
Impact of linker in polypyrrole/quinone conducting redox polymers†
Christoffer Karlssona,
Hao Huanga,
Maria Strømmea,
Adolf Gogollb and
Martin Sjödin*a
aNanotechnology and Functional Materials, Department of Engineering Sciences, The Ångström Laboratory, Uppsala University, Box 534, SE-751 21 Uppsala, Sweden. E-mail: Martin.Sjodin@Angstrom.uu.se
bDepartment of Chemistry – BMC, Biomedical Centre, Uppsala University, Box 576, SE-751 23 Uppsala, Sweden
First published on 9th January 2015
Abstract
Organic conducting redox polymers are being investigated as the active component for secondary battery applications, as they have the potential to solve two of the main problems with small molecule-based organic electrodes for electrical energy storage, viz dissolution of the active compound in the electrolyte, and slow charge transport through the material. Herein we report the synthesis of a series of conducting redox polymers based on polypyrrole with hydroquinone pendant groups that are attached to the backbone via different linkers, and we investigate the impact of the linker on the interaction between the backbone and the pendant groups. For the directly linked polymer, oxidation of the pendant groups leads to a decrease of bipolaron absorbance, as well as a decrease in mass of the polymer film, both of which are reversible. The origin of these effects is discussed in light of the influence of the linker unit, electrolyte polarity, and electrolyte salt. For the longest linkers in the series, no interaction was observed, which was deemed the most beneficial situation for energy storage applications, as the energy storage capacity of the pendant groups can be utilized without disturbing the conductivity of the polymer backbone.
1. Introduction
The world's demand for electrical energy storage (EES) is steadily increasing and puts focus on raw material accessibility, sustainability, and environmental impact of existing EES technologies.1,2 Based on their superior performance in terms of specific energy and power, cycling stability and life time, lithium ion batteries (LIBs) constitute the most promising technology to meet future technical demands.2,3 The prospective usage of LIB technology is, however, not without concern. Due mainly to the cathode materials used, contemporary LIBs are responsible for large carbon emissions in their production, the raw materials are far from renewable, and recycling is complicated and expensive, partly due to the high temperatures required.2,4 The development of alternative battery materials based on renewable and readily accessible raw materials could reduce, or even eliminate, the demand for the limited mineral resources that today are associated with LIBs.2,4 Research efforts have therefore been invested in the development of organic matter based battery materials, and several molecular motifs, including quinones,5,6 carboxylates,7 tetraketopiperazines,8 dithiols9 and nitroxides10,11 have been tested. Although significant progress has been accomplished to identify stable redox chemistries6,12 and systematic trends in redox potentials,13–16 thus providing a rich flora of materials to combine into full battery devices, problems with limited material conductivity as well as dissolution of the redox center remain to be solved.4,7 One strategy to render the active material conductive and at the same time decrease the solubility is to combine well-defined redox centers with conducting polymers (CPs) to form a conducting redox polymer.17 Many conducting metallopolymers, i.e. conducting redox polymers with metal containing pendant groups, have been studied for EES, but due to the large molar mass of the charge carrying unit they are not optimal for this purpose.18 There have also been a number of reports of all-organic conducting redox polymers (OCRPs), albeit much fewer than of pure CPs, redox polymers or conducting metallopolymers. The most common OCRPs are based on polypyrrole (PPy), polythiophene or polyaniline CP backbones, functionalized with quinones,19–26 other carbonyl compounds,26,27 nitroxides,28 pyridines,29 phenothiazine30 or sulfur compounds.26 Very few of these polymers exhibit overlapping redox active potential regions of the pendant group and the CP backbone, which is necessary to benefit from the electronic conduction of the CP when cycling the pendant groups (primarily only quinone and phenothiazine OCRPs have this feature). Moreover, the interaction between the pendant groups and the CP backbone is often overlooked in the study of these polymers, and little information is thus available to aid rational design of OCRPs for EES.We have previously reported on the synthesis and electrochemical, spectroelectrochemical and electrogravimetric behaviour of an OCRP with hydroquinone (HQ) pendant groups on a PPy backbone.17,31,32 That polymer was found to exhibit some unique properties, including loss of bipolaron absorption and contraction of the polymer film as a result of HQ oxidation, which indicated significant influence of the pendant group on the CP backbone.17,31 For EES purposes this interaction is disadvantageous, as the separate uninterrupted properties of the pendant group and the CP backbone are sought. The influence of the pendant group redox chemistry on the backbone in this polymer was assumed to partly originate from the direct attachment of pendant groups to the PPy backbone, causing large strain on the backbone upon π-stacking of the quinone pendant groups. In this report, we have investigated how the backbone–pendant group interaction is affected by introducing various linker units between the PPy backbone and the pendant groups and, in view of the results herein, we discuss the origin of bipolaron absorption loss as well as of polymer contraction during pendant group oxidation.
