DOI:
10.1039/C4RA15392H
(Paper)
RSC Adv., 2015,
5, 10275-10289
Using halogen⋯halogen interactions or C/N–H⋯Cl hydrogen bonding to direct crystal packing in tetrachlorophthalic acid with N-heterocyclic compounds†
Received
28th November 2014
, Accepted 24th December 2014
First published on 24th December 2014
Abstract
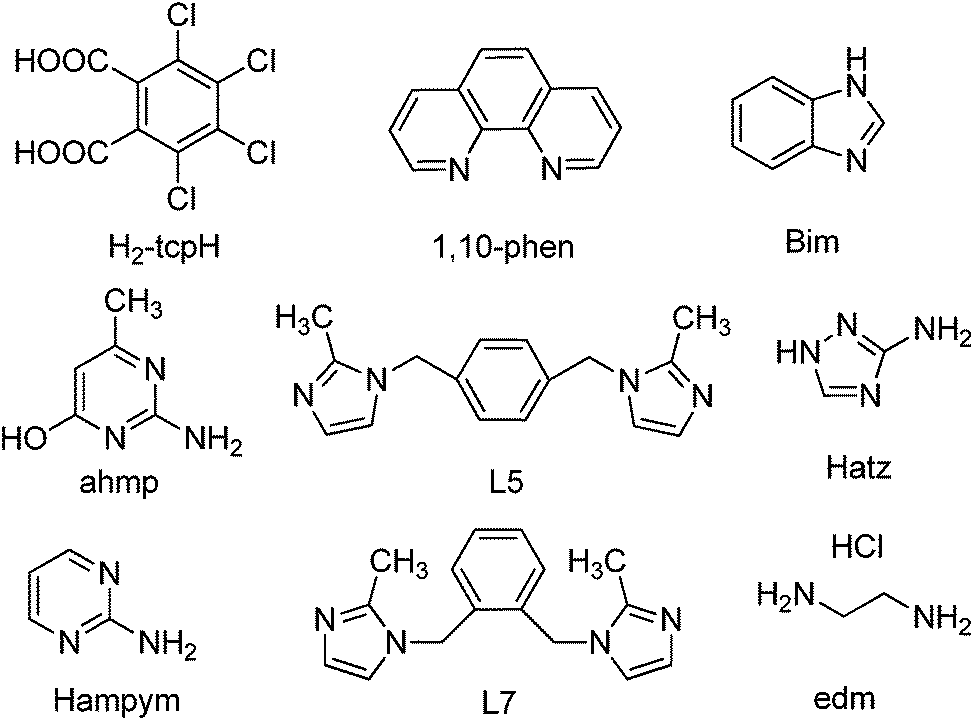
In exploring tetrachlorophthalic acid (H2-tcpH) with a series of N-heterocyclic compounds, eight different types of supramolecular complexes have been obtained, namely, [(H-1,10-phen)·(H-tcpH)·(H2-tcpH)·(CH3OH)] (1), [(H-Hatz)·(H-tcpH)] (2), [(H2-L5)·(H-tcpH)2] (3), [(H-Hampym)·(H-tcpH)] (4), [(H2-edm)·(H-tcpH)2] (5), [(H-ahmp)2·(tcpH)·2(H2O)] (6), [(H2-L7)·(tcpH)·2(H2O)] (7), and [(H-Bim)·(H-tcpH)·(H2O)] (8) (in which 1,10-phen = 1,10-phenanthroline, Hatz = 3-amino-1,2,4-triazole, L5 = 1,4-bis[(2-methylimidazole-1-yl)methyl]benzene, Hampym = 2-aminopyrimidine, edm = ethylenediamine, ahmp = 2-amino-4-hydroxy-6-methylpyrimidine, L7 = 1,2-bis[(2-methylimidazole-1-yl)methyl]benzene, Bim = benzimidazole). These eight complexes displayed amusing structural characteristics and a large amount of hydrogen bonding. Of these, crystals 1–5 generated a 3D supramolecular structure through Cl⋯Cl interactions, whilst C–H⋯Cl and N–H⋯Cl hydrogen bonding exists in the 3D construction of complexes 6–8. In addition, all the complexes were fully characterized by single crystal X-ray diffraction analysis, elemental analysis, infrared spectroscopy (IR), and thermogravimetric analysis (TGA).
Introduction
Non-covalent interactions, such as hydrogen bonding, π–π interactions, van der Waals forces, and other weak interactions1 are responsible for the construction of supramolecular architectures in solid-state materials. In the past few years, tremendous efforts have been made to explore the new types of intermolecular interactions to synthesize multi-component supramolecules.2 In particular, halogen bonding has grown from a scientific curiosity to one of the most intriguing non-covalent interactions in chemists' eyes.3 The term “halogen bonding” (XB) refers to any non-covalent interaction involving halogens as acceptors of electron density.4
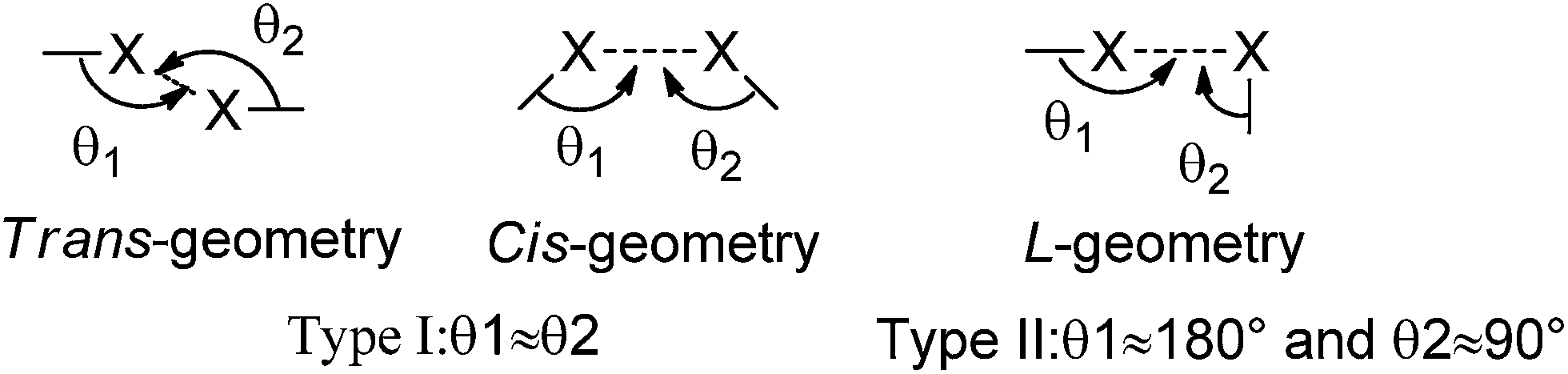
Allen and co-workers showed that the XB manifests as a directional attractive interaction through statistical analysis, which is based on intermolecular contract distances shorter than 1.26 times the sum of the van der Waals radii of two interacting atoms.5 Halogen⋯halogen interactions and C/N–H⋯halogen interactions are one of the most important parts of non-covalent interactions.6 Such halogen⋯halogen interactions have been termed Type I (θ1 ≈ θ2) and Type II (θ1 ≈ 180°, θ2 ≈ 90°) depending on the angular approach of the halogens toward each other (Scheme 1). In many molecular crystals without strong hydrogen bonding (O/N–H⋯O and O/N–H⋯N), the X⋯X and C/N–H⋯X interactions dominate the molecular arrangement as an effective directional tool.7 For example, the presence of C–H⋯O/Cl, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Cl, and Cl⋯Cl interactions in two concomitant conformational polymorphs of 1,3,5-tris(4-chlorobenzoyl) benzene were communicated by Pigge and co-workers.8 In addition, the Jin group first prepared a novel organic phosphorescent co-crystal from 1,4-diiodotrafluorobenzene and carbazole using a C–I⋯π XB based on C–I⋯N XB and π–hole⋯F bonds.4e,9
O⋯Cl, and Cl⋯Cl interactions in two concomitant conformational polymorphs of 1,3,5-tris(4-chlorobenzoyl) benzene were communicated by Pigge and co-workers.8 In addition, the Jin group first prepared a novel organic phosphorescent co-crystal from 1,4-diiodotrafluorobenzene and carbazole using a C–I⋯π XB based on C–I⋯N XB and π–hole⋯F bonds.4e,9
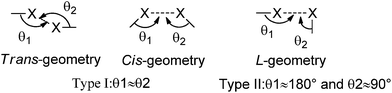 |
| | Scheme 1 Geometrical classification of halogen⋯halogen interactions as Type I and Type II. | |
In fact, to date, rationally controlling the desired structures of solid-state materials still remains a great challenge.10 For this, as Desiraju pointed out, crystal structures are not related to molecular structures (functional groups) in a simple manner: molecules come to form crystal structures which is an emergent property.11 Fortunately, supramolecular synthons appeared.12 Supramolecular synthons are structural units of a supramolecules, which are assembled via intermolecular forces.13 In a paper by Khavasi and Tehrani, Br⋯Br and N⋯Br halogen bonding synthons were displayed in N-(3-bromophenyl)-2-pyrazinecarboxamide molecules.14 Synthons R22(6) and R12(5) were formed between neighboring 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20) molecules and between CL-20 and conformer molecules in a paper, which was composed by Zhang and co-workers in 2014.15 The supramolecular synthon is a simple and popular method to understand intermolecular interactions.15,16 The identification of useful supramolecular synthons is an important aspect of current crystal engineering strategies and it is helpful in the design of materials with predetermined properties.15,17
Carboxylic acids are capable of functioning as hydrogen bonding donors and/or acceptors resulting in supramolecular frameworks by intermolecular interactions and carboxylic acids aggregate in the solid state as dimers, catemers, and bridged motifs,18 thus they are frequently chosen as building blocks for crystal engineering.19 Furthermore, halogenated compounds have received widespread attention due to their applications in life science.6b,20 In view of the above mentioned interests, we selected tetrachlorophthalic acid (H2-tcpH) to explore the nature of intermolecular interactions with a series of N-heterocyclic compounds as a continuation of our work on organic crystals.21
Herein, we report the synthesis and the crystal structures of eight crystals, namely, [(H-1,10-phen)·(H-tcpH)·(H2-tcpH)·(CH3OH)] (1), [(H-Hatz)·(H-tcpH)] (2), [(H2-L5)·(H-tcpH)2] (3), [(H-Hampym)·(H-tcpH)] (4), [(H2-edm)·(H-tcpH)2] (5), [(H-ahmp)2·(tcpH)·2(H2O)] (6), [(H2-L7)·(tcpH)·2(H2O)] (7), and [(H-Bim)·(H-tcpH)·(H2O)] (8) (Scheme 2).
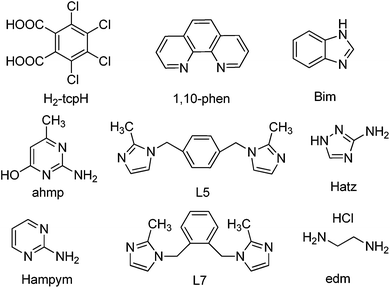 |
| | Scheme 2 Molecular structures in this work. | |
Experimental
Starting materials
All the chemicals and solvents used for synthesis were obtained from commercial sources and were used as received, without further purification, except the ligands L5 and L7. They were prepared by the procedure given in the literature.22
Physical Measurements
Infrared spectra were recorded with a Nicolet Impact 410 FTIR spectrometer in the range of 4000–400 cm−1, and samples were prepared as KBr pellets. Absorptions are denoted as follows: strong (s), medium (m), and weak (w) in the synthesis section. Carbon, hydrogen, and nitrogen contents were performed with a Perkin-Elmer 2400 elemental analyzer. Thermogravimetric analysis (TGA) was performed from room temperature to 900 °C using a Perkin-Elmer TGA-7 TG analyzer with a heating rate of 10 °C min−1 in an N2 atmosphere.
General procedures for the preparation of single crystals
Synthesis of [(C12H9N2+)·(C8HO4Cl4−)·(C8H2O4Cl4)·(CH3OH)] (1). A solution of 1,10-phen (99 mg, 0.05 mmol) in 5 mL of distilled water was mixed with H2-tcpH (31.8 mg 0.10 mmol) in 5 mL of methanol. The reaction mixture was stirred for 15 min and a clear homogeneous solution was obtained. Then, the solution was allowed to stand at room temperature for slow evaporation. Colorless, block crystals were obtained after two weeks. The obtained crystals were separated from the mother solution by filtration, washed with a methanol–distilled water solution (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and dried under vacuum. Yield: 64%. Anal. calcd for C29H16Cl8N2O9: C, 42.44; H, 1.95; N, 3.41%. Found: C, 44.92; H, 2.07; N, 3.06%. Infrared spectrum (KBr disc, cm−1): 3450s, 3102s, 2920s, 2584s, 1757w, 1615m, 1563m, 1511s, 1471s, 1433s, 1343m, 1265w, 1123m, 1045m, 916m, 851w, 811s, 773m, 721m, 683m, 657m, 617m, 591m, 451s, 476m, 398m.
:1), and dried under vacuum. Yield: 64%. Anal. calcd for C29H16Cl8N2O9: C, 42.44; H, 1.95; N, 3.41%. Found: C, 44.92; H, 2.07; N, 3.06%. Infrared spectrum (KBr disc, cm−1): 3450s, 3102s, 2920s, 2584s, 1757w, 1615m, 1563m, 1511s, 1471s, 1433s, 1343m, 1265w, 1123m, 1045m, 916m, 851w, 811s, 773m, 721m, 683m, 657m, 617m, 591m, 451s, 476m, 398m.
Synthesis of [(C2H5N4+)·(C8HO4Cl4−)] (2). To a tetrahydrofuran–distilled water solution (v/v = 1:1, 10 mL) containing Hatz (16.8 mg, 0.20 mmol) H2-tcpH (31.8 mg, 0.10 mmol) was added with constant stirring for 15 min. The clear and homogeneous solution was slowly evaporated at room temperature, and block colorless crystals were obtained three weeks later. The crystals were picked up from the mother liquor and washed with a tetrahydrofuran–distilled water solution (v/v = 1:1), and dried under vacuum. Yield: 67%. Anal. calcd for C10H6Cl4N4O4: C, 30.93; H, 1.55; N, 14.43%. Found: C, 32.04; H, 1.87; N, 14.06%. Infrared spectrum (KBr disc, cm−1): 3430w, 3258w, 3161m, 2946m, 2733m, 2566m, 1913s, 1692w, 1576w, 1561w, 1543w, 1427m, 1408m, 1344w, 1244w, 1202w, 1137s, 1120m, 1055s, 1008s, 956m, 932s, 904m, 865m, 839s, 824s, 795s, 756s.
