
Protective effect of total saponins of Aralia elata (Miq) Seem on lipopolysaccharide-induced cardiac dysfunction via down-regulation of inflammatory signaling in mice
Rong-Chang Chena,
Jian Wangb,
Ying-Li Yua,
Gui-Bo Sun*a and
Xiao-Bo Sun*a
aInstitute of Medicinal Plant Development, Chinese Academy of Medical Science, Peking Union Medical College, No 151, North Road Malianwa, Haidian District, Beijing 100094, China. E-mail: Sunguibo@126.com.cn; sunxiaoboyzs@163.com
bResearch Center of Life Science and Environment Science, Harbin University of Commerce, No 1, Xuehai Street, Songbei District, Harbin, Heilongjiang 150028, China
First published on 17th February 2015
Abstract
Aralia elata (Miq) Seem is widely used in folk medicine for treating various types of diseases, including diabetes, gastric ulcers, hepatitis and rheumatoid arthritis. The present study investigates the therapeutic effects and possible mechanisms of the total saponins of A. elata (Miq) Seem (TAS) against lipopolysaccharide (LPS)-induced septic cardiac dysfunction and inflammation in mice. Mice were intragastrically administrated with TAS (35, 70 and 140 mg kg−1) for one week before LPS challenge (10 mg kg−1, i.p.). Cardiac injury was evaluated 6 h after LPS induction. Six hours of LPS administration deteriorated cardiac function which was attenuated by TAS pretreatment. TAS attenuated LPS-induced the increase of LDH, CK, AST, and cTnI activities in mice. TAS also ameliorated the imbalance between iNOS and eNOS, as well as preventing NF-κB activation and the subsequent myocardial inflammatory responses in endotoxemic mice. TAS could significantly downregulate LPS-mediated NOX2 expression and ROS production, even though TAS had no effect on LPS-activated TLR-4. The effects of TAS were closely associated with PI3K/AKT and MAPK signaling pathways, as characterized by TAS-induced activation in phospho-Akt and inhibition in phospho-ERK1/2, phospho-JNK, and phospho-P38. Besides, TAS also extended the lifespan of the toxemic mice. These results showed that TAS significantly attenuated LPS-induced cardiac dysfunction and production of inflammatory mediators by inhibiting NF-κB activation, indicating TAS as a potential therapeutic agent for septic cardiac dysfunction.
Introduction
Sepsis is a complex pathophysiological state resulting from a harmful or devastating host response to infection,1 which is associated with a high degree of mortality in hospitalised patients.2 As a well-documented feature of endotoxemia, myocardial contractile dysfunction can exist in both experimental and human septic shock, which increases mortality in patients with sepsis.2,3 Although precise mechanisms of myocardial dysfunction in sepsis remain undefined, previous studies suggest that production of several inflammatory cytokines, including tumour necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and cytokine-inducible nitric oxide synthase (iNOS), plays a key role in cardiac dysfunction.2,4Lipopolysaccharide (LPS) is an important structural component of the outer membrane of Gram-negative bacteria. As a major trigger of sepsis, LPS can stimulate multiple cells to release several pro-inflammatory mediators and other soluble factors which ultimately lead cells to a pro-inflammatory state.2,4 Studies show that LPS can act on cardiomyocytes by binding to its receptor, toll-like receptor 4 (TLR4), which will activate several intracellular signalling pathways.2,4,5 Among these signalling pathways, NF-κB, one of the major LPS signalling targets, is an important signal integrator that controls the production of many pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6 and MCP-1.6 Increasing evidence suggests that NF-κB activation contributes to the inflammation response and functional depression of myocardium; inhibition of myocyte NF-κB also benefits myocardial inflammation.7–9 Importantly, inhibition of NF-κB activation prevents LPS-induced cardiac dysfunction.7 Accordingly, NF-κB signalling pathway may play an important role in treating myocardial dysfunction in sepsis. Besides, activation of NF-κB is regulated by protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) pathways which include p38 MAPK, extracellular-signal-regulated kinase (ERK), and c-Jun N-terminal kinases (JNK).10,11
Aralia elata (Miq) Seem, also named Longya Aralia chinensis L. in china, belongs to Araliaceae. This species is widely distributed in China and also found in Russia, Japan and South Korea.12 Its bark and root bark are traditionally used for treating diabetes, gastric ulcer, hepatitis and rheumatoid arthritis in Chinese folk medicine.13 Major components identified in A. elata (Miq) Seem include saponins, aetherolea, alkaloids, tannins, coumarins and other components, such as amino acids and mineral elements.14 The main active ingredients of A. elata are saponins,12 which have various pharmacological activities, such as anti-inflammatory,15 liver-protecting,16 anti-oxidative,17 and hypolipidemic functions.18 Previous studies also indicated that the total saponins of A. elata (Miq) Seem (TAS) have strong cardiac protective effects in various diseases. For example, TAS could strengthen heart activity12,19 and prevent myocardial injury in ischemic cardiomyopathy.20,21 Long Ya Guan Xin Kang Jiao Nang (Chinese name) is a Chinese pharmaceutical drug, and its main pharmacodynamic ingredient is TAS. Long Ya Guan Xin Kang Jiao Nang has shown a conspicuous effect on treating angina pectoris.22 However, whether TAS processed an anti-inflammatory property on cardiac inflammation and the possible molecular mechanisms remain unknown. In the present study, we evaluated the anti-inflammatory effects of TAS on LPS-induced acute cardiac inflammation in a mouse model.
Materials and methods
Material
Roots of A. elata (Miq) Seem were collected from Jilin Province of China in September 2010 and were identified by Professor Zhongkai Yan (Academy of Chinese Medical Sciences of Jilin Province). A voucher specimen (no. 20100920) was deposited in the same department. Extraction, purification and quality control of TAS have been described in our previous study.19 All antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All chemicals were purchased from Sigma (St. Louis, MO, USA).Experimental animals
Male C57BL/6 mice (18 g to 20 g) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China. The mice were acclimatized one week before the experiment. Food and water were freely available, and environment was controlled at 24 °C to 25 °C room temperature, 55% humidity and light conditions (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light:dark cycle). All the procedures were approved by the Laboratory Animal Ethics Committee of the Institute of Medicinal Plant Development, Peking Union Medical College with the (registration number: #IMPLAD2012112207).
:12 h light:dark cycle). All the procedures were approved by the Laboratory Animal Ethics Committee of the Institute of Medicinal Plant Development, Peking Union Medical College with the (registration number: #IMPLAD2012112207).
Experimental design
The mice were randomly divided into 5 groups, each group contained 15 mice, including Cont group, LPS group (10 mg kg−1, normal saline dissolved), LPS + TAS groups (35, 70 and 140 mg kg−1; distilled water dissolved with 0.5% Tween 80). Cont group, LPS group, and LPS + TAS groups were dosed intragastrically with distilled water or TAS (35, 70, and 140 mg kg−1) for 7 days. At 1 h after the last administration, LPS group and LPS + TAS groups were intraperitoneally injected with LPS (10 mg kg−1; 200 μL per mouse). The Cont group received sterile saline only. At 6 h after LPS administration, heart tissues were fixed in 4% buffered paraformaldehyde for histology and immunohistochemistry or were frozen at −80 °C for protein analyses. Another experiment was conducted and survival was monitored once every 2 hours for up to 24 h.Echocardiographic measurements
As previously described, M-mode echocardiography was performed using Vevo 770™ High Resolution Imaging System (VisualSonics Inc., Canada).23 About 6 h after LPS administration, the mice were anaesthetised and their chests were shaved. The mice were then placed in recumbent position. Left ventricular (LV) internal diameter in systole (LVIDs) and diastole (LVIDd), LV posterior wall thickness in systole (LVPWs) and diastole (LVPWd) were measured using M-mode echocardiography. LV end-diastolic volume (LVVd), LV end-systolic volume (LVVs), fractional shortening (FS), and ejection fraction (EF) were automatically calculated using an ultrasound machine.Serum analysis of LDH, CK, AST, and cTnI
Activities of serum myocardial enzymes, including lactate dehydrogenase (LDH), creatine kinase (CK), and aspartate transaminase (AST), were measured with the corresponding detection kit according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering, China). Serum levels of cardiac troponin I (cTnI) was quantified by ELISA according to the manufacturer's instructions (R&D Systems, Wiesbaden, Germany).Histological and immunohistochemical analysis
Heart tissues were fixed in 4% buffered paraformaldehyde, dehydrated in graded ethanol and then embedded in paraffin max. The heart apex was sectioned and stained with hematoxylin and eosin. The structure was then examined under a light microscope (CKX41, 170 Olympus, Tokyo, Japan) by a pathologist blinded to the groups under study.For immunohistochemical analysis, Slides were deparaffinaged and hydrated, and endogenous peroxydase were blocked by hydrogen dioxide. Sections were incubated with goat anti CD 68 monoclonal antibody and then stained using DAB kit. Finally, slides were re-stained with hematoxylin, mounted and observed by light microscopy. Samples were examined in a blind fashion. Briefly, myocardial CD68-positive cells were counted on nine random fields on each slide with a magnification of 200×. The infiltration of myocardial leukocytes or CD 68-positive cells was expressed as the average number of leukocytes or CD 68-positive cells per field (n = 15 per group).24
Measurement of TNF-α, IL-1β, MCP-1, and IL-6 by ELISA
Serum levels of TNF-α, IL-1β, IL-6 and MCP-1 were quantified by ELISA according to the manufacturer's instructions (R&D Systems, Wiesbaden, Germany).Measurement of ROS level in heart tissue
Heart tissues were added with saline at a ratio of 1:9 (mg μL−1) to form homogenate. After centrifugation at 1000 rpm for 10 min, supernatant was used to measure the activity of ROS with the corresponding detection kit according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering, China).