2. Experimental
All reagents were purchased from Sigma Aldrich and were used without further purification. An Autolab PGSTAT302N potentiostat (Ecochemie, the Netherlands) was used for all electrochemical measurements. The organic electrolyte used was 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAHFP) in dry MeCN. The aqueous electrolyte consisted of 1.0 M NaCl, NaNO3 or NaClO4 buffered with phosphate, borate and acetate ions (10 mM each) and the pH was tuned with the respective acid and NaOH (s). All potentials measured in aqueous solutions are reported vs. the standard hydrogen electrode (SHE) and all potentials measured in organic solutions are reported vs. the ferrocene redox couple (Fc0/+). A polished glassy carbon (GC) disk electrode (3.0 mm diameter (BASi, USA)) was used as working electrode, and a Pt wire was used as counter electrode. In organic solutions the reference electrode consisted of a Ag wire (10 mM AgNO3, 0.1 M TBAHFP, −0.096 V vs. Fc0/+) that was kept in a separate compartment. In aqueous solution a Ag/AgCl reference electrode (3 M NaCl, 0.192 V vs. SHE17) was immersed directly into the electrolyte solution. The electrolyte was thoroughly degassed with solvent saturated N2 (g) and kept under N2 (g) atmosphere throughout the measurements. Electrochemical measurements were performed at room temperature (22 °C).2.1 Polymer synthesis
Functionalized pyrrole monomers 1–7 (Scheme 1) were synthesized as described previously.17,31,33 PPy, P1 and P2 were synthesized as reported before,17,31 and the monomers 3–5 were electropolymerized with a similar method: polymerization onto a GC electrode was achieved by cyclic voltammetry (CV) at 0.1 V s−1 for 5 scans, from a 10 mM monomer solution in MeCN with 0.1 M TBAHFP (Scheme 1). The polymer films were washed with EtOH to remove residual monomers and short oligomer chains from the film. The di-O-methyl-protected monomers were deprotected by reaction with BBr3 (1.0 M in DCM). Details regarding polymerization conditions and demethylation times for each specific monomer can be found in Table 1.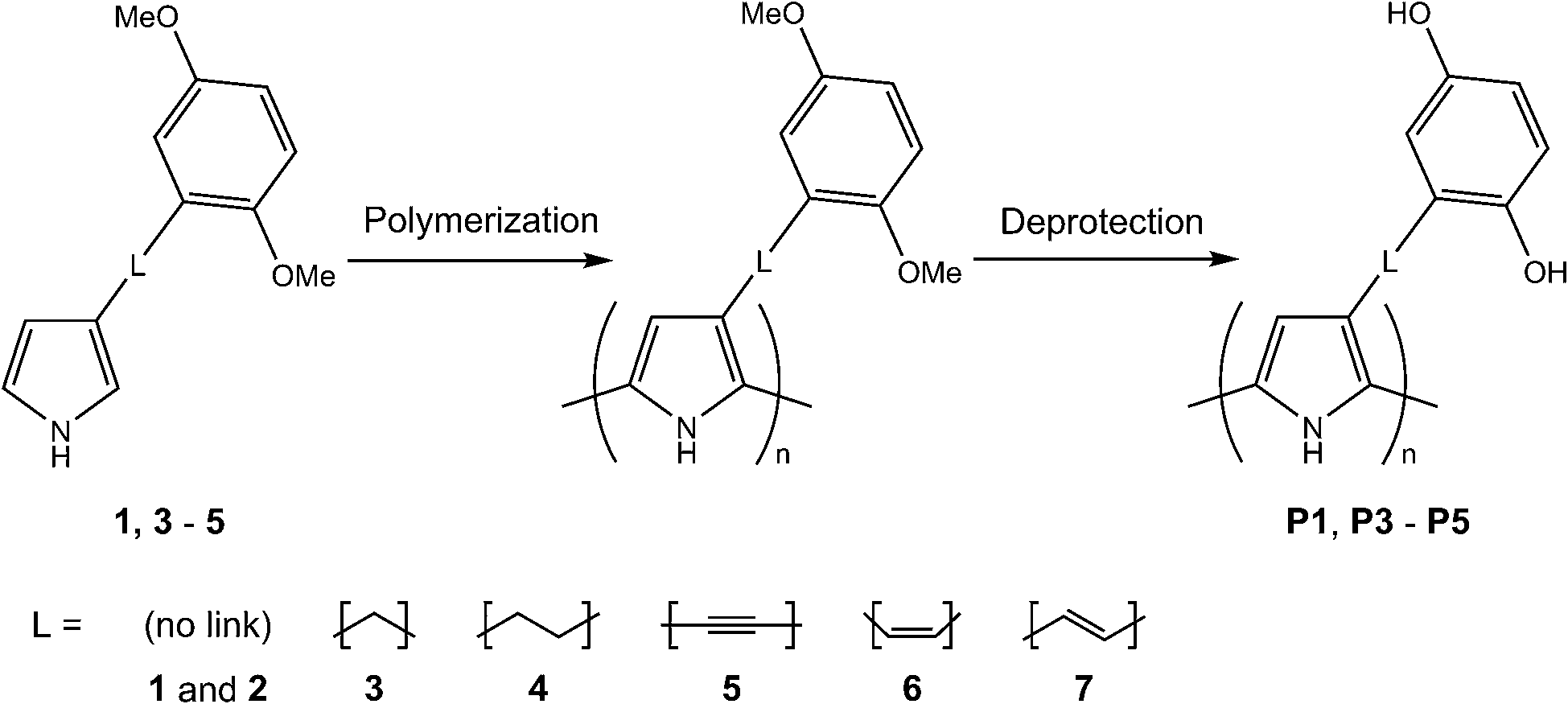 | ||
| Scheme 1 Synthesis of the PPy-HQ polymers. P2 was synthesized by electropolymerization of the non-methylated monomer 2.31 6 and 7 did not form conducting polymer films. | ||
| Linker unita | Protected pendant group?b | [Monomer] (mM) | Epol (V vs. Fc0/+)c | Deprotection reaction time (min)b | EQCM Epol (V vs. Fc0/+)c | |
|---|---|---|---|---|---|---|
| a L in Scheme 1.b Monomer oxygens protected by methyl groups that are removed after polymerization.c Upper vertex potential in the CV polymerization procedure, set to ∼200 mV higher than the pyrrole oxidation onset potential. | ||||||
| 1 | — | Yes | 10 | 1.0 | 15 | 0.625 |
| 2 | — | No | 20 | 1.0 | — | 0.625 |
| 3 | –CH2– | Yes | 10 | 0.8 | 20 | 0.925 |
| 4 | –CH2–CH2– | Yes | 10 | 1.1 | 45 | 0.925 |
| 5 | –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– C– |
Yes | 10 | 0.8 | 45 | 0.925 |
| 6 | cis-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– CH– |
Yes | (10–40) | (0.8–1.4) | — | — |
| 7 | trans-CHCH– |
Yes | (10) | (0.8–1.4) | — | — |
The quinone redox potential (E0Q′) was determined as the average of the CV oxidation and reduction peak potentials, and the PPy doping onset potential (EPPy) was determined from the maximum slope in the cyclic voltammograms.