Synthesis of [(C16H20N42+)·(C8HO4Cl4−)2] (3). A mixture of L5 (15.1 mg, 0.05 mmol), H2-tcpH (31.8 mg, 0.10 mmol), Ni(NO3)2 (29.8 mg, 0.01 mmol), NMP (5 mL), and distilled water (5 mL) was sealed in a Teflon-lined stainless steel vessel (15 mL), which was heated at 120 °C for three days and cooled to room temperature at a rate of 5 °C h−1. Colorless, block shaped crystals of 3 were collected in 38% yield. Anal. calcd for C32H22Cl8N4O8: C, 43.93; H, 2.52; N, 6.41%. Found: C, 44.42; H, 2.66; N, 7.52%. Infrared spectrum (KBr disc, cm−1): 3395w, 3127m, 2991s, 2634s, 2455s, 1946s, 1633w, 1600w, 1526w, 1429w,1448w, 1338w, 1289m, 1230m, 1195m, 1142m, 1120m, 1049m, 979s, 924m, 905m, 843m, 795w, 742w, 651m, 616m, 570m, 465m, 428m.
Synthesis of [(C4H6N3+)·(C8HO4Cl4−)] (4). 5 mL of a distilled water solution of Hampym (9.5 mg, 0.10 mmol) was mixed with 5 mL of an ethanol solution of H2-tcpH (31.8 mg, 0.10 mmol), and the colorless solution was stirred for 15 min and placed for slow evaporation at room temperature. Colorless block shaped crystals in about an 80% yield were obtained after three weeks and separated from the mother liquor. The crystals were washed with an ethanol–distilled water solution (v/v = 1:1) and dried under vacuum. Anal. calcd for C12H7Cl4N3O4: C, 36.10; H, 1.75; N, 10.53%. Found: C, 37.88; H, 1.84; N, 10.48%. Infrared spectrum (KBr disc, cm−1): 3415m, 3323m, 3218s, 3131s, 3092s, 2961s, 2891s, 2836s, 2789m, 2737m, 2686m, 1907s, 1694w, 1682w, 1655w, 1542w, 1459m, 1414w, 1344w, 1287m, 1226w, 1067m, 990m, 898m, 872s, 797w, 742w, 720m, 666w, 638w, 603w, 577m, 541m, 479s, 437m, 423m.
Synthesis of [C2H10N22+)·(C8HO4Cl4−)2] (5). A solution of H2-tcpH (31.8 mg, 0.10 mmol) was prepared in 5 mL of acetone. 5 mL of a distilled water solution of edm (4.9 mg, 0.05 mmol) was added to the above prepared solution. The resulting solution was stirred for 15 min and maintained at room temperature for crystallization. Colorless, block crystals were obtained after two weeks. The obtained crystals were separated from the mother solution by filtration, washed with an acetone–distilled water solution (v/v = 1:1), and dried under vacuum. Yield: 68%. Anal. calcd for C9H6Cl4NO4: C, 32.34; H, 1.80; N, 4.19%. Found: C, 34.01; H, 1.98; N, 4.68%. Infrared spectrum (KBr disc, cm−1): 3436s, 3174m, 3031m, 2944m, 2616s, 2459s, 1716w, 1571w, 1532w, 1478s, 1414s, 1343w, 1239w, 1198m, 1125m, 1037s, 1011s, 933s, 902s, 670m, 648m, 605s, 456s, 415s.
Synthesis of [(C5H8ON3+)2·(C8O4Cl42−)·2(H2O)] (6). H2-tcpH (31.8 mg, 0.10 mmol) and ahmp (25.0 mg, 0.20 mmol) were taken in a 1:2 molar ratio and dissolved in an ethanol–distilled water solution (v/v = 1:1 10 mL), the solution was stirred for 15 min until a colorless solution was obtained. Good quality crystals, suitable for diffraction, were obtained after one week as the solution evaporated slowly at room temperature. The obtained crystals were separated from the mother liquor by filtration, washed with the ethanol–distilled water solution (v/v = 1:1), and dried in a vacuum desiccator. Yield: 62%. Anal. calcd for C18H20Cl4N6O8: C, 36.60; H, 3.39; N, 14.23%. Found: C, 36.88; H, 3.64; N, 15.45%. Infrared spectrum (KBr disc, cm−1): 3408w, 3133m, 2756m, 1706w, 1686w, 1631w, 1591w, 1533w, 1479m, 1416w, 1370m, 1342w, 1250m, 1190 s, 1127 s, 1086m, 1045s, 951m, 911s, 840m, 669m, 649m, 619m, 582w, 539w, 508m.
Synthesis of [(C16H20N42+)·(C8O4Cl42−)·2(H2O)] (7). The same synthetic procedure as that for 6 was used except that ahmp was replaced by L7, affording colorless block crystals of 7 in 62% yield. Anal. calcd for C24H24Cl4N4O8: C, 47.50; H, 3.96; N, 9.24%. Found: C, 49.42; H, 4.05; N, 10.20%. Infrared spectrum (KBr disc, cm−1): 3481w, 3363w, 3138m, 3042m, 2974s, 2528s, 1967s, 1647w, 1602w, 1531m, 1457m, 1412m, 1381w, 1361w, 1339w, 1303m, 1250m, 1176m, 1156m, 1126s, 1092s, 1047s, 970s, 933m, 910m, 782m, 739m, 684m, 655w, 618m, 595m, 539m, 421s.
Synthesis of [(C7H7N2+)·(C8HO4Cl4−)·H2O] (8). Bim (11.8 mg, 0.10 mmol) was dissolved in 5 mL of distilled water, and H2-tcpH (31.8 mg, 0.10 mmol) was dissolved in 5 mL of ethanol. Both the solutions were mixed and stirred at room temperature. After 15 min a homogeneous solution was obtained, which was allowed to stand at room temperature for slow evaporation. Three weeks later crystals started to form and the yield was about 53%. These were further washed with ethanol–distilled water (v/v = 1:1), and dried in vacuum desiccators. Anal. calcd for C15H10Cl4N2O5: C, 40.90; H, 2.27; N, 6.36%. Found: C, 42.45; H, 2.45; N, 7.52%. Infrared spectrum (KBr disc, cm−1): 3435m, 3150, 3075 s, 2818m, 2752m, 2569m, 1909s, 1718w, 1622w, 1594m, 1532m, 1451m, 1406m, 1338w, 1265w, 1244w, 1126s, 1108s, 1004s, 921m, 901m, 873m, 816m, 790 s, 745w, 720w, 671m, 646m, 618w, 609m, 551m, 467s, 422m, 411m.
