
Half-sandwich complexes of ruthenium, osmium, rhodium and iridium with DL-methionine or S-methyl-L-cysteine: a solid state and solution equilibrium study†
János Patalenszkia,
Linda Bíróa,
Attila Csaba Bényeib,
Tereza Radosova Muchovacd,
Jana Kasparkovac and
Péter Buglyó*a
aDepartment of Inorganic and Analytical Chemistry, University of Debrecen, H-4010 Debrecen, P.O. Box 21, Hungary. E-mail: buglyo@science.unideb.hu
bDepartment of Physical Chemistry, University of Debrecen, H-4010 Debrecen, P.O. Box 7, Hungary
cDepartment of Biophysics, Faculty of Science, Palacky University, 17. Listopadu 12, 77146 Olomouc, Czech Republic
dDepartment of Biophysics, Centre of the Region Haná for Biotechnological and Agricultural Research, Palacky University, Slechtitelu 11, 78371 Olomouc, Czech Republic
First published on 23rd December 2014
Abstract
[(η6-p-cym)Ru(H2O)3]2+ binding strength of the thioether ligands DL-methionine (H2met+) or S-methyl-L-cysteine (H2mecys+) was studied with the combined use of pH-potentiometry, NMR and mass spectrometry in aqueous solution. Both ligands were found to form stable [(η6-p-cym)RuA]+ complexes with [S, NH2, COO−] coordination of the amino acids over a wide pH-range. A log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) βRuA value of 17.2(2) was obtained from NMR titrations for the met system by a competition reaction with hydroxide ions. Comparison of the [(η6-p-cym)Ru(H2O)3]2+ binding strength of the [O, O, O] (citrate), [O, N, O] (isoserine) and [S, N, O] (met) donor sets at pH = 7.4 reveals the exclusive formation of a [S, N, O] chelated metal complex. Synthesis and characterisation of [(η6-p-cym)Ru(L)]X, [(η6-p-cym)Os(L)]Cl or [(η5-Cp*)M′(L)]Cl (L = mecys, DL-met, L-met; X = Cl, NO3, CF3SO3; M′ = Rh, Ir) complexes together with the molecular structures of the [(η6-p-cym)Ru(mecys)]NO3 (10) and [(η6-p-cym)Ru(met)]NO3 (11) obtained by the X-ray diffraction method are reported. Both tridentate ligands form chiral-at-metal complexes in which the configuration of the metal centre is determined by the ligands containing a stereogenic centre (αC) as well. Diffraction data indicate that 10 contains the RRuRαCSS stereoisomer while 11 consists of the RRuSαCRS and SRuRαCSS enantiomers in the lattice. Owing to the labile configuration of the thioether sulfurs epimerisation in both systems was detected and the ratio of the diastereomers was estimated. 11 together with [(η6-p-cym)Ru(L-met)]Cl (3), [(η5-Cp*)Rh(DL-met)]Cl (7) and [(η5-Cp*)Ir(DL-met)]Cl (9) were tested for their in vitro cytotoxicity using human-derived ovarian cancer lines (A2780) and showed no anti-proliferative activity in the concentration range (0–50 μM) studied.
βRuA value of 17.2(2) was obtained from NMR titrations for the met system by a competition reaction with hydroxide ions. Comparison of the [(η6-p-cym)Ru(H2O)3]2+ binding strength of the [O, O, O] (citrate), [O, N, O] (isoserine) and [S, N, O] (met) donor sets at pH = 7.4 reveals the exclusive formation of a [S, N, O] chelated metal complex. Synthesis and characterisation of [(η6-p-cym)Ru(L)]X, [(η6-p-cym)Os(L)]Cl or [(η5-Cp*)M′(L)]Cl (L = mecys, DL-met, L-met; X = Cl, NO3, CF3SO3; M′ = Rh, Ir) complexes together with the molecular structures of the [(η6-p-cym)Ru(mecys)]NO3 (10) and [(η6-p-cym)Ru(met)]NO3 (11) obtained by the X-ray diffraction method are reported. Both tridentate ligands form chiral-at-metal complexes in which the configuration of the metal centre is determined by the ligands containing a stereogenic centre (αC) as well. Diffraction data indicate that 10 contains the RRuRαCSS stereoisomer while 11 consists of the RRuSαCRS and SRuRαCSS enantiomers in the lattice. Owing to the labile configuration of the thioether sulfurs epimerisation in both systems was detected and the ratio of the diastereomers was estimated. 11 together with [(η6-p-cym)Ru(L-met)]Cl (3), [(η5-Cp*)Rh(DL-met)]Cl (7) and [(η5-Cp*)Ir(DL-met)]Cl (9) were tested for their in vitro cytotoxicity using human-derived ovarian cancer lines (A2780) and showed no anti-proliferative activity in the concentration range (0–50 μM) studied.
Introduction
Biotransformation reactions of half-sandwich type platinum metal complexes with potential antiproliferative activity1–5 are of great importance for a deeper understanding of their in vivo activity and to develop even more selective and effective compounds as potential anticancer drugs. During the biotransformation processes the half-sandwich complexes with potential antiproliferative activity, depending upon kinetic characteristics and thermodynamic stability, may undergo aquation and subsequent ligand substitution with the involvement of large (albumin, transferrin, immunoglobulin etc.) or low molecular weight components (e.g. carbonate, lactate, citrate, glutamate, cysteine, glutathione etc.) of the serum.1,2To understand the most important factors influencing the stability of these complexes a systematic study has been started in our laboratories. Regarding the donor atom preference of the half-sandwich type metal core comparison of the [(η6-p-cym)Ru(H2O)3]2+ binding strengths of various bidentate and tridentate ligands with different type of donor atoms were explored in aqueous solution. It was found that low basicity (O, O) chelators (oxalate, cyclobutane-dicarboxylate) are effective metal ion binders under acidic conditions only.6 Lactate with the mixed [COO−, O−] binding set cannot hinder the metal ion from hydrolysis at pH = 7.4 either. Complementing, however, the donors with a further carboxylate, the low molecular mass (l.m.m.) serum components, citrate or malate, were found to bind [(η6-p-cym)Ru(H2O)3]2+ very efficiently even in the basic pH-range in the form of monomeric complexes with different protonation degree.7,8 The role of the alcoholate group in effective binding was proven with the comparison of tricarballate ligand as it was found that for the [COO−, COO−, COO−] binding set the metal ion has only very little affinity at neutral pH.7
Replacement of one carboxylate in citrate or malate by an amino group results in an effective [O−, NH2, COO−] chelating set in serine or isoserine. It was also demonstrated that among the chelates that can be formed with these amino acid ligands the one incorporating five-membered [COO−, O−] binding site in isoserine is more stable in terms of ruthenium binding than the six-membered one with serine.9
Substitution of the hard alcoholate by a soft sulfur donor may result in a more favourable environment for these type metal ions. Although the thiolate containing cysteine or penicillamine systems10–15 are more explored especially in the solid state for the thioethers methionine (H2met+) or S-methyl-cysteine (H2mecys+) (with one CH2 unit shorter side chain, see Scheme 1) less information is available on the interaction with half-sandwich type platinum metal ions. In an earlier study synthesis and characterization of [(η5-Cp*)RuII(met)] (Cp* = pentamethylcyclopentadienyl) with tridentate coordination of the ligand was reported16 while X-ray structure of the analogous [(η5-Cp*)IrIII(mecys)]CF3SO3·0.5H2O was shown to feature identical (S, N, O) coordinating ligand with RIr and SS configurations in the molecule.17 Adding a strongly coordinating monodentate ligand to the system in [(η5-Cp)RuII(PPh3)(met)] (N, O) coordination of the amino acid was suggested according to the IR data.18 Based on the formation of stable complexes sequence-selective detection of Met-containing oligopeptides was also demonstrated with the use of the [(η5-Cp*)RhIII type building block as chemosensor.19 In a recent work a combined NMR and MS study on the interaction of [(η6-p-cym)RuCl2]2 and methionine in aqueous solution indicated the formation of a (S, N, O) coordinated major complex 48 h being necessary to reach equilibrium in solution. Samples obtained by the dilution of the DMSO solution of [(p-cym)RuCl2]2 and 6–10 fold excess of met with water were found to exhibit strong cytotoxicity against human ovarian cancer cell lines (A2780 and A2780CisR) with IC50 values of 0.54(7) and 3.53(39) μM, respectively.20
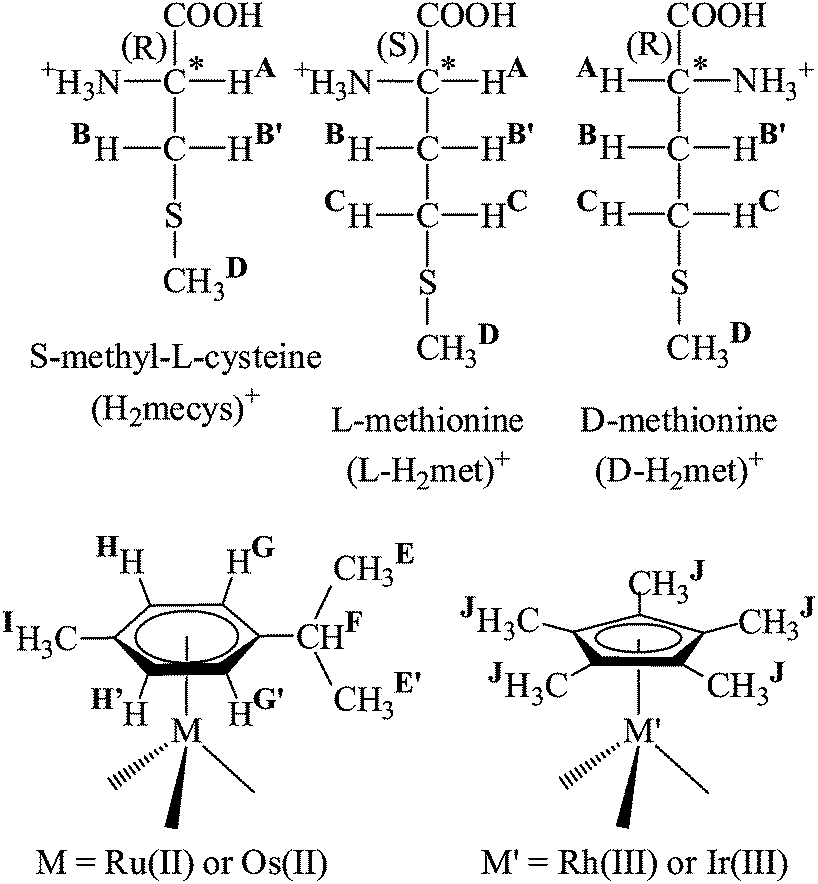 | ||
| Scheme 1 | ||
Upon coordination of a non-symmetrical chelating21 or three different monodentate ligands in these half-sandwich complexes the metal ion becomes a stereogenic centre and in the case of met or mecys further stereogenic centres (αC and thioether S) are also present resulting in the formation of diastereomers.22,23 Dissolution of this type of solid complexes, depending upon kinetic or thermodynamic control of the reactions under a given set of experimental conditions, the configuration of the metal and the sulfur (but not the αC) atoms of the chiral-at-metal complexes can change and may therefore result in the interconversion of the diastereomers and makes also challenging the interpretation of the NMR data.24
Since there appear to exist no detailed solution equilibrium results in the literature on the interaction between half-sandwich type [(η6-p-cym)M(H2O)3]2+ (p-cym = 1-isopropyl-4-methylbenzene; M = Ru, Os) or [(η5-Cp*)M′(H2O)3]2+ (M′ = Rh, Ir) metal ions and thioether-containing amino acids (Scheme 1) we have undertaken a combined study with the aid of pH-potentiometry, NMR, ESI-MS methods to explore the most likely solution structures and to estimate the stability constants of the complexes formed with [(η6-p-cym)Ru(H2O)3]2+ that was selected as model. We have also synthesized and characterized the appropriate complexes in the solid state and studied their transformation reactions and stereochemical features upon dissolving the solids. This paper summarizes the obtained results.