2.2 Spectroelectrochemistry
Electropolymerization was performed on a working electrode consisting of an indium tin oxide (ITO) coated quartz slide (8 × 50 × 1 mm, 20 Ω □−1, Präzisions Glas & Optik, Germany), and the counter electrode was kept in a separate compartment. The conducting ITO substrate was coated with acetate by immersing the glass into a solution of potassium acetate (KOAc, 1.0 M, aq) prior to polymerization.17 Polymerization was performed by CV as described above, but with only three scans at 20 mV s−1 sweep rate for P5. The spectroelectrochemical measurements were performed in a quartz cuvette with 1 cm path length, using buffered aqueous electrolyte as described above. CV was performed at 20 mV s−1 and UV/vis/NIR spectra between 225 and 1100 nm were recorded every 0.5 s on an Agilent 8453 spectrophotometer. The spectra were correlated with the potential by determining the turning points of the absorbance at 1000 nm, which were assumed to correspond to the CV turning potentials.2.3 Electrochemical quartz crystal microbalance
Electrochemical quartz crystal microbalance (EQCM) measurements were performed using an Autolab potentiostat equipped with a Metrohm EQCM module (Eco Chemie, the Netherlands). The working electrode consisted of a gold coated AT-cut quartz EQCM crystal (6 MHz, 13.6 mm overall diameter, 6.7 mm diameter of the electroactive area), the counter electrode was a Pt wire, and a Ag/AgCl (saturated aqueous KCl gel, 0.173 V vs. SHE in water, −0.475 V vs. Fc0/+ in MeCN) electrode was used as reference. All electrodes were kept directly in the electrolyte. Polymerization was performed as described above, but with only three scans for P3, and only one scan for P5, to achieve polymer films of comparable masses. The aqueous electrolyte was as described above, but with only phosphate as buffer. By diluting the 1.0 M electrolytes with water, and then adjusting the pH with the corresponding acid, 0.10 M electrolytes were made. Electrodes covered with polymer were used with different electrolytes, and thorough rinsing with water and new electrolyte was employed between each experiment. In each electrolyte, the polymer was first cycled in a low potential region (below the HQ oxidation onset potential) several times to ensure that negligible amounts of other ions were present in the film.3. Results
3.1 Electrochemical properties
All monomers showed an irreversible oxidation reaction that for 1–5 resulted in a build-up of capacitance due to polymer formation on the electrode surface upon cycling, thus enabling creation of polymer modified electroactive surfaces through electropolymerization (Fig. 1).17,31,33 Monomers 6 and 7, which exhibited the same redox reactions, did however not form electroactive polymers, even at higher concentrations or at different potential intervals. Instead, the monomer redox response decreased for each scan, most likely due to formation of a blocking layer on the electrode. For 3 and 5 the onset potential for polymer formation was significantly lower than for the other compounds studied, and only one oxidation peak was observed. For the other monomers, i.e. for 1, 2, 4, 6, and 7, a peak originating from the dimethoxyphenyl moiety was observed at potentials below the polymer formation potential.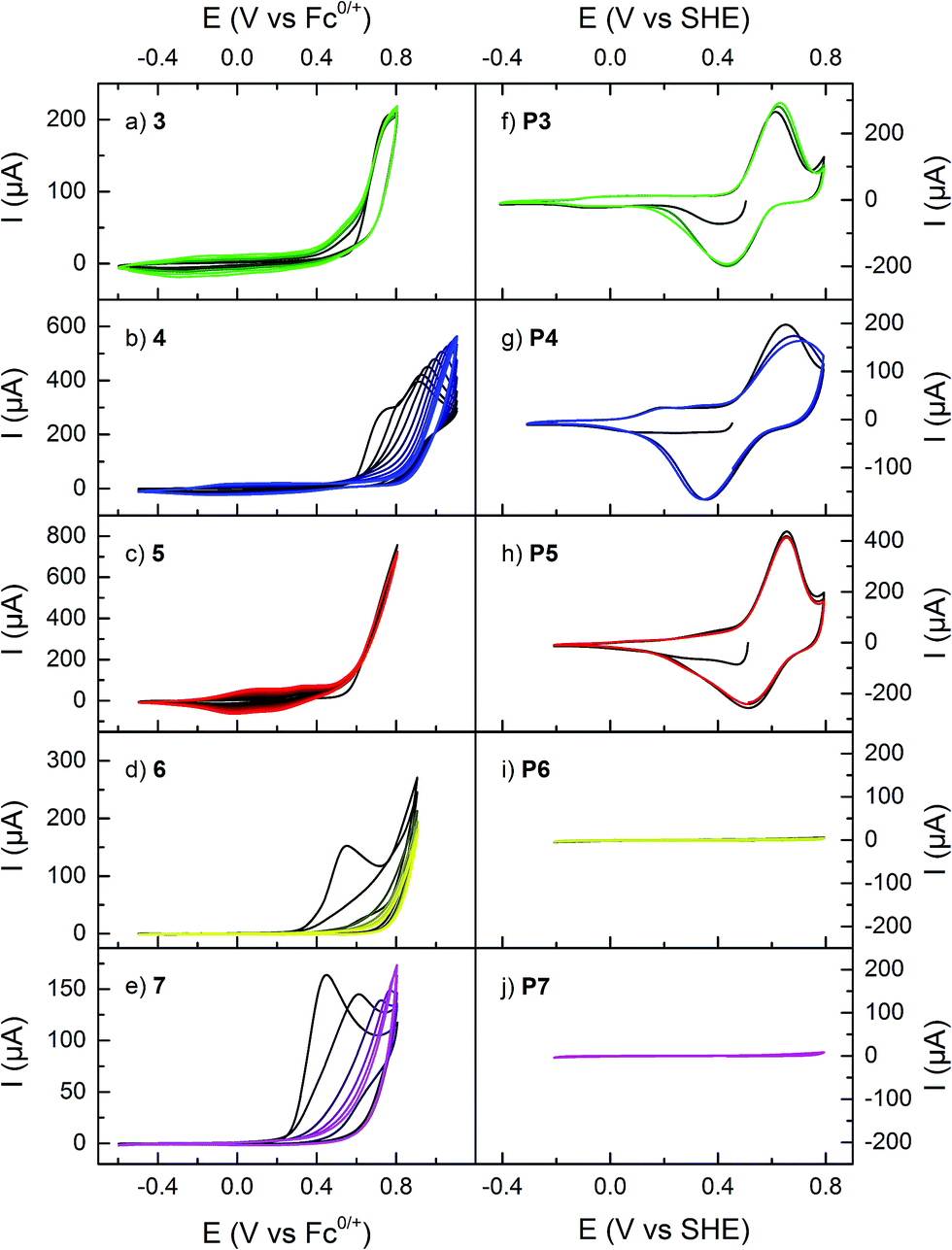 | ||
| Fig. 1 (a–e) Cyclic voltammograms of monomers in organic electrolyte, and (f–j) of the polymers in aqueous electrolyte buffered at pH 2.00. The horizontal axes are the same for all plots in the two respective columns. First scan in black and subsequent scans in lighter color. See Scheme 1 or Table 1 for numbering legend. | ||
The polymers were characterized by CV in aqueous electrolyte buffered at pH 2.00 (Fig. 1). All polymers P1–P5 have the same electrochemical features: a reversible redox reaction at 0.5–0.6 V, corresponding to the pendant group redox conversion between HQ and p-benzoquinone (Q), and capacitive charging originating from the PPy backbone, with an onset potential around −0.2–0.0 V. The quinone redox reaction occurs at approximately the same potential for all polymers, but P3 has a lower onset of PPy doping than the other HQ functionalized polymers (Table 2).17