X-ray crystal structure analyses
The samples for single crystal diffraction of complexes 1–8 were synthesised through the manner described above and selected to be glued at the top of a thin glass fiber with epoxy glue in air for data collection, and the crystallographic data were collected on a Siemens Smart CCD diffractometer equipped with a normal-focus, 2.4 kW sealed-tube X-ray source (graphite-monochromatic MoKa radiation (λ = 0.71073 Å)) operating at 50 kV and 40 mA. There was no evidence of crystal decay during data collection. All crystal structures were solved by direct methods and refined on F2 by full-matrix least squares methods using the SHELXL program package.23 Crystallographic details are summarized in Table 1 and pertinent intermolecular interactions parameters in the structures of 1–8 are listed in Tables 2 and 3.
Table 1 Crystallographic Information of compounds 1–8
| Compound |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
| R1 = ∑ΔFoΔ − ΔFcΔ/∑ΔFoΔ. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Empirical formula |
C29H16Cl8N2O9 |
C10H6Cl4N4O4 |
C32H22Cl8N4O8 |
C12H7Cl4N3O4 |
C9H6Cl4NO4 |
C18H20Cl4N6O8 |
C24H24Cl4N4O8 |
C15H10Cl4N2O5 |
| M |
820.04 |
387.99 |
874.14 |
399.01 |
333.95 |
590.20 |
606.27 |
440.05 |
| Crystal system |
Monoclinic |
Orthorhombic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P21/n |
Pbca |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
C2/c |
P |
C2/c |
P |
| a/Å |
18.1486(4) |
9.4206(3) |
8.5055(6) |
12.614(3) |
36.8502(17) |
7.4149(4) |
12.6127(5) |
7.047(4) |
| b/Å |
8.7692(2) |
12.0251(3) |
9.8663(7) |
11.255(2) |
5.5385(2) |
8.9129(6) |
23.7989(9) |
8.497(5) |
| c/Å |
22.0638(5) |
24.0488(9) |
11.0901(7) |
10.900(2) |
12.5520(6) |
18.7130(11) |
9.5324(4) |
15.269(9) |
| α/° |
90 |
90 |
88.177(1) |
90 |
90 |
97.470(5) |
90 |
84.240(9) |
| β/° |
111.758(2) |
90 |
73.768(1) |
106.969(3) |
105.115(6) |
97.712(5) |
98.590(4) |
83.995(10) |
| γ/° |
90 |
90 |
87.108(1) |
90 |
90 |
98.484(5) |
90 |
72.863(9) |
| V/Å3 |
3261.26(13) |
2724.34(15) |
892.27(11) |
1480.1(5) |
2473.2(2) |
1197.87(13) |
2829.23(19) |
866.5(9) |
| Z |
4 |
8 |
1 |
4 |
8 |
2 |
4 |
2 |
| T/K |
293 |
293 |
293 |
293 |
293 |
293 |
293 |
293 |
| Dc/g cm−3 |
1.670 |
1.892 |
1.627 |
1.791 |
1.794 |
1.636 |
1.423 |
1.687 |
| μ/mm−1 |
0.748 |
0.892 |
0.689 |
0.822 |
0.960 |
0.552 |
0.463 |
0.714 |
| F(000) |
1648.0 |
1552.0 |
442.0 |
800.0 |
1336.0 |
604.0 |
1248.0 |
444.0 |
| h, k, lmax |
22, 10, 27 |
12, 15, 30 |
10, 11, 13 |
14, 13, 12 |
46, 7, 16 |
8, 10, 22 |
15, 29, 11 |
8, 10, 18 |
| R1a |
0.0383 |
0.0306 |
0.0653 |
0.0554 |
0.0337 |
0.0497 |
0.0607 |
0.0891 |
| wR2b (all data) |
0.0991 |
0.0705 |
0.1787 |
0.1498 |
0.0897 |
0.1262 |
0.1551 |
0.2354 |
| S(GOF on F2) |
1.043 |
1.055 |
1.043 |
1.077 |
1.041 |
0.977 |
1.039 |
0.951 |
Table 2 Intermolecular Interactions of 1–8
| D–H⋯A (Å) (symmetry code) |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
∠D–H⋯A (deg) |
| 1O3–H3⋯O7 (x,−1 + y,z) |
0.820 |
1.753 |
2.571 |
175.0 |
| O6–H6⋯O2 (−x,1 − y,−z) |
0.820 |
1.772 |
2.585 |
171.1 |
| N2–H2⋯O9 (x,−1 + y,z) |
0.854 |
1.939 |
2.721 |
151.5 |
| O9–H9⋯O5 (x,−1 + y,z) |
0.887 |
1.949 |
2.798 |
159.7 |
| C36–H36⋯O4 (x,2 − y,z) |
0.930 |
2.568 |
3.491 |
171.8 |
| 2O3–H3⋯O2 (−1 − x,−1 − y,−z) |
0.820 |
1.778 |
2.586 |
168.0 |
| N4–H4⋯O1 (−1 − x,−1 − y,−z) |
0.860 |
1.898 |
2.714 |
157.8 |
| N1–H1B⋯N2 (−x,−y,−z) |
0.860 |
2.233 |
3.058 |
160.5 |
| N3–H3A⋯O2 (x,y,−1 + z) |
0.860 |
2.336 |
2.933 |
126.8 |
| 3O1–H1⋯O1 (x,y,z) |
1.237 |
1.237 |
2.245 |
180.0 |
| N2–H2⋯O4 (1 + x,y,2 + z) |
0.860 |
2.025 |
2.763 |
143.4 |
| C21–H21A⋯O3 (1 + x,2 + y,2 + z) |
0.970 |
2.420 |
3.218 |
139.2 |
| 4O8–H8⋯O2 (1 − x,−y,−z) |
0.818 |
1.667 |
2.483 |
174.4 |
| O3–H3⋯O5 (−1 + x,y,z) |
0.823 |
1.691 |
2.490 |
163.2 |
| N5–H5⋯O6 (2 − x,2 − y,−z) |
0.860 |
1.829 |
2.640 |
156.5 |
| N3–H3A⋯O1 (1 − x,−y,−z) |
0.859 |
1.835 |
2.649 |
157.5 |
| C38–H38⋯O7 (−1 + x,y,z) |
0.930 |
2.617 |
3.256 |
126.4 |
| C35–H35⋯O4 (−1 + x,y,z) |
0.930 |
2.629 |
3.270 |
126.6 |
| 5O4–H4⋯O1 (x,−1 + y,z) |
0.820 |
1.763 |
2.560 |
163.4 |
| N1–H1A⋯O2 (x,−2 + y,z) |
0.890 |
1.900 |
2.743 |
157.2 |
| N1–H1B⋯O2 (x,2 + y,z) |
0.890 |
1.969 |
2.847 |
168.5 |
| N1–H1C⋯O1 (x,−1 + y,z) |
0.890 |