Western blot analysis
Heart tissues were added with saline at a ratio of 1:9 (mg μL−1) to form homogenate. After centrifugation at 7000 rpm for 5 min, precipitation was lysed on ice with tissue-protein extraction reagent containing 0.1 mM dithiothreitol and proteinase inhibitor cocktail. Protein concentration was determined using a BCA kit (Pierce Corporation, Rockford, USA). Equal amounts of protein fractions were separated by 12% SDS-PAGE and then transferred onto nitrocellulose membranes (Millipore Corporation, USA) in tris-glycine buffer at 100 V for 55 min. The membranes were blocked with 5% (w/v) non-fat milk powder in tris-buffer that containing 0.05% (v/v) Tween-20 (TBST) for 2 h at room temperature. After overnight incubation with appropriate primary antibodies at 4 °C, the membranes were washed thrice with TBST and incubated with secondary antibodies for 2 h at room temperature and then washed again with TBST thrice. The protein blots were developed using an enhanced chemiluminescence solution. Protein expression levels were visualised with Image Lab Software (Bio-Rad, USA).
Statistical analysis
Results from at least three independent experiments were expressed as mean ± SE. Statistical comparisons between different groups were measured with Student's t-test or ANOVA using Prism 5.00 software. Statistical significance was considered at p < 0.05.Results
Effects of TAS on survival rate in LPS-treated mice
To understand the protective role of TAS in the inflammatory setting, we evaluated the effect of TAS in sepsis-induced mortality. The mice were pretreated with TAS (35, 70 or 140 mg kg−1, i.g.) or vehicle and then challenged with lethal doses of LPS. Animal survival was monitored for up to 24 h. As shown in Fig. 1, the mice began to die at 8 h after the LPS-treatment. However, the mice treated with TAS exhibited significantly prolonged survival compared with the animals pretreated with the vehicle, and a larger protection was conveyed by TAS at the higher dose relative to the lower dose. No mouse died within 6 h. Therefore, the mice treated with LPS for 6 h were used in the next experiments. As shown in Fig. 2, the mice in control group looked perfectly healthy with smooth hair and quick action. However, 6 h after LPS administration, the mice were keeping camponotus state with sparse hair and less dynamic. TAS pretreatment could improve the health status of mice in a dose-dependently manner.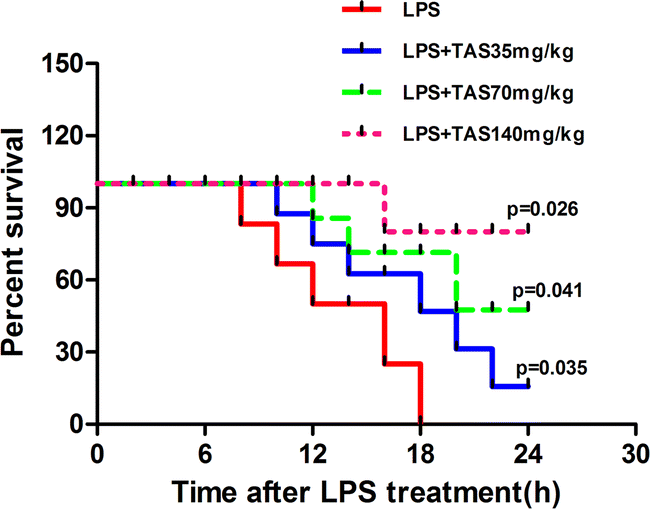 | ||