| E0Q′ (V vs. SHE) | EPPy (V vs. SHE) | |
|---|---|---|
| PPy | — | −0.22 |
| P1 | 0.55 | 0.04 |
| P2 | 0.55 | 0.04 |
| P3 | 0.52 | −0.19 |
| P4 | 0.52 | −0.03 |
| P5 | 0.59 | −0.05 |
3.2 Spectroelectrochemistry
UV/vis/NIR spectra were recorded in situ on polymer films during redox cycling (spectra in Fig. 2a and S1,† absorbance maxima in Table S1†). All polymers exhibit similar spectra,17,31 with four bands characteristic of the PPy backbone, corresponding to the transitions from the valence band to the mid-gap states and to the conduction band (Fig. 2c).17,34,35 Compared to unsubstituted PPy, the band-gap transition (PPy3 in Fig. 2c; 214 nm for PPy) for the HQ functionalized polymers is blueshifted, indicating shorter conjugated segments. This blueshift of the band gap transition is approximately the same for P1–P4 and persists upon polymer doping. In P3 and P4, where the pendant group aromatic system is disconnected from PPy by aliphatic linkers, the anti-bonding polaron state shows approximately the same blueshift relative to the valence band compared to PPy, as judged from the difference in absorbance maxima for the PPy2 transition (Fig. 2 and Table S1†). In P1, P2 and P5 this transition is instead redshifted compared to PPy, indicating that the polaron state is affected by the extended conjugation as a result of substitution, whilst the band gap is uniformly affected in all polymers, and not specifically dependent on a conjugated connection between polymer and pendant group. There are also two transitions originating from the n–π* and π–π* transitions of the pendant groups that are blueshifted upon oxidation.17 The most significant pendant group absorbance band is the n–π* transition in the Q state, which occurs at 250 nm for all polymers in the series. There were no significant spectral differences upon changing the supporting electrolyte salt (NaCl, NaNO3 or NaClO4 at 0.1 M or 1 M).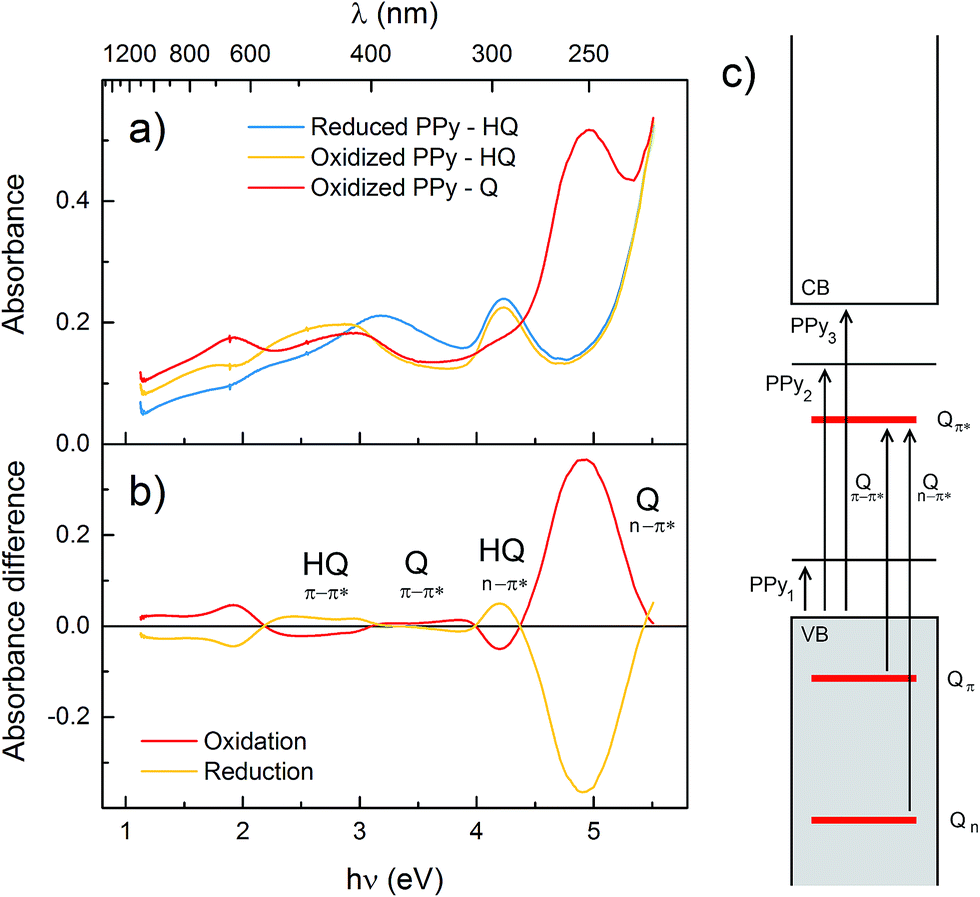 | ||
| Fig. 2 (a) UV/vis/NIR spectra of P3 in aqueous electrolyte (1.0 M NaCl at pH 2.00) in the reduced PPy state (blue), oxidized PPy state (yellow) and with oxidized quinone pendant groups (red). (b) Difference spectra over the quinone oxidation (red) and reduction sweep (yellow). The horizontal axis is the same in both graphs, with wavelength scale above and energy scale below the panels. (c) Energy diagram showing the PPy transitions between the valence band (VB), conduction band (CB) and the two mid-gap states, and the pendant group transitions between the n, π and π* levels (cf. Table S1†). | ||
The absorbances at 250 nm (Abs250 nm, Q transition) and 1000 nm (Abs1000 nm, PPy1 bipolaron transition) were monitored during redox cycling of the polymers (Fig. 3). There is a sigmoidal increase of Abs250 nm during the oxidation of the pendant groups, following a Nernstian behaviour (although broadened according to the CV oxidation peak broadening). The polymer backbone has no absorbance at 250 nm, but the broad band at NIR wavelengths (PPy1) increases with the oxidation state of the PPy backbone, as more bipolaron states are formed in the band gap. Abs1000 nm increases approximately linearly for all polymers at potentials where the quinone pendant groups are not redox active. Hence, unsubstituted PPy exhibits a uniform increase of Abs1000 nm in the entire stable potential region. For P3, on the other hand, Abs1000 nm has a constant value during the quinone redox process, as previously observed for P1,17 and P2 even exhibits a decrease of Abs1000 nm upon oxidation of the pendant groups.31 Abs1000 nm for P4 and P5 is also affected by the pendant group oxidation, but to a lesser extent, and Abs1000 nm continues to increase with potential for these polymers, but the rate is slowed at the HQ oxidation. The inflection points of Abs250 nm coincide with the minima of dAbs1000 nm/dE for all polymers, and also with the oxidation peak potential (cyclic voltammograms in Fig. 1). The absorbances during a reduction sweep follow the same trends reversibly, but are shifted to more negative potentials, analogous to the electrochemical response.