1.989 |
2.819 |
154.6 |
| 6N1–H1⋯O2 (x,y,z) |
0.860 |
1.763 |
2.622 |
176.4 |
| O7–H7B⋯O6 (x,y,z) |
0.855 |
1.883 |
2.701 |
159.8 |
| N4–H4⋯O4 (x,y,z) |
0.860 |
1.884 |
2.668 |
150.9 |
| N2–H2⋯O7 (x,y,−1 + z) |
0.860 |
1.888 |
2.747 |
177.0 |
| O6–H6C⋯O3 (x,y,z) |
0.850 |
1.904 |
2.743 |
168.7 |
| O7–H7A⋯O8 (x,−1 + y,z) |
0.850 |
1.921 |
2.693 |
150.4 |
| N5–H5⋯O1 (x,y,z) |
0.860 |
1.938 |
2.774 |
163.7 |
| O6–H6D⋯O5 (x,1 + y,z) |
0.850 |
1.950 |
2.783 |
166.1 |
| N6–H6B⋯Cl4 (−1 + x,y,z) |
0.860 |
1.991 |
3.399 |
122.1 |
| 7N1–H1⋯O1 (−1 + x,y,−1 + z) |
0.928 |
1.740 |
2.658 |
169.3 |
| O3–H3B⋯O2 (−1 + x,y,z) |
0.850 |
1.904 |
2.753 |
176.8 |
| O3–H3A⋯O1 (x,y,z) |
0.850 |
2.032 |
2.872 |
169.3 |
| C15–H15⋯Cl2 (−1 + x,y,z) |
0.930 |
3.118 |
4.017 |
163.0 |
| 8O3–H3⋯O2 (x,y,1 + z) |
0.820 |
1.779 |
2.568 |
160.8 |
| N1–H1⋯O1 (2 + x,y,z) |
0.860 |
1.831 |
2.684 |
170.7 |
| N2–H2⋯O5 (x,y,z) |
0.860 |
1.838 |
2.666 |
161.1 |
| O5–H5A⋯O2 (−1 + x,y,z) |
0.850 |
1.985 |
2.799 |
160.1 |
| O5–H5B⋯O1 (x,y,z) |
0.850 |
2.475 |
3.108 |
131.9 |
| C19–H19⋯Cl2 (x,y,z) |
0.930 |
2.933 |
3.640 |
133.9 |
Table 3 Halogen⋯halogen short contact
| Crystal |
C–X⋯Y–C |
X⋯Y (Å) |
∠C–X⋯Y (°) |
∠C–Y⋯X (°) |
Symmetry code |
Type |
| 1 |
C7–Cl6⋯Cl5–C14 |
3.513 |
161.1 |
122.0 |
(0.5 − x,−0.5 + y,0.5 − z)/(1 + x,y,z) |
II |
| 2 |
C6–Cl4⋯Cl3–C11 |
3.280 |
77.7 |
172.4 |
(−x,0.5 + y,0.5 − z)/(0.5 + x,1 + y,0.5 − z) |
II |
| C73–Cl2⋯Cl1–C7 |
3.280 |
98.2 |
144.7 |
(−x,−1 − y,−z)/(x,−0.5 − y,−0.5 + z) |
II |
| 3 |
C11–Cl2⋯Cl4–C15 |
3.399 |
165.8 |
123.4 |
(−1 + x,y,z)/(1 − x,3 − y,1 − z) |
II |
| 4 |
C31–Cl4⋯Cl3–C18 |
3.372 |
156.9 |
95.0 |
(1 − x,1 − y,−z)/(1 − x,1 − y,−z) |
II |
| C5–Cl5⋯Cl8–C32 |
3.373 |
94.4 |
157.0 |
(1 − x,2 − y,−z)/(1 − x,3 − y,−z) |
II |
| 5 |
C12–Cl2⋯Cl2–C12 |
3.707 |
126.8 |
126.8 |
(0.5 − x,1.5 − y,1 − z)/(0.5 + x,1.5 − y,0.5 + z) |
I |
| C11–Cl3⋯Cl4–C15 |
3.632 |
122.1 |
175.4 |
(0.5 − x,−0.5 + y,0.5 − z)/(0.5 + x,1.5 − y,0.5 + z) |
II |
Results and discussions
Crystal structure analysis of complex 1
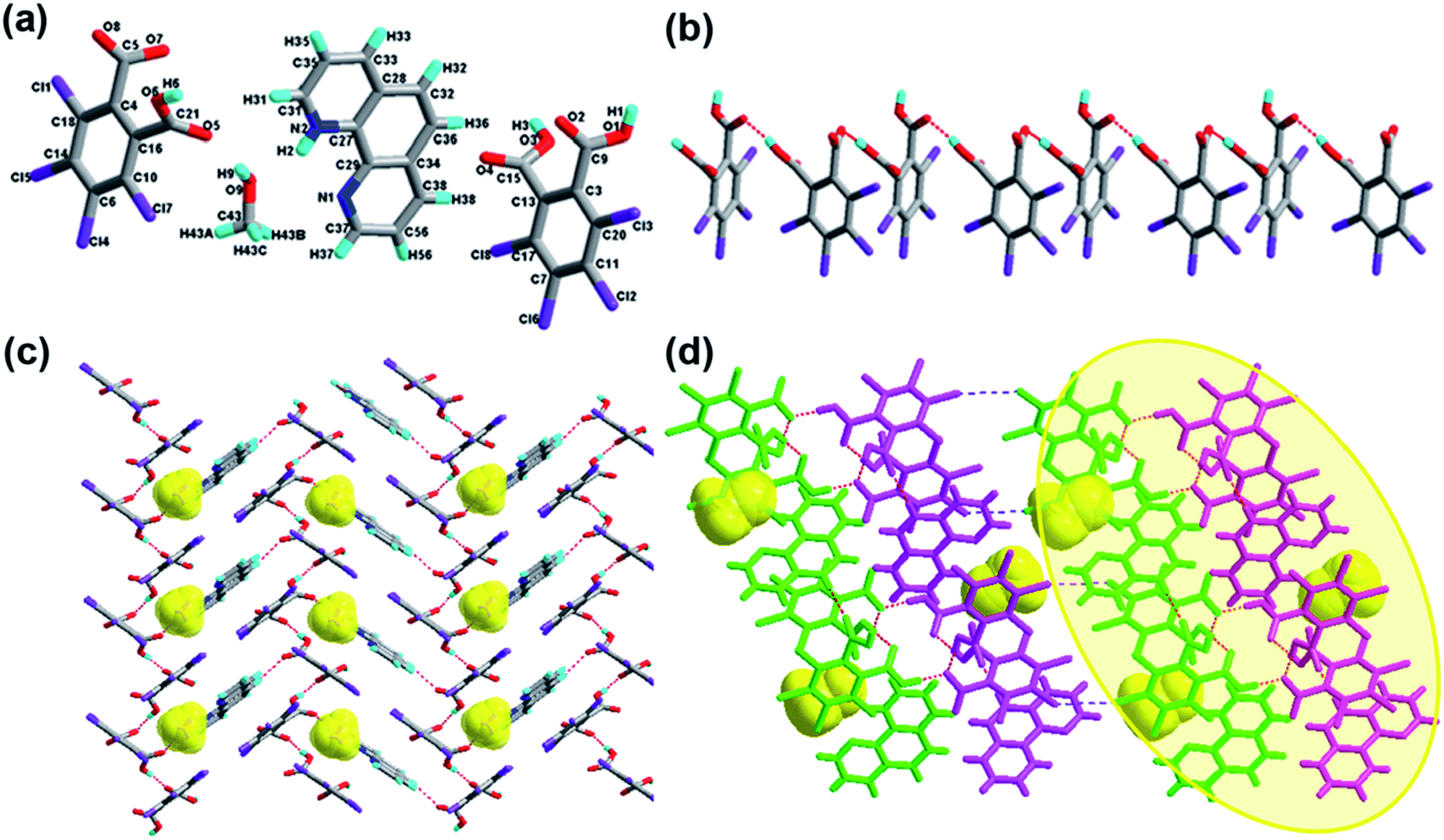
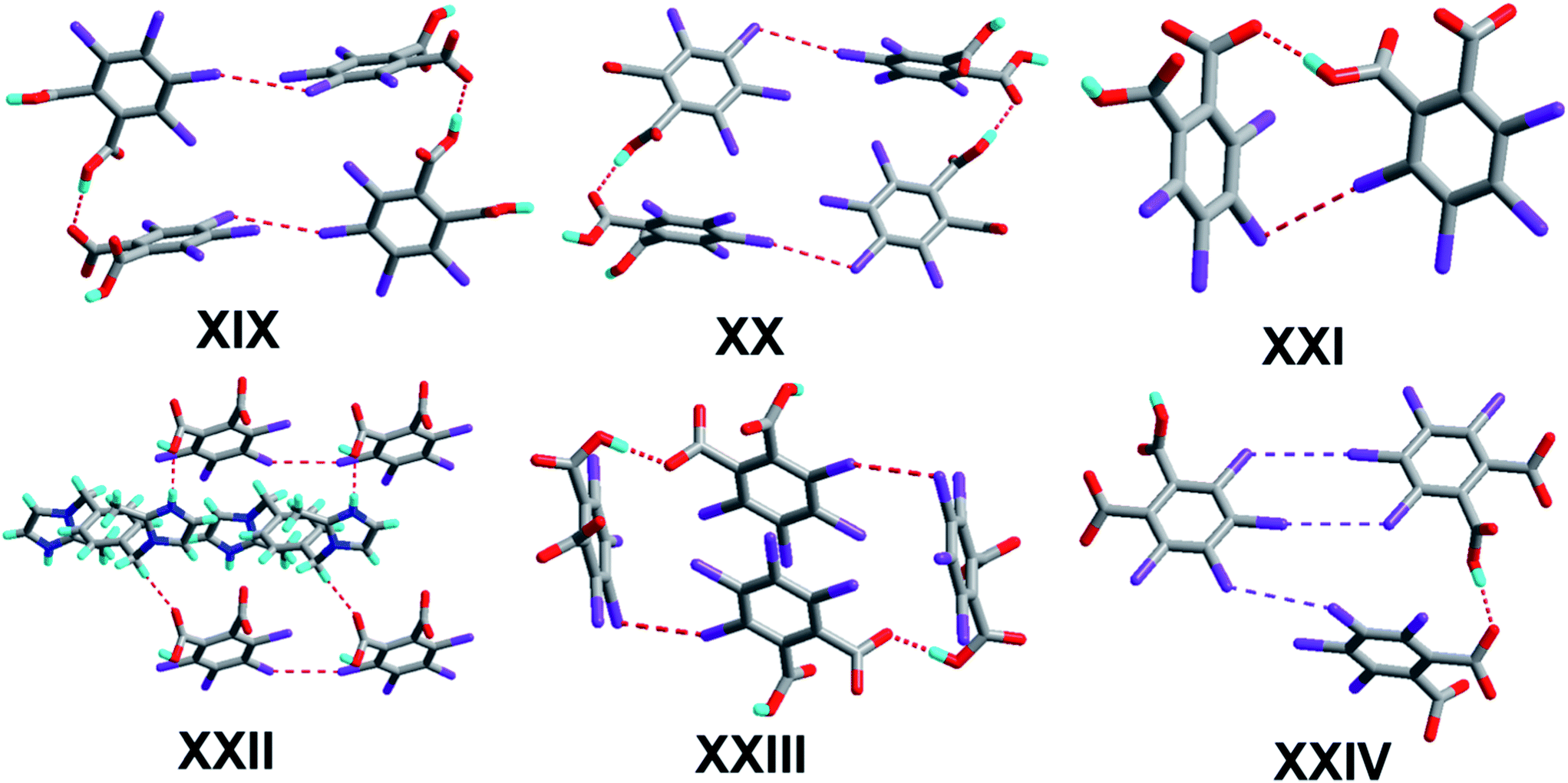
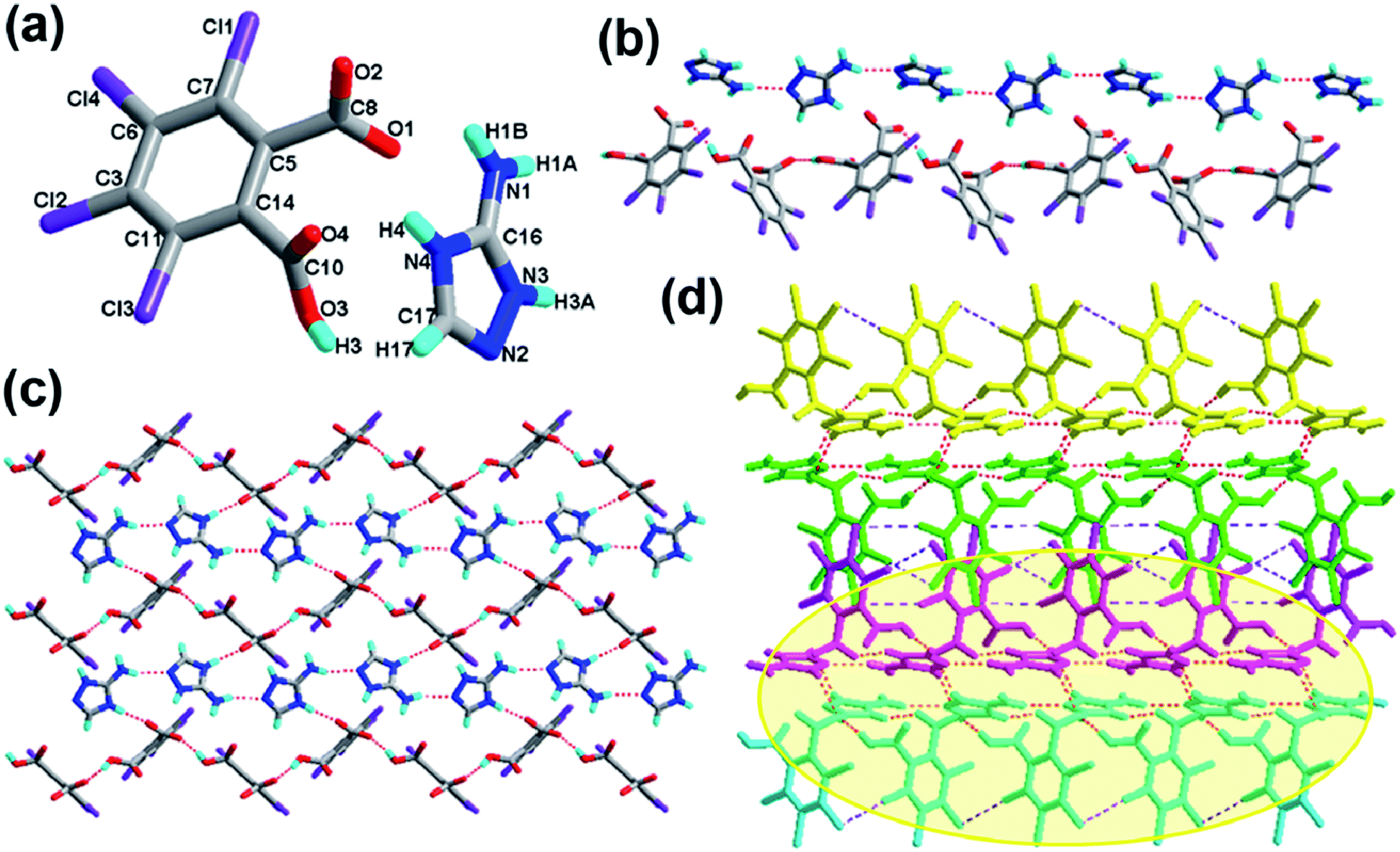
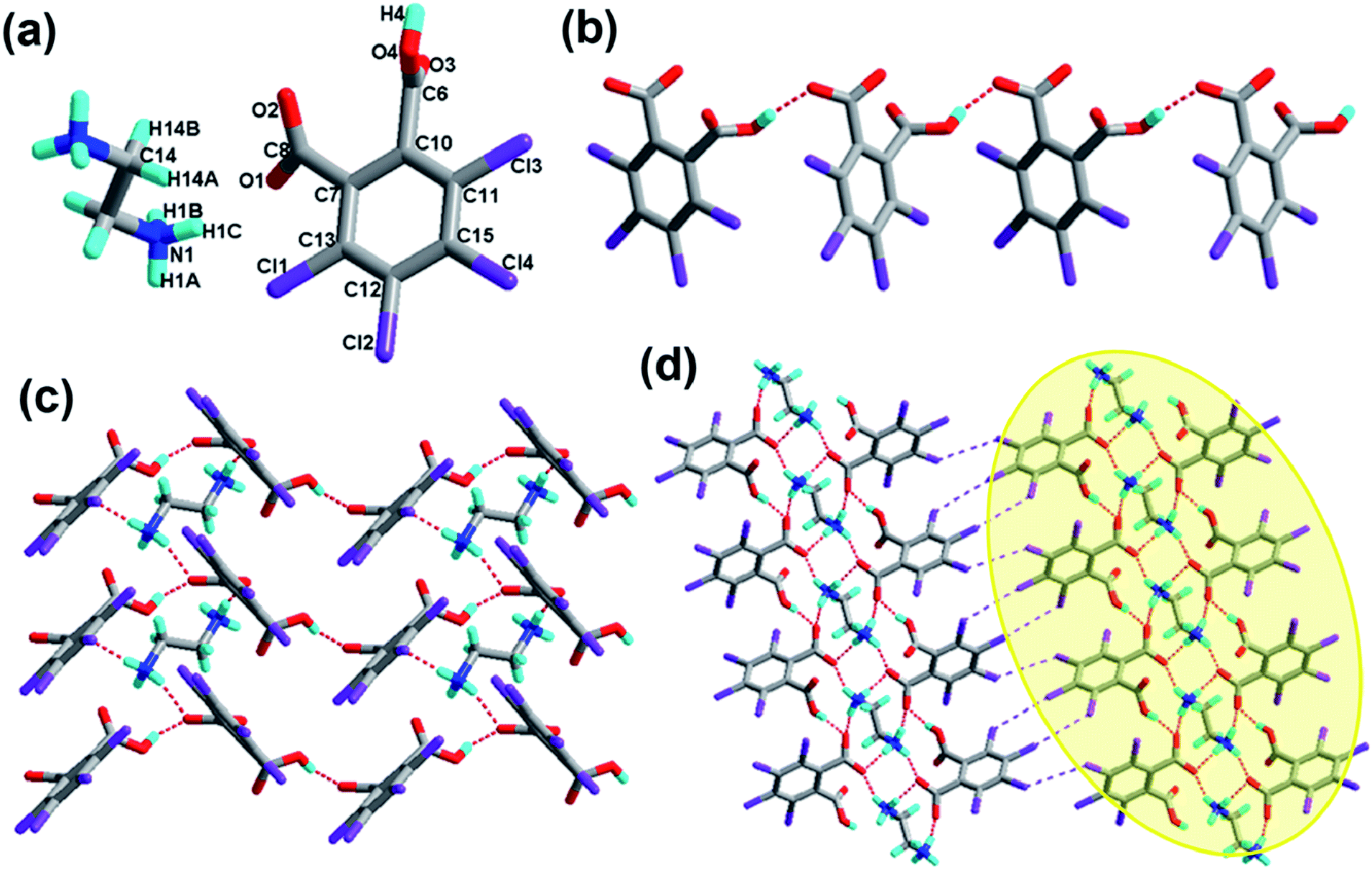
The crystal of 1 crystallizes in the monoclinic system with the P21/n space group with one molecule of tetrachlorophthalic acid (H2-tcpH), one tetrachlorophthalic acid anion (H-tcpH−), one 1,10-phen cation and one molecule of methanol in the asymmetric unit in Fig. 1a, and the dihedral angle between the two benzene rings of the acid molecules is 4.5°. Within each 1,10-phen component, the dihedral angle between the two pyridyl rings is 1.1°, and they make dihedral angles of 1.0° and 0.7°, respectively, with the central benzene ring, revealing the favourable coplanar character. One of the H atoms of the H2-tcpH acid transfers to one of the N atoms of the 1,10-phen molecule. The analysis of the intermolecular interactions in 1 reveals that the adjacent H2-tcpH molecules form a one-dimensional (1D) chain via the strong hydrogen bonding O–H⋯O (2.571 and 2.585 Å), which is shown in Fig. 1b. In Fig. 1c, the adjacent 1D chains, which are assembled with 1,10-phen and the CH3OH solvent via both strong hydrogen bonding O–H⋯O, N–H⋯O and weak C–H⋯O interactions, produce a 2D wavelike structure. In this structure, the solvent molecules are shown in the space-filling model in yellow colour. The hydrogen bonding O1–H1⋯O7 connect the adjacent 2D layers to form 2D double sheets, which are displayed in Fig. 1d and are colour coded in yellow. The analysis again suggests that the 2D double layers are connected to form a 3D structure in the ac plane by the unique type II Cl⋯Cl interactions (distance 3.513 Å, θ1 = 161.1°, θ2 = 122.0°, which is shown in Table 3). Through the Cl5⋯Cl6 interactions, unique synthons XIX and XX (displayed in Scheme 4) can be found in the 3D network. To show more clearly, we display each 2D sheet in different colours, and the solvent molecules are shown in yellow in the space-filling model. Further analysis of the crystal packing indicates that two –COOH groups in an acid unit connect three adjacent acid molecules to form synthon I R44(22) [O1–H1⋯O7 and O6–H6⋯O2]. Additionally, synthon II R24(18) (see Scheme 3) and synthons XV R33(14), XVI R88(50) (see ESI†) cannot be neglected, because they connect the neighbouring layers and can be found in the 3D network.
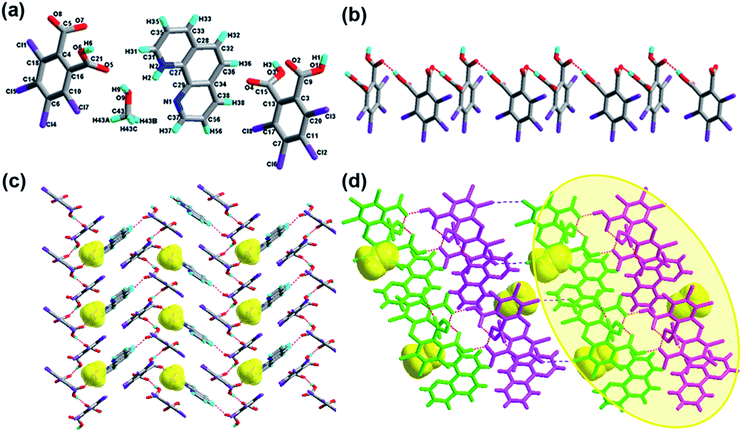 |