Experimental section
Materials and methods
RuCl3·xH2O, α-terpinene, OsO4, [(η5-Cp*)RhCl2]2, [(η5-Cp*)IrCl2]2, AgNO3, AgCF3SO3, S-methyl-L-(−)-cysteine, L-(+)-methionine, DL-methionine were commercial products (Merck, Acros Organics, W. C. Heraeus, Strem Chemicals, Reanal) and used as received. CH3OH, acetone, CH2Cl2, C2H5OH, CH3CN, diisopropyl ether and NaOMe used for the preparation of solid complexes were produced by Sharlau, WVR and Sigma Aldrich. Methanol was dried and distilled according to standard methods.25 [(η6-p-cym)RuCl2]2 or [(η6-p-cym)OsCl2]2 were synthesized and purified according to a literature procedures.26,27 Aqueous solution of [(η6-p-cym)Ru(acetone)3](NO3)2 or [(η6-p-cym)Ru(acetone)3](CF3SO3)2 were obtained from [(η6-p-cym)RuCl2]2 by the removal of chloride ion using equivalent amounts of appropriate silver salts. 1H, 13C NMR, COSY and HSQC spectra were recorded on a Bruker Avance 360 or 400 NMR instrument at room temperature in D2O or CD3OD and referenced to 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (TSP) or to resonances of the solvents. The low temperature spectra (10 °C and 0 °C) were registered on a Bruker Avance 400 or a Bruker Avance DRX-500 spectrometer in D2O. ESI-TOF MS analysis of the prepared complexes in the positive mode was carried out on a Bruker micrOTOFQ instrument. The measurements were performed in water at 0.3 mM metal ion concentration. Temperature of drying gas (N2) was 180 °C. The pressure of the nebulizating gas (N2) was 0.3 bar. The flow rate was 3 μL min−1. The spectra were accumulated and recorded by a digitalizer at a sampling rate of 2 GHz. DataAnalysis (version 3.4) was used for the calculations. IR spectra were recorded on an Perkin Elmer FTIR Paragon 1000 PC instrument. Elemental analyses were conducted on Elementar Vario MICRO CUBE instrument at the Department of Organic Chemistry, University of Debrecen, Hungary.Crystal structure analysis
Diffraction intensity data collection was carried out at 293(2) K on a Bruker-Nonius MACH3 diffractometer equipped with a point detector using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The structure was solved by SIR-92 program28 and refined by full-matrix least-squares method on F2, with all non-hydrogen atoms refined with anisotropic thermal parameters using the SHELXL-97 package.29 Publication material was prepared with the WINGX-suite.30 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were treated with a mixture of independent and constrained refinement. C–H hydrogen atoms were refined using a riding model. Crystallographic and experimental details are summarized in Table 1. The remaining peaks at the difference electron density maps are located close to the ruthenium atoms.†| Compound | 10 | 11 |
|---|---|---|
| a GOF = [∑w(Fo2 − Fc2)2/(n − p)]1/2.b R1 = ∑∣∣Fo∣ − ∣Fc∣∣/∑∣Fo∣, wR2 = [∑w(Fo2 − Fc2)2/∑wFo2]1/2. | ||
| Formula | C14H22NO2RuS·NO3 | C15H24NO2RuS·NO3 |
| Formula weight | 431.46 | 445.49 |
| Crystal system | Monoclinic | Monoclinic |
| Space group (no.) | P21 (4) | P21/n (14) |
| a/Å | 12.891(1) | 12.591(1) |
| b/Å | 7.611(1) | 7.029(1) |
| c/Å | 18.041(1) | 19.817(1) |
| α/° | 90.00 | 90.00 |
| β/° | 99.51(1) | 105.50(1) |
| γ/° | 90.00 | 90.00 |
| V/Å3 | 1745.7(3) | 1690.1(5) |
| Z | 4 | 4 |
| Dc/g cm−3 | 1.642 | 1.751 |
| F(000) | 880 | 912 |
| μ/mm−1 | 1.04 | 1.08 |
| Reflections collected | 4194 | 3527 |
| Unique reflections | 3395 | 3317 |
| Reflections with I > 2σ(I) | 1992 | 2696 |
| Parameters refined | 418 | 221 |
| GOFa on F2 | 1.04 | 1.25 |
| Rb [F2 > 2σ(F2)] | 0.074 | 0.078 |
| Rint | 0.072 | 0.041 |
| wR(F2)b | 0.168 | 0.204 |
| Δρmax, Δρmin/e Å−3 | 0.96, −1.02 | 1.62, −2.28 |
Synthesis of complexes
[(η6-p-cym)Ru(mecys)]Cl·0.3H2O (1). S-methyl-L-(−)-cysteine (54.1 mg, 0.40 mmol), [(η6-p-cym)RuCl2]2 (122.6 mg, 0.20 mmol), NaOMe (21.1 mg, 0.39 mmol). Yield: 66.0 mg, 40.2% (yellow solid). Found: C, 40.15; H, 5.27; N, 3.35; S, 7.85. Calc. for C14H22ClNO2RuS·0.3H2O (Mr 410.32): C, 40.98; H, 5.55; N, 3.41; S, 7.81. IR (KBr): νmax/cm−1 = 3433 br and 3087 s (NH); 1651 vs. (CO). δH (400 MHz, D2O, T = 25 °C, TSP): 1.27 (d, 3J = 7.5 Hz, 3H, cym-CH(CH3)2), 1.29 (d, 3J = 7.5 Hz, 3H, cym-CH(CH3)2), 2.22 (s, 3H, cym-CH3), 2.39 (s, 3H, S-CH3), 2.40 (d, 2J = 14.6 Hz, 1H, β-CH2), 2.79 (h, 1H, cym-CH(CH3)2), 2.80 (dd, 2J = 14.6 Hz, 3J = 4.4 Hz, 1H, β-CH2), 3.84 (d, 3J = 4.1 Hz, 1H, α-CH), 5.74 (d, 3J = 6.0 Hz, 1H, cym-H), 5.89 (d, 3J = 6.1 Hz, 1H, cym-H), 5.92 (d, 3J = 6.2 Hz, 1H, cym-H), 6.08 (d, 3J = 6.0 Hz, 1H, cym-H). Minor isomer: 1.32 (d, 3J = 6.8 Hz, 3H, cym-CH(CH3)2), 2.27 (s, 3H, cym-CH3); 2.85 (s, 3H, S-CH3); 3.03 (d, 2J = 13.3 Hz, 1H, β-CH2), 3.76 (d, 3J = 4.2 Hz, 1H, α-CH), 5.43 (d, 3J = 5.9 Hz, 1H, cym-H). δC (100 MHz, D2O, TSP): 20.3 (cym-CH3), 24.2 (cym-CH(CH3)2), 24.3 (cym-CH(CH3)2), 24.8 (S-CH3), 30.1 (β-CH2), 33.6 (cym-CH(CH3)2), 61.2 (α-CH), 84.9 (cym-CH), 85.2 (cym-CH), 86.0 (cym-CH), 86.5 (cym-CH), 102.3 (cym-C), 109.0 (cym-C), 182.9 (mecys-COO−). Minor isomer: 20.6 (cym-CH3), 29.1 (S-CH3), 37.7 (β-CH2), 59.7 (α-CH), 81.2 (cym-CH), 85.1 (cym-CH), 89.0 (cym-CH). MS (ESI-TOF): m/z (%) = 370.033 (100) [ML]+ (simulated: 370.041).
[(η6-p-cym)Ru(DL-met)]Cl·0.5H2O (2). DL-methionine (60.2 mg, 0.40 mmol), [(η6-p-cym)RuCl2]2 (123.1 mg, 0.20 mmol), NaOMe (21.3 mg, 0.39 mmol). Yield: 26.0 mg, 15.2% (yellow solid). Found: C, 41.75; H, 5.85; N, 3.34; S, 7.54. Calc. for C15H24ClNO2RuS·0.5H2O (Mr 427.95): C, 42.10; H, 5.89; N, 3.27; S, 7.49. IR (KBr): νmax/cm−1 = 3433 br and 3051 s (NH); 1644 vs. (CO). δH (360 MHz, D2O, T = 25 °C, TSP): 1.28 (d, 3J = 6.8 Hz, 6H, cym-CH(CH3)2), 1.80 (br, 1H, β-CH2), 2.16 (br, 3H, cym-CH3 + 1H, β-CH2 + 1H, γ-CH2), 2.72 (m, 3H, S-CH3 + 1H, cym-CH(CH3)2), 3.05 (m, 1H, γ-CH2), 3.61 (br, 1H, α-CH), 5.73–5.94 (m, 4H, cym-H). Minor isomer: 2.21 (s, 3H, S-CH3), 5.46 (br, 1H, cym-H). MS (ESI-TOF): m/z (%) = 384.052 (100) [ML]+ (simulated: 384.057).
[(η6-p-cym)Ru(L-met)]Cl·0.4H2O (3). L-(+)-methionine (60.0 mg, 0.40 mmol), [(η6-p-cym)RuCl2]2 (122.8 mg, 0.20 mmol), NaOMe (21.7 mg, 0.40 mmol). Yield: 87.7 mg, 51.4% (pale orange solid). Found: C, 42.31; H, 5.92; N, 3.31; S, 7.60. Calc. for C15H24ClNO2RuS·0.4H2O (Mr 426.15): C, 42.28; H, 5.87; N, 3.29; S, 7.52. IR (KBr) νmax/cm−1: 3465 br, 3114 s and 3031 s (NH); 1660 vs. (CO). δH (500 MHz, D2O, T = 25 °C, TSP): 1.28 (d, 3J = 6.8 Hz, 6H, cym-CH(CH3)2), 1.80 (br, 1H, β-CH2), 2.17 (br, 3H, cym-CH3 + 1H, β-CH2 + 1H, γ-CH2), 2.71 (m, 3H, S-CH3 + 1H, cym-CH(CH3)2), 3.05 (m, 1H, γ-CH2), 3.62 (br, 1H, α-CH), 5.72–5.93 (m, 4H, cym-H). Minor isomer: 2.21 (s, 3H, S-CH3), 5.46 (br, 1H, cym-H). δH (500 MHz, D2O, T = 0 °C, TSP): 1.28 (m, 6H, cym-CH(CH3)2), 1.79 (m, 1H, β-CH2), 2.16 (m, 3H, cym-CH3 + 1H, β-CH2 + 1H, γ-CH2), 2.72 (m, 3H, S-CH3 + 1H, cym-CH(CH3)2), 3.08 (m, 1H, γ-CH2), 3.60 (br, 1H, α-CH), 5.71 (d, 2J = 6.1 Hz, 1H, cym-H), 5.79 (d, 2J = 6.1 Hz, 1H, cym-H), 5.89 (d, 2J = 6.0 Hz, 1H, cym-H), 5.95 (d, 2J = 6.1 Hz, 1H, cym-H). Minor isomer: 2.19 (s, 3H, cym-CH3), 2.69 (s, 3H, S-CH3), 2.84 (m, 1H, cym-CH(CH3)2), 3.71 (br, 1H, α-CH), 5.44 (d, 2J = 5.8 Hz, 1H, cym-H), 5.77 (d, 2J = 5.7 Hz, 1H, cym-H). MS (ESI-TOF): m/z (%) = 384.053 (100) [ML]+ (simulated: 384.057).
[(η6-p-cym)Os(mecys)]Cl (4). S-methyl-L-(−)-cysteine (54.7 mg, 0.40 mmol), [(η6-p-cym)OsCl2]2 (159.7 mg, 0.20 mmol), NaOMe (21.9 mg, 0.41 mmol). Yield: 117.2 mg, 59.3% (pale yellow solid). Found: C, 33.64; H, 4.63; N, 2.85; S, 6.46. Calc. for C14H22ClNO2OsS (Mr 494.08): C, 34.03; H, 4.49; N, 2.83; S, 6.49. IR (KBr) νmax/cm−1: 3465 br and 3083 s (NH); 1659 vs. (CO). δH (360 MHz, D2O, T = 25 °C, TSP): 1.25 (d, 3J = 6.7 Hz, 3H, cym-CH(CH3)2), 1.27 (d, 3J = 6.7 Hz, 3H, cym-CH(CH3)2), 2.29 (s, 3H, cym-CH3), 2.39 (d, 2J = 14.9 Hz, 1H, β-CH2), 2.43 (s, 3H, S-CH3), 2.70 (h, 3J = 6.9 Hz, 1H, cym-CH(CH3)2) 2.87 (dd, 2J = 14.6 Hz, 3J = 4.2 Hz, 1H, β-CH2), 4.05 (d, 3J = 3.8 Hz, 1H, α-CH), 5.94 (d, 3J = 5.6 Hz, 1H, cym-H), 6.05 (d, 3J = 6.1 Hz, 1H, cym-H), 6.13 (d, 3J = 5.6 Hz, 1H, cym-H), 6.27 (d, 3J = 5.5 Hz, 1H, cym-H). Minor isomer: 2.35 (s, 3H, cym-CH3); 2.53 (dd, 2J = 13.1 Hz, 3J = 4.5 Hz, 1H, β-CH2), 2.93 (s, 3H, S-CH3); 2.97 (d, 2J = 13.4 Hz, 1H, β-CH2), 3.90 (d, 3J = 3.9 Hz, 1H, α-CH), 5.66 (d, 3J = 5.6 Hz, 1H, cym-H). δH (360 MHz, CD3OD, T = 25 °C, solvent residual): 1.30 (d, 3J = 6.7 Hz, 3H, cym-CH(CH3)2), 1.32 (d, 3J = 6.6 Hz, 3H, cym-CH(CH3)2), 2.31 (s, 3H, cym-CH3), 2.40 (d, 2J = 14.3 Hz, 1H, β-CH2), 2.48 (s, 3H, S-CH3), 2.71 (h, 3J = 6.9 Hz, 1H, cym-CH(CH3)2) 2.85 (dd, 2J = 14.4 Hz, 3J = 4.1 Hz, 1H, β-CH2), 3.92 (d, 3J = 3.5 Hz, 1H, α-CH), 5.93 (d, 3J = 5.5 Hz, 1H, cym-H), 6.02 (d, 3J = 6.1 Hz, 1H, cym-H), 6.13 (d, 3J = 5.5 Hz, 1H, cym-H), 6.25 (d, 3J = 5.6 Hz, 1H, cym-H). Minor isomer: 2.38 (s, 3H, cym-CH3), 2.56 (dd, 2J = 13.0 Hz, 3J = 4.3 Hz, 1H, β-CH2), 2.91 (d, 2J = 13.4 Hz, 1H, β-CH2), 2.98 (s, 3H, S-CH3), 3.78 (d, 3J = 3.8 Hz, 1H, α-CH), 5.62 (d, 3J = 5.5 Hz, 1H, cym-H), 6.29 (d, 3J = 5.4 Hz, 1H, cym-H). MS (ESI-TOF): m/z (%) = 460.095 (100) [ML]+ (simulated: 460.098).