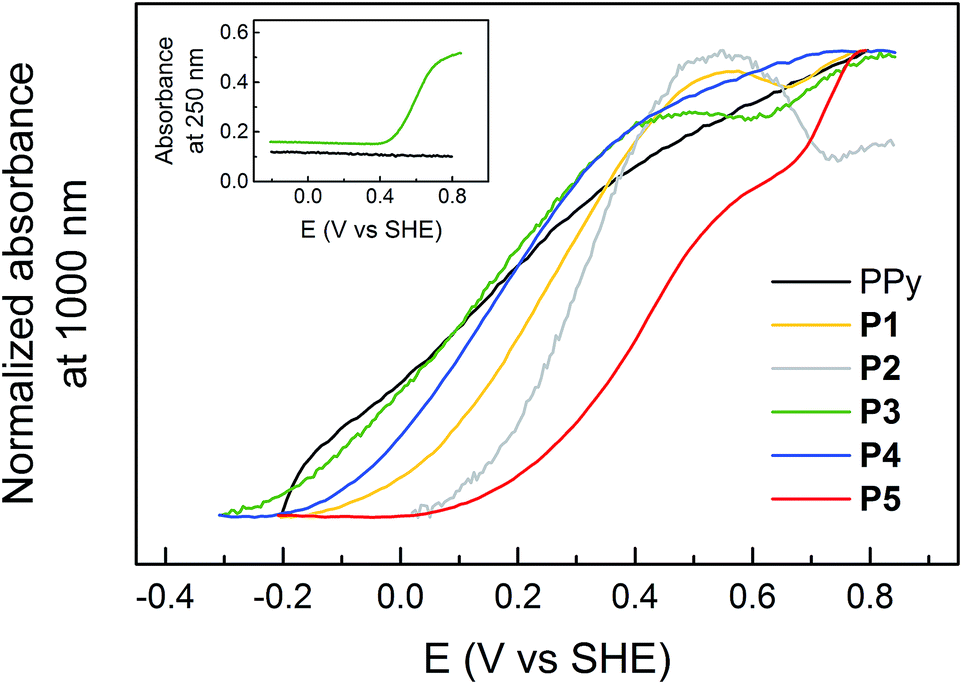 | ||
| Fig. 3 Normalized absorbance at 1000 nm during a CV oxidation sweep for the polymers, corresponding to the bipolaron concentration in the polymers. Absorbance at 250 nm for PPy and P3 are displayed in the inset, showing sigmoidal increase as the pendant groups are oxidized. | ||
3.3 Electrochemical quartz crystal microbalance
EQCM was used to measure mass changes (Δm) of the polymers, in order to gain further insight into the redox reactions of the polymers. The mass deposited on the electrode during polymerization, and the mass changes during polymer cycling, was calculated using the Sauerbrey equation, which has been used successfully on similar systems.31,36 The electropolymerization proceeded in a similar fashion for all monomers (except 6 and 7, vide supra), with a polymer build-up when cycling over the pyrrole oxidation peak (Fig. S2†). The mass increase and decrease due to anion inclusion and expulsion during PPy oxidation and reduction, respectively, are observed in both organic and aqueous electrolytes (Fig. 4 and S3–S8†), and during this process, the mass changes are proportional to the amount of elemental charges passed in the CV experiment (Δn, Fig. 5, S4, S6 and S8†). When the pendant groups are oxidized there is a concurrent mass decrease, which is restored reversibly upon reduction. In general, the rate of the decrease during this process is affected less for the linked polymers (P3–P5) than for P1 and P2, and the effect is smallest for P5. The mass decrease is highly dependent on the polarity of the electrolyte,31 and for the least polar electrolyte (1 M NaClO4) there is almost no mass effect at all during the pendant group redox conversion.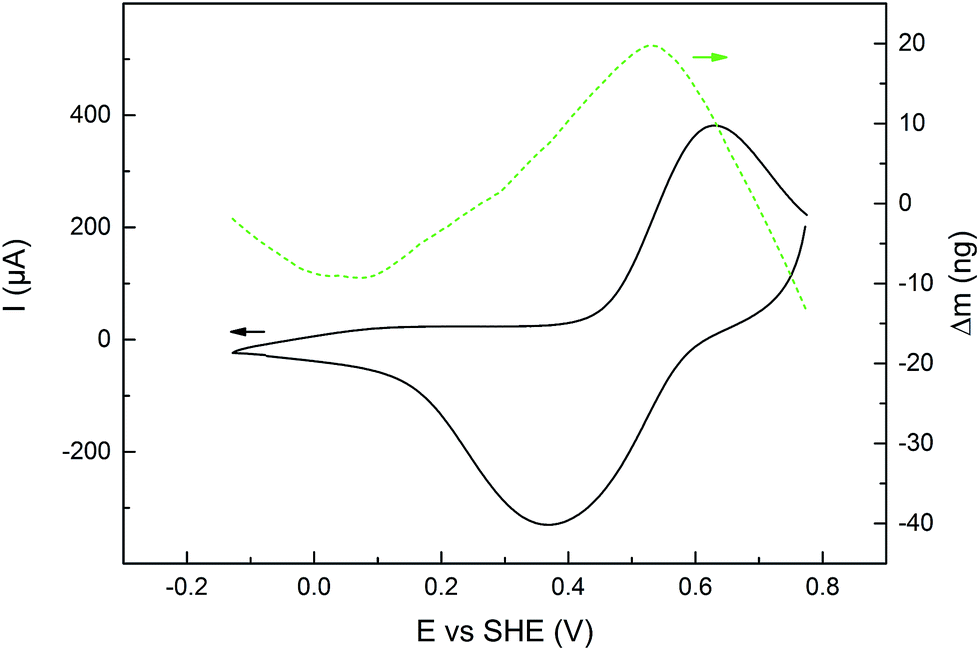 | ||
| Fig. 4 CV current (black solid line, left y-axis) during an EQCM experiment on P3, with the corresponding mass changes (dashed green line, right y-axis), in NaCl (1 M aq) electrolyte buffered at pH 2.00. Mass changes of all polymers and electrolytes can be found in Fig. S3, S5 and S6.† | ||
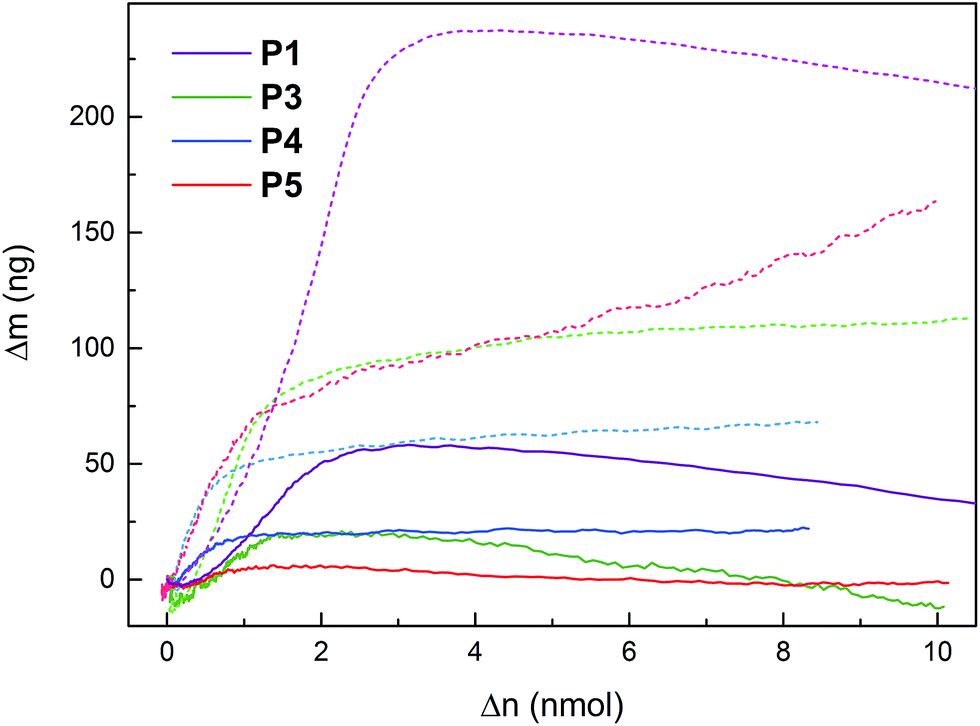 | ||
| Fig. 5 Mass changes during EQCM experiments of P1, P3, P4 and P5 in NaCl (dark solid lines) and NaClO4 (light dashed lines) electrolytes (1 M aq) buffered at pH 2.00, plotted vs. the amount of electrons withdrawn in the CV. Mass changes of all electrolytes can be found in Fig. S4, S6 and S8.† | ||
4. Discussion
4.1 Monomer electrochemistry and polymerization
The monomers 1, 2, 4, 6, and 7 all show an irreversible oxidation of the pendant groups in the potential interval 0.4–0.7 V (Fig. 1).17,31 As previously reported this oxidation leads to partial loss of the methyl protecting groups.17 The peak potential for the pendant group oxidation varies between different monomers, and for 6 and 7 (that have a vinylene linker between the pyrrole and the quinone moieties) it is lower than for the other monomers. Above 0.6 V, all monomers show an irreversible oxidation process that for 1–5 leads to the formation of a conducting polymer on the electrode surface, as judged by the characteristic capacitive build-up around 0.0 V. For 6 and 7 the corresponding process leads to blocking of the electrode due to formation of a resistive layer, in analogy to vinylene substituted thiophene monomers, which have been reported to form electroinactive polymer films.37,38 This problem seems to be related to the extended conjugation of the thiophene or pyrrole ring, decreasing its radical density, and possibly also leading to irreversible side reactions on the vinylene fragment.38 As a general observation, the onset potential for the polymerization reaction is lower for the linked monomers than for 1 and 2,17,31 which is likely due to the less electron withdrawing effect of the linked pendant groups, making them easier to oxidize. Since the rate of polymer degradation increases with potential,39 the possibility of lower polymerization potential improves the quality of the formed polymers.4.2 Polymer electrochemistry