| | Fig. 1 (a) Molecule structure of 1 with atom labels of the asymmetric unit; (b) 1D supramolecular chain via hydrogen bonds (the hydrogen bonds are indicated as broken lines in this and the subsequent figures); (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via Cl⋯Cl interactions, the solvent molecules in this structure are displayed in the space-filling model, and the 2D double sheets are colour coded in yellow. (O, red; N, blue; C, gray; H, turquoise; Cl, violet in this and the subsequent figures). | |
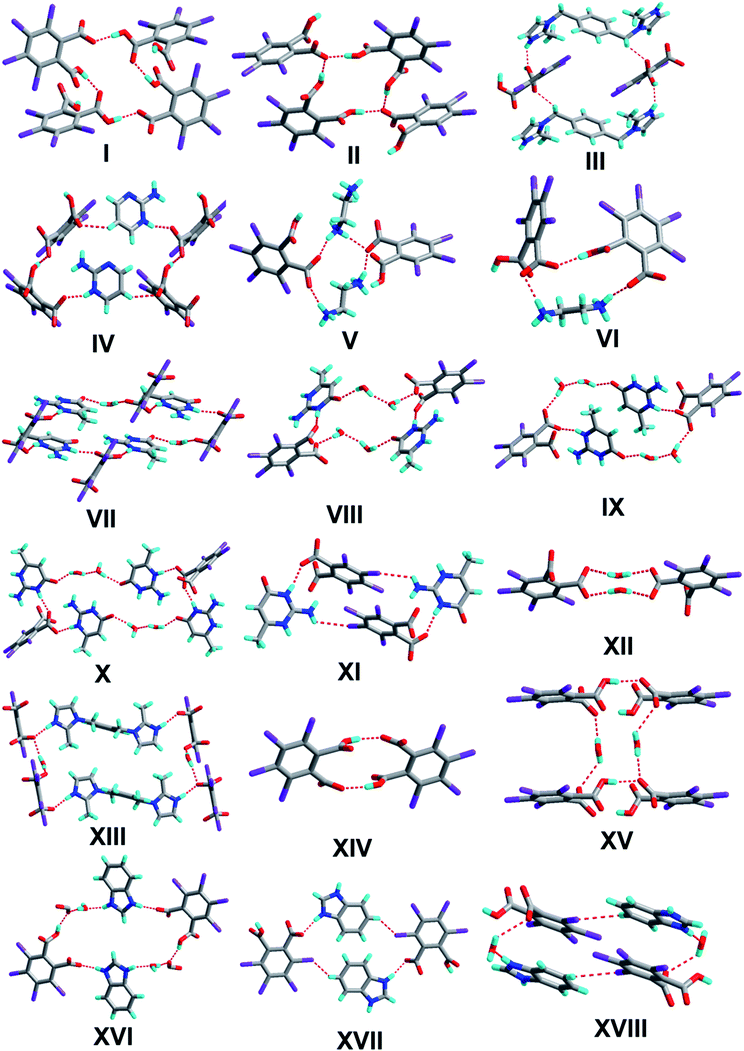 |
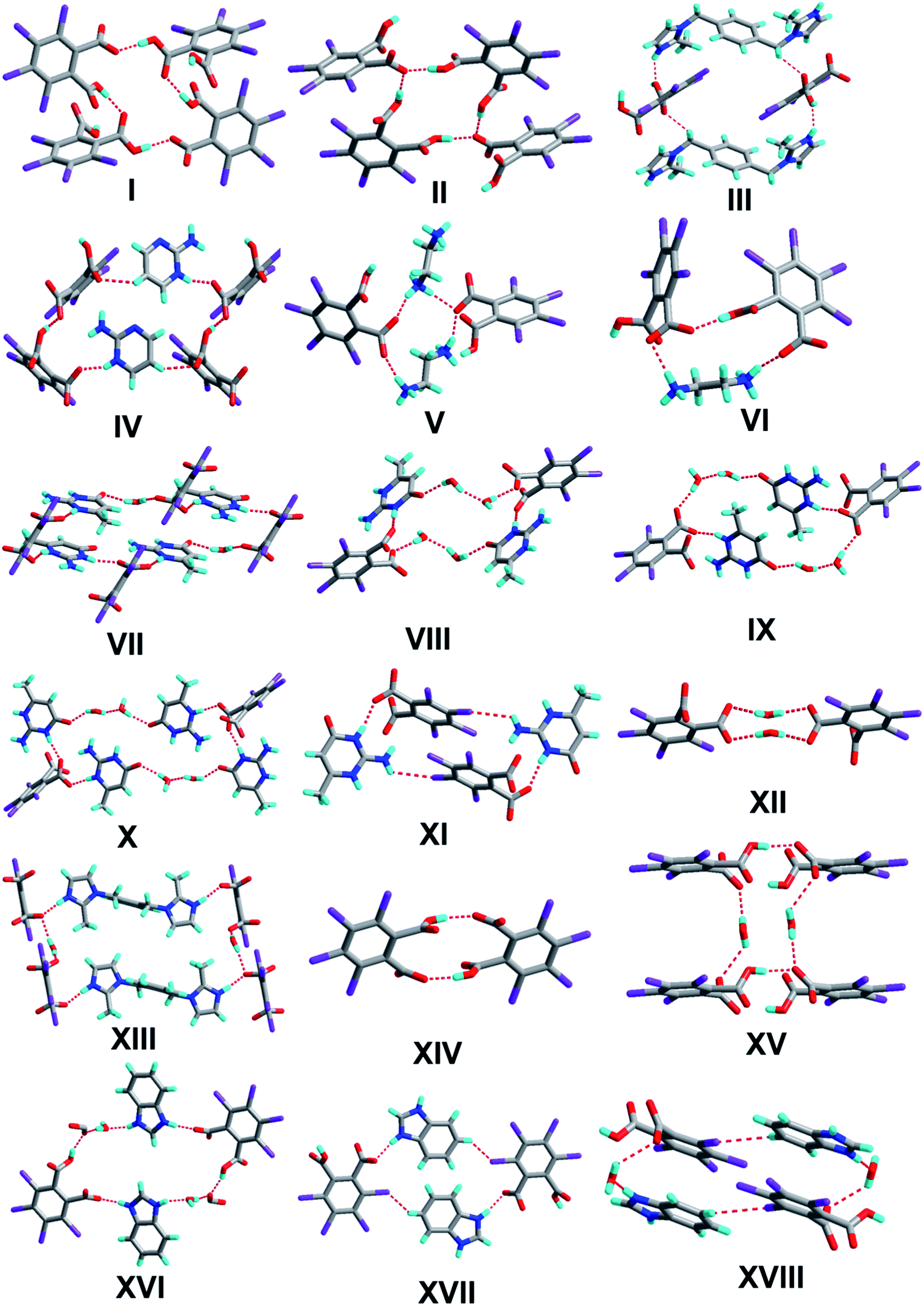
| | Scheme 3 Supramolecular synthons of complexes 1–8. | |
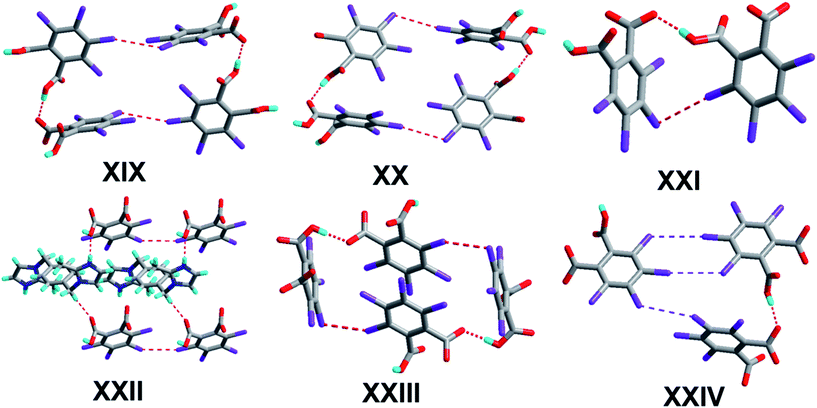 |
| | Scheme 4 Synthons (containing Cl⋯Cl interactions) of complexes 1–5. | |
Crystal structure analysis of complex 2
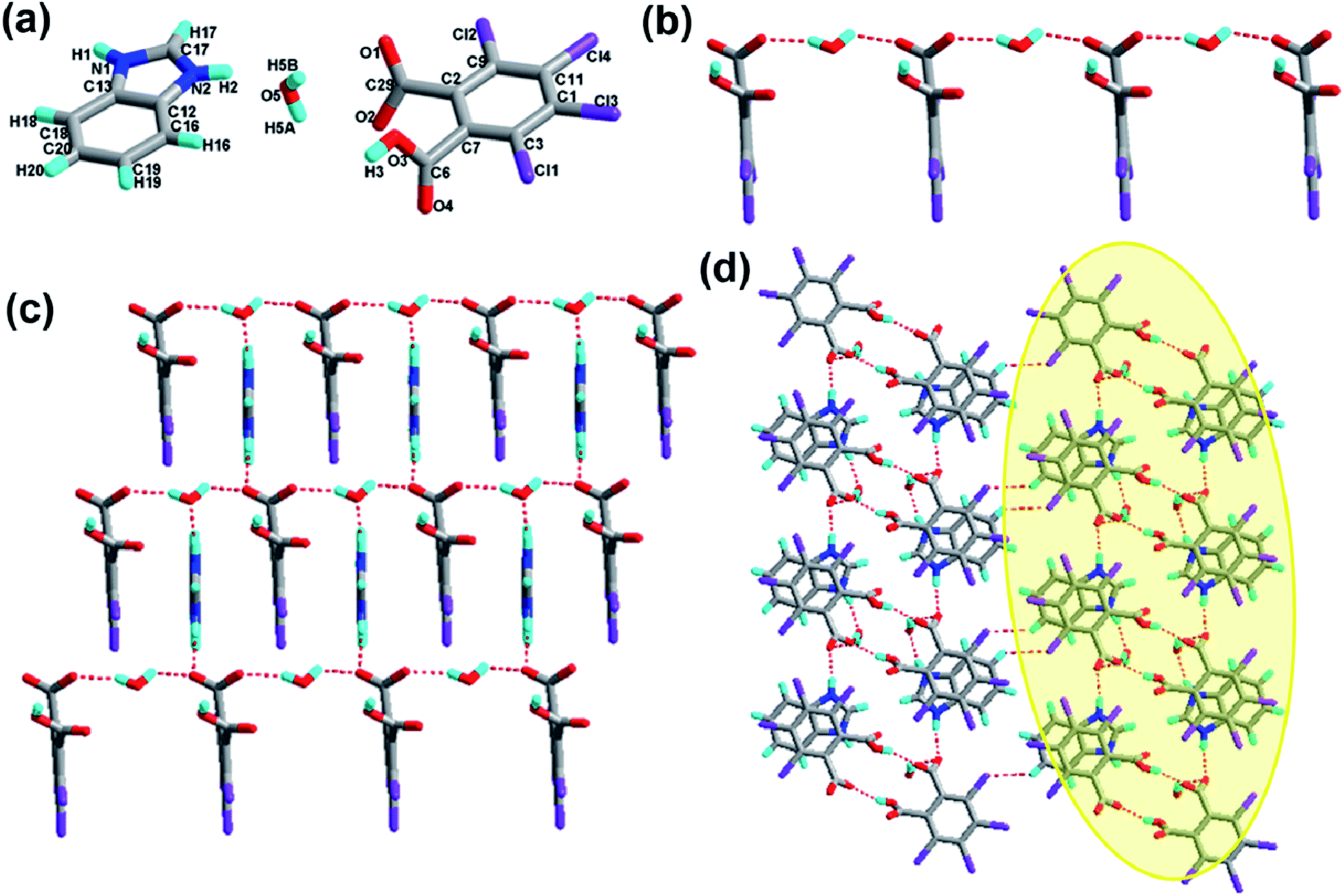
The crystal structure of 2 crystallizes in the orthorhombic system with the space group Pbca (Z = 8, in Table 1). As depicted in Fig. 2a, the asymmetric unit contains one molecule of H-tcpH− anion and one molecule of H-Hatz+ cation. The dihedral angle between the triazole ring of Hatz and benzene ring of H2-tcpH is 71.6°. The amino group and the triazole ring of Hatz are almost in the same plane, and the dihedral angle is 1.3°. In the crystal of 2, one of the H atoms of the H2-tcpH acid transfers to an N atom of the triazole ring of Hatz. For the H2-tcpH acid components, the adjacent crystallographically independent carboxylic groups are almost vertical to each other. As shown in Fig. 2b, the acid molecules arrange each other, in order to exhibit a 1D chain via the strong hydrogen bonding O3–H3⋯O2 (2.586 Å) and the base molecules form a 1D chain through the strong hydrogen bonding N1–H1B⋯N2 (3.058 Å). Through the strong N4–H4⋯O1 interactions, adjacent acid chains and base chains are combined into a 2D sheet, which is displayed in Fig. 2c. The adjacent 2D layers connect to form a 2D double layer and we display the 2D double layer colour coded in yellow. Furthermore, the involvement of type II Cl⋯Cl (Cl3⋯Cl4 distance 3.280 Å, θ1 = 77.7°, θ2 = 172.4° and Cl1⋯Cl2 distance 3.280 Å, θ1 = 98.2°, θ2 = 144.7°) and strong hydrogen bonding N3–H3A⋯O2 between 1,2,4-triazole and acid units generate the synthon XXVII R66(31), which is shown in the ESI.† As a result, the adjacent 2D layers connect to each other to generate a 3D network architecture. In this architecture, the unique synthon XXI can be found, which contains Cl⋯Cl interactions. For clarity, we show the 3D network in four colours, and each colour presents a 2D layer in Fig. 2d.
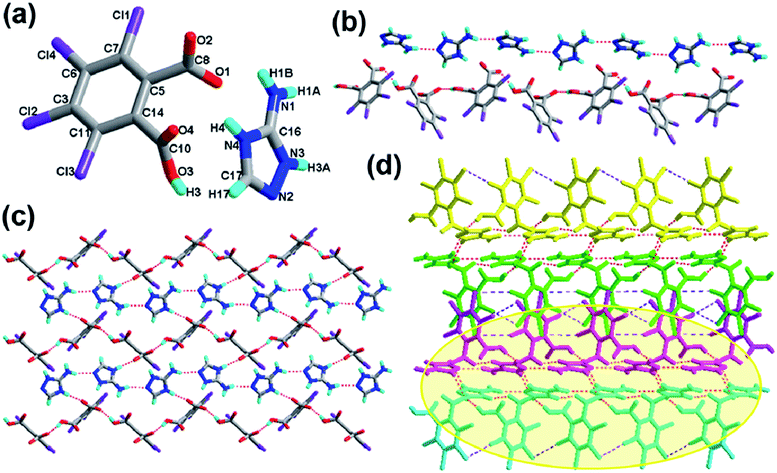 |
| | Fig. 2 (a) Molecular structure of 2 with atom labels of the asymmetric unit; (b) 1D acid chain and base chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via Cl⋯Cl interactions, and the 2D double sheet is colour coded in yellow. | |
Crystal structure analysis of complex 3
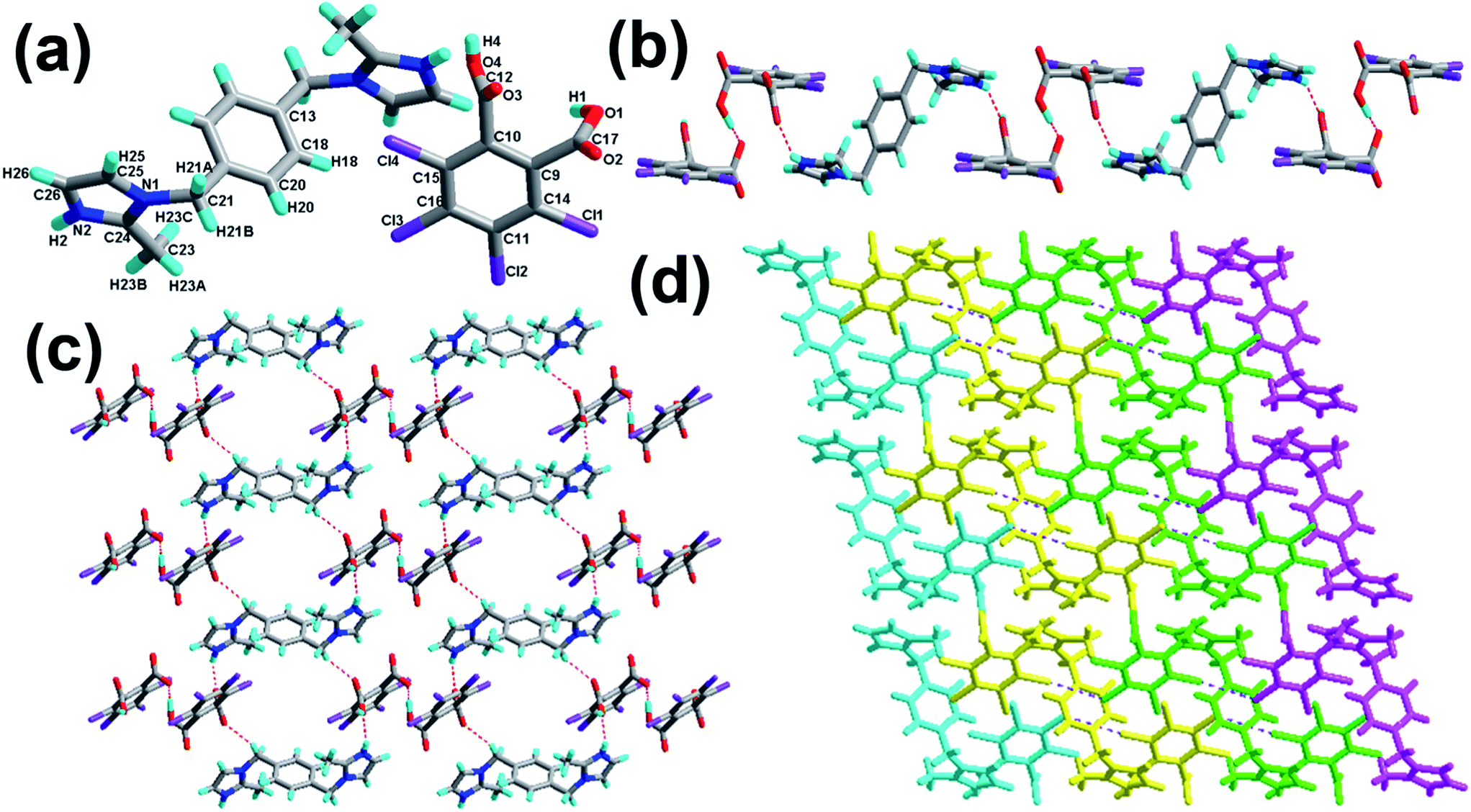
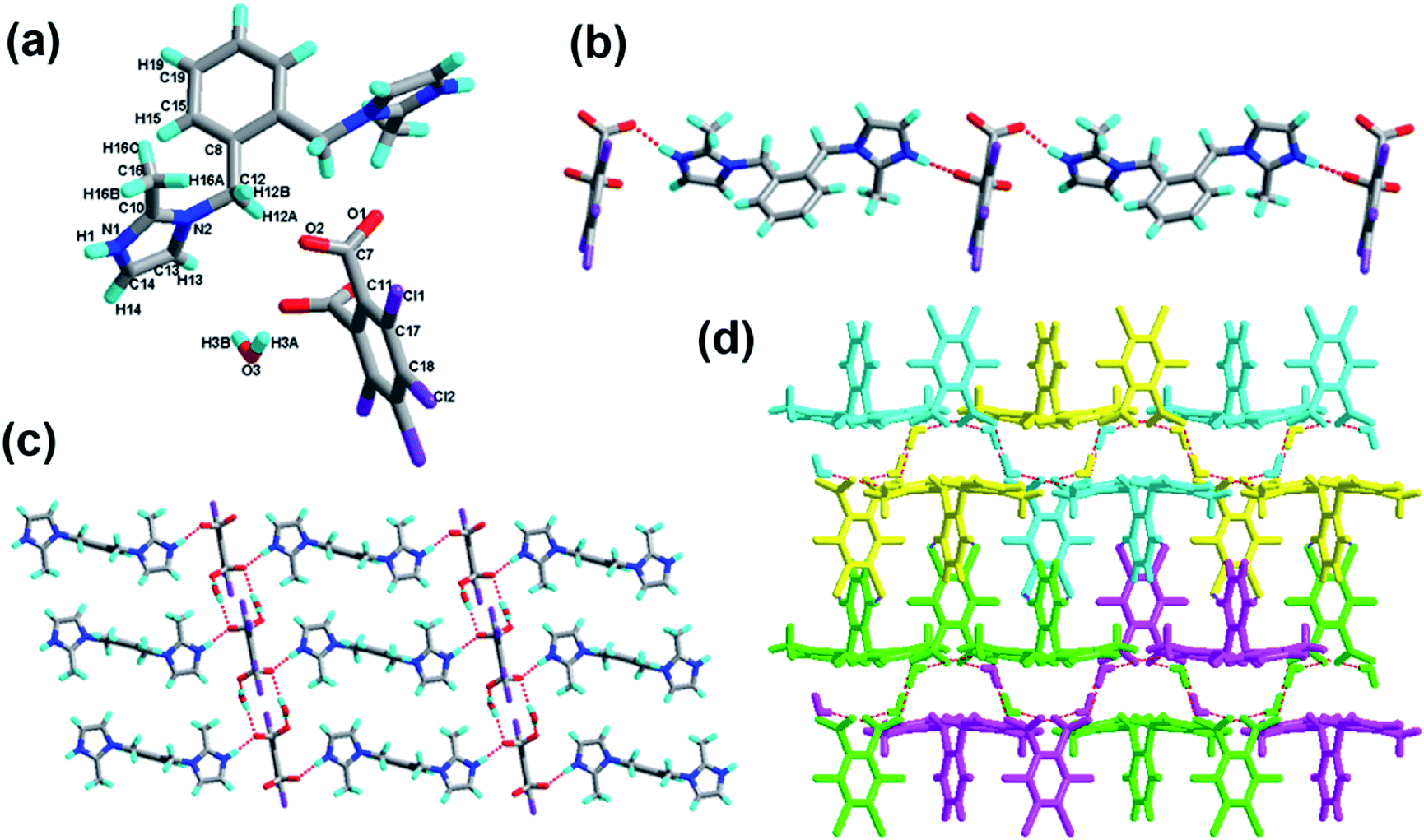
The crystal of 3 crystallizes in the triclinic system with the space group P with Z = 1 (Table 1). There is one H-tcpH− acid anion and one half H2-L5+ independent molecule in the asymmetric unit, which is shown in Fig. 3a. Within each L5 component, the dihedral angle between the imidazole ring and benzene ring is 89.2°. In the crystal asymmetric unit the dihedral angle is 25.1° between the imidazole ring and the benzene ring of H2-tcpH acid. Furthermore, the dihedral angle between the benzene rings of L5 and H2-tcpH is 66.3°. It is notable that the H2-tcpH acid molecules here form intermolecular interactions with L5, including strong N2–H2⋯O4 hydrogen bonding and strong O1–H1⋯O1 hydrogen bonding. As shown in Fig. 3b, the acid and base subunits arrange alternately to exhibit a 1D chain. Due to the presence of the hydrogen bonding C21–H21A⋯O3, these adjacent 1D chains are connected to afford a 2D architecture, which is illustrated in Fig. 3c. In this 2D architecture, there are synthons III R44(28) via N2–H2⋯O4 and C21–H21A⋯O3 interactions. (in Scheme 2) and XXVIII R66(38) (in ESI†) via N2–H2⋯O4, C21–H21A⋯O3 and O1–H121B⋯O1 interactions. Moreover, through type II Cl2⋯Cl4 interactions (distance 3.399 Å, θ1 = 165.8°, θ2 = 123.4°), the neighbouring layers are further extended into a 3D cross-link network. In addition, the synthon XXII [N2–H2⋯O4, C21–H21A⋯O3, Cl2⋯Cl4] is formed in this 3D network. For clarity, we show the 2D sheet in different colours in Fig. 3d. Further analysis of the crystal packing suggests that Cl⋯Cl interactions are very important in expanding the structure dimension.
