaleva, C. Conz, E. Deuerling, D. Boyd, A. Zelazny, E. Bibi, N. Ban and M. Ehrmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4246–4251 CrossRef CAS PubMed.
- A. Pauli, F. Althoff, R. A. Oliveira, S. Heidmann, O. Schuldiner, C. F. Lehner, B. J. Dickson and K. Nasmyth, Dev. Cell, 2008, 14, 239–251 CrossRef CAS PubMed.
- M. J. Adams, J. F. Antoniw and F. Beaudoin, Mol. Plant Pathol., 2005, 6, 471–487 CrossRef CAS PubMed.
- M. C. Wehr, R. Laage, U. Bolz, T. M. Fischer, S. Grunewald, S. Scheek, A. Bach, K. A. Nave and M. J. Rossner, Nat. Methods, 2006, 3, 985–993 CrossRef CAS PubMed.
- M. S. Djannatian, S. Galinski, T. M. Fischer and M. J. Rossner, Anal. Biochem., 2011, 412, 141–152 CrossRef CAS PubMed.
- N. Audet, I. Charfi, O. Mnie-Filali, M. Amraei, A. J. Chabot-Dore, M. Millecamps, L. S. Stone and G. Pineyro, J. Neurosci., 2012, 32, 4827–4840 CrossRef CAS PubMed.
- C. J. van Koppen and K. H. Jakobs, Mol. Pharmacol., 2004, 66, 365–367 CrossRef CAS PubMed.
- P. A. Patel, D. G. Tilley and H. A. Rockman, J. Mol. Cell. Cardiol., 2009, 46, 300–308 CrossRef CAS PubMed.
- S. Rajagopal, J. Kim, S. Ahn, S. Craig, C. M. Lam, N. P. Gerard, C. Gerard and R. J. Lefkowitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 628–632 CrossRef PubMed.
- I. A. A. E. H. Ibrahim and H. Kurose, J. Pharmacol. Sci., 2012, 118, 408–412 CrossRef CAS.
- S. R. George, B. F. O'Dowd and S. P. Lee, Nat. Rev. Drug Discovery, 2002, 1, 808–820 CrossRef CAS PubMed.
- M. A. Ayoub and K. D. Pfleger, Curr. Opin. Pharmacol., 2010, 10, 44–52 CrossRef CAS PubMed.
- E. Urizar, H. Yano, R. Kolster, C. Galés, N. Lambert and J. A. Javitch, Nat. Chem. Biol., 2011, 7, 624–630 CrossRef CAS PubMed.
- P. A. Vidi and V. J. Watts, Mol. Pharmacol., 2009, 75, 733–739 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |