DOI:
10.1039/C4RA16458J
(Paper)
RSC Adv., 2015,
5, 16422-16432
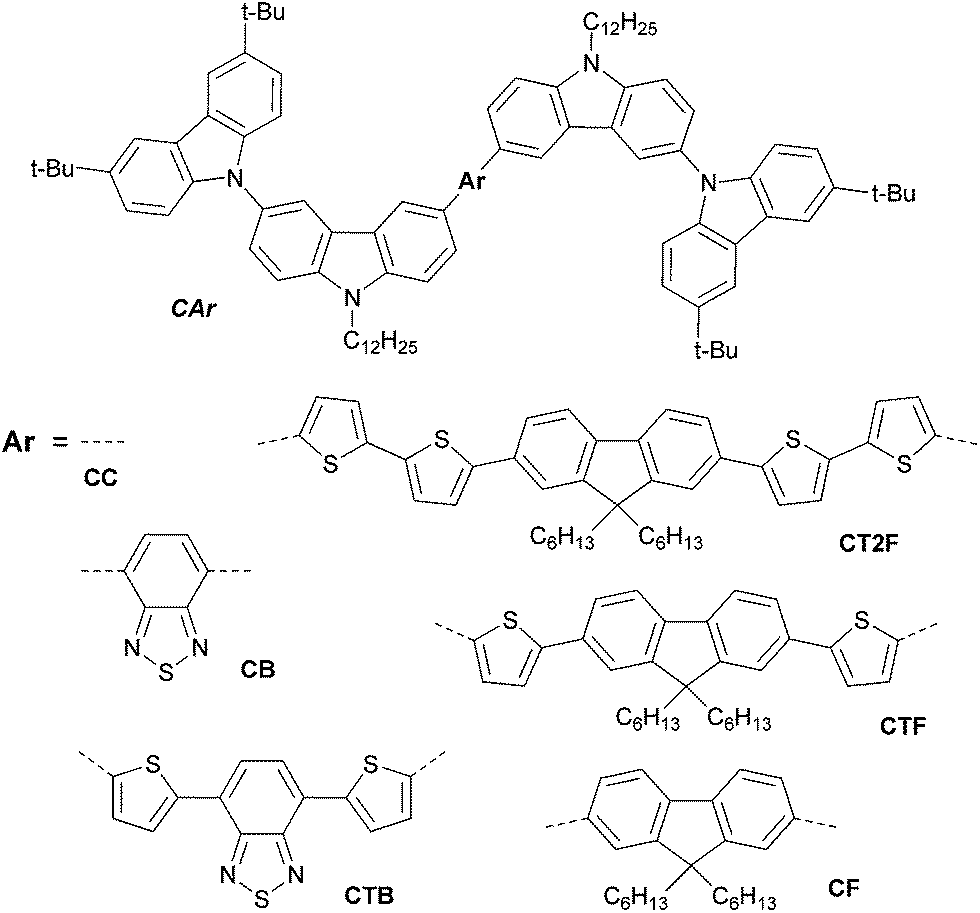
Oligoarylenes end-capped with carbazol-N-yl-carbazole as color tunable light-emitting and hole-transporting materials for solution-processed OLEDs†
Received
16th December 2014
, Accepted 27th January 2015
First published on 28th January 2015
Abstract
A series of oligoarylenes end-capped with carbazol-N-yl-carbazole (CAr) were synthesized and characterized as color tunable light-emitting materials. Their photophysical, thermal, electrochemical, and electroluminescent properties as non-doped solution processed emitters and hole-transporters for OLEDs were investigated. They exhibited a bright blue to red fluorescence with hole-transporting properties and could form morphologically stable amorphous thin films. Simple structured solution-processed CAr-based OLEDs emitted stable colors spanning the entire visible spectrum (blue, light blue, green, yellow and red) with luminance efficiencies up to 13.73 cd A−1. The CIE coordinates of the emitted colors were (0.67, 0.33) for red, (0.30, 0.61) for green and (0.19, 0.18) for blue, which are close to the pure red, green and blue colors. A solution-processed white OLED fabricated with a mixture of CAr radiated a white light with a luminance efficiency of 1.49 cd A−1 at 4.7 mA cm−2 and CIE coordinates of (0.33, 0.35).
Introduction
In recent years, organic light-emitting diodes (OLEDs) have arisen as potential candidates for next-generation displays and solid-state lighting due to their low driving voltage, high brightness and high efficiency.1 An important focus of current OLED research is on improving device structures and new efficient emitting materials. In terms of device fabrication, it is well-established that solution processing is the most favourable fabrication method of OLEDs for low cost and large-area applications.2 Using small molecule emitters also has great advantages, because the materials used are easy to synthesize and to purify.3 The efficient tunability of the emission spectra of OLEDs to a desired color is an important consideration when designing materials. Obviously the key point of materials development for full color-flat displays is to find materials emitting pure colors of red, green and blue with excellent emission efficiency and high stability, however, rationally tuning emission color over the entire visible range has also emerged as an important ongoing research task.4 A simple and efficient way to achieve full emission color tuning OLEDs is to change the molecular structure of the emitting materials. Color tuning has been achieved in conjugated polymers and small molecules by systematic modification of the chemical structure for example polythiophenes,5 poly(p-phenylene vinylene) derivatives,6 1,6-bis(N-phenyl-p-(R)-phenylamino)pyrenes,7 bis-dipolar diphenylamino-end capped oligoarylfluorenes,8 bis(3,6-di-tert-butylcarbazol-9-yl-phenyl)aniline end-capped oligoarylenes,9 tris(5-aryl-8-quinolinolate)Al(III) complexes10 and iridium complexes.11 However, there are some limitations in these materials including wide range of luminance efficiencies and their emission colors and EL spectra of the OLEDs do not span the entire visible spectrum. In term of material development, it is worth to explore new group of organic materials that can emit at wavelengths across the whole visible spectrum with all round good luminance efficiency, while possessing similar physical properties and processability. In this work, we therefore design a group of new solution processed non-doped emitters based on oligoarylenes end-capped with 3,6-di-tert-butylcarbazol-N-yl-carbazole, namely CAr (Fig. 1). The design involves the use of the 3,6-di-tert-butylcarbazol-N-yl-carbazole moieties as end-capping groups aiming to realize the following purposes: (1) to form nonplanar molecules for suppression of aggregation-induced fluorescence quenching of the planar conjugated core as well as improve solubility, preventing intermolecular interaction and inducing formation of amorphous state, (2) to increase the hole transporting capability so as to make the target molecules suitable as non-doped emitters, and (3) to provide sufficient molecular weight and viscosity to make the products suitable for solution process. Many of carbazole derivatives have been employed as excellent hole-transporting materials with superior thermal stability and glass-forming ability.12 While the structural modifications of oligoarylene fluorescent cores by either varying the degree of π-conjugation or using electron affinitive of the aryl cores such as fluorene, oligothiophene-fluorenes, 2,1,3-benzothiadiazole and 4,7-dithien-2-yl-2,1,3-benzothiadiazole will allow the fine-tuning of the emission colors of the materials. With this strategy simple solution processed hole-transporting non-doped emitters could be realized and simple structure full emission color tuning non-doped OLEDs including white OLED (WOLED) could be also fabricated. Herein, we describe a detailed synthesis, characterization, physical properties, and a study on OLED fabrication and performance of CAr.
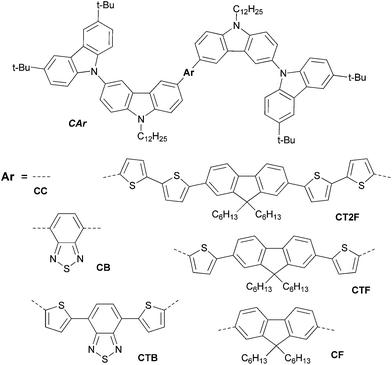 |
| | Fig. 1 Molecular structures of the designed oligoarylenes CAr. | |
Results and discussion
Materials synthesis
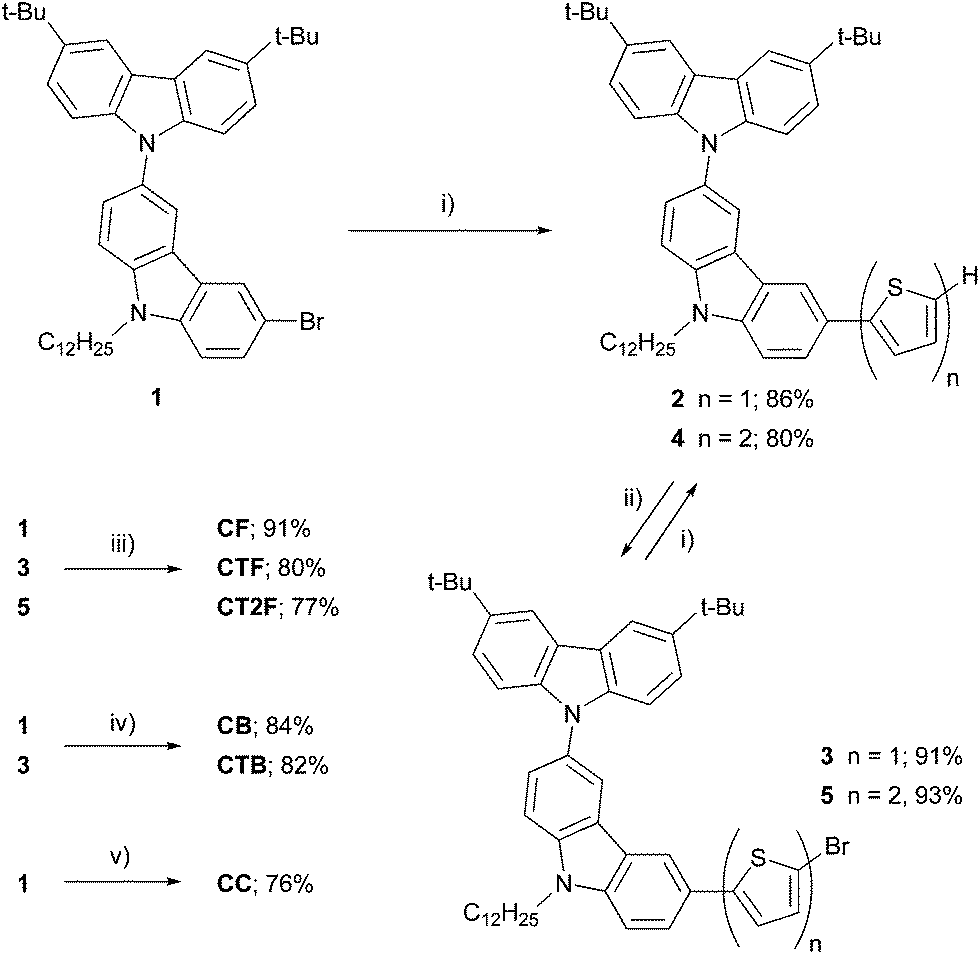
Scheme 1 shows the synthetic procedure for a series of carbazol-N-yl-carbazole end-capped oligoarylenes CAr (CC, CF, CTF, CT2F, CB and CTB). Compound 1 prepared by Ullmann coupling between 3,6-di-tert-butylcarbazole and 3,6-dibromo-N-dodecylcarbazole was first coupling with 2-thiophene boronic acid catalyzed by Pd(PPh3)4–Na2CO3 to give 2, whose the terminal thiophene then was brominated with NBS to give bromide 3 in a good yield. The resultant 3 was allowed to undergo the same sequence of reactions to provide bromide 5 in good yield over two steps. Cross-coupling reactions of these bromide intermediates 1, 3 and 5 with 9,9-dihexylfluororene-2,7-bis(trimethylene borate) in the presence of Pd(PPh3)4–Cs2CO3 gave CF, CTF and CT2F in moderate to good yields, while reactions of 1 and 3 with 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester) catalyzed by Pd(dppf)Cl2–K2CO3 provided CB and CTB in 82–84% yields. Stille coupling of 1 with hexabutylditin afforded dimer CC in 76% yield. The structures and purities of these compounds were analyzed by 1H NMR, 13C NMR and high resolution MALDI-TOF MS and found to be in good agreement with the structures. Colors of the solid products spread from white for CC to pale yellow for CF, yellow for CTF, yellow-orange for CT2F, orange for CB and purple for CTB, indicating a wide range in optical properties. These materials show high solubility in most organic solvents allowing their thin films could be fabricated by solution casting processes, which are desirable for low-cost processing for device fabrication.