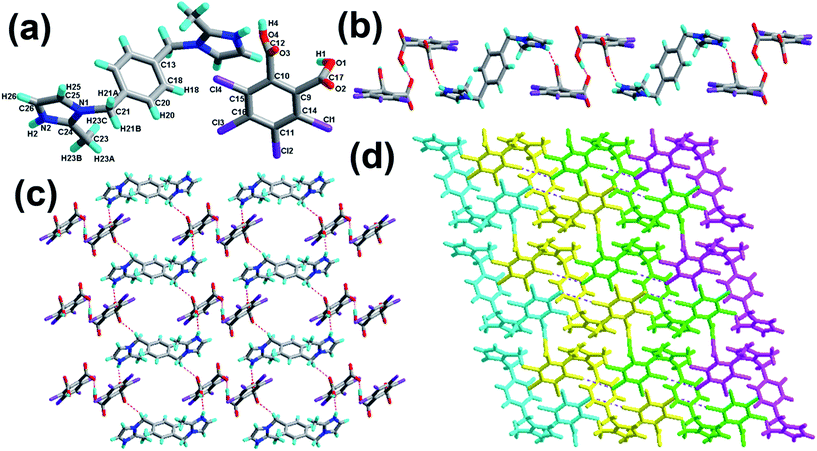 |
| | Fig. 3 (a) Molecular structure of 3 with atom labels of the asymmetric unit; (b) 1D chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via Cl⋯Cl interactions. | |
Crystal structure analysis of complex 4
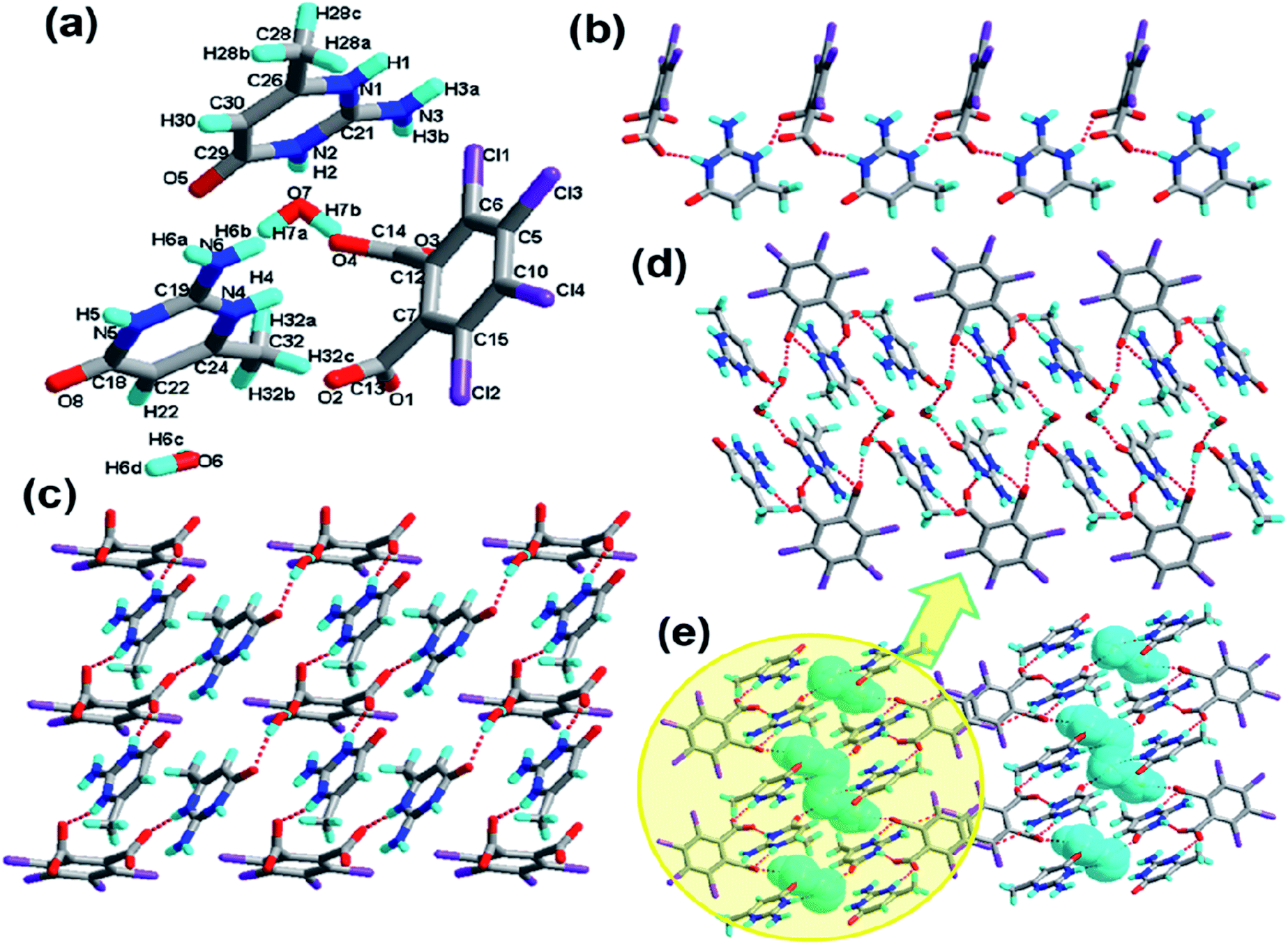
Complex 4 crystallizes in the monoclinic system with the space group P21/c (Z = 4, in Table 1). In the molecular structure of 4 (Fig. 4a), the local asymmetric unit contains two molecules of H-tcpH− acid anion and two molecules of Hampym+ base cation. Within the unit components the dihedral angle is 77.6° between the two benzene rings, and the dihedral angle between the pyrimidine rings is 37.3°. The dihedral angle between the amino group and pyridine ring is 2.7°. It is notable that the arrangement of the carboxylic groups of H2-tcpH in 4 is different from that in 1. In crystal of 1H2-tcpH molecules and H-tcpH− acid anion are alternately connected to each other, while in the crystal of 4, the H-tcpH− acid anions are connected to from 1D chains via the strong hydrogen bonding O3–H3⋯O5 in the Fig. 4b. Moreover, the carboxylic acid chains join two new patterns of supramolecular synthons IV R66(30), via strong N3–H3A⋯O1, N5–H5⋯O6, and O8–H8⋯O2 interactions and weak hydrogen bonding C35–H35⋯O4 and C38–H38⋯O7 along the c axis to form a 2D layer, which is shown in Fig. 4c. The adjacent 2D sheets afford a 2D double layer through the hydrogen bonding C35–H35⋯N1 and C38–H38⋯N2. Significantly, there are an additional unique type II Cl⋯Cl interactions (Cl3⋯Cl4 distance 3.372 Å, θ1 = 156.9°, θ2 = 95.0° and Cl5⋯Cl8 distance 3.373 Å, θ1 = 94.4°, θ2 = 157.0°, shown in Table 3), and as a consequence, 2D double sheets are entangled to build up a unique 3D architecture, which is shown in Fig. 4d and it is colour coded in yellow. Additionally, in this network, there is the unique synthon XXIII, which is illustrated in Scheme 4. This synthon is connected through the Cl⋯Cl interactions and strong hydrogen bonding O–H⋯O.
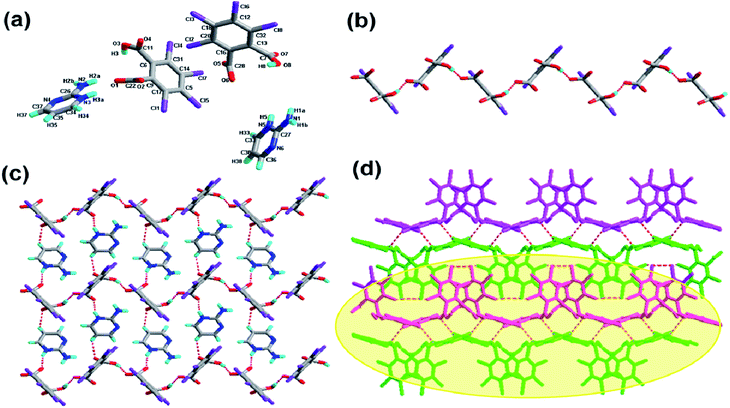 |
| | Fig. 4 (a) Molecular structure of 4 with atom labels of the asymmetric unit; (b) 1D acid chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via Cl⋯Cl interactions, and the 2D double sheet is colour coded in yellow. | |
Crystal structure analysis of complex 5
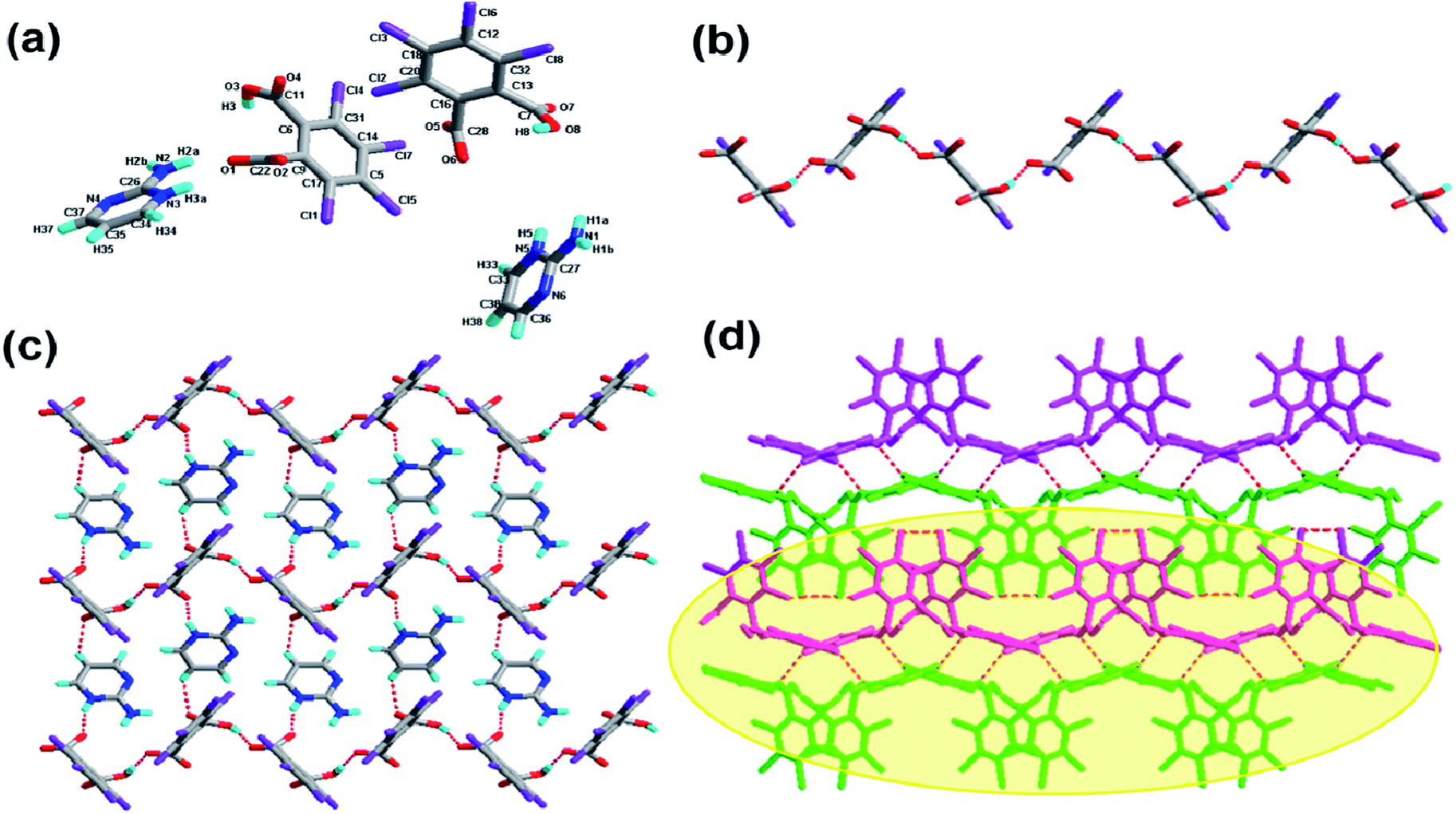
The crystallization of H2-tcpH acid with edm yields the proton-transfer organic salt 5, and it crystallizes in the centrosymmetric monoclinic space group C2/c with Z = 8 (Table 1). The asymmetric unit contains one half H-edm+ cation molecule and one deprotonated H-tcpH− anion in Fig. 5a. In this asymmetric unit, one of the H atoms of the H2-tcpH acid molecules transfers to an N atom of the edm molecule. Due to the deprotonation effect, the usual intramolecular interaction O4–H4⋯O1 hydrogen bonding exists to form a 1D chain along the b axis, which is displayed in Fig. 5b. As illustrated in Fig. 5c, each H-edm+ cation connects two H-tcpH− subunits via synthon VI R33(16) [O4–H4⋯O1, N1–H1A⋯O2 and N1–H1B⋯O2] and N1–H1C⋯O1 hydrogen bonding to form a 2D layer. Moreover, each H-edm+ molecule is also surrounded by three H-tcpH− anion subunits and they act as a bridge to the neighbouring 1D chain. In this structure, the adjacent 2D sheets connect to afford 2D double sheets (displayed in Fig. 5d and colour coded in yellow) through the hydrogen bonding N1–H1B⋯O2 (2.847 Å). Due to the N1–H1B⋯O2 interactions, the 2D double sheets contain the unique synthon V R34(13) (Scheme 3). Additionally, there are unique type I Cl2⋯Cl2 (Cl2⋯Cl2 distance 3.707 Å, θ1 = θ2 = 126.8°) and type II Cl3⋯Cl4 (Cl3⋯Cl4 distance 3.632 Å, θ1 = 122.1°, θ2 = 175.4°, which is shown in Table 3) interactions between each H2-tcpH group. Moreover, the unique synthon XXIV Cl4 [Cl3⋯Cl4] (Scheme 4) can be found in this network, and it is almost identical to the synthon Cl4 in the article composed by Desiraju.25 As a consequence, these layers are further extended into a 3D network, which is shown in Fig. 5d.