[(η6-p-cym)Os(DL-met)]Cl·0.4H2O (5). DL-methionine (60.46 mg, 0.41 mmol), [(η6-p-cym)OsCl2]2 (159.1 mg, 0.20 mmol), NaOMe (21.6 mg, 0.40 mmol). Yield: 43.1 mg, 20.9% (yellow solid). Found: C, 34.51; H, 4.88; N, 2.71; S, 6.31. Calc. for C15H24ClNO2OsS·0.4H2O (Mr 515.31): C, 34.96; H, 4.85; N, 2.72; S, 6.22. IR (KBr) νmax/cm−1: 3434 br and 3053 s (NH); 1650 vs. (CO). δH (500 MHz, D2O, T = 25 °C, TSP): 1.27 (d, 3J = 6.9 Hz, 6H, cym-CH(CH3)2), 2.03 (br, 1H, β-CH2), 2.23 (s, 3H, cym-CH3), 2.45 (m, 1H, β-CH2 + 1H, γ-CH2), 2.67 (br, 1H, cym-CH(CH3)2), 2.81 (s, 1H, S-CH3), 3.15 (br, 1H, γ-CH2), 3.78 (br, 1H, α-CH), 5.91 (m, 2H, cym-H), 6.11 (m, 2H, cym-H). Minor isomer: 2.94 (br, 1H, γ-CH2), 3.90 (br, 1H, α-CH), 5.75 (br, 1H, cym-H). δH (500 MHz, D2O, T = 0 °C, TSP): 1.27 (m, 6H, cym-CH(CH3)2), 2.02 (m, 1H, β-CH2), 2.21 (s, 3H, cym-CH3) 2.43 (m, 1H, β-CH2 + 1H, γ-CH2), 2.65 (h, 2J = 6.7 Hz, 1H, cym-CH(CH3)2), 2.82 (s, 3H S-CH3) 3.18 (m, 1H, γ-CH2), 3.76 (br, 1H, α-CH), 5.89 (d, 3J = 6.1 Hz, 1H, cym-H), 5.90 (d, 3J = 5.9 Hz, 1H, cym-H), 6.11 (m, 2H, cym-H). Minor isomer: 2.24 (s, 3H, cym-CH3), 2.32 (m, 1H, β-CH2), 2.55 (m, 1H, β-CH2), 2.75 (h, 2J = 6.8 Hz, 1H, cym-CH(CH3)2), 3.91 (br, 1H, α-CH), 5.72 (d, 3J = 6.0 Hz, 1H, cym-H), 6.15 (d, 3J = 5.1 Hz, 1H, cym-H), 6.17 (d, 3J = 5.6 Hz, 1H, cym-H). MS (ESI-TOF): m/z (%) = 474.111 (100) [ML]+ (simulated: 474.114).
[(η5-Cp*)Rh(mecys)]Cl (6). S-methyl-L-(−)-cysteine (35.6 mg, 0.26 mmol), [(η5-Cp*)RhCl2]2 (80.0 mg, 0.13 mmol), NaOMe (14.1 mg, 0.26 mmol). Yield: 4.2 mg, 4.0% (hygroscopic orange solid). δH (400 MHz, D2O, T = 25 °C, TSP): 1.83 (s, 15H, Cp*-CH3), 2.38 (s, 3H, S-CH3), 2.64 (d, 2J = 14.7 Hz, 1H, β-CH2), 3.01 (dd, 2J = 14.6 Hz, 3J = 3.6 Hz, 1H, β-CH2), 3.96 (br, 1H, α-CH). Minor: 1.75 (s, 15H, Cp*-CH3). δH (400 MHz, D2O, T = 0 °C, TSP): 1.83 (s, 15H, Cp*-CH3), 2.38 (s, 3H, S-CH3), 2.65 (d, 2J = 14.8 Hz, 1H, β-CH2), 2.99 (dd, 2J = 14.7 Hz, 3J = 4.0 Hz, 1H, β-CH2), 3.96 (br, 1H, α-CH). Minor isomer: 1.76 (s, 15H, Cp*-CH3), 2.72 (s, 3H, S-CH3), 3.28 (d, 2J = 13.4 Hz, 1H, β-CH2), 3.86 (br, 1H, α-CH).
[(η5-Cp*)Rh(DL-met)]Cl·0.3H2O (7). DL-methionine (19.7 mg, 0.13 mmol), [(η5-Cp*)RhCl2]2 (40.2 mg, 0.065 mmol), NaOMe (14.3 mg, 0.26 mmol). Yield: 29.1 mg, 52.4% (orange solid). Found: C, 41.73; H, 6.08; N, 3.26; S, 7.53. Calc. for C15H25ClNO2RhS·0.3H2O (Mr 427.19): C, 42.17; H, 6.04; N, 3.28; S, 7.60. IR (KBr) νmax/cm−1: 3473 s, 3414 s, 3116 s, 3053 s (NH); 1645 vs. (CO). δH (400 MHz, D2O, T = 25 °C, TSP): 1.72 (s, 15H, Cp*-CH3), 2.11 (br, 1H, β-CH2), 2.27 (m, 1H, β-CH2), 2.40 (br, 1H, γ-CH2), 2.58 (s, 3H, S-CH3), 3.10 (br, 1H, γ-CH2), 3.86 (s, 1H, α-CH). δH (400 MHz, D2O, T = 0 °C, TSP): 1.72 (br, 15H, Cp*-CH3), 1.98–2.44 (m, 2H, β-CH2 + 1H, γ-CH2), 2.62 (s, 3H, S-CH3), 3.07 (m, 1H, γ-CH2), 3.88 (br, 1H, α-CH). Minor isomer: 2.52 (s, 3H, S-CH3), 3.20 (m, 1H, γ-CH2), 3.81 (br, 1H, α-CH). MS (ESI-TOF): m/z (%) = 380.060 (100) [ML]+ (simulated: 380.066).
[(η5-Cp*)Ir(mecys)]Cl·0.4CH3CN·0.5H2O (8). S-methyl-L-(−)-cysteine (27.1 mg, 0.20 mmol), [(η5-Cp*)IrCl2]2 (79.5 mg, 0.10 mmol), NaOMe (21.6 mg, 0.40 mmol). Yield: 28.9 mg, 27.7% (yellow solid). Found: C, 33.53; H, 4.91; N, 3.85; S, 6.06. Calc. for C14H23ClIrNO2S·0.4CH3CN·0.5H2O (Mr 522.50): C, 34.02; H, 4.86; N, 3.75; S, 6.14. IR (KBr) νmax/cm−1: 3424 br, 3221 br and 2984 s (NH); 1666 vs. (CO). δH (360 MHz, D2O, T = 25 °C, TSP): 1.85 (s, 15H, Cp*-CH3), 2.42 (s, 3H, S-CH3), 2.67 (d, 2J = 14.7 Hz, 1H, β-CH2), 3.03 (dd, 2J = 14.7 Hz, 3J = 4.2 Hz, 1H, β-CH2), 4.31 (d, 3J = 3.8 Hz, 1H, α-CH). Minor isomer: 1.83 (s, 15H, Cp*-CH3), 2.79 (s, 3H, S-CH3), 3.25 (d, 2J = 13.2 Hz, 1H, β-CH2), 4.11 (d, 3J = 3.8 Hz, 1H, α-CH). MS (ESI-TOF): m/z (%) = 426.104 (100) [ML]+ (simulated: 426.107).
[(η5-Cp*)Ir(DL-met)]Cl·0.5H2O (9). DL-methionine (30.1 mg, 0.20 mmol), [(η5-Cp*)IrCl2]2 (79.7 mg, 0.10 mmol), NaOMe (21.8 mg, 0.40 mmol). Yield: 52.7 mg, 50.7% (yellow solid). Found: C, 34.48; H, 5.09; N, 2.70; S, 6.15. Calc. for C15H25ClIrNO2S·0.5H2O (Mr 520.11): C, 34.64; H, 5.04; N, 2.69; S, 6.17. IR (KBr) νmax/cm−1: 3483 s, 3421 s, 3095 br and 3033 s (NH); 1666 vs. (CO). δH (400 MHz, D2O, T = 25 °C, TSP): 1.70 (s, 15H, Cp*-CH3), 2.22–2.61 (m, 1H, γ-CH2 + 2H, β-CH2), 2.74 (s, 3H, S-CH3), 3.27 (m, 1H, γ-CH2), 4.17 (m, 1H, α-CH). Minor isomer: 1.73 (s, 15H, Cp*-CH3), 2.53 (s, 3H, S-CH3), 4.10 (m, 1H, α-CH). δH (400 MHz, D2O, T = 0 °C, TSP): 1.71 (s, 15H, Cp*-CH3), 2.21–2.60 (m, 1H, γ-CH2 + 2H, β-CH2), 2.75 (s, 3H, S-CH3), 3.28 (m, 1H, γ-CH2), 4.16 (m, 1H, α-CH). Minor isomer: 1.73 (s, 15H, Cp*-CH3), 2.54 (s, 3H, S-CH3), 4.09 (m, 1H, α-CH). MS (ESI-TOF): m/z (%) = 476.116 (100) [ML]+ (simulated: 476.123).
[(η6-p-cym)Ru(mecys)]NO3 (10). [(η6-p-cym)RuCl2]2 (122.7 mg, 0.20 mmol), AgNO3 (135.0 mg, 0.79 mmol), (S-methyl-L-(−)-cysteine (53.6 mg, 0.40 mmol), NaOMe (21.4 mg, 0.40 mmol). Yield: 76.4 mg, 88.5% (orange solid). Found: C, 38.55; H, 5.23; N, 6.55; S, 7.58. Calc. for C14H22N2O5RuS (Mr 431.47): C, 38.97; H, 5.14; N, 6.49; S, 7.43. IR (KBr) νmax/cm−1: 3442 br and 3062 s (NH); 1656 vs. (CO); 1384 vs. and 1302 vs. (NO). δH (400 MHz, D2O, T = 25 °C, TSP): 1.26 (d, 3J = 7.6 Hz, 3H, cym-CH(CH3)2), 1.28 (d, 3J = 7.5 Hz, 3H, cym-CH(CH3)2), 2.21 (s, 3H, cym-CH3), 2.38 (s, 3H, S-CH3), 2.40 (d, 2J = 14.5 Hz, 1H, β-CH2), 2.78 (h, 1H, cym-CH(CH3)2), 2.78 (dd, 2J = 14.5 Hz, 3J = 2.9 Hz, 1H, β-CH2), 3.84 (d, 3J = 3.6 Hz, 1H, α-CH), 5.73 (d, 3J = 5.9 Hz, 1H, cym-H), 5.88 (d, 3J = 5.9 Hz, 1H, cym-H), 5.91 (d, 3J = 6.2 Hz, 1H, cym-H), 6.07 (d, 3J = 5.9 Hz, 1H, cym-H). Minor isomer: 1.32 (d, 3J = 6.5 Hz, 3H, cym-CH(CH3)2), 2.26 (s, 3H, cym-CH3); 2.83 (s, 3H, S-CH3); 3.04 (d, 2J = 12.6 Hz, 1H, β-CH2), 3.76 (d, 3J = 3.2 Hz, 1H, α-CH), 5.42 (d, 3J = 5.3 Hz, 1H, cym-H). MS (ESI-TOF): m/z (%) 370.033 (100) [ML]+ (simulated: 370.041).
[(η6-p-cym)Ru(DL-met)]NO3·0.1EtOH (11). [(η6-p-cym)RuCl2]2 (122.5 mg, 0.20 mmol), AgNO3 (135.3 mg, 0.80 mmol), DL-methionine (59.7 mg, 0.40 mmol), NaOMe (21.4 mg, 0.40 mmol). Yield: 146.8 mg, 81.5% (orange solid). Found: C, 40.75; H, 5.65; N, 6.14; S, 7.14. Calc. for C15H24N2O5RuS·0.1EtOH (Mr 450.10): C, 40.56; H, 5.51; N, 6.22; S, 7.12. IR (KBr) νmax/cm−1: 3435 br, 3151 br and 3080 br (NH); 1655 vs. (CO); 1383 vs., 1341 vs. and 1322 vs. (NO). δH (500 MHz, D2O, T = 25 °C, TSP): 1.28 (d, 3J = 6.8 Hz, 6H, cym-CH(CH3)2), 1.81 (br, 1H, β-CH2), 2.17 (br, 3H, cym-CH3 + 1H, β-CH2 + 1H, γ-CH2), 2.72 (m, 3H, S-CH3 + 1H, cym-CH(CH3)2), 3.05 (m, 1H, γ-CH2), 3.62 (br, 1H, α-CH), 5.72–5.93 (m, 4H, cym-H). Minor isomer: 2.21 (s, 3H, S-CH3), 5.46 (br, 1H, cym-H). δH (500 MHz, D2O, T = 0 °C, TSP): 1.28 (m, 6H, cym-CH(CH3)2), 1.79 (m, 1H, β-CH2), 2.17 (m, 3H, cym-CH3 + 1H, β-CH2 + 1H, γ-CH2), 2.72 (m, 3H, S-CH3 + 1H, cym-CH(CH3)2), 3.07 (m, 1H, γ-CH2), 3.59 (br, 1H, α-CH), 5.71 (d, 2J = 6.0 Hz, 1H, cym-H), 5.79 (d, 2J = 6.2 Hz, 1H, cym-H), 5.88 (d, 2J = 6.1 Hz, 1H, cym-H), 5.95 (d, 2J = 6.1 Hz, 1H, cym-H). Minor isomer: 2.19 (s, 3H, cym-CH3), 2.69 (s, 3H, S-CH3), 2.83 (m, 1H, cym-CH(CH3)2), 3.70 (br, 1H, α-CH), 5.44 (d, 2J = 5.9 Hz, 1H, cym-H), 5.77 (d, 2J = 5.6 Hz, 1H, cym-H). δC (100 MHz, D2O, T = 0 °C, TSP): 19.8 (cym-CH3), 24.0 (cym-CH(CH3)2), 24.3 (cym-CH(CH3)2), 29.0 (β-CH2), 29.4 (S-CH3), 32.9 (γ-CH2), 33.4 (cym-CH(CH3)2), 56.5 (α-CH), 85.7 (cym-CH), 87.6 (cym-CH), 88.1 (cym-CH), 102.6 (cym-C), 107.5 (cym-C), 186.3 (met-COO−). Minor isomer: 20.1 (cym-CH3), 24.4 (cym-CH(CH3)2), 24.5 (cym-CH(CH3)2), 25.0 (S-CH3), 26.8 (β-CH2), 30.2 (γ-CH2), 57.2 (α-CH), 82.1 (cym-CH), 87.0 (cym-CH), 89.5 (cym-CH), 104.0 (cym-C), 108.4 (cym-C), 185.6 (met-COO−). MS (ESI-TOF): m/z (%) 384.049 (100) [ML]+ (simulated: 384.057).