For all studied polymers, the electrochemical response in aqueous electrolyte is very similar (Fig. 1). The doping process of the PPy backbone is clearly present, evident as an increase in capacitance at potentials above EPPy. The doping onset occurs at −0.2 V for P3, which is similar to unsubstituted PPy, while it is about 0.2 V higher for the other polymers. The fact that it is harder to oxidize P1, P2, P4 and P5 suggests that the conjugation length is shorter in these polymers. This in turn indicates that the polymer chain is shorter or more twisted due to the steric bulk of the pendant groups, which is further supported by spectroscopic data (vide infra). The polaron conjugation length is thus shorter, increasing its energy and the doping potential.Overlaid on the PPy capacitive response is a reversible redox process at 0.5–0.6 V, characteristic of the quinone redox reaction.17,40 It is interesting to note that, while EPPy varies with the nature of the linker, E0Q′ is rather constant. This shows that there is little difference in the electronic influence of the PPy backbone on the quinone units.31 All polymers except P2 are treated with the strong oxidant BBr3 to achieve the deprotected HQ units, and there is a risk of degradation of the polymer, which is manifested as a decrease in quinone charge at too long reaction times.17 For the linked polymers, it was possible to prolong the demethylation reaction time without loss of quinone charge, making it possible to achieve higher yields and hence more redox active pendant groups (Table 1). However, the need for the post-polymerization deprotection step does introduce variation in the polymer composition, and reproducible deprotection yields were difficult to obtain for these polymers.
4.3 Spectroelectrochemistry of polymers
All the polymers displayed very similar UV/vis/NIR spectra to unsubstituted PPy in all redox states, with only small variations in wavelengths (Fig. 2), which shows that attachment of pendant groups has little influence on the energetics of the PPy backbone.17,31,34,35 However, there is a small blueshift of the band-gap transition for the substituted polymers compared to PPy, indicating a somewhat shorter conjugation length, in line with the electrochemical results. The Nernstian increase of the Q and HQ absorbance bands upon oxidation and reduction, respectively, of the pendant groups can be extracted from difference spectra (Fig. 2b and Table S1†). The most interesting analysis of the spectroelectrochemical data is to compare the evolution of the absorbances at 250 and 1000 nm during an oxidation sweep (Fig. 3). Unsubstituted PPy shows only small changes in Abs250 nm in the investigated potential range, as do the substituted polymers at potentials sufficiently far from E0Q′. Abs250 nm can thus be used to probe the redox state of the pendant groups. Since the pendant groups have no absorbance band at 1000 nm, where one of the PPy bipolaron absorbance bands is located (PPy1 in Fig. 2c), Abs1000 nm can be used to probe the bipolaron concentration of the polymer. For unsubstituted PPy, Abs1000 nm increases steadily over the whole potential range, with a slightly higher rate at 0.2 V (where there is also a broad current peak in the CV). The substituted polymers also have the same feature at potentials sufficiently far from E0Q′ (Fig. 3). During the pendant group redox conversion, however, deviations from this behaviour are observed. As seen before for P1, P3 experiences a constant bipolaron absorbance during the HQ to Q oxidation (Fig. 3), and it continues to increase again after the pendant group reaction has been completed. As for P1, this means that the amount of bipolaron states does not increase during the pendant group oxidation, indicating that the doping level is constant during this process despite the increasing applied potential. For the polymers with longer linker units, the effect is still noticeable, but smaller than for P1 and P3. The rate of the doping process decreases, but it is not constant at the HQ/Q conversion potential. For P2, which has a higher concentration of redox active pendant groups, the absorbance at 1000 nm actually decreases during the HQ to Q conversion.31 The effect of the pendant groups on the PPy bipolaron absorption decreases in the following order:| P2 ≫ P1 ≥ P3 > P5 > P4 > PPy |
Qualitatively the effect of pendant group oxidation on the bipolaron absorption thus follows the same order as the length of the linker unit for the HQ functionalized polymers. As this phenomenon has not been reported for other polymers, we have speculated as to the cause of this phenomenon in our previous studies of P1 (ref. 17) and P2,31 and spectroscopic as well as EQCM data on the new linked polymers P3–P5 gives additional insights, which are discussed in section 4.5.