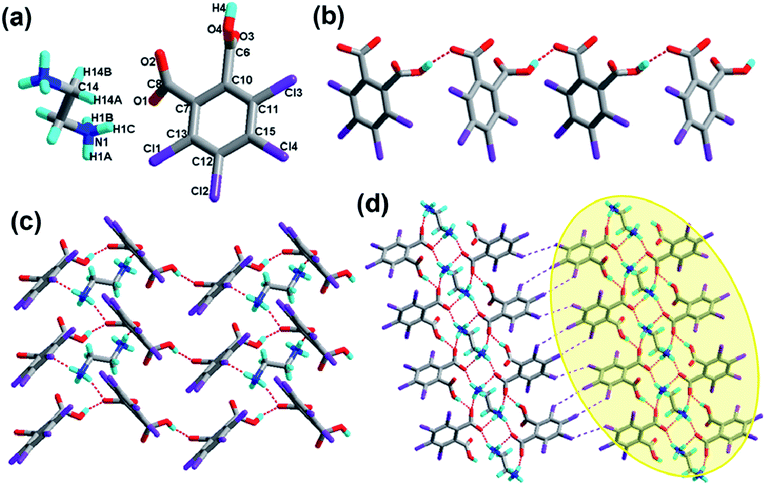 |
| | Fig. 5 (a) Molecular structure of 5 with atom labels of the asymmetric unit; (b) 1D acid chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via Cl⋯Cl interactions, and the 2D double sheet is colour coded in yellow. | |
Crystal structure analysis of complex 6
The crystal of 6 crystallizes in the triclinic system with the space group P (Z = 2, in Table 1). The asymmetry unit of 6 (shown in Fig. 6a) contains a pair of ahmp+ cation molecules, one tcpH2− dianion molecule, and two water molecules. The two ahmp+ base molecules crystallize nearly parallel with the incline angle of only 2.0°. The dihedral angles between the pyrimidine ring and benzene ring are 71.8° and 69.8°, respectively. The adjacent acid and base components are linked to form a linear tape via N4–H4⋯O4 (2.668 Å) and N5–H5⋯O1 (2.774 Å) hydrogen bonding, which is shown in Fig. 6b. Both N atoms in the pyrimidine ring take part in the N4–H4⋯O4/N5–H5⋯O1 intermolecular interactions between base and acid units. Furthermore, as shown in Fig. 6c, one of the water moieties behave as a hydrogen-bonding 2-connector [O6–H6C⋯O3 and O6–H6D⋯O5] with acid and base groups to result in a 2D sheet. In this 2D sheet, there is the larger synthon VII R1010(46), which is shown in Scheme 3. Through further O7–H7A⋯O8 and O7–H7B⋯O6 intermolecular interactions, the neighboring 2D layers via synthon VIII R88(30) [N5–H5⋯O1, O6–H6C⋯O3, O7–H7B⋯O6 and O7–H7A⋯O8], synthon IX R88(28) [N4–H4⋯O4, O6–H6C⋯O3, O7–H7A⋯O8 and O7–H7B⋯O6] and synthon X R1010(36) [N1–H1⋯O2, N5–H5⋯O1, O6–H6D⋯O5, O7–H7A⋯O8 and O7–H7B⋯O6] form a 2D double layer, which is displayed in Fig. 6d and is colour coded in yellow in Fig. 6e. From another side view, we can clearly see that it is different from complexes 1–5. In structure 6, which forms N6–H6B⋯Cl4 hydrogen bonding between the base units and the carboxylic acid units between the adjacent 2D double layers, result in a 3D architecture, as illustrated in Fig. 6e. Moreover, the unique synthon XI R44(22) (in Scheme 3) was obtained, which concludes the weak hydrogen bonding N6–H6B⋯Cl4. The water moieties in the 3D network are displayed in a turquoise colour in the space-filling model.
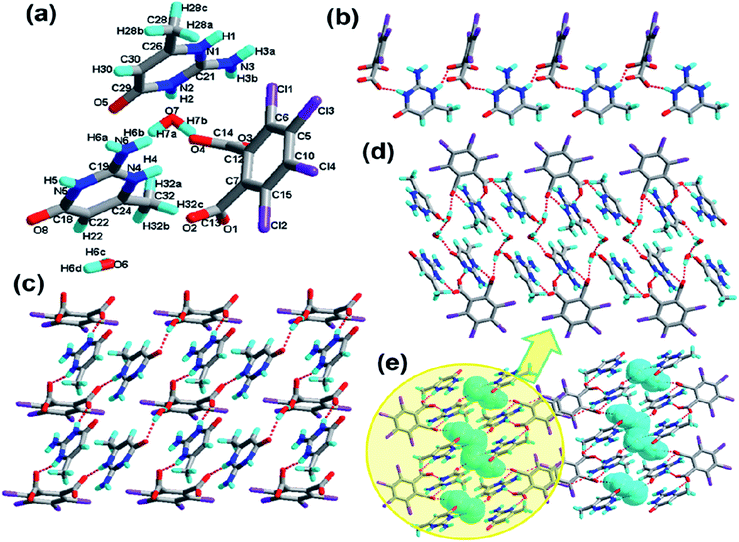 |
| | Fig. 6 (a) Molecular structure of 6 with atom labels of the asymmetric unit; (b) 1D acid chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 2D double layer via water molecules; (e) 3D network via N–H⋯Cl hydrogen bonding, and the 2D double sheet is colour coded in yellow. | |
Crystal structure analysis of complex 7
Crystallization of H2-tcpH acid with L7 yields a proton-transfer organic salt 7, which crystallizes in the monoclinic system with the space group C2/c (Z = 2, in Table 1). The asymmetric unit of 7 consists of one half of the tcpH2− dianion molecule and half of one corresponding monoprotonated L7 dication, as well as two water molecules (see Fig. 7a). In the cationic moiety, the imidazole ring forms the dihedral angle of 81.6° with the central benzene ring. The dihedral angle between the benzene ring of the H2-tcpH acid and the imidazole ring of L7 is 89.1°, and the dihedral angle between the two benzene rings is 64.5°. In structure 7, the proton transfer from the carboxylic group to the imidazole ring of L7, leads to the formation of charge-assistant N+–H⋯O− hydrogen bonding. Particularly, the acid and base components display an alternate disposition along the [001] direction, that is generated by the b axis to constitute a distinct 1D chain, which is displayed in Fig. 7b. Notably, one water of the H3A–O3–H3B forms a pair of O3–H3A⋯O1 and O3–H3B⋯O2 strong hydrogen bonds with the carboxylic groups of the H2-tcpH acid and the other of N1–H1⋯O1 hydrogen bonding to define the R44(12) and R68(44) motifs (synthons XII and XIII shown in Scheme 3), which connect the adjacent 1D chains to generate a 2D sheet and the water molecules are shown in the space-filling model in a bright green colour (see Fig. 7c). The adjacent layers are further connected through the weak C15–H15⋯Cl2 interactions between the acid and base components, and neighbouring sheets are connected to result in a 3D network along the [001] direction as seen in Fig. 7d, and for displaying more clarity, we show the 2D layers in different colours.
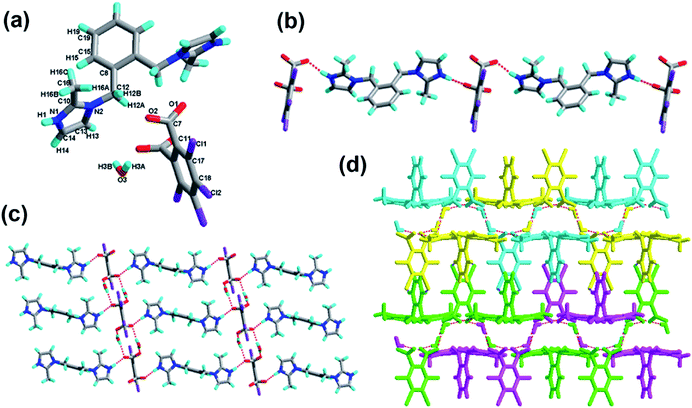 |
| | Fig. 7 (a) Molecular structure of 7 with atom labels of the asymmetric unit; (b) 1D chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via C–H⋯Cl hydrogen bonding. | |
Crystal structure analysis of complex 8
The structure of 8 is composed of one H2-tcpH acid anion, one Bim cation, and two lattice water molecules in the asymmetric unit (see Fig. 8a), and the crystal crystallizes in the triclinic space group P with Z = 2 (Table 1). The dihedral angle between the two benzene rings of H2-tcpH acid and Bim is 29.2°. The imidazole ring of Bim deviates by 0.7° from the benzene ring plane, it is almost coplanar. In this case, an unusual chain is produced along the c axis via strong O5–H5A⋯O2 and O5–H5B⋯O1 interactions from the water bridges, which is displayed in Fig. 8b. Furthermore, the H atoms of imidazole ring here lead to the formation of N1–H1⋯O1 and N2–H2⋯O5 hydrogen bonding between the water molecule and H2-tcpH acid moieties from adjacent 1D chains (synthon XXX R68(24) in the ESI†), generating a nearly planar 2D supramolecular sheet (see Fig. 8c). Eventually, interlayer O3–H3⋯O2 hydrogen bonding between the acid molecules extend the 2D patterns to 2D double layers, and these interactions define synthons XIV R22(14) [O3–H3⋯O2], XV R66(24) [O3–H3⋯O2 O5–H5A⋯O2 and O5–H5B⋯O1] and XVI R68(30) [N1–H1⋯O1, N2–H2⋯O5, O3–H3⋯O2 and O5–H5A⋯O2], whereas the other weak hydrogen bond between C19–H19⋯Cl2 interlink the 2D double layers to construct a 3D hydrogen-bonded network (see Fig. 8d). Due to the weak hydrogen bonding C19–H19⋯Cl2, the unique synthon XVII R44(24) (Scheme 3) appears in the 3D architecture.
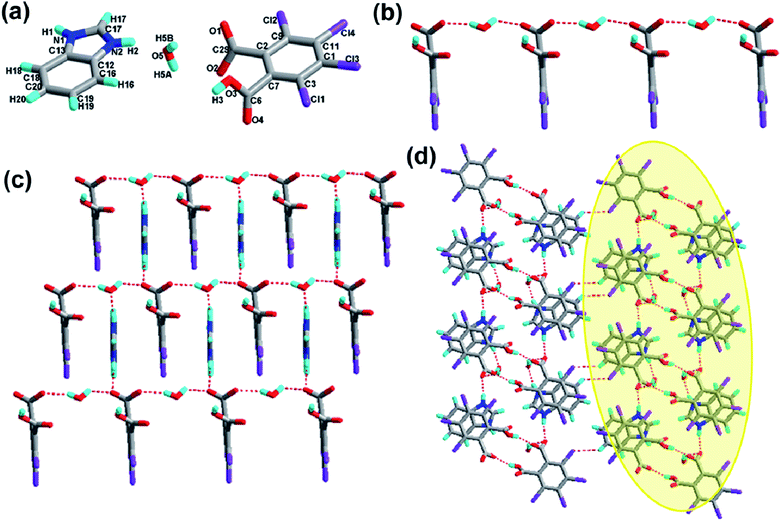 |
| | Fig. 8 (a) Molecular structure of 8 with atom labels of the asymmetric unit; (b) 1D acid chain via hydrogen bonds; (c) perspective view of the 2D hydrogen-bonded layer; (d) 3D network via C–H⋯Cl hydrogen bonding, and the 2D double sheet is colour coded in yellow. | |
Thermal stability analysis
All of the complexes, 1–8, are stable in air and can maintain their structural integrity at ambient conditions for a long time. In order to examine the thermal stability of all complexes, TGA and DSC were carried out between room temperature and 900 °C in nitrogen atmosphere. The DSC traces and TGA data for the crystals are presented in the ESI.† TGA experiments were implemented to investigate their thermal stability. As for complex 3, the TGA results indicate that they remain intact until 205 °C, and then there is a sharp weight loss ending at 280 °C (peaks: 265.1 °C for crystal 3). The weight loss of complex 6 up to 60.30% to 260 °C corresponds to the loss of an acid molecule (calculated: 70.48%). The second weight loss of 39.74% (calculated: 29.53%) can be detected from 280 to 527 °C, which is due to the decomposition of a base molecule. The TGA curve of 5 is very similar to complex 3, which indicates the two consecutive weigh losses of acid and base molecules from 135 to 260 °C (peaking at 210.18 °C) and 310 to 472 °C (peaking at 433.53 °C). The other two consecutive weight losses are from the acid and base components. The TGA curves of 7 and 8 are very similar, which indicate that there are two consecutive weight losses of the two samples. Complex 7 decomposes from 61 to 115 °C (peaking at 90.41 °C), which corresponds to the water molecules. While in complex 8, the loss of water continues to 160 °C. When it reaches 163 °C and 165 °C, respectively, the decomposition of the framework begins in complex 7 and 8 (peaking at 319.4 °C and 218.6 °C, respectively). As for 1, the first weight loss of 6.97% from 78 to 168 °C (calculated: 3.90%) corresponds to the loss of one methanol molecule per formula. The second weight loss of 58.86% (calculated: 75.12%) can be detected from 168 to 242 °C, which is due to the decomposition of acid molecules, and the second weight loss represented the loss of base components (calculated: 24.15%, found: 36.96%). Compared with complex 1, the TGA measurement of 2 shows a weight loss of 11.90% in the temperature range 165 to 256 °C, which corresponds to the loss of a 3-amino-1,2,4-triazole molecule, and the second weight loss represents the loss of acid components (calculated: 79.38%, found: 66.55%). The TGA measurement of 4 indicates that the complex does not melt and is stable up to 166 °C, at which temperature the crystal begins to decompose. The ligand benzimidazole decomposes at 166 to 178 °C (peaking at 165.18 °C), the second weight loss of 85.55% from 178 to 380 °C, which corresponds to the acid molecules. Moreover, the theoretical values and practical values differ within a reasonable range. Broadly speaking, the eight frameworks have a remarkably thermal stability.
Conclusions
In this article, we have successfully synthesized and characterized eight new complexes using the H2-tcpH acid ligand along with a series of N-heterocyclic compounds. X-ray analysis revealed that these salts exhibit different intriguing architectures in which various hydrogen bonding and halogen⋯halogen interactions are also found. The eight complexes all have the H2-tcpH acid molecule, and the H atom of the carboxylate group transfers to the base molecule. Furthermore, structural analysis of complexes 1–8 found that only one H atom transfers in complexes 1–5 and 8, while the proton transfer occurs entirely in complexes 6 and 7. Different from the other crystals, complex 1 contains a methanol solvent molecule in the asymmetry unit. Moreover, complexes 1–8 display intriguing 3D structures, Furthermore, they form a 2D double layer by the different hydrogen bonding, expect for complex 5. In addition, it is interesting to note that in crystals 1–5, they demonstrate 3D architectures via Cl⋯Cl interactions, while in complexes 7–8 the adjacent 2D organic layers are further connected through the weak hydrogen bonding C–H⋯Cl, generating a 3D network. According to the above structural description of our complexes, we find that the H2-tcpH acid ligand adopts a variety of connected modes, and it is a good candidate for constructing supramolecular chemistry.