| Fig. 1 Effects of TAS on mice survival rate. | ||
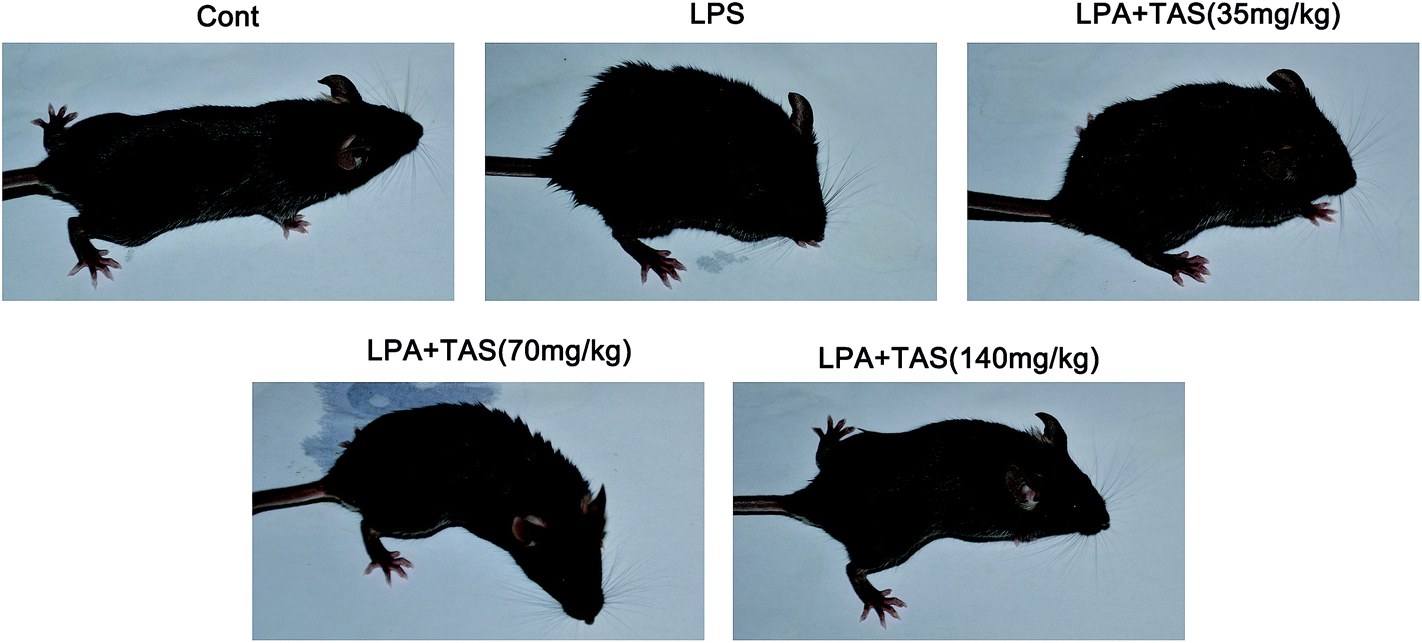 | ||
| Fig. 2 Effects of TAS on health status of mice at 6 h after LPS treatment. | ||
Effects of TAS on heart function in LPS-treated mice
To investigate the effect of TAS on heart function of LPS-treated mice, M-mode echocardiography was used to measure cardiac parameters. LPS administration significantly decreased the cardiac function in mice as evidenced by the decrease in EF, FS, LVVd and the increase in LVVs compared with saline-treated controls. TAS pretreatment prevented the changes of EF, FS, and LVVs but had no effect on LVVd (Fig. 3).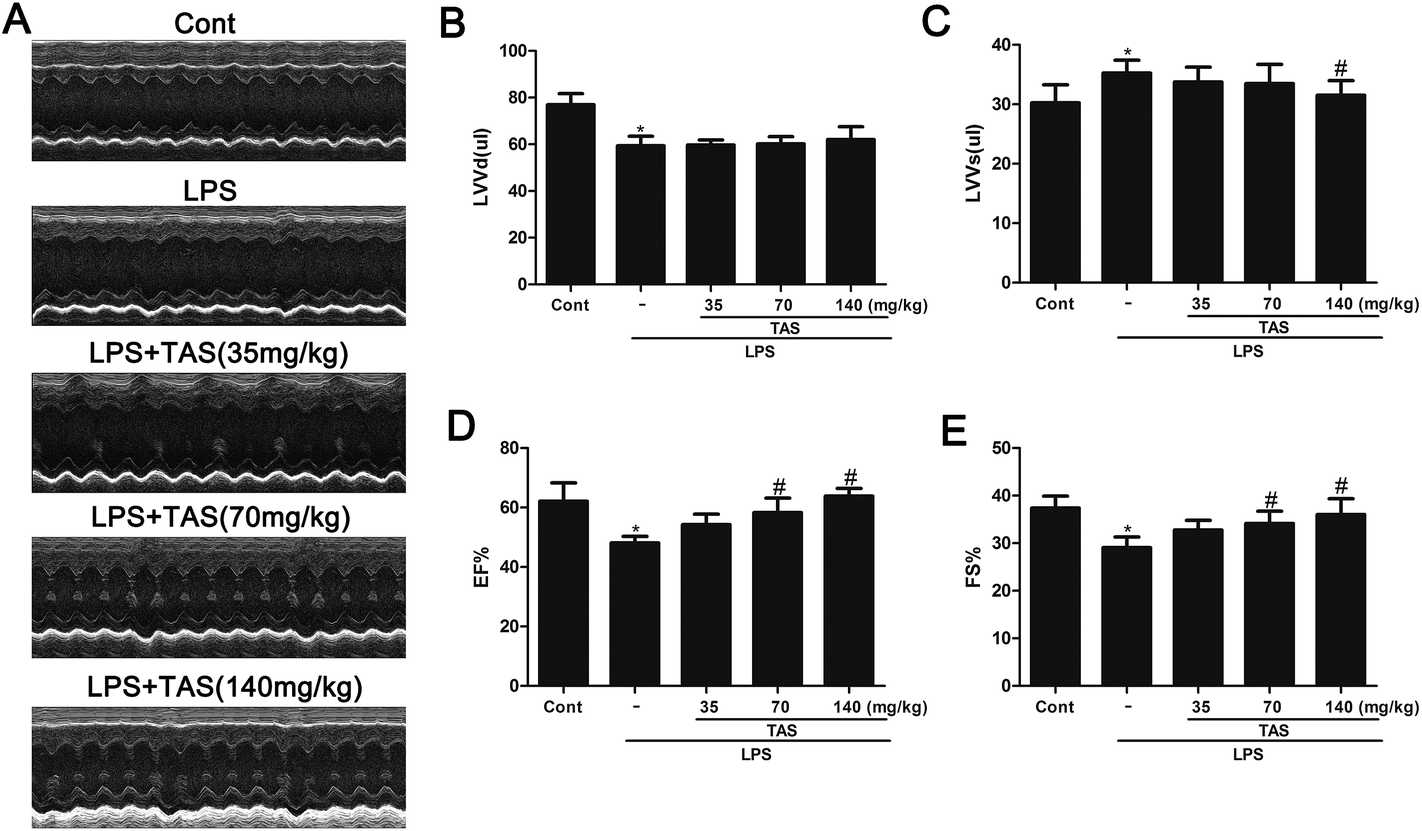 | ||
| Fig. 3 Effects of TAS on left ventricular functions in mice. (A) Representative images of M-mode echocardiogram. Echocardiography values are expressed as mean ± SD. (B) EF, ejection fraction; (C) FS, fractional shortening; (D) LVVd, left ventricular end-diastolic volume; (E) LVVs, left ventricular end-systolic volume. Each bar was represented as mean ± SD (n = 10); *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
Effects of TAS on LDH, CK, AST, and TnT activities in LPS-treated mice
LDH, CK and AST are three important cardiac function indicators. As shown in Fig. 4, LPS significantly increased activities of LDH, CK and AST in the serum of mice, which indicated a severe cardiac injury. Pretreatment with different doses of TAS could depress these elevations induced by LPS at varying degrees.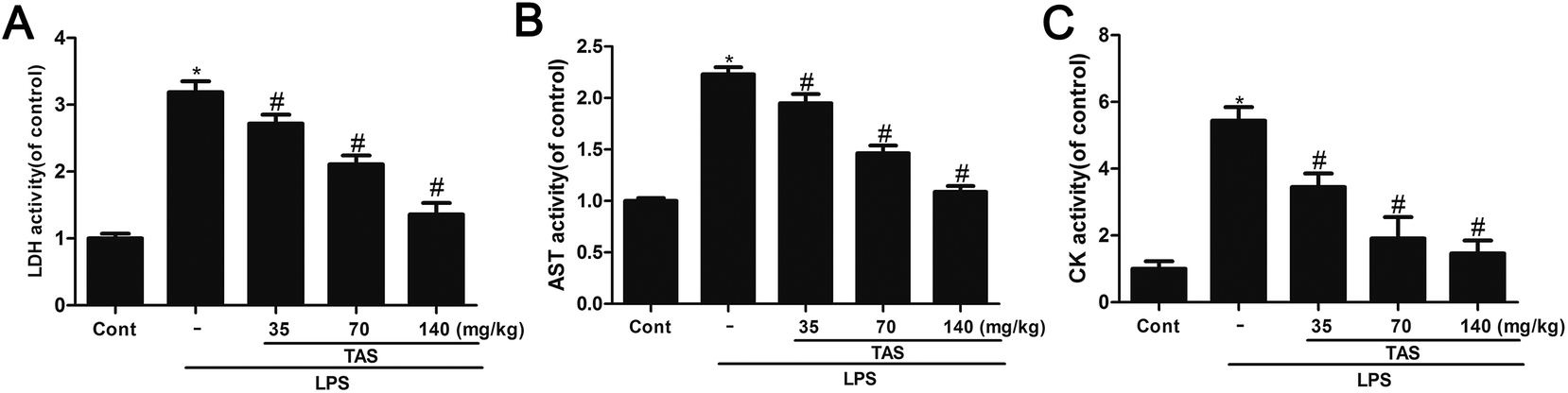 | ||
| Fig. 4 Effects of TAS on myocardial enzyme activities. (A) The effect of TAS on LDH level. (B) The effect of TAS on AST level. (C) The effect of TAS on CK level. Each bar was represented as mean ± SD (n = 10); *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
cTnI is a protein that controls the interaction between actin and myosin, which mediated by calcium. The release of cTnI in the blood positively correlates with myocardial damage.25 In order to further understand the protective effect of TAS on LPS-induced cardiac injury in mice, serum levels of cTnI were measured by ELISA. LPS treatment significantly increased the serum levels of cTnI compared with saline-treated control group, which was significantly attenuated by TAS pretreatment (Fig. 5).