4.4 Electrochemical quartz crystal microbalance
The mass changes in P3–P5 observed by EQCM follow the same qualitative behavior as for P1 and P2 reported before, i.e. linear mass increase during the PPy doping at low potentials in a CV oxidation sweep, a decrease in the rate of mass uptake or even a decreased mass upon pendant group oxidation at higher potentials, and complete reversibility on the reduction sweep and subsequent scans (Fig. 4 and 5).31 The mass increase at low potentials is dependent on the electrolyte anion molar mass, while the mass decrease at high potentials does not show such a correlation. The mass decrease for P2 instead has a clear correlation with the electrolyte polarity.31 For the other polymers, which have undergone post-polymerization deprotection, the variation due to that reaction step makes the correlation with electrolyte polarity non-significant. In general, however, the effect on the mass decrease (as measured by the slope in the Δm vs. Δn plot at the quinone oxidation potential31) is much smaller in 1 M NaClO4 than in all other electrolytes (1 M NaClO4 differs the most in polarity from the other electrolytes). Comparing the polymers in the series, the linked polymers have smaller mass effects than the directly linked polymers, and the effect decreases in the following order:| P2 ≈ P1 > P3 > P4 > P5 |
There are several processes occurring that in principle could be causing the mass decrease. The loss of protons upon HQ to Q conversion only corresponds to about 1% of the polymer mass, and cannot directly be the cause for the mass decrease. If the loss of absorbance at 1000 nm, observed most pronouncedly for P2, is interpreted as a doping reversal phenomenon (as discussed below), this would lead to a mass decrease during the pendant group oxidation.31 However, in that case the mass decrease should depend on the electrolyte anion molar mass, and not on electrolyte polarity, as observed. Furthermore, if the bipolaron absorptivity is directly interpreted as the doping level of the polymer, the spectroelectrochemical results would suggest that the doping rate is merely slowed or halted during the HQ to Q redox conversion in the other polymers, i.e. not reversed. The fact that the mass decreases to less than the value in the completely reduced state, which is observed in some cases, is hence difficult to account for only by doping reversal.
As opposed to the effects discussed above, considering the pendant groups' interaction with each other and with the solvent, and its change upon oxidation, yields a consistent explanation for the observed mass decrease. The reduced HQ pendant groups have hydrogen bonding opportunities with the water solvent, and have limited interaction with each other. When the number of oxidized Q moieties increase, the π interaction between pendant groups increases, as quinhydrone (HQ–Q) complexes can form, as well as π–π stacking of Q units.41 The decreased polarity of the polymer as the hydrophobic Q units are formed would force the electrolyte out of the polymer film, and this effect is expected to depend on the electrolyte polarity. Pendant group packing could also lead to a contraction of the polymer film, expelling even more electrolyte from the polymer. The latter effect would explain the differences between the polymers, which should otherwise be nearly identical, as the polarity of the pendant groups does not depend on the linker unit: a more flexible linker allows for sufficient overlap between pendant groups without the need for a contraction of the polymer film, since they would have sufficient freedom to come in close contact without disturbing the PPy backbone.
4.5 A model for the processes occurring during pendant group redox conversion
A hypothesis that we initially suggested to explain the constant bipolaron concentration during pendant group oxidation in P1 was a change in pendant group electronegativity upon oxidation.17 However, the fact that the effect is almost identical in P1 and P3 clearly shows that this cannot be the case. The introduction of a –CH2– linker decreases the extent of electronic communication severely,33 and the pendant groups' effect on the bipolaron concentration would be much lower for P3 than for P1 if an electronegativity change was the cause. Moreover, this means that the electronic communication is very limited also in the directly linked polymers, which is due to the lack of possibility for resonance between the pyrrole and HQ moieties since the pendant groups are twisted out of plane.31 Only induction can communicate the change in electronegativity of the pendant groups, and this effect is evidently much too small to be the main reason for the constant bipolaron concentration in these polymers (especially P3).Increased pendant group interaction and contraction of the polymer film, as evidenced by EQCM, does give a rational explanation for the spectroscopic data. The decrease in bipolaron formation rate upon pendant group oxidation means that the energy for the charged bipolaron state is higher when the pendant groups are in the oxidized state. The decreased polarity in the polymer, induced by the oxidation of the pendant groups, would definitely have this effect as the charges would be destabilized in the less polar polymer matrix. However, the decreased polarity alone is expected to yield the same effect on the bipolaron concentration for all polymers, since the polarity of the polymer film changes to the same degree. A longer linker between the pendant groups and the polymer chain could allow for some shielding by the solvent, thus decreasing the influence, but this effect should be minor since the distances are short and the environment is rather nonpolar. The other effect of the pendant group oxidation, i.e. the increased π interaction, does account for the difference in the pendant groups' influence on the bipolaron concentration as a function of linker unit. To allow the pendant groups to pack efficiently, the PPy backbone must be twisted, an effect that is more pronounced for the polymers with shorter links. For all polymers in the series except for P4, the extent of PPy twisting is expected to be major, since there is very little flexibility to accommodate the pendant group packing. This is in excellent agreement with the spectroscopic results. Twisting pyrrole subunits relative to each other breaks the conjugation and forces the extended bipolaron charges to localize on the chain, accounting for the loss of bipolaron absorption. Since the energy of these localized states is very high in energy, this is also expected to lead to a dedoping of the CP. The energy gain of packing the oxidized pendant groups seemingly outweighs the energy penalty of breaking the PPy conjugation. Hence, both the polarity decrease and the concurrent PPy twisting leads to the loss of bipolaron states and decrease in doping rate of these polymers.
It should be noted that the loss of bipolaron states, together with their decreased mobility on the polymer chain, is expected to decrease the conductivity of the OCRPs drastically as the HQ pendant groups are oxidized. For this reason, P4 is proposed as the most advantageous polymer in the series to use in secondary batteries, since it has the least interaction between the pendant groups and the backbone, and should hence experience the smallest conductivity decrease.