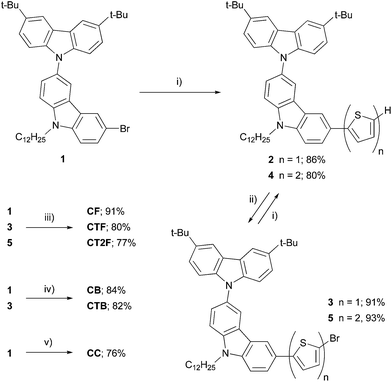 |
| | Scheme 1 Synthesis of the oligoarylenes CAr. Reagents and conditions: (i) 2-thiophene boronic acid, Pd(PPh3)4, 2 M Na2CO3, THF, heat; (ii) NBS, THF; (iii) 9,9-dihexylfluororene-2,7-bis(trimethylene borate), Pd(PPh3)4, Cs2CO3, THF–H2O, heat; (iv) benzothiadiazole-4,7-bis(boronic acid pinacol ester), Pd(dppf)Cl2, 2 M K2CO3, THF, heat; (v) hexabutylditin, Pd(PPh3)4, toluene, heat. | |
Quantum chemical calculation
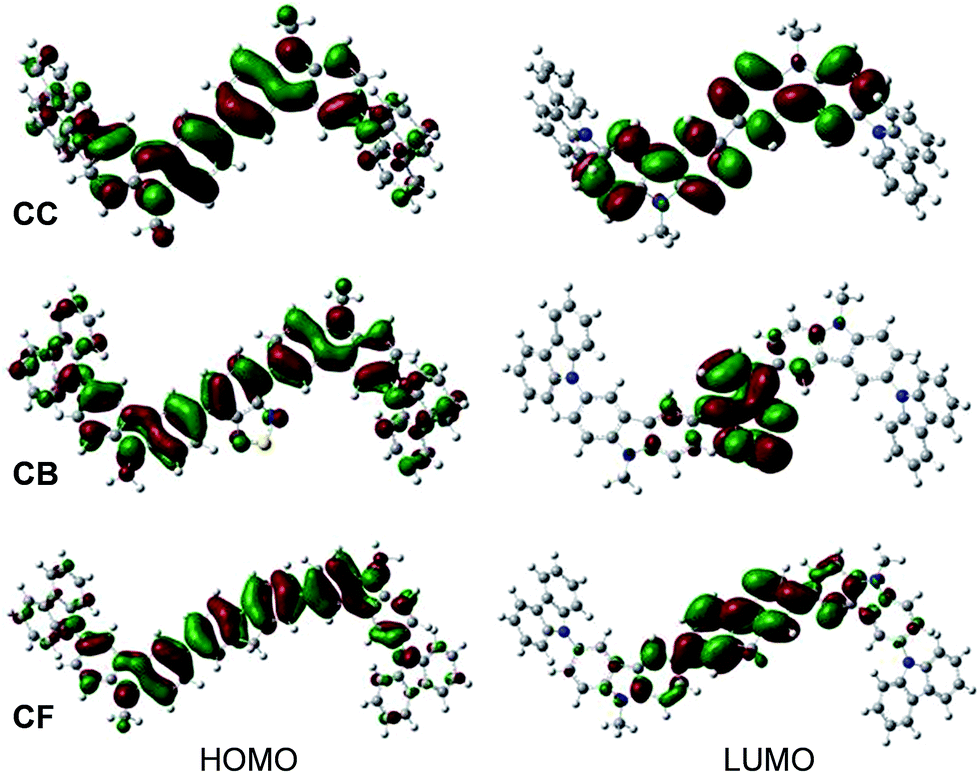
To understand the geometries and the electronic properties of CAr series, quantum chemical calculations were performed using TD-B3LYP/6-31G (d,p) basic set.13 Their optimized structures reveal that the 3,6-di-tert-butylcarbazol-N-yl-carbazole terminal moieties adopt a non-planar geometry around the planar oligoarylene cores (Fig. S1†), which could help to prevent the close π–π contact between the molecules and enhance their thermal properties.14 In the HOMOs of CAr, π-electrons are delocalized over the entire oligoarylene core and end-capping moieties. In the LUMOs of CTnF (CF, CTF and CT2F) having fluorene as a core, the excited electrons delocalize over the quinoid-like fluorene and oligothiophene fluorene planes, while in the LUMOs of CB and CTB having an electron deficient 2,1,3-benzothiadiazole as a central aryl core, these electrons localize mainly on the quinoid-like 2,1,3-benzothiadiazole plane, creating a donor–acceptor (D–A) characteristic (Fig. 2 and S2†). The HOMO–LUMO energy gaps (Ecalg) of CAr series were calculated and are summarized in Table 1. These Ecalg values (1.91–3.47 eV) are slightly diverged from those estimated from the optical onset (Eoptg = 2.03–3.31 eV). There are several factors responsible for the errors in the calculated results because the orbital energy difference between HOMO and LUMO is still an approximate estimation to the transition energy since the transition energy also contains significant contributions from some two electron integrals. The real situation is that an accurate description of the lowest singlet-excited state requires a linear combination of a number of excited configurations.
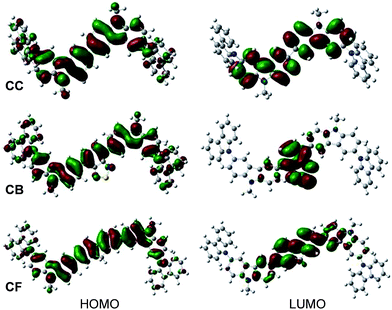 |
| | Fig. 2 The HOMO and LUMO orbitals of CC, CB and CF calculated by the B3LYP/6-31G(d,p) method in CH2Cl2. | |
Table 1 Physical and photophysical data for CAr
| Compd |
λabs(log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε)a (nm M−1 cm−1) ε)a (nm M−1 cm−1) |
λsolema (nm) |
λfilmemb (nm) |
Stokes shiftc (nm) |
ΦFd |
Tg/Tde (°C) |
E1/2 vs. Ag/AgClf (V) |
Eoptg/Ecalg/Eelegg (eV) |
HOMO/LUMOh (eV) |
| Measured in CH2Cl2. Measured as thin film coated on quartz substrate. Calculated from the difference between λmax of the absorption and emission spectra. Measured in CH2Cl2 with quinine sulfate and coumarin 6 as standards. Obtained from DSC/TGA measured at 10 °C min−1 under N2. Obtained from CV measured in CH2Cl2–n-Bu4NPF6 (0.1 M) at scan rate of 50 mV s−1. Obtained from Eoptg = 1240/λonset, Ecalg from the TD-B3LYP/6-31G(d,p) calculation in CH2Cl2, Eeleg = Ereonset − Eoxonset. Calculated from HOMO = −(4.44 + Eoxonset); LUMO = HOMO + Eoptg. sh means shoulder peak. |
| CC |
351 (3.15) |
396 |
410 |
45 |
0.30 |
41/318 |
1.03, 1.51 |
3.31/3.47/NA |
−5.39/−2.08 |
| CF |
350 (3.70) |
395, 415 |
412, 423sh |
45 |
0.72 |
100/382 |
0.99, 1.29, 1.39 |
3.19/3.35/NA |
−5.35/−2.16 |
| CTF |
405 (3.86) |
454, 482 |
468, 494 |
49 |
0.70 |
109/366 |
0.87, 0.96, 1.15, 1.49 |
2.74/2.81/NA |
−5.33/−2.59 |
| CT2F |
445 (4.17) |
490, 515sh |
526, 557sh |
45 |
0.75 |
117/320 |
0.83, 1.06, 1.17, 1.33 |
2.41/2.50/NA |
−5.19/−2.78 |
| CB |
350 (2.90), 452 (3.31) |
556 |
561 |
104 |
0.12 |
110/350 |
−1.50, 0.99, 1.40 |
2.38/2.37/2.33 |
−5.35/−2.97 |
| CTB |
354 (3.70), 525 (3.56) |
625 |
646 |
100 |
0.06 |
126/320 |
−1.25, 0.71, 1.02, 1.15, 1.25 |
2.03/1.91/1.85 |
−5.10/−3.07 |
Optical, thermal and electrochemical properties
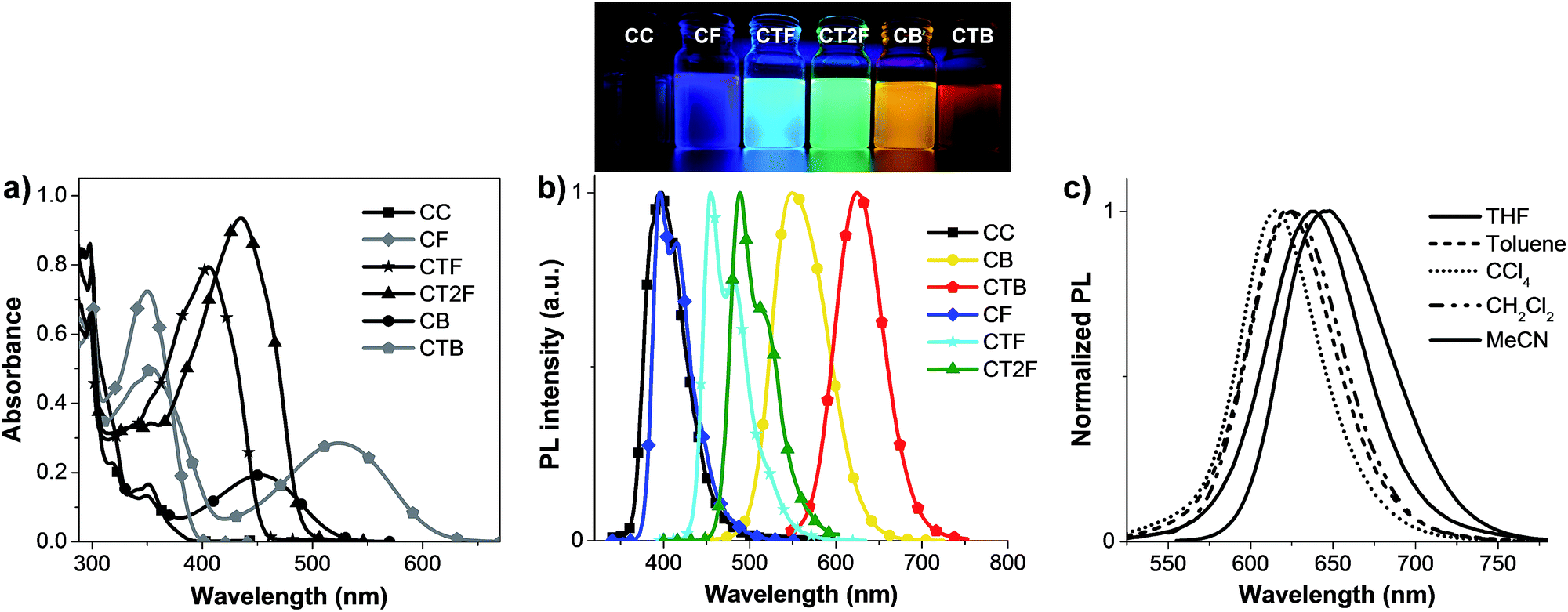