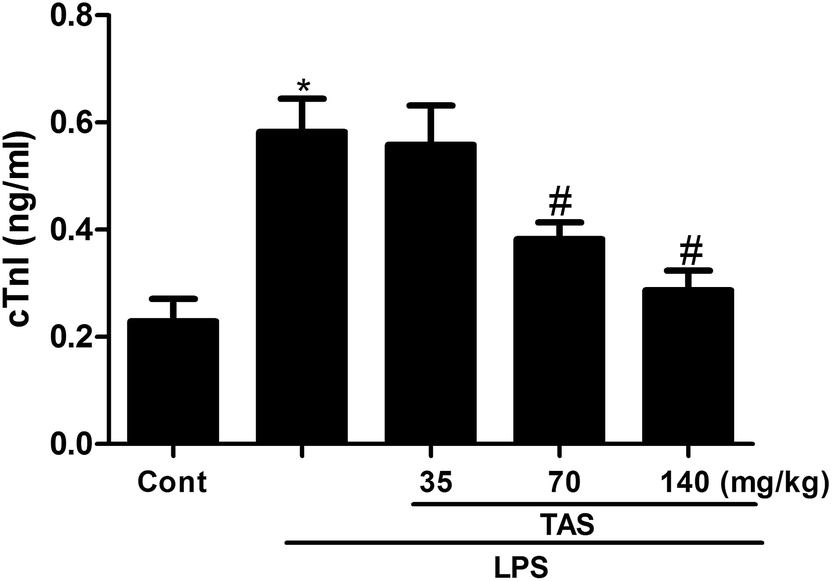 | ||
| Fig. 5 Effects of TAS on serum level of cTnI. Each bar was represented as mean ± SD (n = 10); *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
Effects of TAS on LPS-induced heart damage
As shown in Fig. 6A, LPS administration significantly increased erythrocyte leakage and leukocyte infiltration into the cardiac interstitium, as observed by using H&E staining. Besides, CD68-positive cells, representing monocyte/macrophage in an activated state, were increased in the heart tissues after LPS challenge (Fig. 6B). In contrast, TAS pretreatment obviously attenuated LPS-induced neutrophil/leukocyte infiltration.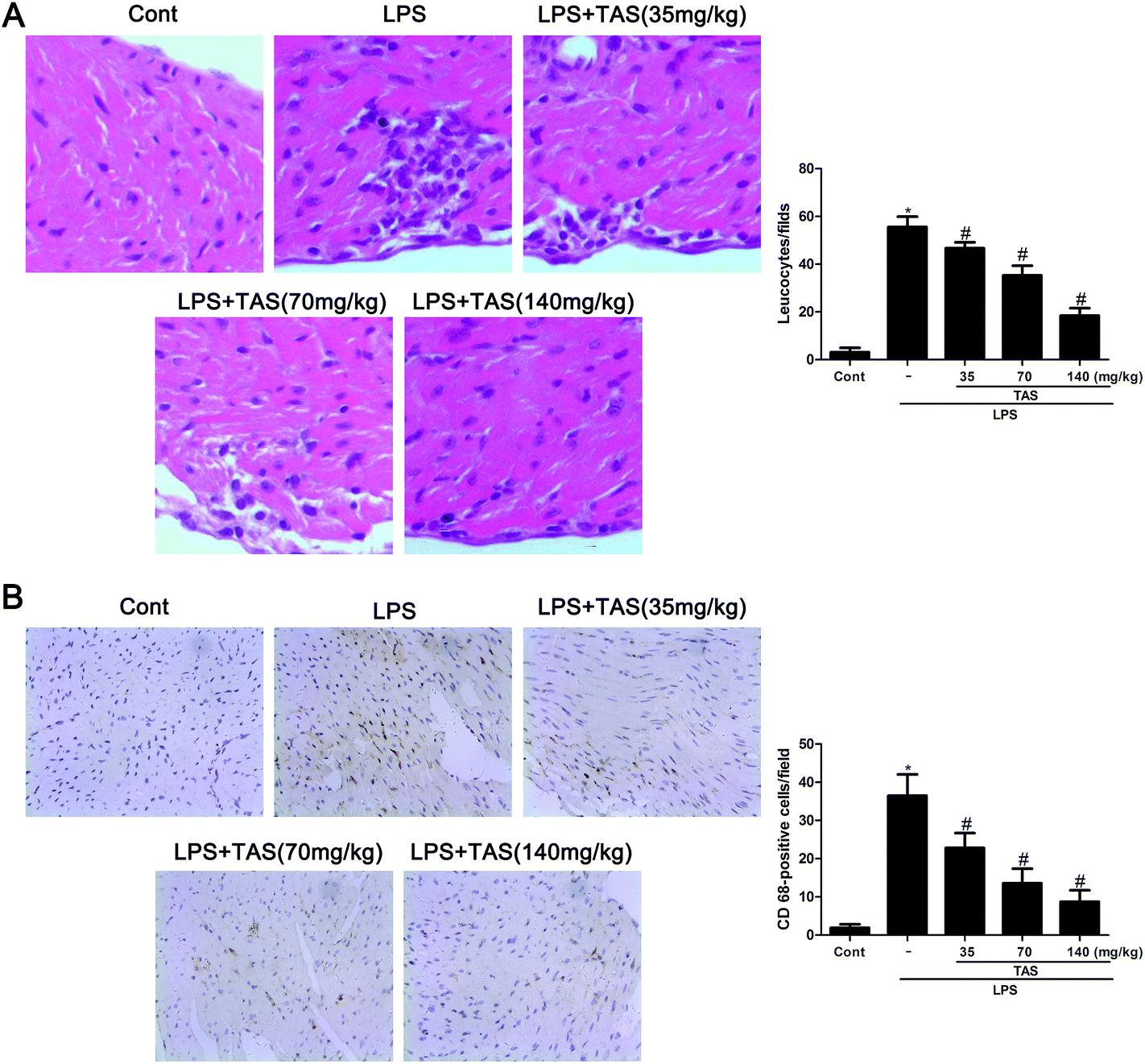 | ||
| Fig. 6 Effects of TAS on neutrophil/leukocyte infiltration. After 6 h LPS administration, hearts were harvested and sectioned for HE counterstaining (A) or immunohistochemistry (B). Infiltrated leukocytes or CD68-positive cells were calculated. Arrowheads in A indicate infiltrated leukocytes; arrowheads in B indicate 68-positive cells. Each bar was represented as mean ± SD (n = 10); *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
Effects of TAS on inflammatory cytokine production in LPS-treated mice
The protein levels of TNF-α, IL-1β and IL-6 in heart tissues of mice significantly increased after 6 h of LPS exposure compared with those of saline-treated controls (Fig. 7A and B). This increase was significantly attenuated by TAS-pretreatment, suggesting that TAS pretreatment leads to the suppression of myocardial inflammatory responses during endotoxemia.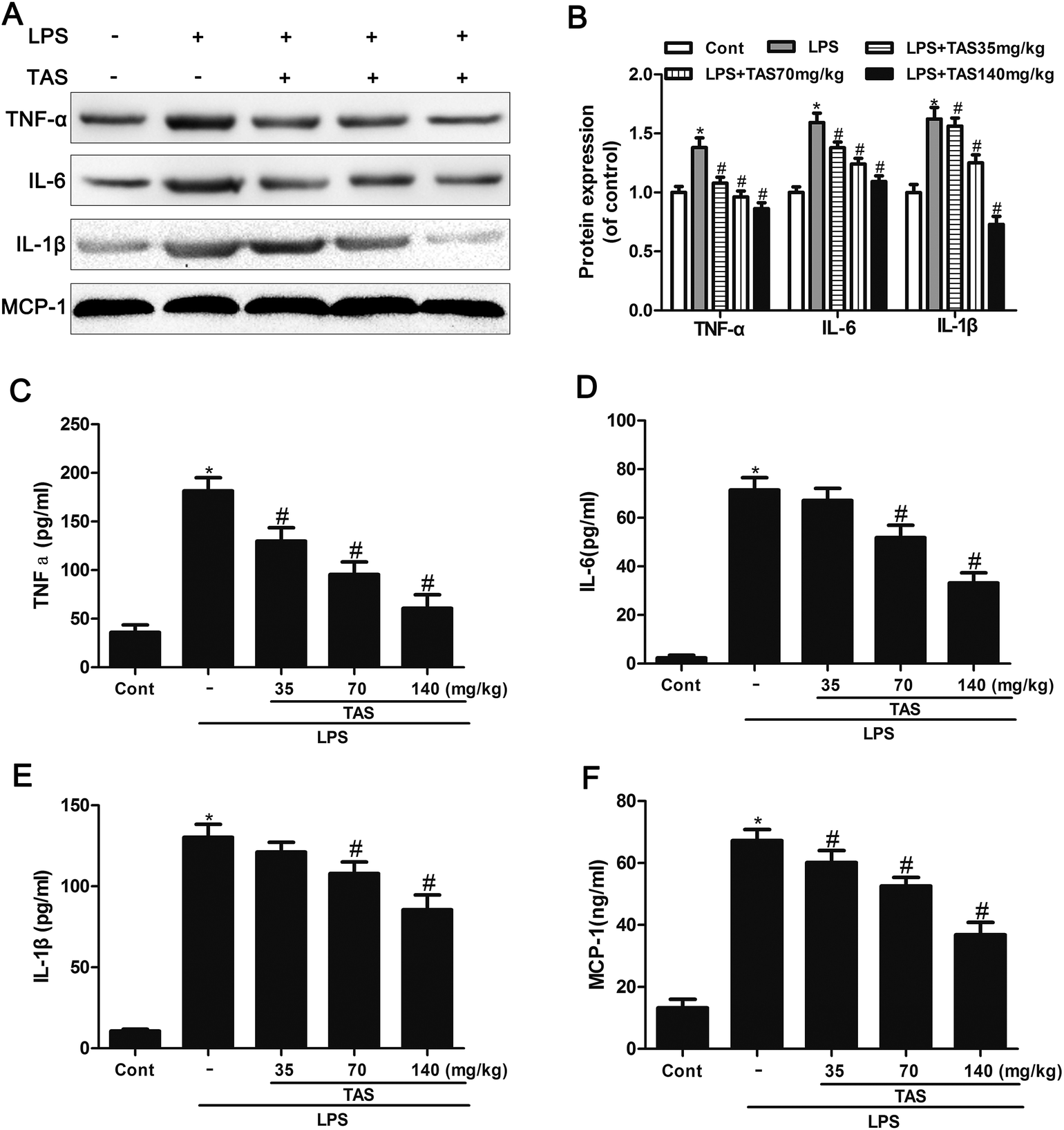 | ||
| Fig. 7 Effects of TAS on the levels of inflammatory cytokines in LPS treated mice. (A) Myocardial TNFα, IL-1β, and IL-6 expression was assayed by western blot analysis. (B) Quantification of protein expression. (C–F) The serum levels of TNF-α, IL-1β, IL-6, and MCP-1 were measured by ELISA. Each bar was represented as mean ± SD (n = 10); *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
ELISA was used to determine the inflammatory cytokine levels in response to LPS stimulation. Serum levels of IL-1β, IL-6, TNF-α, and MCP-1 in the LPS treated mice increased significantly compared with those in the control group. TAS pretreatment prevented the increase of these cytokines in a dose-dependent manner (Fig. 7C–F).