[(η6-p-cym)Ru(mecys)]CF3SO3 (12). [(η6-p-cym)RuCl2]2 (62.0 mg, 0.10 mmol), AgCF3SO3 (102.8 mg, 0.40 mmol), (S-methyl-L-(−)-cysteine (27.2 mg, 0.20 mmol), NaOMe (21.9 mg, 0.41 mmol). Yield: 39.4 mg, 38.0% (yellow solid). Found: C, 34.12; H, 4.20; N, 2.61; S, 12.41. Calc. for C15H22F3NO5RuS2 (Mr 518.53): C, 34.74; H, 4.28; N, 2.70; S, 12.37. IR (KBr) νmax/cm−1: 3462 br, 3246 s, 3207 s and 3088 s (NH); 1666 vs. (CO); 1256 s, 1226 s and 1165 s (CF); 1031 s and 638 s (SO). δH (360 MHz, D2O, T = 25 °C, TSP): 1.26 (d, 3J = 7.2 Hz, 3H, cym-CH(CH3)2), 1.28 (d, 3J = 7.2 Hz, 3H, cym-CH(CH3)2), 2.20 (s, 3H, cym-CH3), 2.38 (s, 3H, S-CH3), 2.40 (d, 2J = 14.5 Hz, 1H, β-CH2), 2.77 (h, 1H, cym-CH(CH3)2) 2.77 (dd, 2J = 14.4 Hz, 3J = 4.8 Hz, 1H, β-CH2), 3.83 (d, 3J = 3.8 Hz, 1H, α-CH), 5.73 (d, 3J = 6.0 Hz, 1H, cym-H), 5.88 (d, 3J = 6.1 Hz, 1H, cym-H), 5.91 (d, 3J = 6.5 Hz, 1H, cym-H), 6.07 (d, 3J = 5.9 Hz, 1H, cym-H). Minor isomer: 1.32 (d, 3J = 6.6 Hz, 3H, cym-CH(CH3)2), 2.26 (s, 3H, cym-CH3); 2.83 (s, 3H, S-CH3); 3.04 (d, 2J = 13.2 Hz, 1H, β-CH2), 3.75 (d, 3J = 3.6 Hz, 1H, α-CH), 5.41 (d, 3J = 6.1 Hz, 1H, cym-H). δH (400 MHz, CD3OD, solvent residual): 1.30 (d, 3J = 7.0 Hz, 3H, cym-CH(CH3)2), 1.33 (d, 3J = 7.0 Hz, 3H, cym-CH(CH3)2), 2.22 (s, 3H, cym-CH3), 2.40 (d, 2J = 14.4 Hz, 1H, β-CH2), 2.43 (s, 3H, S-CH3), 2.74 (d, 2J = 14.5 Hz, 3J = 4.0 Hz, 1H, β-CH2), 2.79 (h, 3J = 6.9 Hz, 1H, cym-CH(CH3)2), 3.72 (d, 3J = 3.7 Hz, 1H, α-CH), 5.74 (d, 3J = 6.0 Hz, 1H, cym-H), 5.86 (d, 3J = 6.0 Hz, 1H, cym-H), 5.91 (d, 3J = 5.8 Hz, 1H, cym-H), 6.07 (d, 3J = 6.0 Hz, 1H, cym-H). Minor isomer: 2.27 (s, 3H, cym-CH3); 2.86 (s, 3H, S-CH3); 2.99 (d, 2J = 13.1 Hz, 1H, β-CH2), 3.64 (d, 3J = 3.8 Hz, 1H, α-CH), 5.37 (d, 3J = 5.8 Hz, 1H, cym-H), 5.95 (d, 3J = 6.0 Hz, 1H, cym-H), 6.12 (d, 3J = 5.8 Hz, 1H, cym-H). MS (ESI-TOF): m/z (%) = 370.033 (100) [ML]+ (simulated: 370.041).
[(η6-p-cym)Ru(DL-met)]CF3SO3 (13). [(η6-p-cym)RuCl2]2 (61.2 mg, 0.10 mmol), AgCF3SO3 (101.5 mg, 0.39 mmol), DL-methionine (30.2 mg, 0.20 mmol), NaOMe (21.3 mg, 0.39 mmol). Yield: 66.38 mg, 62.3% (orange solid). Found: C, 35.94; H, 4.63; N, 2.68; S, 12.18. Calc. for C16H24F3NO5RuS2 (Mr 532.56): C, 36.08; H, 4.54; N, 2.63; S, 12.04. IR (KBr) νmax/cm−1: 3446 br, 3222 s and 3072 s (NH); 1647 vs. (CO); 1285 s, 1246 s and 1154 s (CF); 1028 s and 638 s (SO). δH (360 MHz, D2O, T = 25 °C, TSP): 1.28 (d, 3J = 6.8 Hz, 6H, cym-CH(CH3)2), 1.80 (br, 1H, γ-CH2), 2.16 (br, 3H, cym-CH3 + 1H, β-CH2 + 1H, γ-CH2), 2.71 (m, 3H, S-CH3 + 1H, cym-CH(CH3)2), 3.04 (br, 1H, γ-CH2), 3.61 (br, 1H, α-CH), 5.72–5.93 (m, 4H, cym-H). Minor isomer: 2.21 (s, 3H, S-CH3), 5.46 (br, 1H, cym-H). MS (ESI-TOF): m/z (%) 384.049 (100) [ML]+ (simulated: 384.057).
Solution studies
For solution studies doubly deionised and ultra-filtered water was obtained from a Milli-Q RG (Millipore) water purification system. pH-potentiometric measurements were carried out at an ionic strength of 0.20 M KNO3 and at 25.0 ± 0.1 °C. Carbonate-free KOH solutions of known concentrations (ca. 0.2 M) were used as titrant. HNO3 stock solutions were prepared from concentrated HNO3 and their concentrations were determined by potentiometric titrations using the Gran's method.31 A Mettler Toledo T50 titrator with a Metrohm double junction electrode (type 6.0255.100) was used for the pH-metric measurements. The electrode system was calibrated according to Irving et al.,32 the pH-metric readings could therefore be converted into hydrogen ion concentration. The water ionization constant, pKw, was 13.75 ± 0.01 under the conditions employed. Titrations were performed in the pH range 2.0–11.0. Initial volume of the samples was 15.00 mL. The metal ion concentrations were varied in the range 1.0–4.0 mM and 1:1, 1:2 and 2:1 metal to ligand ratios were titrated. The reproducibility of the equilibrium titration points included in the evaluation was within 0.005 pH unit. The samples were in all cases completely deoxygenated by bubbling purified argon for ca. 20 min prior the measurements.
1H NMR titrations were carried out on a Bruker Avance 360 or 400 NMR instrument at room temperature in D2O (99.8%) or in H2O (containing ∼15% D2O in a sealed capillary) at cRu = 10–20 mM to register the pH dependence of the chemical shifts belonging to the hexahapto bonded p-cym, the pentahapto Cp* or the thioether ligands. Water saturation was achieved by low power irradiation during the relaxation delay. Chemical shifts are reported in ppm (δH) from 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (TSP) as internal reference. pH was set up with NaOD or DNO3 in D2O and with KOH or HNO3 in H2O. Individual samples were equilibrated at least for two hours before measurements. pH* values (direct pH-meter readings in a D2O solution of a pH-meter calibrated in H2O according to Irving et al.)32 were converted to pH values measureable at an ionic strength of 0.20 M using the following equation: pH = 0.936pH* + 0.412.33
For the ESI-MS analysis of the solutions the measurements were performed in water at 1.0 mM [(η6-p-cym)Ru(H2O)3]2+ concentration at different pH values and at 1:1 metal ion to ligand ratio.
Calculation of the stability constants (βp,q,r = [RupAqHr]/[Ru]p[A]q[H]r; where “Ru” stands for [(η6-p-cym)Ru]2+ and “A” represents the mecys− and met− anions) of the complexes using the titration curves was attempted by the PSEQUAD or SUPERQUAD computer programs.34,35 During the calculations hydrolysis of [(η6-p-cym)Ru(H2O)3]2+ was taken into consideration and the following species was assumed: [{(η6-p-cym)Ru}2(μ2-OH)3] + (logβ2,0,−3 = −9.16).36 Ratios of the areas belonging to the corresponding NMR resonances of the free and complexed metal ion or ligand provided access to the stability constant of the [(η6-p-cym)Ru(met)]+ complex.
Cytotoxicity tests
Human ovarian carcinoma A2780 cells, kindly supplied by Professor B. Keppler, University of Vienna (Austria), were grown in RPMI 1640 medium (PAA; Pasching, Austria) supplemented with streptomycin (100 μg L−1), penicillin (100 U mL−1) (both Sigma) and 10% heat inactivated fetal bovine serum (PAA; Pasching, Austria). The cells were cultured in a humidified incubator at 37 °C in a 5% CO2 atmosphere and subcultured 2–3 times a week with an appropriate plating density.Cytotoxic effect of the compounds 3, 7, 9 and 11 were evaluated by using assays based on the tetrazolium compound MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] metabolization and neutral red (NR) uptake. The adherent cells A2780 were plated out 16 h prior to testing in 96-well tissue culture plates at a density of 104 cells per well in 100 μL of medium. The cells were treated for 72 h with the compounds at the final concentrations in the range of 0 to 50 μM in a final volume of 200 μL per well. Concentrations of the compounds in the medium during treatment were verified by flameless atomic absorption spectrometry. Thereafter, a viability of the cells was tested by both MTT and NR assays as described previously.37 Briefly, after the treatment period, 10 μL of a freshly diluted MTT solution (2.5 mg mL−1) was added to each well, and the plate was incubated at 37 °C in a humidified 5% CO2 atmosphere for 4 h. At the end of the incubation period, the medium was removed, and the formazan product was dissolved in 100 μL of dimethyl sulfoxide. Cell viability was evaluated by measurement of the absorbance at 570 nm, using an absorbance reader Synergy MX (Biotek, VT, USA); the reading values were converted to the percentage of the untreated control (percentage cell survival). Alternatively, after the treatment period, 20 μL of a 0.33% solution of neutral red in phosphate buffered saline (PBS) was added to each well with adherent cells and the plate was incubated at 37 °C in a humidified 5% CO2 atmosphere for 2 h. Afterwards, the dye containing medium was carefully removed and the cells were quickly rinsed with PBS. The incorporated dye was then solubilized in 200 μL of 1% acetic acid in 50% ethanol, allowed to stand for 10 min at room temperature and the absorbance was measured at λ = 540 nm with absorbance reader Synergy MX (Biotek, VT, USA). The background absorbance of the plates at 690 nm was also measured and subtracted from 540 nm measurement. The reading values were converted to the percentage of the control (percentage cell survival). All experiments were repeated at least three times, each repetition made in triplicate.
Results and discussion
To explore the solution behaviour of the half-sandwich type platinum metal ions with the title ligands, as a model, [(η6-p-cym)Ru(H2O)3]2+ was chosen and the interaction with DL-methionine (met) and S-methyl-L-cysteine (mecys) was studied in aqueous solution by the combined use of potentiometry, NMR and MS.pH-potentiometric results
pH-potentiometric titrations assisted in checking the purity and exact concentration of the ligands and in redetermining their protonation constants. The estimated values appear in Table 2 and are in good agreement with those previously obtained under our experimental conditions.38K) of the ligands and stability constants (logβ) of the [(η6-p-cym)Ru]2+ complexes at 25.0 °C and I = 0.20 M (KNO3)a
Representative titration curves obtained for the [(η6-p-cym)Ru(H2O)3]2+-containing samples with met (Fig. 1) indicate that the complex formation is already intensive at pH = 2.1. At 1:1 ratio the one equivalent of extra base consumption by pH = 6.0 clearly supports the exclusive formation of a 1:1 complex. Lack of any hydrolysis above pH = 9.0 is consistent with the tridentate coordination of the ligand. The exactly half equivalent of extra base consumption at 1:2 ratio indicates that here a 1:1 species is present too. From the 2:1 sample it is clear that the ligand is not capable of binding a second metal ion as the 1.5 equivalent of base consumption starting at pH ∼3.5 is consistent with the hydrolysis of this excess of metal ion resulting in the formation of [{(η6-p-cym)Ru}2(μ2-OH)3]+.36 Very similar titration curves and trends were obtained for the mecys system too (see Fig. S1†). Due to the high stability of the 1:1 complexes pH-potentiometry turned to be not suitable for the estimation of their stability constants.