The absorption and photoluminescence (PL) spectra of the compounds are presented in Fig. 3 and the optical data are summarized in Table 1. The absorption at λmax ∼ 299 nm is attributed to π–π* transition of carbazole units, and the bands at λmax ∼ 351 nm for CC, 350 nm for CF, 405 nm for CTF, 445 nm for CT2F, 350 nm for CB and 354 nm for CTB are likely due to π–π* transition of conjugated backbones of the carbazole oligothiophene fluorene moieties. As the conjugation length of the backbone increases by increasing number of thiophene unit, these absorption bands are more intense and red-shifted. Donor–acceptor (D–A) characteristics in CB and CTB can also be recognized as an intramolecular charge-transfer (ICT) band at a longer wavelength at 452 and 525 nm, respectively. Generally, the ICT absorption band of the D–A molecule red-shifts when the strength of either the electron acceptor or donor increases. In our case, given that the electron-acceptor moiety in both CB and CTB is the same, the observed strong red-shift of ICT band of CTB compared to CB can be understood to arise from the stronger ICT with increasing electron donor strength. The reason for the observed red-shift with increasing electron donor strength is the greater degree of ICT (increasing ground-state dipole moments) in going from CB to CTB as observed in the TD calculation results (Table S2†).15 These ICT bands also show weak solvatochromism in different solvents. The PL spectra of CAr in dilute CH2Cl2 solutions gradually red shift with increasing π-conjugation length and electron accepting strength in the molecules. Thus, emission colors span the entire visible region starting from blue to deep red. All the compounds except for CC are strongly emissive in CH2Cl2 (Fig. 3b). As illustrated in Fig. S4,† the emission wavelengths of CB and CTB increase as the solvent polarity increases (λem (CCl4) < λem (toluene) < λem (CH2Cl2) < λem (THF) < λem (CH3CN)). This clearly points out that the excited state in these molecules possesses strong ICT character. Additional evidence supporting the stronger ICT interaction in CTB than CB with increasing strength of the electron donor in the molecules comes from their PL quantum yields (ΦF) in CH2Cl2 solution. It is well-known that the ΦF value of a D–A molecule decreases with increasing ICT character.16 We found that the ΦF values of the D–A molecules decreased with increasing strength of the acceptor block (Table 1). High ΦF values of 0.50–0.75 were obtained for the π–π* transition molecules CC and CTnF. The ΦF values decreased to 0.11 in CB to as low as 0.06 in CTB. This decreasing trend in PL quantum yield values in going from CTB to CB correlates very well with the estimated increasing ground-state dipole moments. Thus, although one can effectively tune the emission colors to achieve long wavelength emission with a D–A molecule, it invariably comes at the expense of reduced charge-transfer fluorescence efficiencies in the red region, which could affect the ultimate OLED performance. The π–π* transition molecules CC and CTnF show low Stokes shifts (45–49 nm in CH2Cl2) in the emission spectra. The relatively higher Stokes shifts (100–104 nm) of D–A molecules CB and CTB may result in partial bleaching of emission and lower the quantum yields. The solid-state fluorescence spectra are more bathochromic and broad compared with those in CH2Cl2 due to the intermolecular interactions (Fig. 6b). Although the solid-state PL quantum yields were not measured, it was clear from the measured fluorescence intensity of thin films that these materials could be used as non-doped emissive layer for OLEDs.
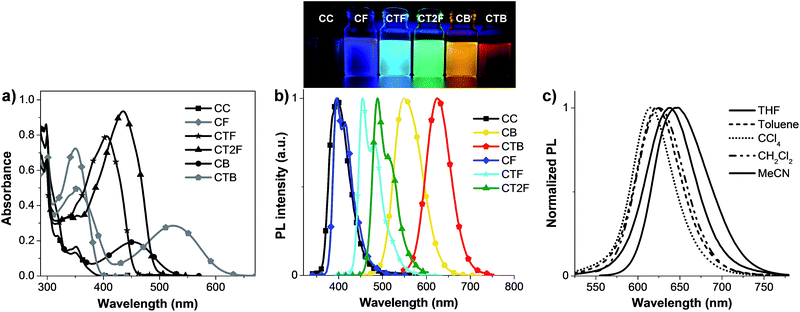 |
| | Fig. 3 Plots of (a) UV-vis absorption, (b) PL spectra of CAr in CH2Cl2 and their emission colors upon illumination and (c) PL spectra of CTB in different solvents. | |
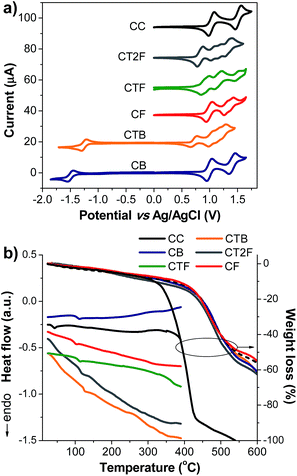 |
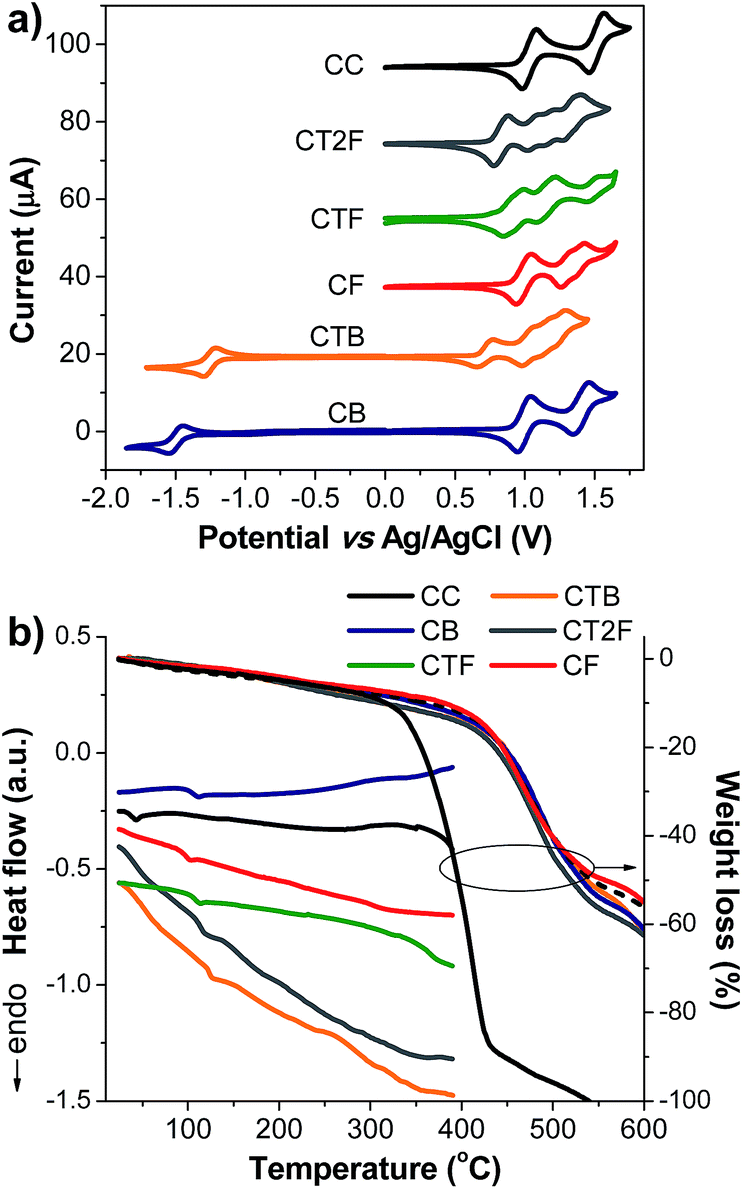
| | Fig. 4 Plots of (a) CV traces of CAr measured in CH2Cl2–n-Bu4NPF6 at a scan rate of 50 mV s−1 under Ar. and (b) DSC (2nd heating scan) and TGA thermograms of CAr measured at a heating rate of 10 °C min−1 under N2. | |
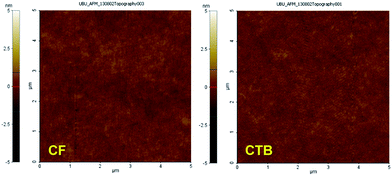 |