Effects of TAS on the LPS-induced imbalance between eNOS and iNOS
LPS administration induced a significant decrease in the eNOS levels and increase in the iNOS levels compared with saline-treated control group, which was significantly attenuated by TAS pretreatment (Fig. 8A).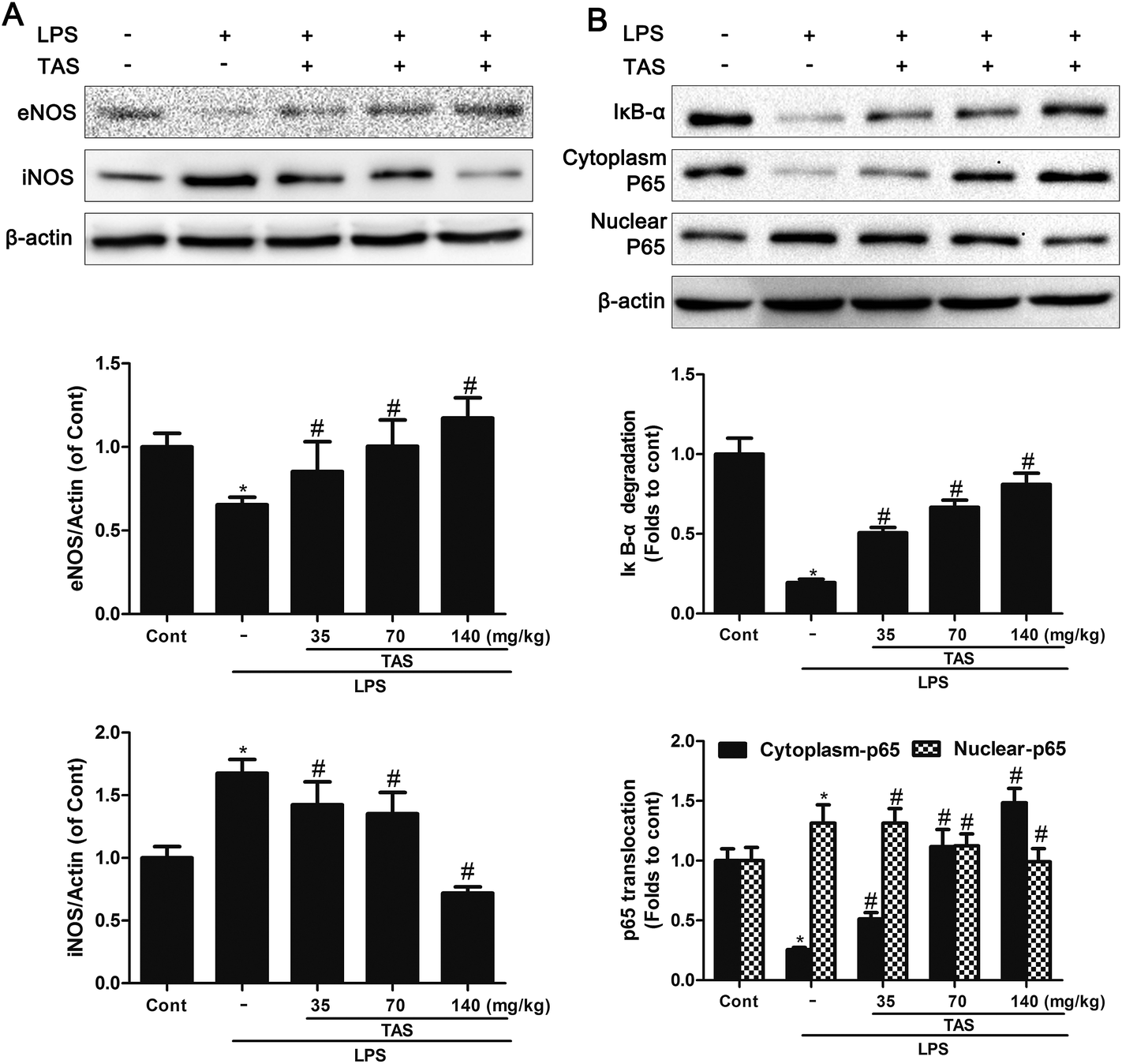 | ||
| Fig. 8 Effects of TAS on the levels of eNOS, iNOS and NF-κB activation. (A) Protein levels of eNOS and iNOS in the myocardium examined by western blot analysis. (B) Protein levels of IκB and NF-κB in the myocardium examined by western blot analysis. Each bar was represented as mean ± SD from three independent experiments; *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
Effects of TAS on NF-κB singling pathway
NF-κB singling pathway in myocardial is activated after cardiac dysfunction during sepsis/septic shock.26 The LPS group showed a significant increase in IκB-α degradation. Besides, NF-κB p65 decreased in the cytoplasm protein extracts but increased in nuclear after LPS-treatment. By contrast, TAS significantly inhibited translocation of p65 from cytoplasm to nucleus and decreased IκB-α degradation as opposed to the LPS group (Fig. 8B).Effects of TAS on NOX2 and ROS formation
Here, we investigated the effect of LPS on endothelial ROS production. Results showed that LPS treatment induced significant increase in ROS formation, which could be inhibited by TAS (Fig. 9A). NOX2, an important member of NADPH oxidase isoforms, has been shown to play a key role in ROS production in cardomyocytes.27 In LPS-treated mice, NOX2 protein expression was increased dramatically. However, pretreatment with TAS markedly reduced LPS-induced NOX1 expression (Fig. 9B).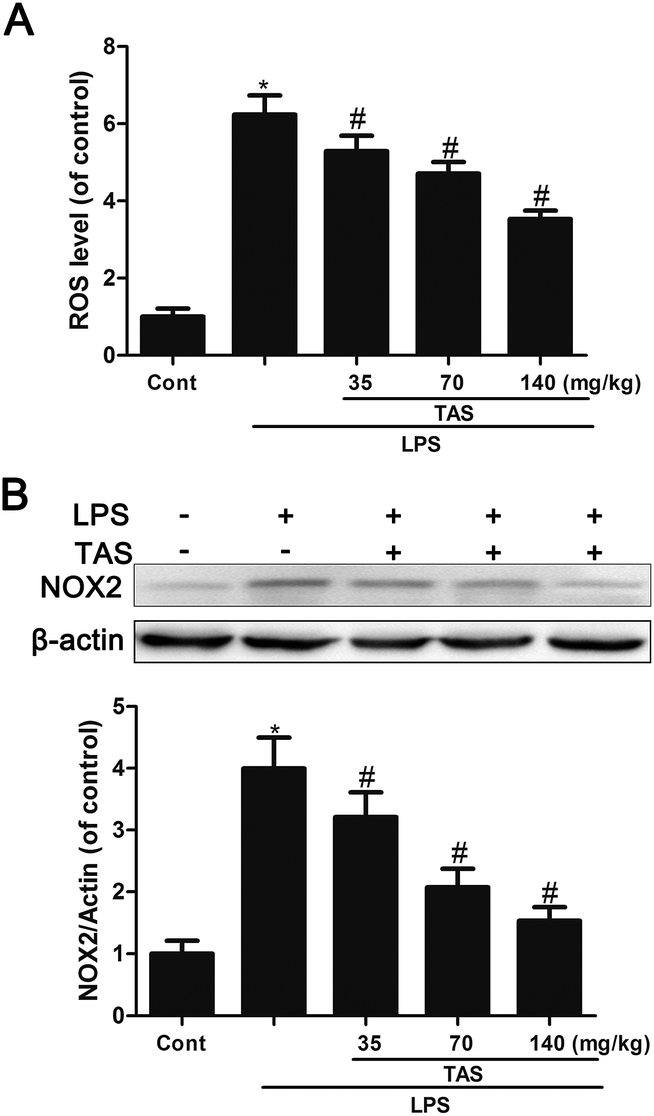 | ||
| Fig. 9 Effects of TAS on the expression of NOX1 and ROS level in heart tissue of mice. (A) Protein level of NOX1 in the myocardium examined by western blot analysis. (B) ROS levels in heart tissues of mice. Each bar was represented as mean ± SD from three independent experiments; *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
Effects of TAS on AKT, TLR-4 and MAPK signaling pathway
AKT and MAPK signaling play an important role in modulating the NF-κB signaling pathway.10 As shown in Fig. 10, LPS stimulation significantly increased protein expression of TLR-4 and phosphorylation of JNK, ERK, p38 MAPK, and AKT in comparison to the control group. TAS pretreatment dramatically inhibited the phosphorylation of JNK, ERK, p38 MAPK, and AKT. These results demonstrated that TAS inhibited the activity of MAPK and AKT pathways in LPS-induced endotoxemia. On the other hand, TAS had no obvious effect on the LPS-induced up-regulation of TLR-4.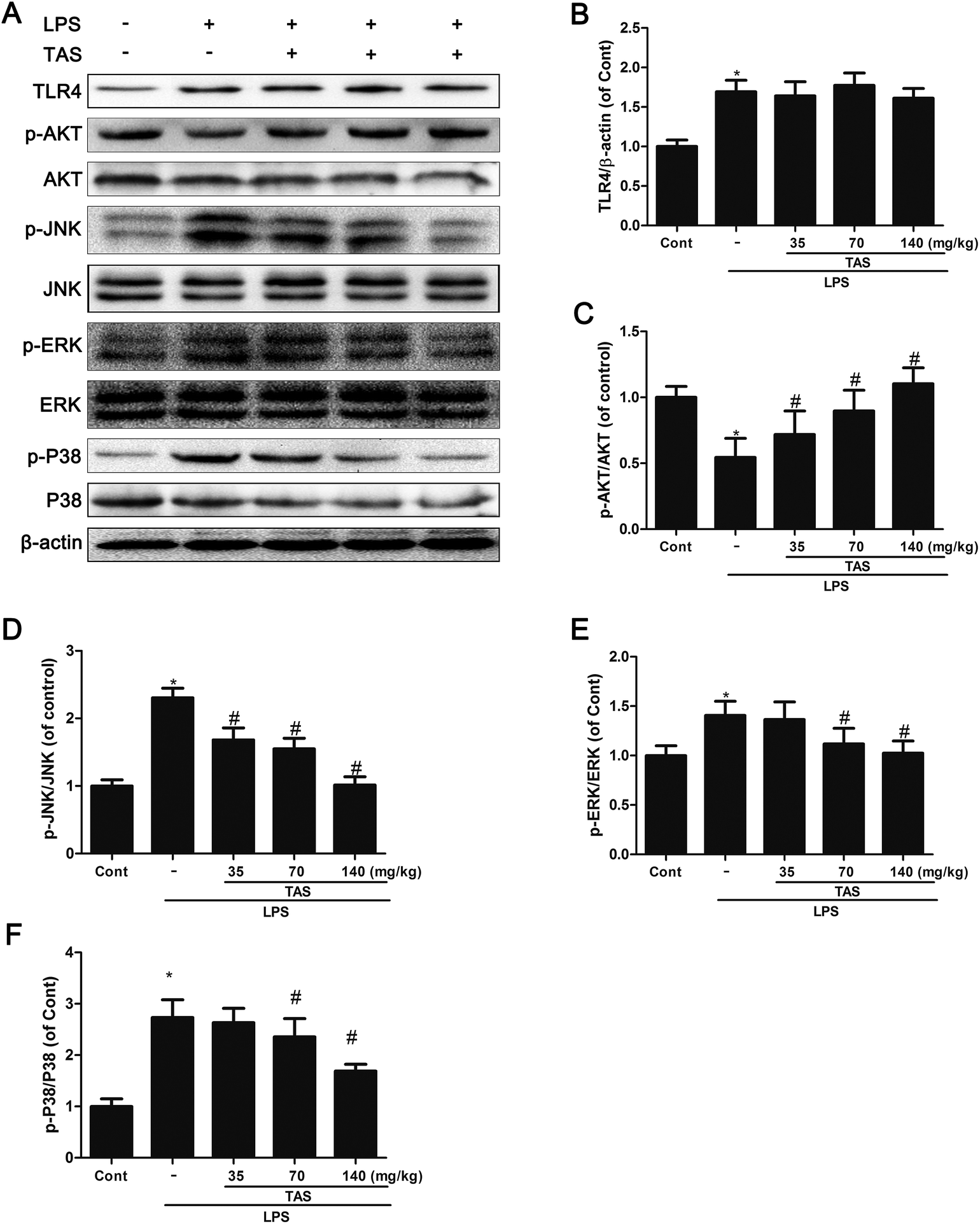 | ||