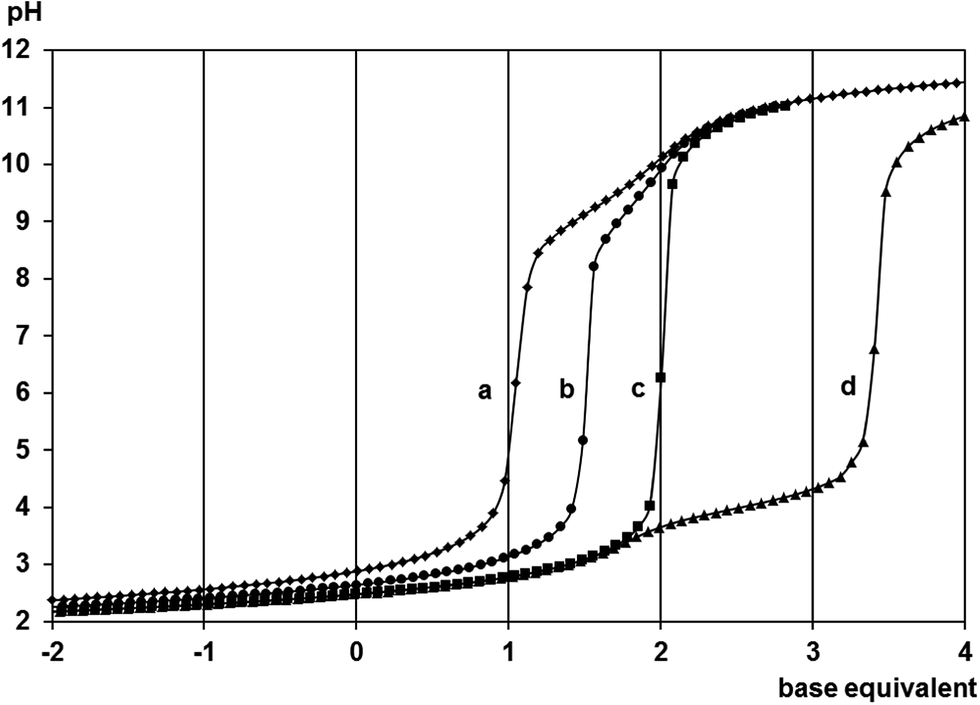 | ||
| Fig. 1 Titration curves with DL-methionine for the H+–ligand system (a) and the [(η6-p-cym)Ru(H2O)3]2+–ligand systems at 1:2 (b), 1:1 (c) and 2:1 (d) ratios. Negative base equivalent refers to an excess of acid in the sample. | ||
NMR results
To identify the resonances in the NMR spectra of the [(η6-p-cym)Ru(H2O)3]2+-ligand samples, spectra of the free ligands in the function of the pH were also registered. The assigned δ values together with the J values appear in Table 3. For both ligands the β-CH2 hydrogens are magnetically non-equivalent as α-CH is a stereogenic centre. For mecys the multiplicity of these resonances is a distorted doublet of doublets (roof effect) as a consequence of geminal and vicinal spin–spin couplings while in the case of met a multiplet can be observed for β-CH2 due to another vicinal couplings. For both ligands the multiplicity of the resonances of the hydrogens on α-C is also a doublet of doublets. On increasing the pH the δ values for the α-C hydrogens change the most while the methyl hydrogens being furthest from the deprotonation sites exhibit the smallest change.| Ligand | pH | Species | δ(S-CH3) D | δ(β-CH2) B | δ(β-CH2) B′ | δ(γ-CH2) C | δ(α-CH) A | 2JBB′ | 3JAB | 3JAB′ | 3JBC = 3JB′C |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Labelling of the hydrogens is shown in Scheme 1.b Cannot be determined due to the multiplicity of these resonances. | |||||||||||
| Mecys | 0.83 | [H2A]+ | 2.17 (s) | 3.09 (dd) | 3.20 (dd) | — | 4.34 (dd) | 15.0 | 7.6 | 4.5 | — |
| 11.96 | [A]− | 2.11 (s) | 2.74 (dd) | 2.83 (dd) | — | 3.42 (dd) | 13.6 | 6.9 | 5.2 | — | |
| Met | 1.02 | [H2A]+ | 2.12 (s) | 2.20 (m) | 2.31 (m) | 2.70 (t) | 4.25 (dd) | —b | 6.6 | 6.2 | 7.3 |
| 12.21 | [A]− | 2.11 (s) | 1.80 (m) | 1.91 (m) | 2.55 (t) | 3.32 (dd) | —b | 7.2 | 5.6 | 7.8 | |
NMR spectra registered in the range 0.99 < pH < 10.12 for the [(η6-p-cym)Ru(H2O)3]2+−mecys system at 1:1 ratio (Fig. 2) indicate that the resonances of the p-cymene unit of the metal ion belong to a single major species. Neither signals of the uncomplexed [(η6-p-cym)Ru(H2O)3]2+ (2.24 ppm, cym-CH3) nor that of [{(η6-p-cym)Ru}2(μ2-OH)3]+ (2.08 ppm, cym-CH3)36 can be detected in the range 1 < pH < 10 in accordance with the potentiometric results. Downfield shift of the α-CH (3.84 ppm) and S-CH3 (2.39 ppm) protons of the ligand in the metal ion containing samples compared to those of the free deprotonated ligand (Table 3), furthermore, lack of pH-dependent shift of the resonances clearly supports the formation of [RuA]+ over a wide pH-range with tridentate coordination of mecys. Very similar trends were found with met too (see Fig. S2†) with the only exception that the signals exhibit broadening. This is due to the frequency of some interconversion reactions in the complex (vide infra) being comparable to the frequency difference between the resonances. Identical stability trends and NMR behaviour were found for the [(η5-Cp*)Ir(H2O)3]2+ containing systems too (not shown).
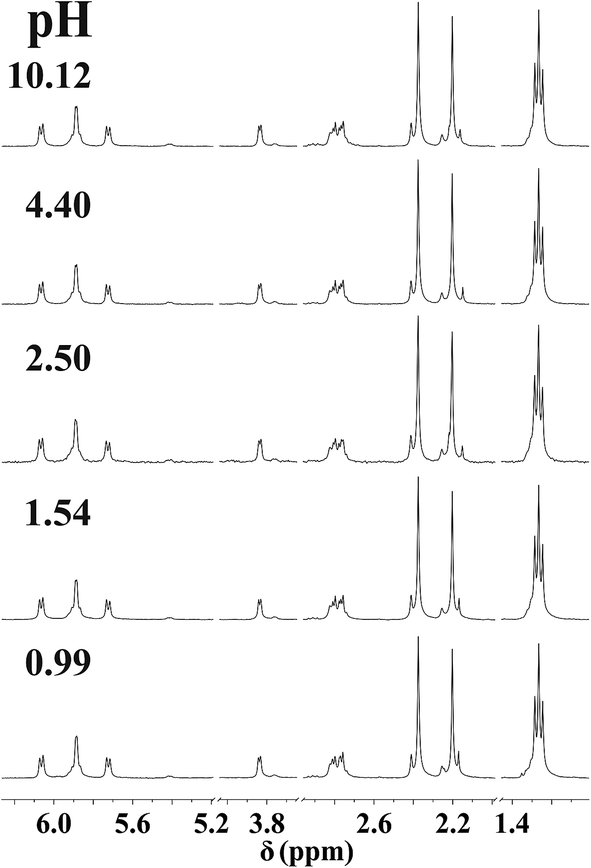 | ||
| Fig. 2 Dependence on pH of the 1H NMR spectra of the [(η6-p-cym)Ru(H2O)3]2+–mecys system at 1:1 ratio and at 298 K in D2O (I = 0.20 M KNO3, cRu = 10.0 mM). “Ru” stands for the [(η6-p-cym)Ru]2+ entity. | ||
NMR spectra registered in H2O (see Experimental) for the [(η6-p-cym)Ru(H2O)3]2+–met system in the range 12.4 < pH < 12.9 allowed the estimation of the stability constant of [(η6-p-cym)Ru(met)]+ by considering the ratio of the areas of the appropriate signals belonging to the complex and to the free ligand or [{(η6-p-cym)Ru}2(μ2-OH)3]+. As it is shown in Fig. 3 there is no indication for the formation of mixed hydroxido complexes as no new signals appear in the spectra in this pH-range beside those belonging to the uncomplexed ligand or [{(η6-p-cym)Ru}2(μ2-OH)3]+. The obtained stability constant appears in Table 2 while the calculated concentration distribution curves are shown in Fig. 4. As Fig. 4 reveals the concentrations of the [(η6-p-cym)Ru(met)]+ (■) and [{(η6-p-cym)Ru}2(μ2-OH)3]+ (○) obtained from the individual NMR experiments at various pH values are in agreement with the calculated concentrations.
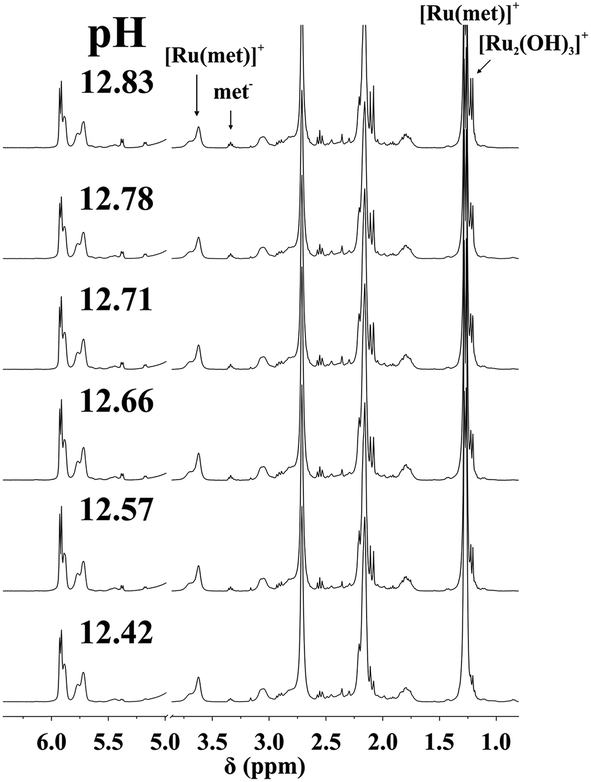 | ||
| Fig. 3 Dependence on pH of the 1H NMR spectra of [(η6-p-cym)Ru(H2O)3]2+–met system at 1:1 ratio and at 298 K in H2O (I = 0.20 M KNO3, cRu = 13.0 mM). “Ru” stands for the [(η6-p-cym)Ru]2+ entity. | ||
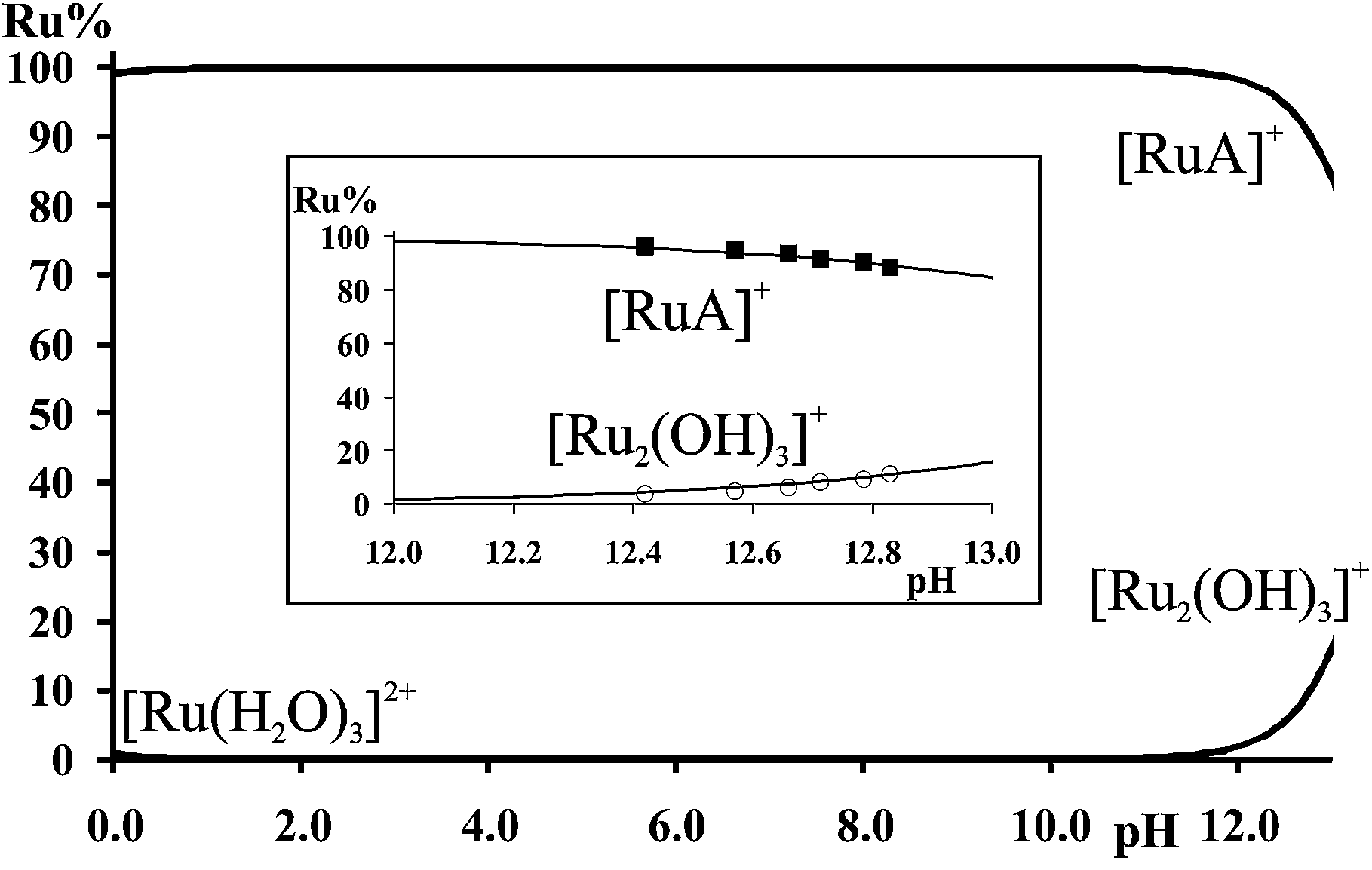 | ||
| Fig. 4 Calculated concentration distribution curves for the [(η6-p-cym)Ru(H2O)3]2+–met system at 1:1 ratio. cRu = 13 mM. Inlet shows the calculated curves together with the percentage of the species estimated by the ratio of the area of the NMR signals belonging to [RuA]+ (■) and [(η6-p-cym)Ru(μ2-OH)3]+ (○). “Ru” refers to [(η6-p-cym)Ru]2+. | ||
Regarding the mecys system, slow colour change, decrease and disappearance of the signals of the analogous [RuA]+ complex together with the appearance of new resonances in very basic solutions suggest irreversible processes making not possible the direct estimation of the stability constant of the [(η6-p-cym)Ru(mecys)]+ complex. Nevertheless, as Fig. 5 indicates, in a [(η6-p-cym)Ru(H2O)3]2+–met–mecys 1:1:1 system the ratio of the NMR resonances of the corresponding met and mecys complexes supports the slightly higher stability of [(η6-p-cym)Ru(met)]+ over the mecys complex and allows an estimation of logβ = 16.4(4) for [(η6-p-cym)Ru(mecys)]+ using the logβ = 17.2(2) of [(η6-p-cym)Ru(met)]+.