5. Conclusions
The interaction between the backbone and pendant groups in a series of OCRPs has been studied, in which HQ was attached covalently to PPy via different linker units. OCRPs are being investigated as the active component in secondary battery applications, and knowledge about the interaction between the two redox systems is crucial for rational design of such OCRPs. The polymers were found to exhibit similar redox chemistry to that of the corresponding directly linked polymer, i.e. separate redox reactions of the PPy backbone and the HQ pendant groups. However, the introduction of the linker allowed for production of higher quality polymer films, as they could be synthesized at lower oxidative potentials. The length and flexibility of the linker unit was found to have a large impact on the degree of interaction, specifically on a decrease in bipolaron absorbance upon pendant group oxidation, and a concurrent mass decrease that is observed for all but the polymers with the longest linker units (–CH2–CH2– and –CC–). The nature of the linker affects these two processes to varying extent, which allowed for new insight into this phenomenon. We propose that both effects are related to the decreased polarity and increased interaction between the pendant groups in the oxidized, p-benzoquinone, state. This forces the polar electrolyte out of the polymer film (decreasing its apparent mass), and packing of the pendant groups is also responsible for twisting of the polymer backbone, leading to localization of charges, loss of bipolaron absorbance, and a decrease of the doping level. The overall effect of the pendant group oxidation on the backbone is detrimental, since it is likely to be accompanied by a loss of conductivity. However, we show that a two-carbon linker such as –CH2–CH2– is sufficient to eliminate this interaction.
Acknowledgements
This work was funded by the Swedish Foundation for Strategic Research (SSF), The Carl Trygger Foundation, The Swedish Energy Agency and the European Institute of Innovation and Technology under the KIC InnoEnergy NewMat and electrical energy storage project.Notes and references
- H. D. Abruña, Y. Kiya and J. C. Henderson, Phys. Today, 2008, 61, 43–47 CrossRef PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J.-M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330–1337 RSC.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J.-M. Tarascon, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- S. Renault, J. Geng, F. Dolhem and P. Poizot, Chem. Commun., 2011, 47, 2414–2416 RSC.
- T. Sarukawa and N. Oyama, J. Electrochem. Soc., 2010, 157, F23–F29 CrossRef CAS PubMed.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751–754 CrossRef CAS PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- M. Stolar and T. Baumgartner, Phys. Chem. Chem. Phys., 2013, 15, 9007–9024 RSC.
- C. Karlsson, E. Jämstorp, M. Strømme and M. Sjödin, J. Phys. Chem. C, 2012, 116, 3793–3801 CAS.
- K. Hernández-Burgos, S. E. Burkhardt, G. G. Rodríguez-Calero, R. G. Hennig and H. D. Abruña, J. Phys. Chem. C, 2014, 118, 6046–6051 Search PubMed.
- C. Karlsson, A. Gogoll, M. Strømme and M. Sjödin, J. Phys. Chem. C, 2013, 117, 894–901 CAS.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- C. Karlsson, H. Huang, M. Strømme, A. Gogoll and M. Sjödin, J. Phys. Chem. C, 2013, 117, 23558–23567 CAS.
- B. J. Holliday and T. M. Swager, Chem. Commun., 2005, 23–36 RSC.
- D. Häringer, P. Novák, O. Haas, B. Piro and M.-C. Pham, J. Electrochem. Soc., 1999, 146, 2393–2396 CrossRef PubMed.
- T. L. Rose, A. B. Kon and F. Wang, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 1995, 209, 311–312 Search PubMed.
- A. B. Kon, J. S. Foos and T. L. Rose, Chem. Mater., 1992, 4, 416–424 CrossRef CAS.
- J. S. Foos, S. M. Degnan, D. G. Glennon and X. Beebe, J. Electrochem. Soc., 1990, 137, 2530–2533 CrossRef CAS PubMed.
- C. P. Andrieux and P. Audebert, J. Electroanal. Chem. Interfacial Electrochem., 1990, 285, 163–175 CrossRef CAS.
- C. P. Andrieux and P. Audebert, J. Electroanal. Chem. Interfacial Electrochem., 1989, 261, 443–448 CrossRef CAS.
- T. Inagaki, M. Hunter, X. Q. Yang, T. A. Skotheim, H. S. Lee and Y. Okamoto, Mol. Cryst. Liq. Cryst. Incorporating Nonlinear Opt., 1988, 160, 79–88 CrossRef.
- J. L. Segura, R. Gómez, R. Blanco, E. Reinold and P. Bäuerle, Chem. Mater., 2006, 18, 2834–2847 CrossRef CAS.
- F. Rosciano, M. M. Salamone, R. Ruffo, M. Sassi and L. Beverina, J. Electrochem. Soc., 2013, 160, A1094–A1098 CrossRef CAS PubMed.
- A. Deronzier, D. Lìmosin and J.-C. Moutet, Electrochim. Acta, 1987, 32, 1643–1647 CrossRef CAS.
- S. Sen, J. Saraidaridis, S. Y. Kim and G. T. R. Palmore, ACS Appl. Mater. Interfaces, 2013, 5, 7825–7830 CAS.
- A. Deronzier, M. Essakalli and J.-C. Moutet, J. Chem. Soc., Chem. Commun., 1987, 773–775 RSC.
- C. Karlsson, H. Huang, M. Strømme, A. Gogoll and M. Sjödin, J. Phys. Chem. C, 2014, 118, 23499–23508 CAS.
- C. Karlsson, H. Huang, M. Strømme, A. Gogoll and M. Sjödin, J. Electroanal. Chem., 2014, 735, 95–98 CrossRef CAS PubMed.
- H. Huang, C. Karlsson, M. Strømme, M. Sjödin and A. Gogoll, Unpublished work, 2014.
- J.-L. Brédas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef.
- R. Čabala, J. Škarda and K. Potje-Kamloth, Phys. Chem. Chem. Phys., 2000, 2, 3283–3291 RSC.
- D. A. Buttry and M. D. Ward, Chem. Rev., 1992, 92, 1355–1379 CrossRef CAS.
- M. A. Vorotyntsev and S. V. Vasilyeva, Adv. Colloid Interface Sci., 2008, 139, 97–149 CrossRef CAS PubMed.
- J. R. Smith, S. A. Campbell, N. M. Ratcliffe and M. Dunleavy, Synth. Met., 1994, 63, 233–243 CrossRef CAS.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- J. Q. Chambers, in The chemistry of the quinonoid compounds, ed. S. Patai, John Wiley & Sons, London, UK, 1974, vol. 1, pp. 737–791 Search PubMed.
- M. Hesse, H. Meier and B. Zeeh, in Spectroscopic Methods in Organic Chemistry, Georg Thieme Verlag, Stuttgart, Germany, 2nd edn, 2008, pp. 23–24 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV/vis/NIR spectra, EQCM polymerization and characterization curves. See DOI: 10.1039/c4ra15708g |
| This journal is © The Royal Society of Chemistry 2015 |