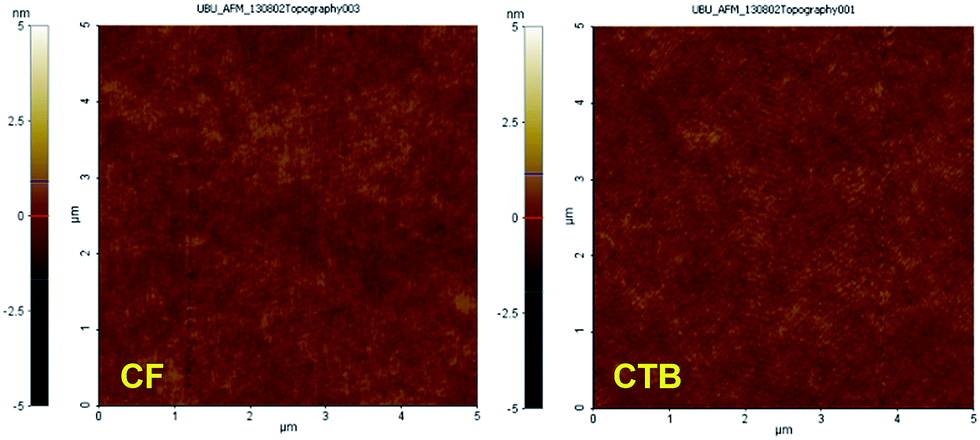
| | Fig. 5 Tapping mode AFM images of spin-coated thin films of CF and CTB. | |
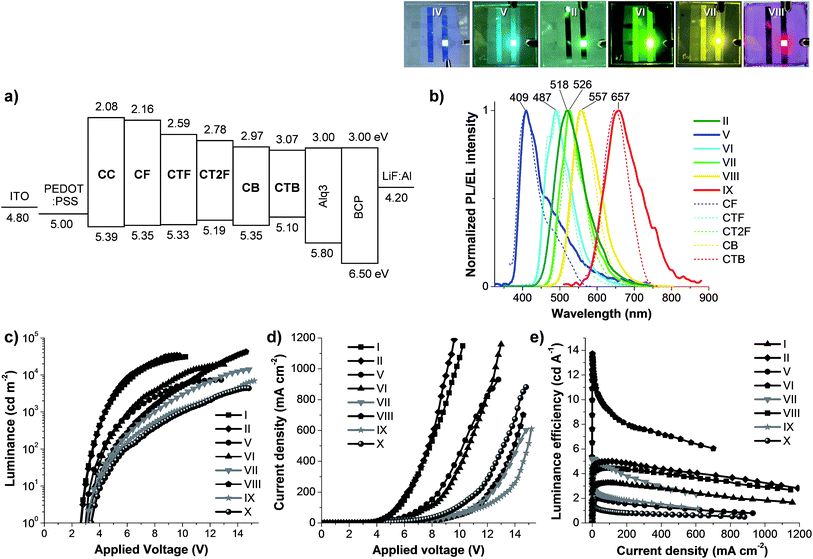 |
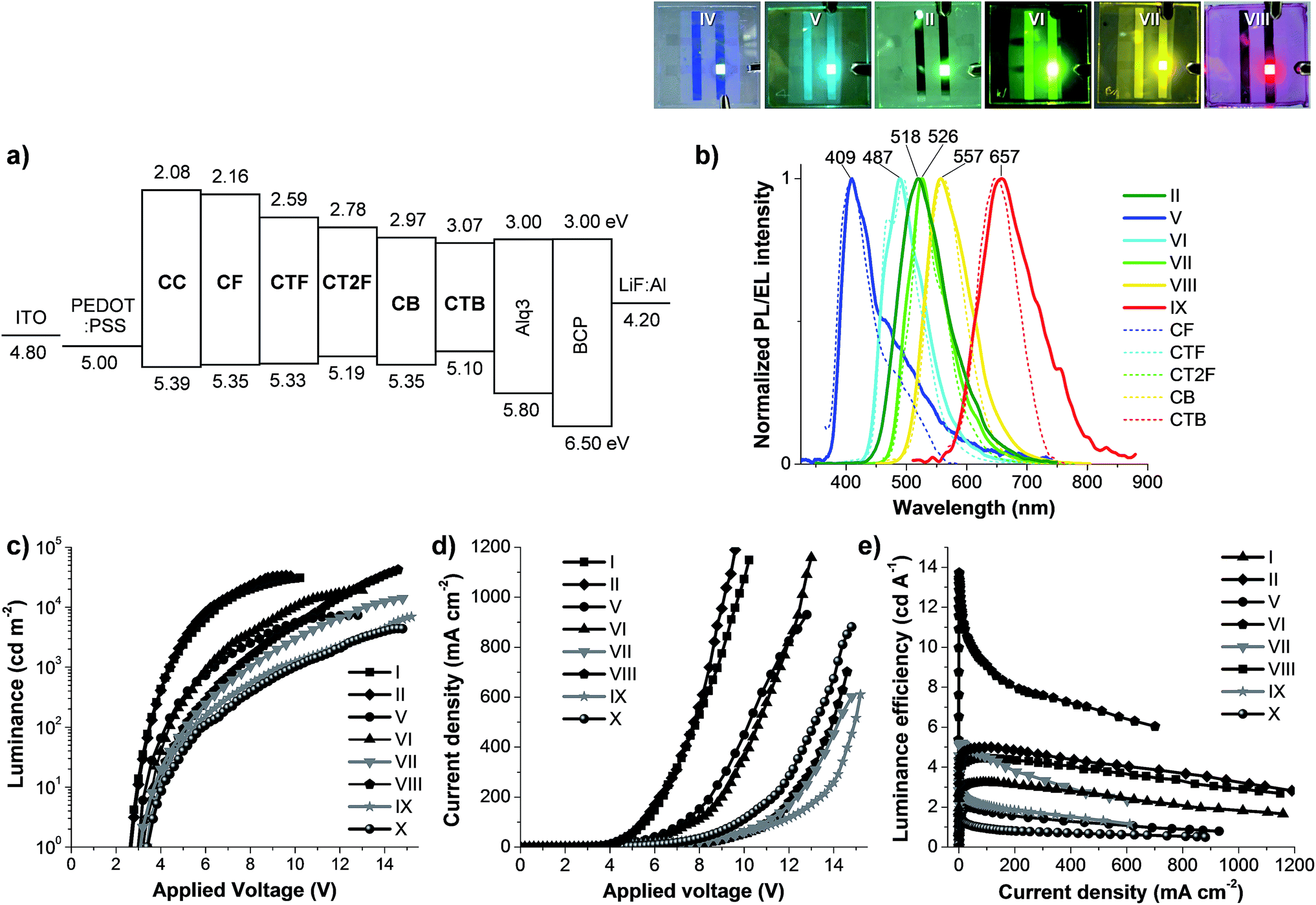
| | Fig. 6 (a) Schematic energy band diagram of each layer of the OLEDs. (b) PL spectra (dotted line) of spin-coated thin films of CAr, EL spectra (solid line) of OLEDs (devices II and V–IX) and their emission colors under applied voltage. Plots of (c) luminance–voltage (L–V), (d) current density–voltage (J–V) and (e) luminance efficiency–current density (η–J) characteristics of OLEDs (I–II and V–X). | |
Electrochemical properties of the new compounds were investigated by cyclic voltammetry (CV), and representative CV plots are shown in Fig. 4a (Table 1). All compounds display multiple quasi-reversible oxidation waves attributable to the successive oxidation of the end-capped carbazol-N-yl-carbazoles and the oligoarylene cores. The first oxidation wave can be ascribed to the removal of electrons from the carbazol-N-yl-carbazole unit. This potential is affected by the conjugation length of the oligoarylene backbones. As in the cases of CTnF series, and CB and CTB, this oxidation potential shifts to lower potentials with increasing number of thiophene unit in the backbone. Similar behavior has been frequently reported in the literature.17 It is also noticed that the potentials of the first oxidation wave of CTnF series, and CB and CTB (Eox1/2 = 0.99–0.71 V) appear at a lower potential than that of N-alkylcarbazole (Eox1/2 = 1.09 V), indicating a strong π-electron conjugation between the carbazole and oligoarylene cores, as observed in the TD calculation results.
A quasi-reversible reduction wave attributable to the reduction of the benzothiadiazole core to form the anion radical is also observed in compounds CB and CTB. The reduction of benzothiadiazole core of CTB (Ere1/2 = −1.25 V) appears at lower potential than that of CB (Ere1/2 = −1.50 V). However, no cathodic reduction waves for CTnF series could be observed with CH2Cl2 as the solvent. Importantly, multiple CV scans of all compounds reveal identical CV curves with no additional peak at lower potential on the cathodic scan (Epc) being observed (Fig. S3†), suggesting no electrochemical oxidative coupling reaction at the 3,6 positions of the peripheral carbazole led to electro-polymerization occurred in the process. The present of 3,6-di-tert-butyl groups could prevent this type of electrochemical coupling reaction which is usually detected in some carbazole derivatives with unprotected 3,6-positions and less hindered.18 All newly synthesized compounds are electrochemically stable molecules, as expected. The HOMO and LUMO energy levels of CAr series calculated from their oxidation onset potentials (Eoxonset) and optical energy gaps (Eoptg) are listed in Table 1. Their HOMO levels are in the range of −5.10 to −5.39 eV, which match quite well with the work function of commonly used hole-injection/transport layers (PEDOT:PSS (−5.00 eV)) and anodes (ITO (−4.80 eV)), indicating that these materials are suitable for applications as hole-transporting light-emitters in OLEDs. Their LUMO levels calculated by subtracting the Eoptg from the HOMO levels are in the range of −2.08 to −3.07 eV. The LUMO levels of CT2F (−2.78 eV), CB (−2.97 eV) and CTB (−3.07 eV) are close to the work function of the LiF:Al anode (4.20 eV) and seem suitable for efficient electron injection. Whereas the LUMO levels of CC (−2.08 eV), CF (−2.16 eV) and CTF (−2.59 eV) are rather high and seems to be not suitable for efficient electron injection. The electron transporting layer or hole-blocking layer do require when they are fabricated as an emissive layer (EML) in the OLEDs. From electrochemical measurements, the energy gaps (Eeleg) of CB and CTB are calculated to be 2.33 and 1.85 eV, respectively, which are close to those estimated from their corresponding optical spectra (Eoptg = 2.38 and 2.03 eV), indicating that the LUMO and HOMO levels obtained by the electrochemical measurements are reliable (Table 1).