 | ||
| Fig. 5 Dependence on pH of the 1H NMR spectra of the [(η6-p-cym)Ru(H2O)3]2+–mecys–met system at 1:1:1 ratio, at 298 K in D2O, with any form of the uncomplexed mecys (a) or met (b). (I = 0.20 M KNO3, cRu = 20.0 mM). “Ru” refers to [(η6-p-cym)Ru]2+. | ||
To compare the [(η6-p-cym)Ru(H2O)3]2+ binding capability of small tridentate bioligands with various [O, O, O] (citrate), [O, N, O] (isoserine) and [S, N, O] (methionine) donor atom sets, the distribution of the metal ion in a hypothetical [(η6-p-cym)Ru(H2O)3]2+–citr–ise–met 1:1:1:1 system, as a function of pH, using the previously determined logβ values7,9 and the data in Table 2, is calculated and shown in Fig. S3.† The speciation indicates that both in the micromolar (Fig. S3a†) and millimolar (Fig. S3b†) concentration range of Ru(II) in the acidic and neutral pH range [(η6-p-cym)Ru(met)]+ is the single species present and the competition of isoserinate becomes significant only in the very basic pH range. Citrate with the alcoholate and carboxylate O donors is found to be the least effective binder among these small chelators, while met, even with the low basicity sulfur, has the highest affinity to the half-sandwich type metal ion.
ESI MS results
ESI-MS provided further information on the composition of the species present in aqueous solution. Spectra, registered in the range 1.1 < pH < 10.5, revealed the exclusive formation of [RuA]+ in both systems with m/z values of 370.031 (mecys) and 384.052 (met). Neither any form of the uncomplexed metal ion under acidic conditions nor [{(η6-p-cym)Ru}2(μ2-OH)3]+ (523.037)36 was detectable in the samples supporting thus the potentiometric and NMR results. As a representative example, MS spectrum of a [(η6-p-cym)Ru]2+–met sample (pH = 10.8) together with the observed and expected spectra for [(η6-p-cym)Ru(met)]+ in the inlet are shown in Fig. 6.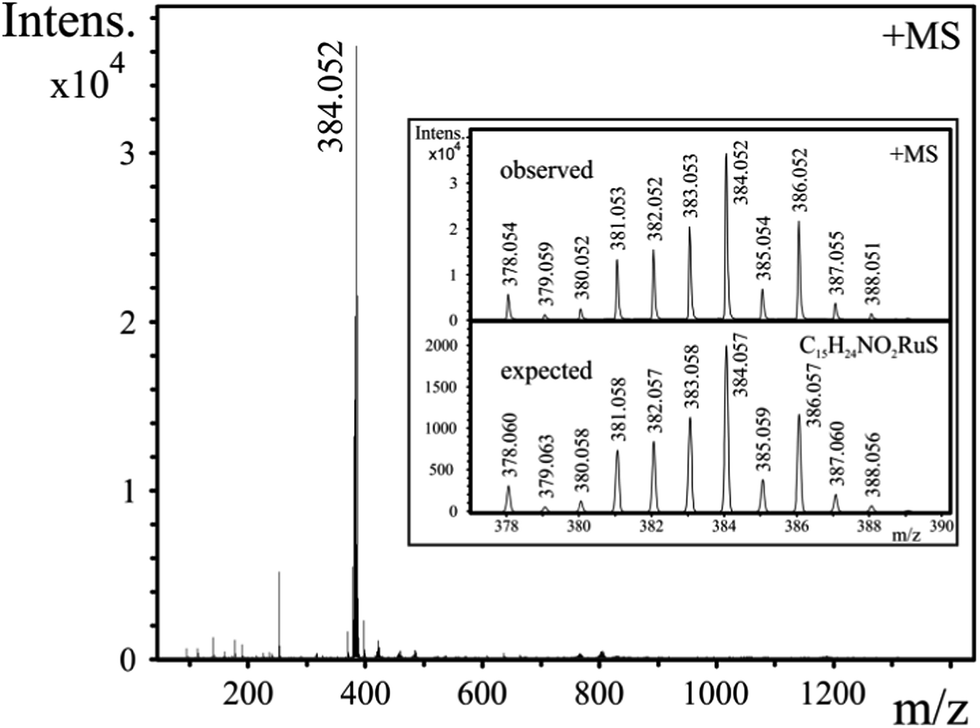 | ||
| Fig. 6 ESI-MS spectrum of an aqueous 1:1 [(η6-p-cym)Ru]2+–met sample at pH = 10.8, cRu = 0.3 mM. Inlet shows representative observed and expected ESI-MS spectra of [(η6-p-cym)Ru(met)]+. “Ru” refers to [(η6-p-cym)Ru]2+. | ||
Solid state studies
Reaction of [(η6-p-cym)MCl2]2 (M = Ru, Os) or [(η5-Cp*)M′Cl2]2 (M′ = Rh, Ir) with DL-met or L-mecys in MeOH afforded pale yellow to orange coloured solids in modest yield via the slow crystallization of the crude complexes from dichloromethane/isopropylether, acetonitrile/isopropylether or ethanol/isopropylether mixtures. Both elemental analysis and IR support the expected [(η6-p-cym)Ru(met/mecys)]X (X = Cl, NO3, CF3SO3), [(η6-p-cym)Os(met/mecys)]Cl or [(η5-Cp*)M′(met/mecys)]Cl general composition of the solids and the tridentate coordination of the ligands. In particular, νCO values being in the range 1644–1666 cm−1 (see Experimental) are consistent with the coordinating carboxylate.15,16,21Single crystals of 10 and 11 were grown by slow diffusion of isopropylether into an ethanolic solution of the complexes at 253 K. ORTEP views of [(η6-p-cym)Ru(mecys)]NO3 (10) and [(η6-p-cym)Ru(DL-met)]NO3 (11) are shown in Fig. 7 and 8 while key bond lengths and angles are summarized in Table 4. Both molecular structures contain tridentate ligands with [S, NH2, COO−] coordination. In 10, the configuration of the metal centre is also determined by the R absolute configuration of S-methyl-L-cysteine, therefore only the RRuRαC stereoisomer is present. Upon coordination, the thioether-S also becomes a stereogenic centre in the molecule; in the lattice of 10 the stereoisomer with the RRuRαCSS absolute configuration can only be detected. Identical behaviour was found previously for [(η5-Cp*)Ir(mecys)]CF3SO3·0.5H2O too.17 Interestingly, the asymmetric unit of 10 contains two slightly different conformational isomers (Fig. S4†).
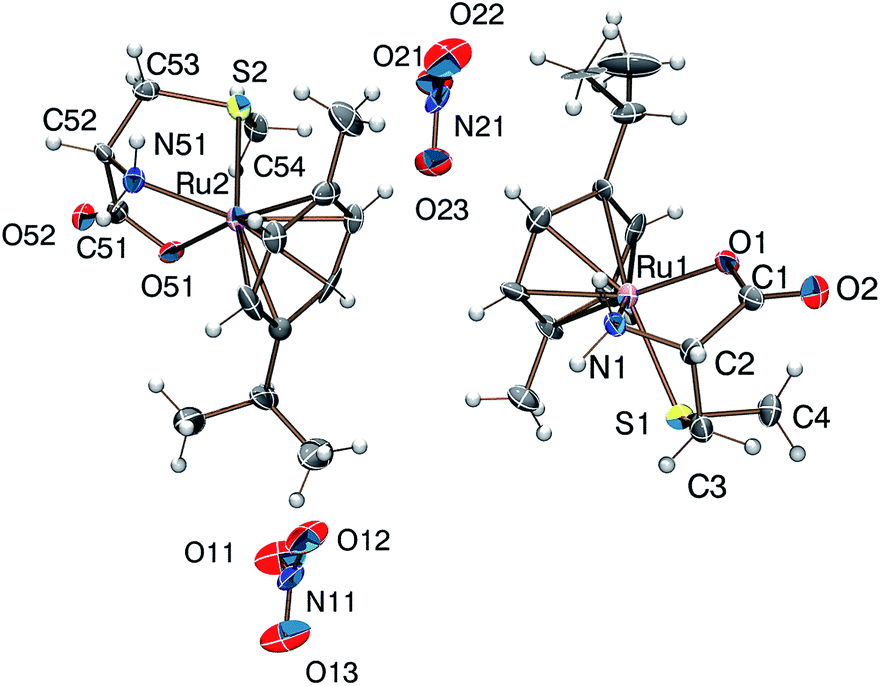 | ||
| Fig. 7 Molecular structure of the RRuRαCSS diastereomer of the [(η6-p-cym)Ru(mecys)]NO3 (10) complex exhibiting two slightly different conformers. | ||
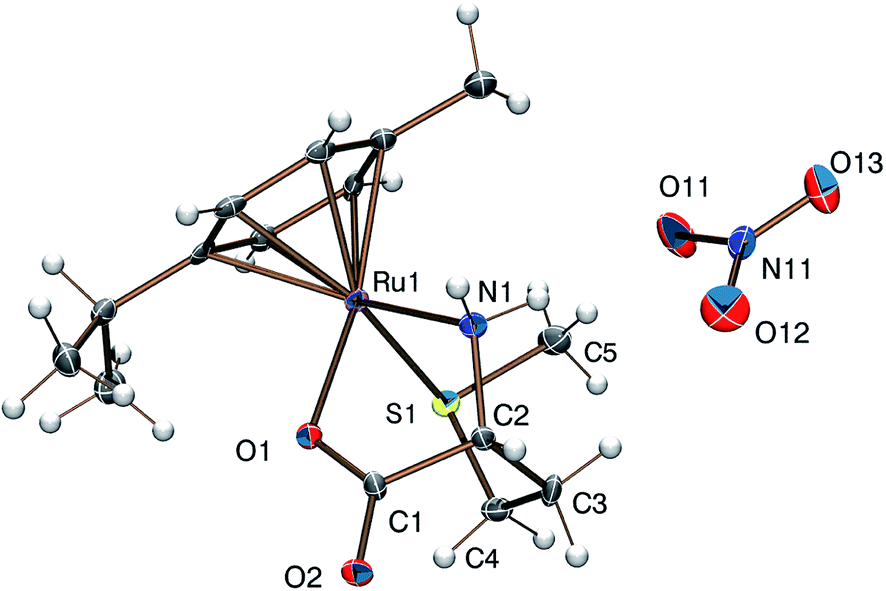 | ||
| Fig. 8 Molecular structure of the SRuRαCSS diastereomer of the [(η6-p-cym)Ru(DL-met)]NO3 (11) complex (the RRuSαCRS enantiomer is not shown). | ||
| 10 | 11 | |
|---|---|---|
| S(1)–Ru(1) | 2.407(6) | 2.390(2) |
| N(1)–Ru(1) | 2.140(19) | 2.113(7) |
| O(1)–Ru(1) | 2.079(12) | 2.095(6) |
| Ru(1)–Ccymene center | 1.667 | 1.660 |
| C(1)–O(1) | 1.21(2) | 1.233(10) |
| C(1)–O(2) | 1.27(2) | 1.280(10) |
| S(2)–Ru(2) | 2.396(6) | — |
| N(51)–Ru(2) | 2.100(18) | — |
| O(51)–Ru(2) | 2.112(13) | — |
| Ru(2)–Ccymene center | 1.685 | — |
| C(51)–O(51) | 1.31(3) | — |
| C(51)–O(52) | 1.27(2) | — |
| O(1)–Ru(1)–S(1) | 85.5(4) | 86.38(18) |
| N(1)–Ru(1)–S(1) | 79.5(5) | 88.7(2) |
| O(1)–Ru(1)–N(1) | 76.4(6) | 76.7(2) |
| C(4)–S(1)–Ru(1) | 108.3(9) | — |
| C(5)–S(1)–Ru(1) | — | 107.6(4) |
| O(51)–Ru(2)–S(2) | 86.2(4) | — |
| N(51)–Ru(2)–S(2) | 80.2(5) | — |
| N(51)–Ru(2)–O(51) | 76.2(6) | — |
| C(54)–S(2)–Ru(2) | 106.3(9) | — |
For 11 with the racemic DL-methionine, two enantiomers, RRuSαCRS and SRuRαCSS in 1:1 ratio in the lattice, are detected (Fig. 8). Identical RMSαCRS configuration was identified previously with L-met in [(η5-Cp*)Co(L-met)]FeCl4.39 Comparison of the structures 10 and 11 reveals that in both complexes the five-membered chelates have an envelope conformation with the N atom in the flap position. For 11 the six-membered chelate shows a distorted chair conformation with axial position of the thioether S donors. In both complexes the S-methyl groups are parallel with the arene ring being closer to the NH2 group in 11 and to the COO− in 10. The appropriate bond length and angle values are comparable with each other (Table 4) while the significantly larger N(1)–Ru(1)–S(1) angle in 11 can be rationalized by the one unit larger size of the (S, N) chelate. The solid state structures are stabilized by strong hydrogen bond networks with the participation of N–H protons and carboxylate or nitrate oxygen atoms as acceptors as well as by weak C–H⋯O hydrogen bonds.