| Fig. 10 Effects of TAS on the expression of TLR-4 and phosphorylation of ERK1/2, JNK, P38, and AKT. The protein levels of phospho-ERK, phospho-JNK, phospho-P38, phospho-Akt, and TLR4 in the myocardium were examined by western blot analysis. Each bar was represented as mean ± SD from three independent experiments; *P < 0.05 vs. control group; #P < 0.05 vs. LPS group. | ||
Discussion
Aralia elata (Miq) Seem is one of the main anti-rheumatics of Chinese herbs, which has been commonly used for expelling wind and removing dampness for thousands of years. Saponins are the main components of these herbal extracts, which possess various pharmacological properties, such as anti-inflammatory, liver-protecting, anti-oxidative and hypolipidemic functions.15–17,28–30 Our previous study revealed the positive inotropic effect of TAS on canine myocardium and isolated rat cardiomyocytes.19 However, no information exists regarding the effects of TAS on LPS-induced heart inflammation. To expand the application of this agent further, anti-inflammatory effects of TAS have been investigated in the current study using LPS-induced sepsis model. The results indicated that TAS prolonged the survival rate and improved the health status of mice in a dose-dependent manner. Further study revealed that TAS prevented LPS-induced heart dysfunction and suppressed secretion of pro-inflammatory mediators by inhibiting of NF-κB pathways.LPS derived from Gram-negative bacteria is the most important pathogen leading to sepsis development and has been used as a common experimental model of sepsis. As an important organ system frequently affected by sepsis, the cardiovascular system and its dysfunction during sepsis are associated with a significantly increased mortality rate compared with septic patients without cardiovascular impairment,31 which has been intensively studied. We found that mice exposure with LPS showed poor health status and high mortality compared with the control group. LPS exposure for 6 h also decreased myocardial contraction function, which is consistent with another study.32 The cardiac dysfunction was evoked shortly after mice exposure to LPS. Therefore, TAS treatment should be preventive or administered as early as possible before LPS administration. The dose of TAS was chosen based on the results of previous studies.19,33 TAS in 35, 70, 140 mg kg−1 could significantly improve the survival rate and myocardial function of mice in a dose dependent manner. To investigate the LPS injury on cardiomyocytes further, myocardial enzyme activities were measured. LPS increased LDH, CK, AST, and cTnI levels in the myocardium tissues, which was attenuated by TAS pretreatment. Our study firstly proved the protective effects of TAS on LPS-induced cardiac depression.