Moreover, in all of these 3D supramolecular complexes the strong hydrogen bonding O–H⋯O, N–H⋯O, N–H⋯N, and the weak C–H⋯O, C–H⋯Cl, N–H⋯Cl, Cl⋯Cl interactions are present. They not only extend these networks from 1D to 2D/3D, but also reinforce their structures because the synthons formed by these interactions are stable in complexes 1–8. The weak hydrogen bonding C–H⋯O can affect the crystal packing in unpredictable manner, and it is widely used in crystal engineering to construct synthons as addressed in previous accounts.24 In complex 3, C–H⋯O hydrogen bonding participate in the synthon formation in III and XXII. Due to the stereochemistry effect, the classical synthons R22(6) and R22(8) have not been found in complexes 1–8. Nevertheless, a number of larger synthons exist in these structures, for example, synthons III, IV, VII, X, XIII, XVI, XVII and XX. Clearly, the presence of the H2-tcpH acid is a common structural feature of these larger synthons, and it is obvious that the “supramolecular synthon” strategy will be frequently applied in the design of novel periodic and predictable superstructures.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51372125, 21371105, and 21203106), and the Scientific and Technical Development Project of Qingdao (no. 13-1-4-184-jch).
References
-
(a) T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed;
(b) M. M. Conn and J. Rebek, Jr., Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed;
(c) A. Jasat and J. C. Sherman, Chem. Rev., 1999, 99, 931–967 CrossRef CAS PubMed;
(d) S. S. Mondal, A. Bhunia, A. Kelling, U. Schilde, C. Janiak and H. J. Holdt, J. Am. Chem. Soc., 2014, 136, 44–47 CrossRef CAS PubMed;
(e) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed;
(f) G. A. Hembury, V. V. Borovkov and Y. Inoie, Chem. Rev., 2008, 108, 1–73 CrossRef CAS PubMed;
(g) B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179–4198 RSC;
(h) L. Wang, L. Zhao, L. Y. Xu, R. X. Chen and Y. Yang, CrystEngComm, 2012, 14, 6998–7008 RSC;
(i) L. Wang, L. Zhao, Y. J. Hu, W. Q. Wang, R. X. Chen and Y. Yang, CrystEngComm, 2013, 15, 2835–2852 RSC;
(j) L. Wang, L. Zhao, W. M. Liu, R. X. Chen, Y. X. Gu and Y. Yng, Sci. China: Chem., 2012, 55, 2381–2387 CrossRef CAS.
-
(a) K. Biradha, A. Nangia, G. R. Desiraju, C. J. Carrell and H. L. Carrell, J. Mater. Chem., 1997, 7, 1111–1122 RSC;
(b) V. R. Thalladi, H. C. Weiss, D. Bläser and R. Boese, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS;
(c) S. R. Perumalla, V. R. Pedireddi and C. C. Sun, Mol. Pharmaceutics, 2013, 10, 2462–2466 CrossRef CAS PubMed;
(d) L. Wang, L. Zhao, M. Liu, F. Q. Liu, R. X. Chen, Y. Yang and Y. X. Gu, Sci. China: Chem., 2012, 55, 2115–2122 CrossRef CAS.
-
(a) M. Pérez-Torralba, M. Ángeles García, C. López, M. C. Torralba, M. R. Torres, R. M. Claramunt and J. Elguero, Cryst. Growth Des., 2014, 14, 3499–3509 CrossRef;
(b) G. Nandi, H. M. Titi and I. Goldberg, Cryst. Growth Des., 2014, 14, 3557–3566 CrossRef CAS;
(c) P. Panini and D. Chopra, Cryst. Growth Des., 2014, 14, 3155–3168 CrossRef CAS;
(d) F. Bertolotti, A. V. Shishkina, A. Forni, G. Gervasio, A. I. Stash and V. G. Tsirelson, Cryst. Growth Des., 2014, 14, 3587–3595 CrossRef CAS;
(e) E. Guzamán-Percástegui, J. G. Alvarado-Rodríguez, J. Cruz-Borbolla, N. Andrade-López, R. A. Vázquez-Garacía, R. N. Nav-Galindo and T. Pandiyan, Cryst. Growth Des., 2014, 14, 3742–3757 CrossRef.
-
(a) P. Metrangolo, H. Neukorch, T. Pilati and G. Resnti, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed;
(b) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC;
(c) H. R. Khavasi and A. A. Tehrani, CrystEngComm, 2013, 15, 5813–5820 RSC;
(d) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed;
(e) H. Y. Gao, X. R. Zhao, H. Wang, X. Pang and W. J. Jin, Cryst. Growth Des., 2012, 12, 4377–4387 CrossRef CAS.
-
(a) J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS;
(b) M. Freytag, P. G. Jones, B. Ahrens and A. K. Fischer, New J. Chem., 1999, 23, 1137–1139 RSC.
-
(a) M. Podsiadlo, A. Olejniczak and A. Katrusiak, CrystEngComm, 2014, 16, 8279–8285 RSC;
(b) R. B. K. Siram, D. P. Karothu, T. N. Guru Row and S. Patil, Cryst. Growth Des., 2013, 13, 1045–1049 CrossRef CAS.
-
(a) M. Baldrighi, G. Cavallo, M. R. Chierotti, R. Gobetto, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Mol. Pharmaceutics, 2013, 10, 1760–1772 CrossRef CAS PubMed;
(b) R. J. Baker, P. E. Colavita, D. M. Murphy, J. A. Platts and J. D. Wallis, J. Phys. Chem. A, 2012, 116, 1435–1444 CrossRef CAS PubMed;
(c) V. R. Hathwar, T. S. Thakur, R. Dubey, M. S. Pavan, T. N. Guru Row and G. R. Desiraju, J. Phys. Chem. A, 2011, 115, 12852–12863 CrossRef CAS PubMed;
(d) D. Chopra, T. S. Cameron, J. D. Ferrara and T. N. Guru Row, J. Phys. Chem. A, 2006, 110, 10465–10477 CrossRef CAS PubMed;
(e) C. F. Matta, N. Castillo and R. J. Boyd, J. Phys. Chem. A, 2005, 109, 3669–3681 CrossRef CAS PubMed;
(f) V. R. Hathwar and T. N. Guru Row, Cryst. Growth Des., 2011, 11, 1338–1346 CrossRef CAS;
(g) T. Shirman, J. Lamere, L. J. M. Shimon, T. Gupta, J. M. L. Martin and M. E. van der Boom, Cryst. Growth Des., 2008, 8, 3066–3072 CrossRef CAS;
(h) S. K. Nayak, M. K. Reddy, T. N. Guru Row and D. Chopra, Cryst. Growth Des., 2011, 11, 1578–1596 CrossRef CAS;
(i) P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278–294 CrossRef CAS PubMed.
- V. S. Senthil Kumar, F. C. Pigge and N. P. Rath, CrystEngComm, 2004, 6, 102–105 RSC.
- H. Wang, R. X. Hu, X. Pang, H. Y. Gao and W. J. Jin, CrystEngComm, 2014, 16, 7942–7948 RSC.
-
(a) M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2014, 114, 1343–1370 CrossRef CAS PubMed;
(b) T. Samanta, L. Dey, J. Dinda, S. K. Chattopadyhyay and S. K. Seth, J. Mol. Struct., 2014, 1068, 58–70 CrossRef CAS PubMed;
(c) A. Zawada, R. W. Góra, M. M. Mikolajczyk and W. Bartkowiak, J. Phys. Chem. A, 2012, 116, 4409–4416 CrossRef CAS PubMed.
-
(a) F. H. Allen, W. D. Samuel Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25–34 RSC;
(b) V. R. Thalladi, B. S. Goud, V. J. Hoy, F. H. Allen, J. A. K. Howard and G. R. Desiraju, Chem. Commun., 1996, 3, 401–402 RSC.
-
(a) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS;
(b) S. Tothadi and G. R. Desiraju, Chem. Commun., 2013, 49, 7791–7793 RSC;
(c) A. Mukherjee, S. Tothadi, S. Chakraborty, S. Ganguly and G. R. Desiraju, CrystEngComm, 2013, 15, 4640–4654 RSC;
(d) M. Du, Z. H. Zhang and X. J. Zhao, Cryst. Growth Des., 2011, 11, 1199–1208 CrossRef;
(e) M. Du, Z. H. Zhang and X. J. Zhao, Cryst. Growth Des., 2005, 5, 1247–120854 CrossRef CAS;
(f) M. Du, Z. H. Zhang, X. J. Zhao and H. Cai, Cryst. Growth Des., 2006, 6, 114–121 CrossRef CAS;
(g) M. Du, X. J. Jiang, X. Tan, Z. H. Zhang and H. Cai, CrystEngComm, 2005, 5, 454–462 Search PubMed;
(h) S. Tothadi, A. Mukherjee and G. R. Desiraju, Chem. Commun., 2011, 47, 12080–12082 RSC.
- K. Merz, M. V. Evers, F. Uhl, R. I. Zubatyuk and O. V. Shishkin, Cryst. Growth Des., 2014, 14, 3124–3130 CAS.
- H. R. Khavasi and M. Esmaeili, CrystEngComm, 2014, 16, 8479–8485 RSC.
- C. Zhang, Z. Yang, X. Q. Zhou, C. H. Zhang, Y. Ma, J. J. Xu, Q. Zhang, F. Nie and H. Z. Li, Cryst. Growth Des., 2014, 14, 3923–3928 CAS.
-
(a) J. A. R. P. Sarma, F. H. Allen, V. J. Hoy, J. A. K. Howard, R. Thaimattam, K. Biradha and G. R. Desiraju, Chem. Commun., 1997, 101–102 RSC;
(b) K. S. Eccles, R. E. Morrison, A. R. Maguire and S. E. Lawrence, Cryst. Growth Des., 2014, 14, 2753–2762 CrossRef CAS;
(c) A. Mukherjee and G. R. Desirju, Chem. Commun., 2011, 47, 4090–4092 RSC;
(d) V. R. Thalladi, S. Brasselet, D. Bläser, R. Boese, J. Zyss, A. Nangia and G. R. Desiraju, Chem. Commun., 1997, 1841–1842 RSC.
-
(a) P. J. Langley, J. Hulliger, R. Thaimattam and G. R. Desiraju, New J. Chem., 1998, 22, 1307–1309 RSC;
(b) S. R. Perumalla, E. Suresh and V. R. Pedireddi, Angew. Chem., Int. Ed., 2005, 44, 7752–7757 CrossRef CAS PubMed;
(c) V. R. Vangala, R. Mondal, C. K. Broder, J. A. K. Howard and G. R. Desiraju, Cryst. Growth Des., 2005, 5, 99–104 CrossRef CAS.
-
(a) S. Kumaresam, P. G. Seethalakshmi, P. Kumaradhas and B. Devipriya, J. Mol. Struct., 2013, 1032, 169–175 CrossRef PubMed;
(b) S. W. Jin, M. Guo, D. Q. Wang and H. Cui, J. Mol. Struct., 2011, 1006, 128–135 CrossRef CAS PubMed;
(c) L. Wang, Y. J. Hu, W. Y. Xu, Y. Y. Pang, F. Q. Liu and Y. Yang, RSC Adv., 2014, 4, 56816–56830 RSC;
(d) L. Wang, L. Zhao, M. Liu, R. X. Chen, Y. Yang and Y. X. Gu, Sci. China: Chem., 2012, 55, 2115–2122 CrossRef CAS;
(e) L. Wang, R. Y. Xue, Y. X. Li, Y. R. Zhao, F. Q. Liu and K. K. Huang, CrystEngComm, 2014, 16, 7074–7089 RSC.
-
(a) A. Muhherjee and G. R. Desiraju, Cryst. Growth Des., 2014, 14, 1375–1385 CrossRef;
(b) S. Tothadi, S. Joseph and G. R. Desiraju, Cryst. Growth Des., 2013, 13, 3242–3254 CrossRef CAS;
(c) Y. B. He, S. C. Xiang and B. L. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed;
(d) P. K. Knacharla, T. Kato and D. Crich, J. Am. Chem. Soc., 2014, 136, 5472–5480 CrossRef PubMed;
(e) P. Li, Y. B. He, H. D. Arman, R. Krishna, H. L. Wang, L. H. Weng and B. L. Chen, Chem. Commun., 2014, 50, 13081–13084 RSC;
(f) F. Guo, F. Wang, H. Yang, X. L. Zhang and J. Zhang, Inorg. Chem., 2012, 51, 9677–9682 CrossRef CAS PubMed.
-
(a) F. Zordan, L. Brammer and P. Sherwood, J. Am. Chem. Soc., 2005, 127, 5979–5989 CrossRef CAS PubMed;
(b) S. K. Nayak, M. K. Reddy, T. N. Guru Row and D. Chopra, Cryst. Growth Des., 2011, 11, 1578–1596 CrossRef CAS.
-
(a) L. Wang, L. Zhao, Y. J. Hu, W. Q. Wang, R. X. Chen and Y. Yang, CrystEngComm, 2013, 15, 2835–2852 RSC;
(b) L. Wang, Y. J. Hu, W. Q. Wang, F. Q. Liu and K. K. Huang, CrystEngComm, 2014, 16, 4142–4161 RSC.
- B. F. Hoskins, R. Robson and D. A. Slizys, J. Am. Chem. Soc., 1997, 119, 2952–2953 CrossRef CAS.
-
(a) G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, SHELXL-97, Programs for X-ray Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
-
(a) T. Steiner, New J. Chem., 1998, 22, 1099–1103 RSC;
(b) P. K. Thallapally, A. K. Katz, H. L. Carrell and G. R. Desiraju, CrystEngComm, 2003, 5, 87–92 RSC;
(c) N. N. Laxmi Madhavi, C. Bilton, J. A. K. Howard, F. H. Allen, A. Nangia and G. R. Desiraju, New J. Chem., 2000, 24, 1–4 RSC;
(d) R. Thaimattam, D. S. Reddy, F. Xue, T. C. W. Mak, A. Nangia and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2., 1998, 1783–1789 RSC.
- R. Banerjee, G. R. Desiraju, R. Mondal, A. S. Batsanov, C. K. Broder and J. A. K. Howard, Helv. Chim. Acta., 2003, 86, 1339–1351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR, TGA, DSC data and synthons XXV–XXXIV about this work. CCDC 1032979–1032986. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15392h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.