Regardless of the counter ion, dissolving the solid complexes in various solvents practically identical NMR spectra could be obtained and these very much resembled to those of the corresponding metal ion–ligand systems previously studied in aqueous solution at different pH values. This, on the one hand, indicates that the tridentate coordination detected in the solid state is retained and, on the other hand, provides further support for the binding mode in aqueous solution, suggested previously. As representative examples, NMR spectra of [(η6-p-cym)Ru(mecys)]NO3 (10) in D2O and [(η6-p-cym)Ru(mecys)]CF3SO3 (12) in CD3OD, respectively, as well as those of [(η6-p-cym)Os(mecys)]Cl (4) in D2O and in CD3OD are shown in Fig. 9. Furthermore, COSY spectra of [(η6-p-cym)Ru(mecys)]Cl (1) in D2O, [(η6-p-cym)Ru(mecys)]CF3SO3 (12) in CD3OD (Fig. S5†) and HSQC spectrum of [(η6-p-cym)Ru(mecys)]Cl (1) in D2O (Fig. S6†) are also presented. In all cases (Fig. 9) the very similar doubling of the resonances of the ring hydrogens (5.7–6.3 ppm) and those of the iPr-methyl protons (1.20–1.35 ppm) of the p-cymene ligand supports the loss of symmetry of the arene because the metal ion also becomes a chirality centre upon coordination of the mecys ligand. COSY (Fig. S5b†) and HSQC (Fig. S6†) also indicate that the methylene hydrogens of the ligand exhibit rather different δ values (2.80 (dd) and 2.40 (d) ppm) due to their non-equivalent environment in the complex. Regarding the ring protons of p-cymene, for example in Fig. 9b, the signals at 5.86 and 5.91 ppm belong to a strongly coupled AB spin system while those at 5.74 and 6.07 ppm to a weakly coupled MX system.
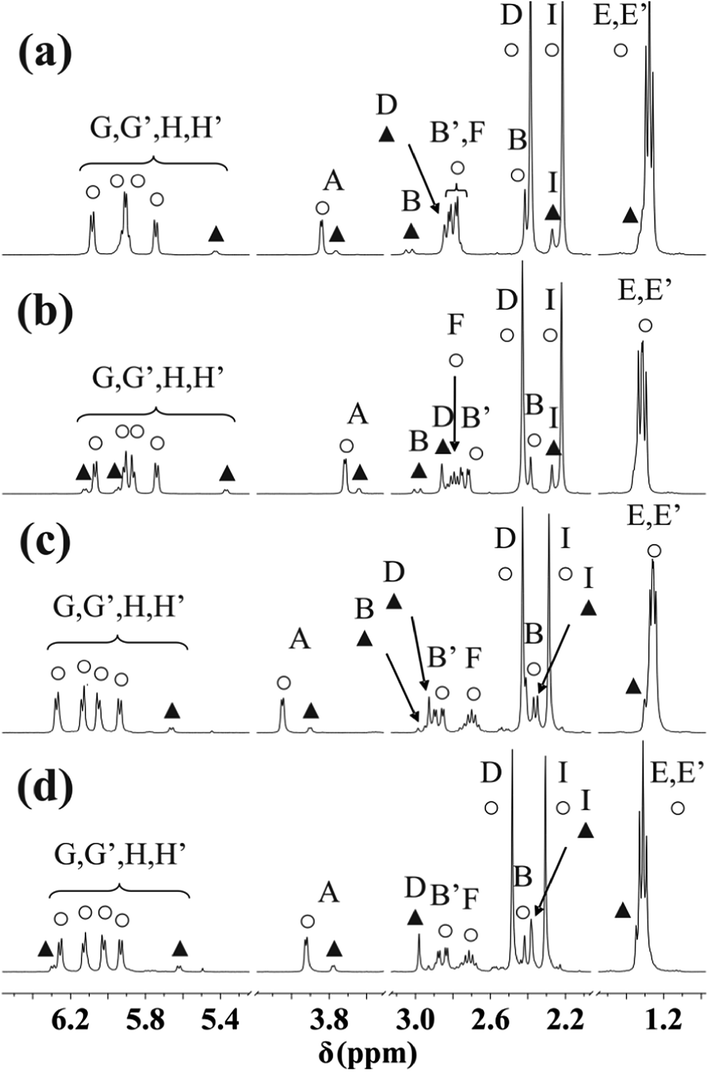 | ||
| Fig. 9 NMR spectra of [(η6-p-cym)Ru(mecys)]NO3 (10) in D2O (a), [(η6-p-cym)Ru(mecys)]CF3SO3 (12) in CD3OD (b), [(η6-p-cym)Os(mecys)]Cl (4) in D2O (c) and in CD3OD (d) together with the notation of the hydrogens according to Scheme 1. Resonances of the major diastereomer are marked with ○, while those of the minor one with ▲. | ||
Careful analysis of the NMR spectra (Fig. 9, S5 and S6†) also reveals that beside the major complex, resonances of a minor species are also present. COSY spectrum (Fig. S5†) indicates that there is no cross-coupling between the signals of the major and minor species, furthermore, analytical information also supports the presence of complexes with identical (i.e. 1:1 metal ion to ligand) stoichiometry only.
With the interaction of L-mecys and [(η6-p-cym)Ru(H2O)3]2+ a “chiral-at-metal” complex is formed and owing to the tridentate coordination of the chiral ligand, the configuration of the metal ion is determined resulting in the exclusive formation of the RRuRαC stereoisomer. When coordinating, the thioether-S also becomes a stereogenic centre in the complex. Unlike the metal ion and αC, the thioether-S has labile configuration in the complexes24 which can rationalize the NMR results. It is very likely that the major resonances in the spectra belong to the thermodynamically preferred RRuRαCSS diastereomer (also supported by the X-ray structure) while the minor ones to RRuRαCRS, formed after dissolution of the solid. With this assumption all the resonances in the spectra (Fig. 9, S5 and S6†) can be assigned (Table 5). As Fig. S5 and S6† reveal, the largest chemical shift differences for the S-methyl (D) and β-CH2 (B) protons as well as carbon atoms can be observed in accordance with the structural differences of the two epimers (Fig. S7†). For all the [(η6-p-cym)Ru(mecys)]X (X = Cl, NO3, CF3SO3), [(η6-p-cym)Os(mecys)]Cl or [(η5-Cp*)M′(mecys)]Cl complexes the calculated amount of the minor diastereomer (%) together with the diastereomeric excess (de %) are summarized in Table 6. As it is seen in Table 6 the solvent has slightly larger influence on the ratio than the counter ion. Comparing the metals, for the Os complexes the epimerization is the most pronounced. For the Rh complex broadening of the signals was observed at ambient temperature, however, for the cooled samples the minor isomer could also be detected (see Fig. S8†). The ratios of the diastereomers (Table 6) did not change either in a longer period of time (days) or by increasing the temperature of the samples; in the latter case faster exchange processes were only observed.
| Complex/solvent | (1) | (12) | (4) | (6)b | (8) | ||
|---|---|---|---|---|---|---|---|
| D2O | CD3OD | D2O | CD3OD | D2O | D2O | ||
| a Notation of the A–J protons can be seen in Scheme 1.b Determined at 0 °C. | |||||||
| D (s) | Major | 2.39 | 2.43 | 2.43 | 2.48 | 2.38 | 2.42 |
| Minor | 2.85 | 2.86 | 2.93 | 2.98 | 2.72 | 2.79 | |
| B (d) | Major | 2.40 | 2.40 | 2.39 | 2.40 | 2.65 | 2.67 |
| Minor | 3.03 | 2.99 | 2.97 | 2.91 | 3.28 | 3.25 | |
| B' (dd) | Major | 2.80 | 2.74 | 2.87 | 2.85 | 2.99 | 3.03 |
| Minor | Overlap | Overlap | 2.53 | 2.56 | Overlap | Overlap | |
| A (d) | Major | 3.84 | 3.72 | 4.05 | 3.92 | 3.96 | 4.31 |
| Minor | 3.76 | 3.64 | 3.90 | 3.78 | 3.86 | 4.11 | |
| E/E′ (d) | Major | 1.27 | 1.30 | 1.25 | 1.30 | — | — |
| 1.29 | 1.33 | 1.27 | 1.32 | — | — | ||
| Minor | 1.32 | 1.36 | 1.29 | 1.33 | — | — | |
| I (s) | Major | 2.22 | 2.22 | 2.29 | 2.31 | — | — |
| Minor | 2.27 | 2.27 | 2.35 | 2.38 | — | — | |
| F (h) | Major | 2.79 | 2.79 | 2.70 | 2.71 | — | — |
| Minor | Overlap | Overlap | Overlap | Overlap | — | — | |
| G/G′/H/H′ (d) | Major | 5.74 | 5.74 | 5.94 | 5.93 | — | — |
| 5.89 | 5.86 | 6.05 | 6.02 | — | — | ||
| 5.92 | 5.91 | 6.13 | 6.13 | — | — | ||
| 6.08 | 6.07 | 6.27 | 6.25 | — | — | ||
| Minor | 5.43 | 5.37 | 5.66 | 5.62 | — | — | |
| Overlap | 5.95 | Overlap | 6.29 | — | — | ||
| Overlap | 6.12 | Overlap | Overlap | — | — | ||
| Overlap | Overlap | Overlap | Overlap | — | — | ||
| J (s) | Major | — | — | — | — | 1.83 | 1.85 |
| Minor | — | — | — | — | 1.76 | 1.83 | |
| Complex | Minor isomer% | de % |
|---|---|---|
| a In CD3OD.b At 0 °C. | ||
| [(η6-p-cym)Ru(mecys)]Cl (1) | 12.7 | 74.6 |
| [(η6-p-cym)Ru(mecys)]NO3 (10) | 10.9 | 78.2 |
| [(η6-p-cym)Ru(mecys)]CF3SO3 (12) | 11.5 | 77.0 |
| 14.3a | 71.4 | |
| [(η6-p-cym)Os(mecys)]Cl (4) | 13.7 | 72.6 |
| 15.9a | 68.2 | |
| [(η5-Cp*)Rh(mecys)]Cl (6) | 4.6b | 90.8 |
| [(η5-Cp*)Ir(mecys)]Cl (8) | 5.4 | 89.2 |
| [(η6-p-cym)Ru(DL-met)]NO3 (11) | 25.6 | 48.8 |
| 25.1b | 49.8 | |
| [(η6-p-cym)Os(DL-met)]Cl (5) | 26.0b | 48.0 |
| [(η5-Cp*)Rh(DL-met)]Cl (7) | 32.0b | 36.0 |
| [(η5-Cp*)Ir(DL-met)]Cl (9) | 41.0 | 18.1 |
| 38.1b | 23.8 | |
For the DL-met containing complexes broadening of the signals in the 1H NMR and COSY spectra (Fig. 10a and S9a†) at room temperature was detected. This is because of the frequency difference between the resonances is comparable to the frequency of the interconversion of the epimers. To support this, as it is seen in Fig. 10, S9 and S10,† on cooling the samples the resonances become sharper owing to slower processes. Analysis of the spectra (see Fig. 10c as a representative example) reveals identical situation for the met containing complexes as it was found for the mecys compounds previously: beside a major species a minor one is also present in solution in every system. Again, for both complexes tridentate binding mode of the ligand with the difference in the configuration of the coordinating thioether S can be assumed. Interestingly, for the met complex, due to the chirality of the sulfur atom, the γ-C protons also become magnetically non-equivalent. The chemical shift difference is larger for these protons than that of the β-C protons (Fig. S10†) supporting the epimerization of sulfur atoms as it was detected in the mecys system. Significant broadening at room temperature in the case of the met complexes over the mecys ones can be rationalized by the formation of the more flexible six-membered (S, N) and five-membered (N, O) joined chelate rings with the former ligand over the more rigid 5 + 5 membered chelates present in the mecys complexes.