The thermal properties of these new compounds were determined by DSC and TGA measurements (Fig. 4b and Table 1). All compounds exhibit a glass transition in the second heating cycle, and no crystallization exotherm and melting endotherm are noticed at higher temperatures, indicating they are amorphous. Obviously, two non-planar 3,6-di-tert-butylcarbazol-N-yl-carbazole segments help to suppress the aggregation of the flat oligoarylene cores and induce the formation of glassy state. The incorporation of tert-butyl groups in the peripheral carbazole ring results could also an increase of the glass transition temperature (Tg) of the molecule.19 The Tgs of CTnF, CB and CTB are in the range of 100 to 126 °C, while the Tg of CC is somewhat lower. The high Tgs and thermal decomposition temperatures (Td) (318–382 °C) of the compounds (Table 1) should be beneficial to the stability of the deposited films.
Electroluminescence (EL) properties
According to the above discussed properties including vary HOMO–LUMO energy levels, good luminescence property, and amorphous morphology, the abilities of CAr as solution processed hole-transporting and non-doped light-emitting materials for OLEDs were investigated. The thin layers of CAr were spin coated from a CHCl3–toluene (1:1) solution with controlled thickness. The morphology of the spin-casting films, which is one of the key factors for realizing high-performance OLEDs, was examined by atomic force microscopy (AFM). The AFM images of all thin films show a pinhole-free surface indicating excellent film-forming properties (Fig. 5 and S4†). The thin films have a very uniform and flat surface. This stable homogeneous amorphous morphology appears favourable for OLEDs with reduced leak currents and improved thermal stability during device operation.
As a hole-transporter for Alq3-based OLED
The hole-transporting properties of the carbazol-N-yl-carbazole end-caps of CAr were investigated by using some of CAr as a hole-transporting materials (HTM) for Alq3-based green OLED. Owning to their suitable HOMO levels (−5.40 eV), amorphous solid, colourless, emission in high energy region and high quality spin coated thin films, CC and CF are the two best candidates. To test this hypothesis, double layer Alq3-based OLEDs (devices I–II) with the structure of ITO/PEDOT:PSS/CC or CF (spin coating)/Alq3/LiF:Al were fabricated and investigated. Where CC and CF layers as a hole-transporting layer (HTL) and tris(8-hydroxyquinoline)aluminum (Alq3) as green light emitting (EML) and electron-transporting layers (ETL) (Fig. 6a). The reference devices (III and VI) fabricated with and without a commercial HTM, N,N′-bis(naphthalen-1-yl)-N,N′-bis(phenyl) benzidine (NPB), as HTL were made for comparison. The detailed EL and J–V–L data are shown in Fig. 6 and summarized in Table 2. Comparison with the reference device (IV) fabricated without HTL reveals that the incorporation of CC and CF in the devices (I, II) as HTL not only increases the maximum luminance (Lmax) and luminance efficiency (η), but also significantly decreases the Von and V100 of the diode, proving a ability of the carbazol-N-yl-carbazole moiety as HTM. The Von and V100 of both diodes are 2.6 V and 3.5 V, respectively, indicating good device performance is achieved. The device (I) fabricated with CC as HTL exhibits comparable performance with the reference device (III) using NPB as HTL, while the device (II) fabricated with CF as HTL show a better performance. The CF-based green OLED (device V) displays a high maximum brightness of 33590 cd m−2 at 9.6 V, a ηmax as high 4.99 cd A−1 at current density of 4.16 mA cm−2 and a maximum external quantum efficiency (EQE) of 1.24%. Under applied voltages, the devices (I, II) emit a bright green emission with peak centred at 518 nm and CIE coordinates of (0.30, 0.54). The EL spectra are matched with the PL spectrum of Alq3, the EL of the reference devices (III and IV) and also other reported EL spectra of Alq3-based devices.20 No emission at the longer wavelength owing to exciplex species formed at the interface of HTL and EML materials, which often occurs in the devices fabricated from HTL with planar molecular structure, is observed.21 A stable emission is obtained with the EL spectra did not change over the entire applied voltages (Fig. S5†). From these results and in view of the fact that a barrier for electron migration at the Alq3/HTL interface (0.84–0.92 eV) is larger than those for hole-migration at the HTL/Alq3 interface (0.41–0.45 eV). Hence, under the present device structure, CC and CF act only as HTL, and Alq3 acts preferably as an electron blocker more than as a hole blocker and charge recombination thus confined to Alq3 layer.
Table 2 Device characteristics of OLED fabricated with CAr as EMLs, and CC and CF as the HTL
| Device |
EML/HTL |
λEL (FWHM) (nm) |
Von/V100c (V) |
Lmax at the voltaged (cd m−2 V−1) |
Jmaxe (mA cm−2) |
ηmax at J/η at L100/η at L1000f (cd A−1, mA cm−2) |
EQEg (%) |
CIE (x, y) |
| ITO/PEDOT:PSS/HTL(spin coating)/Alq3/LiF:Al. ITO/PEDOT:PSS/EML(spin coating)/BCP/LiF:Al. Turn-on voltages at 1 and 100 cd m−2. Maximum luminance at applied voltage. Current density at maximum luminance. Luminance efficiencies at maximum, at luminance of 100 and 1000 cd m−2. Maximum external quantum efficiency. sh means shoulder peak. |
| Ia |
CC |
518 (82) |
2.6/3.5 |
30895 (10.2) |
1148 |
4.45 (84.6)/3.73/4.23 |
1.10 |
0.30, 0.54 |
| IIa |
CF |
518 (81) |
2.6/3.5 |
33590 (9.6) |
1190 |
4.99 (92.3)/4.16/4.74 |
1.24 |
0.30, 0.54 |
| IIIa |
NPB |
519 (92) |
2.5/3.6 |
31857 (9.4) |
1599 |
4.45 (115.1)/4.21/4.30 |
0.54 |
0.30, 0.53 |
| IVa |
— |
518 (90) |
4.2/5.4 |
4961 (10.0) |
693 |
0.91 (366.8)/0.73/0.82 |
0.15 |
0.30, 0.54 |
| Vb |
CF |
409 (77) |
3.0/4.2 |
7361 (12.8) |
930 |
1.99 (28.7)/1.45/1.88 |
0.81 |
0.19, 0.18 |
| VIb |
CTF |
487 (79) |
3.1/4.3 |
19569 (12.8) |
1000 |
3.22 (76.8)/2.13/3.04 |
0.13 |
0.18, 0.37 |
| VIIb |
CT2F |
526 (74) |
3.1/5.1 |
14081 (14.8) |
601 |
5.21 (11.9)/5.06/5.11 |
1.29 |
0.30, 0.61 |
| VIIIb |
CB |
557 (90) |
3.2/5.0 |
42305 (14.6) |
700 |
13.73 (5.8)/11.55/11.54 |
5.90 |
0.45, 0.54 |
| IXb |
CTB |
657 (110) |
3.3/5.4 |
6945 (15.2) |
611 |
2.79 (4.7)/2.52/2.28 |
0.26 |
0.67, 0.33 |
| Xb |
CF:CT2F:CTB |
429sh, 483, 619 |
3.4/5.9 |
4397 (14.6) |
851 |
1.49 (4.7)/1.18/0.95 |
0.14 |
0.33, 0.35 |
As a non-doped emitter
The above results reveal that CAr retain a good luminescence property as well as good hole-transporting abilities, and hence exhibit great potential for use as solution processed non-doped hole-transporting emitters in OLEDs. Simple solution processed double-layer devices using CAr as an emissive layer (EML) with the structure of ITO/PEDOT:PSS/CAr(spin coating) (∼40 nm)/BCP(40 nm)/LiF(0.5 nm):Al(150 nm) were investigated. Analysis of the band–energy diagram of CAr as EML, ITO/PEDOT:PSS anode and LiF:Al cathode reveals that a barrier for electron injection at the EML/LiF:Al interface (∼1.13–2.04 eV) is about five to ten fold higher than that for hole migration at the PEDOT:PSS/EML interface (∼0.1–0.39 eV) (Fig. 6a), indicating that a hole-injection to EML is more efficient than an electron-injection. It has been known that the effective recombination of electrons and holes affects the electroluminescence efficiency of OLEDs.22 Dimethyl-4,7-diphenyl-1,10-phenanthroline (BCP) as an electron injection/hole-blocking layer was integrated in-between the EML layer and LiF:Al electrode in order to balance the charge injection in the device and hence enable the higher performance of the fabricated OLEDs. It has been proved that BCP can improve the performance of multilayer devices fabricated with a predominantly hole-transporting emitter by assisting more electron injection into the EML.23 The EL spectra and J–V–L characteristics of the devices are shown in Fig. 6 and their electrical parameters are summarized in Table 2. Under forward-bias voltage, these devices (V–IX) emit a bright light of various colors covering the entire visible light including blue (device V with CF as EML), light blue (device VI with CTF as EML), green (device VII with CT2F as EML), yellow (device VIII with CB as EML) and red (device IX with CTB as EML). The EL spectra are gradually changed from 409 nm for blue to 487 nm for light blue, 526 nm for green, 557 nm for yellow and 657 nm for red (Fig. 6b). The EL spectra are analogous to their corresponding thin film PL spectra indicating that the EL emission originates from the singlet-excited states of the EML materials. No emission shoulder at a longer wavelength due to excimer and exciplex species formed at the interface of the EML and BCP layers, which often occurs in devices fabricated from EMLs with planar molecular structure, is detected.24 More importantly, a stable emission is obtained from all diodes with the EL spectra and CIE coordinates do not change over the entire applied voltages (Fig. S5†). The device IX fabricated with CTB as EML emits a pure red color with CIE coordinates of (0.67, 0.33), which are match with the National Television System Committee (NTSC) red (0.67, 0.33) standards, while devices V and VII emit blue and green lights with CIE coordinates of (0.19, 0.18) and (0.30, 0.61), which are close to the NTSC blue (0.14, 0.08) and green (0.26, 0.65) standards.25 These double-layer OLEDs fabricated with CAr as EML show moderate to high device luminescence efficiencies. These devices exhibit Lmax of 7361–42305 cd m−2, ηmax of 1.99–13.73 cd A−1 and EQE of 0.26–5.90%. The light turn-on voltages (Von) at 1 cd m−2 for all devices are in the range of 3.0–3.3 V and the operating voltages at 100 cd m−2 (V100) are in the range of 4.2–5.4 V, indicating a decent operating performance is achieved for all diodes (Table 2). Among these devices, the yellow light-emitting OLED (device VIII) exhibits the best device best performance with a high Lmax of 42305 cd m−2 at 14.6 V, a low Von of 3.1 V and an EQE of 5.90%, while the green diode (device VII) show the second best device performance with Lmax of 5.21 cd m−2 at 14.8 V and an EQE of 1.29%. A comparable device performance was observed for the blue, light blue and red diodes (devices V, VI and IX) with EQE values in the range of 0.13–0.81%. These differences in efficiency of CAr-based devices may arise from a combination of the PL quantum yield, the ICT effect as well as well balance of the HOMO and LUMO energy levels of the EML. However, from the view point of simple molecular structure, color quality and device efficiency these materials are among good non doped emitters reported, especially CB for yellow.