Neutrophil/leukocyte infiltration serves an important function in cardiac dysfunction during sepsis/septic shock. Our study indicated that LPS administration significantly increased neutrophil/leukocyte infiltration in the myocardium, which was accompanied by the elevated myocardial expression of a series of pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, and MCP-1. Pro-inflammatory mediators are major contributors to the development of myocardial dysfunction in sepsis.4 TNF-α is an important early mediator of endotoxin-induced shock.34 Studies have shown that TNF-α is also secreted by cardiac myocytes in response to sepsis. Application of anti-TNF-α antibodies improved LV function in patients with septic shock.35,36 Mice with cardiac targeted expression of MCP-1 show inflammatory cell infiltration, iNOS expression, LV hypertrophy, dilation, fibrosis and contractile dysfunction.37 IL-1 plays a crucial role in the systemic immune response and depresses cardiac contractility by stimulating NO synthase.38 Similar to a previous report, our data confirmed the role of myocardial inflammatory activation in myocardial dysfunction after LPS challenge.32 However, TAS pretreatment effectively decreased the neutrophil/leukocyte infiltration in the myocardium and the serum levels of pro-inflammatory mediators. Theses results implied that the anti-inflammatory effect of TAS may be associated with its cardioprotective effects.
As reported in the literature, NO is responsible for direct effects on vascular tone, inhibition of mitochondrial respiration and further release of pro-inflammatory cytokines.39 LPS and pro-inflammatory cytokines consequently up-regulate iNOS activity in cardiomyocytes.40 The expression of iNOS increased in the myocardium during sepsis, thereby producing high-level NO, which importantly contributes to myocardial dysfunction.41 Our study also showed that LPS administration caused an imbalance between eNOS and iNOS in the myocardium, which was significantly inhibited by TAS pretreatment.
TLRs expressed in the heart may trigger immune response and promote functional depression of the heart during sepsis.26 Substantial evidence indicates that LPS induced myocardial inflammation and dysfunction by interacting with its ligand TLR4 on cardiomyocytes, thus activating multiple signalling pathways, such as NF-κB.5 NF-κB significantly controls the production of many pro-inflammatory cytokines.26 In our current study, after stimulated with LPS, NF-κB was activated and translocated into the nucleus as a result of phosphorylation-mediated degradation of IκB proteins in the heart of sepsis mice. However, pre-treatment with TAS decreased the degradation of IκB-α and nuclear translocation of p65 NF-κB. These results demonstrated that TAS specifically attenuated LPS-induced activation of NF-κB and downstream TNF-α, IL-1β, IL-6 and MCP-1 production, even though TAS could not attenuate the activation of TLR-4.
It is well documented that MAPKs family serves an important function in LPS-induced inflammatory reaction that contributes to the development of septic cardiac dysfunction.42 MAPKs activation initiate the induction of downstream transcription factors like NF-κB.43 A previous study suggests that the activation of ERK1/2, JNK, and p38 is required for LPS-induced TNFα expression.42 Western blot assays showed that the nuclear translocation of p65 was accompanied by phosphorylation of JNK, ERK, and P38-MAPK in LPS-treated mice. TAS pretreatment strongly suppressed MAPKs family activation. These results suggested that TAS might suppress LPS-induced acute inflammatory responses by inhibiting MAPKs signaling pathway in mice. MAPK is sensitive to oxidative stress. NAD(P)H oxidase has been shown to be a major source of oxidative stress in the myocardium. A previous study showed that NOX2/NADPH oxidase was documented in LPS-induced activation of MAPKs and corresponding TNF-α expression in cardiomyocytes.44 NOX2/NADPH oxidase may be the downstream of toll-like receptors and upstream of MAPKs in cardiomyocytes. Our study found that LPS treatment increased the expression of NOX2 and ROS formation in heart tissue of mice, which inhibited by TAS pretreatment. Further study is needed to evaluate whether TAS inhibits NOX2 directly or through other signaling pathway.
PI3K/Akt signaling pathway plays a crucial role in negatively regulating LPS-induced acute inflammatory responses in vitro and in vivo.45 Recent studies have demonstrated that the activation of PI3K/Akt will negatively regulate the NF-κB activation pathway and improve survival and cardiac function in septic mice.46 We found that TAS-pretreatment significantly attenuated the decrease of the phosphorylation of Akt in LPS-treated mice, indicating that TAS might inhibit LPS-induced acute inflammatory responses through PI3K/Akt activation. Previous study showed that PI3-K/Akt activation attenuated proinflammatory cytokines production in response to LPS in H9c2 cardiomyocytes by inhibiting the transcription of NF-κB and the phosphorylation of MAPKs family.47 Besides, PI3K/Akt activation can also inhibit activation of NOX2 and ROS formation.48 However, the main downstream targets of PI3K/Akt activation involved in mediating the protective effects remain to be identified. The mechanism of TAS protect against LPS-induced cardiac dysfunction needs further study.
In summary, our results firstly showed that pretreatment with TAS significantly attenuated LPS-induced myocardial inflammatory cytokine production, imbalance between iNOS and eNOS, and NF-κB activation, and improved myocardial dysfunction in septic mice. The mechanisms by which TAS attenuated cardiac dysfunction involve the preserved activation of the PI3K/Akt signaling pathway and inhibition of MAPKs family. These findings demonstrate the potential of TAS for treating endotoxin-induced cardiac dysfunction.
Conflict of interest
None declared.Abbreviations
| Akt | Protein kinase B |
| AST | Aspartate transaminase |
| CK | Creatine kinase |
| cTnI | Cardiac troponin I |
| EF | Ejection fraction |
| ERK1/2 | Extracellular signal-regulated kinase 1 and 2 |
| FS | Fractional shortening |
| I-κBα | Inhibitory-κBα |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| JNK | c-Jun N-terminal kinases |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| LVIDs | Left ventricular internal diameter in systole |
| LVIDd | Left ventricular internal diameter in diastole |
| LVPWs | Left ventricular posterior wall thickness in systole |
| LVPWd | Left ventricular posterior wall thickness in diastole |
| LVVd | Left ventricular end-diastolic volume |
| LVVs | Left ventricular end-systolic volume |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor-κB |
| NOX2 | NADPH oxidase 2 |
| PI3K | Phosphatidlinositide 3-kinase |
| TAS | Total saponins of A. elata (Miq) Seem |
| TLR | Toll-like receptor |
| TNF | Tumour necrosis factor |
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant no. 81173589), the Major Scientific and Technological Special Project for Significant New Drugs Formulation (Grant nos 2010ZX09401-305-47, 2010ZX09401-305-21, and 2012ZX09501001-004), and the Key Scientific and Technological Project of Jilin Province (Grant no. 11ZDGG008).References
- G. Martin, D. Mannino, S. Eaton and M. Moss, N. Engl. J. Med., 2003, 348, 1546–1554 CrossRef PubMed.
- S. L. Zanotti-Cavazzoni and S. M. Hollenberg, Curr. Opin. Crit. Care., 2009, 15, 392–397 CrossRef PubMed.
- M. W. Merx and C. Weber, Circulation, 2007, 116, 793–802 CrossRef CAS PubMed.
- A. Rudiger and M. Singer, Crit. Care Med., 2007, 35, 1599–1608 CrossRef PubMed.
- O. Avlas, R. Fallach, A. Shainberg, E. Porat and E. Hochhauser, Antioxid. Redox Signaling, 2011, 15, 1895–1909 CrossRef CAS PubMed.