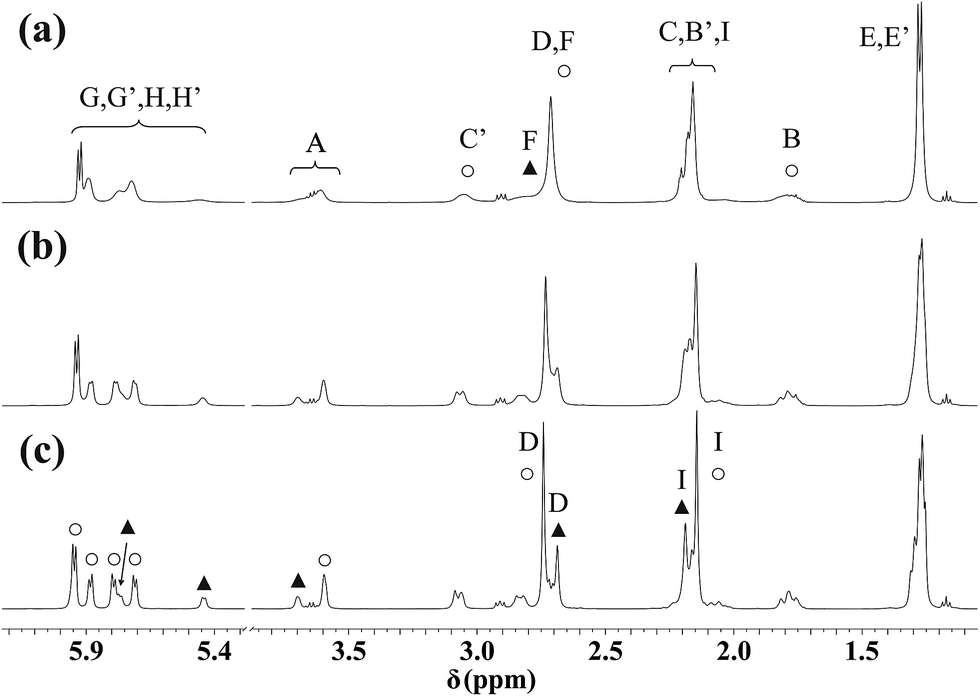 | ||
| Fig. 10 1H NMR spectra of [(η6-p-cym)Ru(mecys)]NO3 (11) in D2O at 298 K (a), 283 K (b) and 273 K (c). Triplets at 1.17 ppm and quartets at 3.65 ppm originate from ethanol while the triplets at 2.91 ppm from TSP. Notation of the A–I protons can be seen in Scheme 1. Resonances of the major diastereomer are marked with ○, while those of the minor one with ▲. | ||
It is worth emphasizing that with the racemic DL-met four stereoisomers can be formed. Tridentate coordination of L-met (with S absolute configuration) or D-met (with R absolute configuration) again will determine the configuration of the metal ion as stereogenic centre in the “chiral-at-metal” complexes resulting in RRuSαC or SRuRαC stereoisomers. Furthermore, with the coordination of the thioether S the following isomers can be expected: RRuSαCRS (1) and RRuSαCSS (2) for the L- and SRuRαCSS (3) and SRuRαCRS (4) for D-met. Although 1 can be distinguished both from 2 and 4 by its different resonances in the NMR spectrum, since 2 and 4 form enantiomers, only two sets of NMR resonances are expected. To confirm this NMR spectrum of [(η6-p-cym)Ru(L-met)]Cl (3) was also registered and found to be identical with that of the DL-met containing spectrum.
The calculated ratio of the diastereomers (%) for all the [(η6-p-cym)Ru(DL-met)]NO3 [(η6-p-cym)Os(DL-met)]Cl or [(η5-Cp*)M′(DL-met)]Cl complexes together with the diastereomeric excess (de %) are summarized in Table 6. As it is seen in Table 6 cooling did not alter significantly the ratios, furthermore, with met the amount of the minor epimer is larger than previously found with mecys (Table 6). This can be explained with the presence of the more flexible jointed six-membered (S, N) and five-membered (N, O) chelates in the case of the met complexes over the mecys ones. Similarly as for mecys, in the case of [(η5-Cp*)Rh(DL-met)]Cl (7) the minor diastereomer can only be detected at lower temperature (Fig. S11†).
Cytotoxicity in cancer cells
The in vitro anti-cancer chemotherapeutic potential of the complexes 3, 7, 9 and 11 towards a human ovarian cancer cell line (A2780) was also tested. Several assays are commonly used for in vitro chemosensitivity testing of tumour cell lines, although use of them has various advantages and limitations. It is so because the principles of these assays are differently dependent on metabolic and other factors and/or some compounds can directly interfere with reactions responsible for conversion of the colourless dye to the coloured product without having any effects on cell viability which may, in turn, substantially affect the quantitation of cell viability. Therefore two independent assays, MTT and NR assays have been employed to determine cytotoxic effect of the compound tested in this work. The MTT assay requires cellular metabolic activity (measures mitochondria dehydrogenase activity as a marker of cell viability) to convert the colourless tetrazolium to the purple-coloured formazan dye. On the other hand, the neutral red uptake assay is based on the ability of viable cells to incorporate and bind the dye in lysosomes by an active metabolic process.The results clearly showed that all complexes (3, 7, 9 and 11) tested in this work have insignificant anti-proliferative properties, since they failed to reduce the viability of the model cell line across both the concentration range (0–50 μM) and incubation period (72 h) studied. In contrast, cisplatin displayed a significant reduction in cellular viability of A2780 cells under the same conditions with mean IC50 values of 3.4 ± 0.3 μM and 2.8 ± 0.1 μM determined by MTT and NR assay, respectively. The IC50 > 50 μM values obtained for 3, 7, 9 and 11 are in contrast with recently published results20 but might be rationalized by the different experimental conditions applied.
Conclusions
In summary, we have demonstrated that the amino acids, S-methyl-L-cysteine and DL-methionine, both bearing a thioether S donor too, are efficient binders for half-sandwich type platinum metal ions over a wide pH-range in aqueous solution. Combined pH-potentiometry, NMR and MS as well as single crystal X-ray diffraction methods indicated the ligands to coordinate via a [S, NH2, COO−] manner in the complexes. The slightly higher stability of [(η6-p-cym)Ru(met)]+ over the mecys analogue was rationalized by the more flexible (6 + 5) membered joined chelates with the former ligand compared to the (5 + 5) membered ones formed with mecys. Solution behaviour of the solid complexes after redissolving them in various solvents showed that with both ligands beside the major stereoisomer a minor one with the different configuration of the coordinating thioether S only is also formed. The extent of epimerization was found to be higher for the met containing complexes due to the less rigid chelate ring formed. This kinetically labile character of the thioether S donor atom may have a significant contribution to the experimental findings, namely, the studied complexes, although they are characterized by high thermodynamic stability, do not show in vitro antiproliferative activity against A2780 human derived cancer cell lines in the concentration range 0–50 μM most likely due to their insufficient kinetic inertness.Acknowledgements
The authors thank members of the EU COST CM1105 for motivating discussions. The research was supported by EU and co-financed by the European Social Fund under the project ENVIKUT (TAMOP-4.2.2.A-11/1/KONV-2012-0043) and by the Hungarian Scientific Research Fund (OTKA K112317).References
- M. Melchart and P. J. Sadler, in Bioorganometallics, ed. G. Jaouen, Wiley-VCH, Weinheim, Germany, 2006, pp. 39–64 Search PubMed.
- P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1–62 CrossRef CAS.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and B. K. Keppler, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, Weinheim, Germany, 2011, pp. 160–164 Search PubMed.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- G. Süss-Fink, J. Organomet. Chem., 2014, 751, 2–19 CrossRef PubMed.
- L. Bíró, E. Farkas and P. Buglyó, Dalton Trans., 2010, 10272–10278 RSC.
- L. Bíró, D. Hüse, A. C. Bényei and P. Buglyó, J. Inorg. Biochem., 2012, 116, 116–125 CrossRef PubMed.
- D. Hüse, L. Bíró, J. Patalenszki, A. C. Bényei and P. Buglyó, Eur. J. Inorg. Chem., 2014, 5204–5216 CrossRef.
- L. Bíró, E. Balogh and P. Buglyó, J. Organomet. Chem., 2013, 734, 61–68 CrossRef PubMed.
- K. Severin, R. Bergs and W. Beck, Angew. Chem., Int. Ed., 1998, 37, 1634–1654 CrossRef.
- F. Wang, H. Chen, J. A. Parkinson, P. Murdoch and P. J. Sadler, Inorg. Chem., 2002, 41, 4509–4523 CrossRef CAS PubMed.
- L. E. H. Paul, B. Therien and J. Furrer, J. Biol. Inorg. Chem., 2012, 17, 1053–1062 CrossRef CAS PubMed.
- W. S. Sheldrick and S. Heeb, J. Organomet. Chem., 1989, 377, 357–366 CrossRef CAS.
- G. Capper, D. L. Davies, J. Fawcett and D. R. Russel, Acta Crystallogr., Sect. C: Struct. Chem., 1995, 51, 578–580 CrossRef.
- W. S. Sheldrick and R. Exner, J. Organomet. Chem., 1990, 386, 375–387 CrossRef CAS.
- W. S. Sheldrick and A. Gleichmann, J. Organomet. Chem., 1994, 470, 183–187 CrossRef CAS.
- M. Scharwitz, T. van Almsick and W. S. Sheldrick, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, 230–232 Search PubMed.
- X. L. Lu, L. Zhang, J.-D. Lou, J. Yan, P.-S. Nong, X.-H. Chen, J.-J. Yang and M. Gao, Transition Met. Chem., 2010, 35, 513–519 CrossRef CAS.
- A. Buryak and K. Severin, Angew. Chem., Int. Ed., 2004, 43, 4771–4774 CrossRef CAS PubMed.
- F. A. Egbewande, L. E. H. Paul, B. Therrien and J. Furrer, Eur. J. Inorg. Chem., 2014, 1174–1184 CrossRef CAS.
- D. Carmona, M. P. Lamata, F. Viguri, E. San José, A. Mandoza, F. J. Lahoz, P. García-Orduña, R. Atencio and L. A. Oro, J. Organomet. Chem., 2012, 717, 152–163 CrossRef CAS PubMed.
- C. Ganter, Chem. Soc. Rev., 2003, 32, 130–138 RSC.
- H. Brunner, Eur. J. Inorg. Chem., 2001, 905–912 CrossRef CAS.
- I. Weber, F. W. Heinemann, W. Bauer and U. Zenneck, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 123–140 CAS.
- D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Pergamon, Oxford, UK, 3rd edn, 1988 Search PubMed.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC.
- A. J. Godó, A. C. Bényei, B. Duff, D. A. Egan and P. Buglyó, RSC Adv., 2012, 2, 1486–1495 RSC.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. Gran, Acta Chem. Scand., 1950, 4, 559–577 CrossRef CAS PubMed.
- H. M. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- A. Krężel and W. Bal, J. Inorg. Biochem., 2004, 98, 161–166 CrossRef PubMed.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. Leggett, Plenum, New York, 1985, pp. 291–299 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, J. Chem. Soc., Dalton Trans., 1985, 1195–1200 RSC.
- L. Bíró, E. Farkas and P. Buglyó, Dalton Trans., 2012, 41, 285–291 RSC.
- V. Novohradsky, L. Zerzankova, J. Stepankova, A. Kisova, H. Kostrhunova, Z. Liu, P. J. Sadler, J. Kasparkova and V. Brabec, Metallomics, 2014, 6, 1491–1501 RSC.
- I. Sóvágó, K. Várnagy and A. C. Bényei, Magy. Kem. Foly., 1986, 92, 114–116 Search PubMed.
- W. S. Sheldrick, E. Hauck and S. Korn, J. Organomet. Chem., 1994, 467, 283–292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Titration curves with S-methyl-L-cysteine for the H+–ligand system and the [(η6-p-cym)Ru(H2O)3]2+–ligand systems at 1:2, 1:1 and 2:1 ratios; dependence on pH of the 1H NMR spectra of the [(η6-p-cym)Ru(H2O)3]2+–met system at 1:1 ratio and at 298 K in D2O; hypothetical concentration distribution curves of the [(η6-p-cym)Ru]2+–met–citr–ise system at 1:1:1:1 ratio; superimposed molecular structures of the two conformers of the RRuRαCSS diastereomer of [(η6-p-cym)Ru(mecys)]NO3 (10); COSY spectra of [(η6-p-cym)Ru(mecys)]Cl (1) in D2O, [(η6-p-cym)Ru(mecys)]CF3SO3 (12) in CD3OD and [(η6-p-cym)Ru(met)]NO3 (11) in D2O at 298 K as well as [(η6-p-cym)Ru(met)]NO3 (11) in D2O at 273 K; Newman projection of the two diastereomers of [(η6-p-cym)Ru(mecys)]+; HSQC spectra of [(η6-p-cym)Ru(mecys)]Cl (1) in D2O at 298 K and [(η6-p-cym)Ru(met)]NO3 (11) in D2O at 273 K; dependence on temperature of the 1H NMR spectra of [(η5-Cp*)Rh(mecys)]Cl (6) and [(η5-Cp*)Rh(met)]Cl (7) in D2O at 298, 283 and 273 K. CCDC 1025973 and 1025974. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15649h |
| This journal is © The Royal Society of Chemistry 2015 |