As an emitter for white OLED
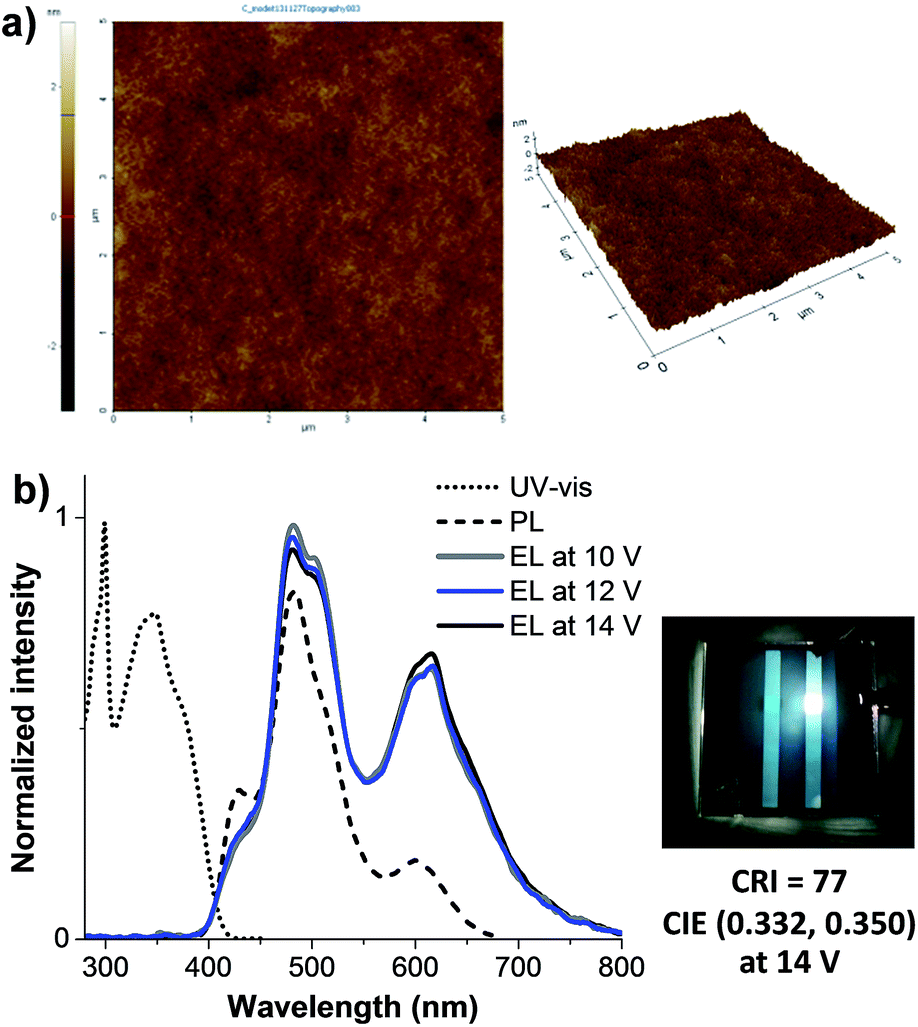
According to the above results showing that the emission of the monochromatic devices (V, VII, IX) based on CF (blue), CT2F (green) and CTB (red) emitters covers the entire visible light spectrum. Therefore, white-light emission can be obtained if emission of a specific color is carefully operated. As emission of these individual colors (red, green and blue) is carefully worked with appropriate blending ratio, white-light emission can be obtained with device structure of ITO/PEDOT:PSS/CF (blue):CT2F (green):CTB (red) (∼50 nm) (spin-coating)/BCP(40 nm)/LiF (0.5 nm)/Al (150 nm). A preliminary study finds that when the blend composition of 100:3.0:0.2 (CF:CT2F:CTB) by weight the white EL spectrum shows very balanced RGB distribution and full coverage of the whole visible range (375–750 nm). The AFM image of a thin film of the blend spin-coated from CHCl3:toluene solution shows a pinhole-free, uniform and smooth surface, indicating a well blending of all chromophores (Fig. 7a). The UV-vis absorption spectrum of the thin film exhibits two main absorption bands at 298 and 347 nm, while its PL spectrum display peaks at 428, 485 and 603 (Fig. 7b). Under forward-bias voltage, the device (X) emits white light. The EL spectra of the WOLED (device X) under biases of 11 and 14 V are illustrated in Fig. 7b, with the three main peaks at ηmax ca. 429sh, 483 and 619 nm corresponding to characteristic blue and green and red emissions from CF, CT2F and CTB, respectively. The peak width at FWHM covers a wide wavelength range of ca. 450–651 nm, which results in a color rendering index (CRI) of 77 and a correlated color temperature (CCT) of 6025 K for the white light emission. Accordingly, this light is suitable for reading light applications. As a bias voltage is increased from 10 to 15 V, the lower energy emission bands (green and red) are fairly changed in relative intensity. The best white emission with CIE coordinates of (0.33, 0.35) is achieved at 14 V and the coordinates locate very close to the standard white light point of (0.33, 0.33). A very small offset of CIE coordinates of emitted light of (0.010, 0.009) are observed under the various biases (10–15 V). These data demonstrate that decent white emission and remarkably good color stability have been achieved using these carbazol-N-yl-carbazole end-capped oligoarylenes. Moreover, it is noticed that at all driving voltages no emission due to the excimer and exciplex emissions at longer wavelengths is observed, indicating the white emission just coming from all three red, green and blue chromophores. This is the benefit of their bulky structures. The fabricated WOLED (device X) exhibits a low turn-on voltage of 3.4 V, a Lmax of 4397 cd m−2 and an EQE of 0.14% is achieved, as shown in Fig. 6c and d and Table 2. The device exhibits a ηmax of 1.49 cd A−1 at 4.7 mA cm−2 (5.0 V). The inset of Fig. 7b is a snapshot of the WOLED at 14.0 V; a suitable white light emission with a uniform emitting area is seen.
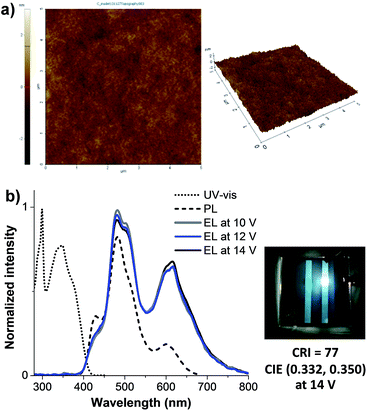 |
| | Fig. 7 (a) Tapping mode AFM images of spin-coated thin film of CF:CT2F:CTB (100:3.0:0.2). (b) Plots of UV-Vis absorption and PL spectra of spin-coated thin film of CF:CT2F:CTB (100:3.0:0.2), and EL spectra and emission color of the WOLED (device X). | |
Experimental
Materials and methods
All chemicals were purchased from Aldrich, Acros or Thai companies and used as received. THF and CH2Cl2 for cyclic voltammetry (CV) measurements were distilled according to the standard methods. 1H and 13C NMR spectra were recorded on a Brüker AVANCE 300 MHz spectrometer. UV-Vis spectra were recorded on a Perkin-Elmer UV Lambda 25 spectrometer. Photoluminescence spectra and fluorescence quantum yields (ΦF) were recorded with a Perkin-Elmer LS 50B Luminescence Spectrometer. Quinine sulfate solution in 0.01 M H2SO4 (ΦF = 0.54) and coumarin 6 in ethanol (ΦF = 0.78) were used as reference standards.26 Differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) were performed on a METTLER DSC823e thermal analyzer and a Rigaku TG-DTA 8120 thermal analyzer, respectively, with heating rate of 10 °C min−1 under N2 atmosphere. Cyclic voltammetry (CV) measurements were performed on an Autolab potentiostat PGSTAT 12 with a three electrode system (platinum counter electrode, glassy carbon working electrode and Ag/AgCl reference electrode) in CH2Cl2 under Ar atmosphere with n-Bu4NPF6 as a supporting electrolyte at scan rate of 50 mV s−1. The concentration of analytical materials and the electrolyte were 10−3 M and 0.1 M, respectively. Melting points were measured using an Electrothermal IA 9100 series of digital melting point instrument and are uncorrected. MALDI-TOF mass spectra were recorded on Bruker Daltonics (Bremen, Germany) Autoflex II Matrix-Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometer (BIFEX). The atomic force microscopy (AFM) analysis was performed on Park System model XE 100 using standard non-contact mode with resonance of 316.17 kHz.
All calculations were performed by Gaussian 09 code in CH2Cl2 solvent by C-PCM model.13 The energy or geometry optimizations were done by B3LYP/6-31G(d,p) method. The ground to excited state excitation energies were calculated by TD-B3LYP/6-31G(d,p) in CH2Cl2.
Materials synthesis
Synthesis of CC. A mixture of 1 (0.46 g, 0.65 mmol), hexa-n-butylditin (0.15 g, 0.27 mmol) and Pd(PPh3)4 (11 mg, 0.01 mmol) in toluene (50 ml) was degassed with N2 and then heated at reflux under N2 for 20 h. Water (30 ml) was added and the mixture was extracted with CH2Cl2 (30 ml × 2). The combined organic phase was with water (50 ml), brine solution (50 ml), dried with anhydrous Na2SO4 and filtered. The solvents were removed to dryness and the crude product was purified by column chromatography eluting with a mixture of CH2Cl2:hexane (1:8) and recrystallization from CH2Cl2:MeOH to give CC as white solids (76%); m.p. 134 °C; 1H NMR (300 MHz, CDCl3) δ 8.18–8.15 (8H, m), 7.60 (3H, dd, J = 8.7 Hz, J = 1.5 Hz), 7.58 (3H, d, J = 9.0 Hz), 7.45 (4H, dd, J = 8.7 Hz, J = 1.8 Hz), 7.36–7.29 (6H, m), 4.36 (4H, t, J = 7.2 Hz), 1.94 (4H, q, J = 7.2 Hz), 1.48–1.26 (72H, m), 0.87 (6H, t, J = 6.9 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 142.51, 140.21, 139.71, 139.57, 129.83, 128.89, 125.73, 124.22, 123.52, 123.33, 123.07, 122.63, 119.30, 116.20, 111.89, 110.48, 109.85, 109.12, 43, 54, 34.74, 32.06, 31.91, 29.61, 29.58, 29.52, 29.40, 29.34, 29.02, 27.33, 22.69, 14.11 ppm. HRMS MALDI-TOF (m/z) (M+) calcd for C88H110N4: 1223.8418, found 1223.4090.