- Z. Guolong and S. Ghosh, J. Endotoxin Res., 2000, 6, 453–457 CrossRef PubMed.
- S. B. Haudek, E. Spencer, D. D. Bryant, D. J. White, D. Maass, J. W. Horton, Z. J. Chen and B. P. Giroir, Am. J. Physiol.: Heart Circ. Physiol., 2001, 280, H962–H968 CAS.
- S. Frantz, K. Hu, B. Bayer, S. Gerondakis, J. Strotmann, A. Adamek, G. Ertl and J. Bauersachs, FASEB J., 2006, 20, 1918–1920 CrossRef CAS PubMed.
- M. Thielmann, Circ. Res., 2002, 90, 807–813 CrossRef CAS.
- C. Y. Lee, J. J. Yang, S. S. Lee, C. J. Chen, Y. C. Huang, K. H. Huang and Y. H. Kuan, J. Agric. Food Chem., 2014, 62, 6337–6344 CrossRef CAS PubMed.
- G. J. Huang, J. S. Deng, C. C. Chen, C. J. Huang, P. J. Sung, S. S. Huang and Y. H. Kuo, J. Agric. Food Chem., 2014, 62, 5321–5329 CrossRef CAS PubMed.
- X. D. Xu, J. S. Yang and Z. Y. Zhu, Yaoxue Xuebao, 1997, 32, 711–720 CAS.
- M. Li and W. Lu, Med. Recapitulate, 2009, 20, 3157–3160 Search PubMed.
- W. C. Dong, Zhongyao Tongbao, 1986, 11, 44–46 CAS.
- S. J. Suh, U. H. Jin, K. W. Kim, J. K. Son, S. H. Lee, K. H. Son, H. W. Chang, Y. C. Lee and C. H. Kim, Arch. Biochem. Biophys., 2007, 467, 227–233 CrossRef CAS PubMed.
- S. Saito, J. Ebashi, S. Sumita, T. Furumoto, Y. Nagamura and K. Nishida, Chem. Pharm. Bull., 1993, 41, 1395–1401 CrossRef CAS.
- M. Zhang, G. Liu, S. Tang, S. Song, K. Yamashita, M. Manabe and H. Kodama, Planta Med., 2006, 72, 1216–1222 CrossRef CAS PubMed.
- M. Yoshikawa, H. Matsuda, E. Harada and E. Elatoside, Chem. Pharm. Bull., 1994, 42, 1354–1356 CrossRef CAS.
- M. Wang, X. Xu, H. Xu, F. Wen, X. Zhang, H. Sun, F. Yao, G. Sun and X. Sun, J. Ethnopharmacol., 2014, 155, 240–247 CrossRef CAS PubMed.
- F. Wen, H. Xu, W. Zhang, T. Ding and X. Sun, Modernization of Traditional Chinese Medicine and Materia Medica-WORLD SCIENCE AND TECHNOLOGY, 2005, 7, pp. 5–8 Search PubMed.
- H. W. Deng, Y. J. Li, N. Shen, X. Chen and Z. C. Zhou, Chin. J. Pharmacol., 1988, 2, 20–23 CAS.
- D. Li, The clinical research of Long Ya Guan Xin Kang Jiao Nang on the treatment of angina pectoris patients with the syndrome of blood stasis due to qi deficiency (in chinese), Beijing University of Chinese Medicine, 2005, pp. 43–71 Search PubMed.
- X. Sun, R. C. Chen, Z. H. Yang, G. B. Sun, M. Wang, X. J. Ma, L. J. Yang and X. B. Sun, Food Chem. Toxicol., 2014, 63, 221–232 CrossRef CAS PubMed.
- Y. L. Wu, L. H. Lian, Y. Z. Jiang and J. X. Nan, J. Pharm. Pharmacol., 2009, 61, 1375–1382 CrossRef CAS.
- M. H. Hessel, D. E. Atsma, E. J. van der Valk, W. H. Bax, M. J. Schalij and A. van der Laarse, Pfluegers Arch., 2008, 455, 979–986 CrossRef CAS PubMed.
- T. Kawai and S. Akira, Trends Mol. Med., 2007, 13, 460–469 CrossRef CAS PubMed.
- B. Scott, Mol. Microbiol., 2015 DOI:10.1111/mmi.12946.
- J. H. Lee and C. S. Jeong, Environ. Toxicol. Pharmacol., 2009, 28, 333–341 CrossRef CAS PubMed.
- S. Xi, G. Zhou, X. Zhang, W. Zhang, L. Cai and C. Zhao, Exp. Mol. Med., 2009, 41, 538–547 CrossRef CAS PubMed.
- J. Zhang, H. Wang, Y. Xue and Q. Zheng, Carbohydr. Polym., 2013, 93, 442–448 CrossRef CAS PubMed.
- R. Bone, R. Balk, F. Cerra, R. Dellinger, A. Fein, W. Knaus, R. Schein and W. Sibbald, Chest, 1992, 101, 1644–1655 CrossRef CAS.
- B. Sun, J. Xiao, X. B. Sun and Y. Wu, Br. J. Pharmacol., 2013, 168, 1758–1770 CrossRef CAS PubMed.
- R. Li, X. Zhang, M. Ren, X. Xiao, H. Li and M. Liu, J. Chongqing Med. Univ., 2011, 36, 148–151 CAS.
- A. Sharma, S. Motew, S. Farias, K. Alden, H. Bosmann, W. Law and J. Ferguson, J. Mol. Cell. Cardiol., 1997, 29, 1469–1477 CrossRef CAS PubMed.
- J. Horton, D. Maass, J. White and B. Sanders, Am. J. Physiol.: Heart Circ. Physiol., 2000, 278, H1955–H1965 CAS.
- J. Vincent, r. J. Bakke, G. Marecaux, L. Schandene, R. Kahn and E. Dupont, Chest, 1992, 101, 810–815 CrossRef CAS.
- P. E. Kolattukudy, T. Quach, S. Bergese, S. Breckenridge, J. Hensley, R. Altschuld, G. Gordillo, S. Klenotic, C. Orosz and J. Parker-Thornburg, Am. J. Pathol., 1998, 152, 101–111 CAS.
- S. Francis, H. Holden, C. Holt and G. Duff, J. Mol. Cell. Cardiol., 1998, 30, 215–223 CrossRef CAS PubMed.
- R. Haque, H. Kan and M. Finkel, Basic Res. Cardiol., 1998, 93, 86–94 CrossRef CAS.
- T. Yamashita, S. Kawashima, Y. Ohashi, M. Ozaki, T. Ueyama, T. Ishida, N. Inoue, K. i. Hirata, H. Akita and M. Yokoyama, Circulation, 2000, 101, 931–937 CrossRef CAS.
- J. C. Preiser, H. Zhang, B. Vray, A. Hrabak and J. L. Vincent, Nitric Oxide, 2001, 5, 208–211 CrossRef CAS PubMed.
- W. J. Frazier, J. Xue, W. A. Luce and Y. Liu, PLoS One, 2012, 7, e50071 CAS.
- H. Nakano, M. Shindo, S. Sakon, S. Nishinaka, M. Mihara, H. Yagita and K. Okumura, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3537–3542 CrossRef CAS.
- T. Peng, X. Lu and Q. Feng, Circulation, 2005, 111, 1637–1644 CrossRef CAS PubMed.
- Y. Zong, L. Sun, B. Liu, Y. S. Deng, D. Zhan, Y. L. Chen, Y. He, J. Liu, Z. J. Zhang, J. Sun and D. Lu, PLoS One, 2012, 7, e44107 CAS.
- J. Downward, Science, 1998, 279, 673–674 CrossRef CAS.
- P. Yang, Y. Han, L. Gui, J. Sun, Y. L. Chen, R. Song, J. Z. Guo, Y. N. Xie, D. Lu and L. Sun, Biochem. Pharmacol., 2013, 85, 1124–1133 CrossRef CAS PubMed.
- J. Qin, Y. Y. Xie, L. Huang, Q. J. Yuan, W. J. Mei, X. N. Yuan, G. Y. Hu, G. J. Cheng, L. J. Tao and Z. Z. Peng, Nephrology, 2013, 18, 690–699 CAS.
| This journal is © The Royal Society of Chemistry 2015 |