Synthesis of CF, CTF and CT2F. A mixture of 1, 3 or 5 (0.46 g, 0.65 mmol), 9,9-dihexylfluororene-2,7-bis(trimethylene borate) (0.11 g, 0.22 mmol), Pd(PPh3)4 (18 mg, 0.015 mmol) and 2 M Cs2CO3 (7 ml) in THF (30 ml) was degassed with N2 and then heated at reflux under N2 for 24 h. Water (30 ml) was added and the mixture was extracted with CH2Cl2 (50 ml × 2). The combined organic phase was washed with water (50 ml), brine solution (50 ml), dried with anhydrous Na2SO4 and filtered. The solvents were removed to dryness and the crude product was purified by column chromatography eluting with a mixture of CH2Cl2:hexane (1:5) and recrystallization from CH2Cl2:MeOH to give:
CF. CF as pale yellow solids (91%); m.p. 110 °C; 1H NMR (300 MHz, CDCl3) δ 8.37 (4H, d, J = 9 Hz), 8.23 (s, 4H), 7.87 (2H, d, J = 8.7 Hz), 7.80 (2H, d, J = 7.8 Hz), 7.72–7.66 (4H, m), 7.62–7.57 (6H, m), 7.49 (4H, d, J = 8.7 Hz), 7.40 (4H, d, J = 8.7 Hz), 4.45–4.42 (4H, m), 2.09–1.99 (8H, m), 1.52 (36H, s), 1.29–0.89 (58H, m), 0.72 (6H, t, J = 6.9 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 151.75, 142.40, 140.51, 140.39, 139.86, 139.56, 133.25, 129.48, 126.04, 125.92, 125.22, 123.90, 123.46, 123.11, 123.05, 121.52, 119.91, 119.44, 118.94, 116.20, 109.69, 109.27, 109.20, 55.34, 43.56, 40.57, 34.75, 32.09, 31.92, 31.49, 29.73, 29.63, 29.56, 29.47, 29.35, 29.15, 27.41, 23.87, 22.69, 22.55, 14.12, 13.96 ppm; HRMS MALDI-TOF (m/z) (M+) calcd for C113H142N4: 1556.3634, found 1556.3822.
CTF. CTF as yellow solids (80%); m.p. 154 °C; 1H NMR (300 MHz, CDCl3) δ 8.34 (2H, d, J = 1.2 Hz), 8.29 (2H, d, J = 1.2 Hz), 8.21 (4H, d, J = 1.5 Hz), 7.86 (2H, dd, J = 8.7 Hz, J = 1.5 Hz), 7.70–7.57 (10H, m), 7.47 (6H, dd, J = 9.0 Hz, J = 1.2 Hz), 7.39–7.34 (8H, m), 4.41 (4H, t, J = 6.9 Hz), 2.06–1.96 (8H, m), 1.52 (36H, s), 1.50–1.27 (50H, m), 1.14–1.06 (12H, m), 0.88 (6H, t, J = 6.9 Hz), 0.74 (6H, t, J = 6.0 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 151.74, 144.50, 143.26, 142.46, 140.64, 140.28, 140.11, 139.81, 133.31, 129.72, 126.04, 125.36, 124.47, 123.84, 123.68, 123.50, 123.09, 123.03, 120.04, 119.70, 119.34, 117.71, 116.21, 109.80, 109.40, 109.20, 55.28, 43.56, 34.76, 32.09, 31.92, 31.46, 29.69, 29.63, 29.55, 29.44, 29.35, 29.14, 27.38, 23.77, 22.70, 22.57, 14.12, 13.98 ppm; HRMS MALDI-TOF (m/z) (M+) calcd for C121H146N4S2: 1720.6107, found 1720.6812.
CT2F. CT2F as yellow-orange solids (77%); m.p. 174 °C; 1H NMR (300 MHz, CDCl3) δ 8.29 (2H, d, J = 1.2 Hz), 8.27 (2H, d, J = 1.2 Hz), 8.20 (4H, d, J = 1.5 Hz), 7.81 (2H, d, J = 8.7 Hz), 7.70 (2H, d, J = 8.1 Hz), 7.65–7.55 (8H, m), 7.50 (6H, d, J = 8.7 Hz), 7.38 (4H, d, J = 8.7 Hz), 7.32 (2H, d, J = 3.9 Hz), 7.28 (2H, d, J = 5.1 Hz), 7.21 (4H, t, J = 3.9 Hz), 4.40 (4H, t, J = 6.9 Hz), 2.00–1.96 (8H, m), 1.50 (36H, s), 1.43–1.27 (50H, m), 1.12–1.06 (12H, m), 0.88 (6H, t, J = 6.9 Hz), 0.75 (6H, t, J = 9.2 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 151.79, 144.35, 143.51, 142.47, 140.69, 140.26, 139.80, 136.76, 135.71, 133.00, 129.76, 125.68, 125.40, 124.61, 124.51, 124.19, 123.63, 123.51, 123.08, 123.05, 122.78, 120.14, 119.75, 119.32, 117.73, 116.20, 109.83, 109.44, 109.18, 55.31, 43.57, 34.75, 32.08, 31.92, 31.45, 29.67, 29.63, 29.60, 29.54, 29.43, 29.35, 29.12, 27.37, 23.75, 22.69, 22.56, 14.11, 13.98 ppm; HRMS MALDI-TOF (m/z) (M+) calcd for C129H150N4S4: 1884.8581, found 1885.1111.
Synthesis of CB and CTB. A mixture of 1 or 3 (0.26 g, 0.33 mmol), 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester) (43 mg, 0.11 mmol) and Pd(dppf)Cl2 (10 mg, 0.012 mmol) and 2 M K2CO3 (5 ml) in THF (25 ml) was degassed with N2 and then heated at reflux under N2 for 24 h. Water (30 ml) was added and the mixture was extracted with CH2Cl2 (30 ml × 2). The combined organic phase was washed with water (50 ml), brine solution (50 ml), dried with anhydrous Na2SO4 and filtered. The solvents were removed to dryness and the crude product was purified by column chromatography eluting with a mixture of CH2Cl2:hexane (1:4) and recrystallization from CH2Cl2:MeOH to give:
CB. CB as orange solids (64%); m.p. 144 °C; 1H NMR (300 MHz, CDCl3) δ 8.69 (2H, d, J = 0.9 Hz), 8.32 (2H, s), 8.20–8.18 (6H, m), 7.90 (2H, s), 7.62 (6H, d, J = 10.8 Hz), 7.48 (4H, dd, J = 8.7 Hz, J = 0.9 Hz), 7.36 (4H, d, J = 11.7 Hz), 4.45 (4H, t, J = 6.9 Hz), 2.02 (4H, q, J = 6.9 Hz), 1.48–1.27 (72H, m), 0.87 (6H, t, J = 6.9 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 154.57, 142.42, 142.39, 141.04, 140.35, 139.86, 133.21, 129.65, 129.24, 128.88, 128.61, 127.96, 127.69, 125.29, 124.00, 123.47, 123.05, 122.96, 121.51, 119.51, 116.18, 109.75, 109.21, 109.12, 43.58, 34.74, 32.08, 31.92, 30.92, 29.63, 29.56, 29.48, 29.35, 29.16, 27.40, 22.69, 14.11 ppm; HRMS MALDI-TOF (m/z) (M+) calcd for C94H112N6S: 1358.0003, found 1357.8565.
CTB. CTB as purple solids (62%); m.p. 150 °C; 1H NMR (300 MHz, CDCl3) δ 8.38 (2H, s), 8.29 (2H, s), 8.20 (4H, d, J = 1.2 Hz), 8.14 (2H, d, J = 3.9 Hz), 7.88 (4H, d, J = 5.4 Hz), 7.65 (3H, d, J = 10.2 Hz), 7.59 (3H, d, J = 8.4 Hz), 7.50 (4H, d, J = 8.4 Hz), 7.44 (2H, d, J = 3.9 Hz), 7.39 (4H, d, J = 8.4 Hz), 4.40 (4H, bs), 1.98 (4H, bs), 1.49–1.26 (72H, m), 0.87 (6H, t, J = 6.6 Hz) ppm; 13C NMR (75 MHz, CDCl3) δ 152.62, 146.73, 142.47, 140.81, 140.26, 139.79, 137.66, 129.79, 128.63, 125.77, 125.64, 125.36, 125.08, 124.60, 123.69, 123.52, 123.10, 123.05, 119.34, 117.93, 116.20, 109.82, 109.41, 109.21, 43.56, 34.76, 32.09, 31.92, 29.71, 29.63, 29.55, 29.44, 29.35, 29.13, 27.37, 22.69, 14.12 ppm; HRMS MALDI-TOF (m/z) (M+) calcd for C102H116N6S3: 1522.2476, found 1522.0153.
OLED fabrication and testing
OLED devices using CAr as a non-doped missive layer (EML) with the device configurations of ITO/PEDOT:PSS/CAr(spin coating)/BCP(40 nm)/LiF(0.5 nm):Al(150 nm), and CC and CF as a hole-transporting layer (HTL) with the device configurations of ITO/PEDOT:PSS/CC or CF(spin coating)/Alq3(50 nm)/LiF(0.5 nm)/Al(150 nm) were fabricated and characterized as followed. Thin films of CAr were coated on top of PEDOT:PSS (50 nm) coated ITO by spin-coating CHCl3:toluene solutions (1:1) of CAr (1–2% w/v) at a spin speed of 3000 rpm for 30 second to get a 30–40 nm thick. The film thickness was measured by using a Tencor α-Step 500 surface profiler. Then either BCP or Alq3 were deposited onto the surface of the CAr films as electron-transporting layer (ETL) or missive layer, respectively, with a thickness of 40–50 nm by evaporation from resistively heated alumina crucibles at evaporation rate of 0.5–1.0 nm s−1 in vacuum evaporator deposition (ES280, ANS Technology) under a base pressure of ∼10−5 mbar. The film thickness was monitored and recorded by quartz oscillator thickness meter (TM-350, MAXTEK). The chamber was vented with dry air to load the cathode materials and pumped back; a 0.5 nm thick LiF and a 150 nm thick aluminum (Al) layers were the subsequently deposited through a shadow mask on the top of BCP or Alq3 films without braking vacuum to from an active diode areas of 4 mm2. The measurement of device efficiency was performed according to M. E. Thomson's protocol and the device external quantum efficiencies were calculated using procedure reported previously.27 Current density–voltage–luminescence (J–V–L) characteristics were measured by the use of a Keithley 2400 source meter and a Keithley 6485 picoammeter equipped with a calibrated UDT Instruments 2153 brightness sensor. The EL spectra were acquired by an Ocean Optics USB4000 multichannel spectrometer. All the measurements were performed under ambient atmosphere at room temperature soon after breaking the chamber.
Conclusions
In summary, we have demonstrated the design strategy and synthesis a series of carbazol-N-yl-carbazole end-capped oligoarylenes CAr as a non-doped solution processed hole-transporting emitters for full emission color tuning OLEDs. By using carbazol-N-yl-carbazole as end-capping groups, we are able to improve hole-transporting property, reduce the crystallization and retain the high emissive ability of a planar fluorescent core in the solid state, as well as improve the amorphous forming ability and solubility of the material. By using different oligoarylene cores, we are able to realize color tunable light-emitters. CAr can form morphologically stable amorphous thin films with Tgs of 100–126 °C. They exhibit a bright fluorescence with hole-transporting property. Their emission spectra in solution and thin film span the entire visible light spectrum (from 415 to 625 nm). As an emitter, the solution processed non-doped CAr-based double-layer OLEDs (ITO/PEDOT:PSS/CAr/BCP/LiF:Al) exhibit moderate to excellent device performance (1.99–13.73 cd A−1) with emission colors spanning the whole visible spectrum (blue to red). The RGB (red, green, blue) OLEDs based on these materials exhibit moderate to high luminescence efficiency and good color purity closed to the pure RGB colors. The solution processable molecular WOLED (ITO/PEDOT:PSS/CF:CT2F:CTB (100:3.0:0.20)/BCP/LiF:Al) shows luminous efficiency of 1.49 cd A−1 and white color stability with a color rendering index (CRI) of 77. This report offers a useful strategy to decorate the highly efficient but planar fluorophores to be suitable for applications in solution-processable and non-doped OLEDs.
Acknowledgements
This work was financially supported by the Thailand Research Fund (DBG 5580001) and Rayong Institute of Science and Technology (RAIST) Foundation. We acknowledge the post-doctoral scholarship support from Suranaree University of Technology and the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission.
Notes and references
- F. So, J. Kido and P. Burrows, MRS Bull., 2008, 33, 663 CrossRef CAS; B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef; M. C. Gather, A. Kohnen and K. Meerholz, Adv. Mater., 2011, 23, 233 CrossRef PubMed.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Mackay, K. Marks, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS; Y. Yang, Y. Zhou, Q. G. He, C. He, C. H. Yang, F. L. Bai and Y. F. Li, J. Phys. Chem. B, 2009, 113, 7745 CrossRef PubMed.
- L. Duan, L. Hou, T.-W. Lee, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, J. Mater. Chem., 2010, 20, 6392 RSC; X.-H. Zhu, J. Peng, Y. Cao and J. Roncali, Chem. Soc. Rev., 2011, 40, 3509 RSC.
- C.-J. Chang, C.-H. Yang, K. Chen, Y. Chi, C.-F. Shu, M.-L. Ho, Y.-S. Yeh and P.-T. Chou, Dalton Trans., 2007, 1881 RSC; F. M. Hwang, H. Y. Chen, P. S. Chen, C. S. Liu, Y. C. Chi, C. F. Shu, F. L. Wu, P. T. Chou, S. M. Peng and G. H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef CAS PubMed; E. Kim and S. B. Park, Chem.–Asian J., 2009, 4, 1646 CrossRef PubMed.
- M. R. Andersson, O. Thomas, W. Mammo, M. Svensson, M. Theander and O. Inganäs, J. Mater. Chem., 1999, 9, 1933 RSC; M. R. Andersson, M. Berggren, O. Inganäs, G. Gustafsson, J. C. Gustaffson-Carlberg, D. Selse, T. Hjertberg and O. Wennerström, Macromolecules, 1995, 28, 7525 CrossRef CAS.
- T. Ahn, M. Sik Jang, H.-K. Shim, D.-H. Hwang and T. Zyung, Macromolecules, 1999, 32, 3279 CrossRef CAS; M. R. An-derson, G. Yu and A. J. Heeger, Synth. Met., 1997, 85, 1275 CrossRef; L. Akcelrud, Prog. Polym. Sci., 2003, 28, 875 CrossRef.
- K.-R. Wee, H.-C. Ahn, H.-J. Son, W.-S. Han, J.-E. Kim, D. W. Cho and S. O. Kang, J. Org. Chem., 2009, 74, 8472 CrossRef CAS PubMed.
- T. Ahn, M. Sik Jang, H.-K. Shim, D.-H. Hwang and T. Zyung, Chem. Mater., 2005, 17, 5032 CrossRef.
- T. Khanasa, N. Prachumrak, R. Rattanawan, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk, T. Tuntulani and V. Promarak, J. Org. Chem., 2013, 78, 6702 CrossRef CAS PubMed.
- R. Pohl, V. A. Montes, J. Shinar and P. Anzenbacher Jr, J. Org. Chem., 2004, 69, 1723 CrossRef CAS PubMed; R. Pohl and P. An-zenbacher Jr, Org. Lett., 2003, 5, 2769 CrossRef PubMed; V. A. Montes, G. Li, R. Pohl, J. Shinar and P. Anzenbacher Jr, Adv. Mater., 2004, 16, 2001 CrossRef.
- P. Coppo, E. A. Plummer and L. D. Cola, Chem.
Commun., 2004, 1774 RSC; S. Lamansky, P. Djarovich, D. Murphy, F. A. Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704 CrossRef CAS PubMed.
- P. Moonsin, N. Prachumrak, R. Rattanawan, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2012, 48, 3382 RSC; V. Promarak, M. Ichikawa, T. Sudyoadsuk, S. Saengsuwan, S. Jungsuttiwong and T. Keawin, Synth. Met., 2007, 157, 17 CrossRef CAS PubMed; J. Li, C. Ma, J. Tang, C.-S. Lee and S. Lee, Chem. Mater., 2005, 17, 615 CrossRef; Q. He, H. Lin, Y. Weng, B. Zhang, Z. Wang, G. Lei, L. Wang, Y. Qiu and F. Bai, Adv. Funct. Mater., 2006, 16, 1343 CrossRef; J.-H. Jou, W.-B. Wang, S.-M. Shen, S. Kumar, I.-M. Lai, J.-J. Shyue, S. Lengvinaite, R. Zostautiene, J. V. Grazulevicius, S. Grigalevicius, S.-Z. Chen and C.-C. Wue, J. Mater. Chem., 2011, 21, 9546 RSC; N. Seidler, S. Reineke, K. Walzer, B. Lüssem, A. Tomkeviciene, J. V. Grazulevicius and K. Leo, Appl. Phys. Lett., 2010, 96, 093304 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Norm, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Z. J. Ning, Y. C. Zhou, Q. Zhang, D. G. Ma, J. J. Zhang and H. Tian, J. Photochem. Photobiol., A, 2007, 192, 8 CrossRef CAS PubMed; N. Prachumrak, S. Pojanasopa, R. Tarsang, S. Namuangruk, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, New J. Chem., 2014, 38, 3282 RSC.
- R. Misra, R. Sharma and S. P. Bhattacharyya, in Atomic and Molecular Nonlinear Optics: Theory, Experiment and Computation, ed. G. Maroulis, T. Bancewicz and B. Champagn, IOS Press, Amsterdam, 2011, pp. 55–71 Search PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef CAS PubMed; K. Bhattacharyya and M. Chowdhury, Chem. Rev., 1993, 93, 507 CrossRef; S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 2001, 34, 7315 CrossRef.
- K. E. Linton, A. L. Fisher, C. Pearson, M. A. Fox, L.-O. Pålsson, M. R. Bryce and M. C. Petty, J. Mater. Chem., 2012, 22, 11816 RSC; V. Promarak, A. Punkvuang, S. Jungsuttiwong, S. Saengsuwan, T. Sudyoadsuk and T. Keawin, Tetrahedron Lett., 2007, 48, 3661 CrossRef CAS PubMed; W. M. Albers, G. W. Canters and J. Reedijk, Tetrahedron, 1995, 51, 3895 CrossRef.
- A. M. Thangthong, N. Prachumrak, S. Namuangruk, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2012, 27, 5263 CrossRef CAS; K. T. Kamtekar, C. Wang, S. Bettington, A. S. Batsanov, I. F. Perepichka, M. R. Bryce, J. H. Ahn, M. Rabinal and M. C. Petty, J. Mater. Chem., 2006, 16, 3823 RSC.
- F. Sanda, T. Kawaguchi, T. Masuda and N. Kobayashi, Macromolecules, 2003, 36, 2224 CrossRef CAS; M. Zhu, Q. Wang, Y. Gu, X. Cao, C. Zhong, D. Ma, J. Qina and C. Yang, J. Mater. Chem., 2011, 21, 6409 RSC.
- Y. K. Kim and S.-H. Hwang, Synth. Met., 2006, 156, 1028 CrossRef CAS PubMed; A. Thaengthong, S. Saengsuwan, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Tetrahedron Lett., 2011, 52, 4749 CrossRef PubMed; A. M. Thangthong, D. Meunmart, N. Prachumrak, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2011, 47, 7122 RSC.
- U. Mitschke and P. BaÈuerle, J. Mater. Chem., 2000, 10, 1471 RSC.
- S. T. Zhang, Z. J. Wang, J. M. Zhao, Y. Q. Zhan, Y. Wu, Y. C. Zhou, X. M. Ding and X. Y. Hou, Appl. Phys. Lett., 2004, 84, 2916 CrossRef CAS PubMed.
- C. Tang, F. Liu, Y.-J. Xia, J. Lin, L.-H. Xie, G.-Y. Zhong, Q.-L. Fan and W. Huang, Org. Electron., 2006, 7, 155 CrossRef CAS PubMed.
- D. Y. Kim, H. N. Cho and C. Y. Kim, Prog. Polym. Sci., 2000, 25, 1089 CrossRef CAS.
- K. Wang, F. Zhao, C. Wang, S. Chen, D. Chen, H. Zhang, Y. Liu, D. Ma and Y. Wang, Adv. Funct. Mater., 2013, 23, 2672 CrossRef CAS; W.-C. Wu, C.-L. Liu and W.-C. Chen, Polymer, 2006, 47, 527 CrossRef PubMed.
- T. Kartens and K. Kobs, J. Phys. Chem., 1980, 84, 1871 CrossRef.
- S. R. Forrest, D. D. C. Bradley and M. E. Thomson, Adv. Mater., 2003, 15, 1043 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1–5, quantum chemical calculation results, multiple scan CV plots, AFM images, EL spectra of the OLEDs and 1H NMR and 13C NMR spectra. See DOI: 10.1039/c4ra16458j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.