
Supramolecular solvent-based dispersive liquid–liquid microextraction of copper from water and hair samples
Funda Aydina,
Erkan Yilmazb and
Mustafa Soylak*b
aYuzuncu Yil University, Faculty of Pharmacy, Department of Basic Sciences, 65080 Van, Turkey
bErciyes University, Faculty of Sciences, Department of Chemistry, 38039-Kayseri, Turkey. E-mail: soylak@erciyes.edu.tr; Tel: +90 3524374933
First published on 16th April 2015
Abstract
A supramolecular solvent based dispersive liquid–liquid microextraction (SM-DLLME) procedure has been established for the separation and preconcentration of Cu(II) before its determination by microsampling flame atomic absorption spectrometry. The proposed method involves the use of a supramolecular solvent in which reverse micelles of 1-decanol are dispersed in tetrahydrofuran. The Cu(II)–pyrrolidinedithiocarbamate complex was formed to increase interactions with the supramolecular phase at pH 6. After the supramolecular solvent was added to the medium, the formation of micelles of nano and molecular size was observed in an ultrasonic bath. The solution was centrifuged, and the metal complex formed was extracted into the supramolecular solvent phase. Some analytical parameters that are important in the experiment were examined in detail. The detection limit (LOD), the quantification limit (LOQ) and the relative standard deviation (RSD) of the developed method were found to be 0.11 μg L−1, 0.34 μg L−1 and 2.2%, respectively. The preconcentration factor was 60. Addition/recovery studies were also performed in water and human hair samples. The accuracy of the proposed method was assessed by analyzing certified reference materials. The procedure was applied for the determination of copper in water and hair samples.
1. Introduction
Heavy metals are widely used in all areas. They are found in substantial quantities around the world because of industrial and agricultural activities.1 Heavy metals enter our bodies through drinking water, food and air. Some trace levels of heavy metals such as copper are necessary to maintain the metabolism of the human body. Living organisms need this metal for the proper functioning of organs and metabolic processes. However, copper may also be toxic when taken in a very high dose and can damage an organism.2The broad application of technologies such as those used in the electrical and electronic, construction, transportation, paint, and chemical industries increases the prevalence of copper as a pollutant. A large amount of copper can cause important environmental problems. Copper ions are present in some surface and ground waters due to the misuse of industrial wastes. This widespread use of copper affects human health adversely.3 Many pollution problems related to heavy metals such as ecological and health problems are increasing.4 Many international organizations have set a permissible limit for the levels of metals in drinking water. For example, the World Health Organization and Council of the European Union have accepted a maximum allowable limit of 2 mg L−1 for copper in drinking water.5,6 The determination of toxic heavy metal ions from various matrices at the trace level is very important. Due to the low concentration of metals and the effects of the matrix, the determination of ultra-trace levels in real samples is difficult. Hence, a separation and preconcentration method is necessary for the analysis of environmental samples.7
Some analytical techniques for preconcentration and separation of metal ions at trace levels, such as liquid–liquid or liquid–solid extraction techniques, have the disadvantage of using a large amount of organic solvent, and such solvents are toxic and pollutants for the environment.8–12 Because of these disadvantages of classical techniques, microextraction techniques, which are simple and miniaturized and consume less chemical solvents, have become widely used in recent years. Single-drop microextraction (SDME),13,14 solid-phase microextraction (SPME),15 dispersive liquid–liquid microextraction (DLLME),16 hollow fiber liquid-phase microextraction (HF-LPME),17 solidified floating organic drop microextraction (SFODME)18,19 and supramolecular solvent-based microextraction (SM-SBM)20–25 are some examples of microextraction techniques.
Supramolecular solvent-based microextraction techniques have been used for organic and inorganic determinations.26–28 Supramolecular solvents (SUPRAS) are nano-structured liquids that consist of assemblies of amphiphiles dispersed in a continuous phase.29,30 SUPRAS are made up of reverse micelle aggregates of alkanols that spontaneously form in tetrahydrofuran (THF)/water solution through self-assembly processes. These solvents consist of aqueous cavities surrounded by the polar groups of alkanols with the hydrocarbon chains dissolved in THF. The size of the aqueous cavities can be tailored by controlling the THF/water ratio in the bulk solution where alkanols self-assemble. THF plays a dual role, not only acting as a dispersing solvent but also causing self-assembly of decanol. Alkanol-based SUPRAS provide an effective medium for isolating and preconcentrating hydrophobic complexes of metals with a ligand and organic compounds. These solvents have different interactions with this kind of structure. SUPRAS involve both dispersion and hydrogen bonding interactions. This nanostructured liquid provides an excellent reaction media for the extraction process.31 Supramolecular-based methods, which have been developed as an alternative to organic solvents, have been widely used as an environmentally friendly solvent for the extraction of hydrophobic compounds from environmental waters. This method allows us to achieve a short extraction time and easy sample preparation and does not require large quantities of toxic substances.
The aim of this work was to develop a suitable microextraction method by using a supramolecular solvent that is rapid, easy and low cost for the separation and preconcentration of copper.
2. Experimental
2.1. Reagents and solutions
All chemicals used were analytical grade reagents, and all aqueous solutions were prepared with ultrapure water. A Millipore water purification device (18.2 MΩ cm) was used. A stock solution of 1000 ppm Cu2+ was prepared from Cu(NO3)2·3H2O. Working solutions were prepared by appropriate dilution of the stock solution. Ethanol was used to prepare 0.1% (w/v) ammonium pyrrolidinedithiocarbamate (ADPC; Sigma-Aldrich). Ammonium acetate/acetic acid solution was used as a buffer to adjust the pH of sample solutions to pH 6. 1-Decanol and THF purchased from Merck were used as extraction solvents. These solutions were added to the solutions by mixing 150 μL of 1-decanol and 600 μL of THF for each microextraction. Solutions of 30% (v/v) H2O2 and 65% HNO3 (Merck) were used for digestion of human hair samples. The TMDA-51.3 and TMDA-64.2 (Water-Trace Elements) and NCS ZC 8100 2b Human Hair certified reference materials were used for validation of the procedure.2.2. Apparatus
A flame atomic absorption spectrometer (Perkin-Elmer Model 3110) with air–acetylene and a copper hollow cathode lamp was used. The instrumental parameters were set as follows: wavelength of 324.8 nm, slit width of 0.7 nm and lamp current of 15.0 mA. A home-made micro-sampling introduction system made of Teflon was connected to the FAAS nebulizer. During the analysis, a 100 μl sample was injected through the space in the middle of the Teflon by micropipette.32A brand mark micropipette was used to transfer reagents and for injections. Measurements were made considering the peak heights. A Sartorius PT-10 pH meter with a combined glass electrode was utilized for pH adjustment. An ultrasonic water bath (Sonorex) was used to acquire a good cloudy solution. A Centrifuge ROTINA 38 centrifuge equipped with an angled rotor (8 × 50 mL, 5000 rpm) from Hettich was used to separate the supramolecular solvent phase from the aqueous phase.
2.3. Supramolecular solvent-based dispersive liquid–liquid microextraction procedure
A schematic presentation of the presented procedure is given in Fig. 1. Studies of the method were carried out with 10 mL model solutions containing 0.10 μg of Cu(II). First, 10 mL of solution containing copper was placed in a 50 mL conical centrifuge tube. Thereafter, 2.5 mL of acetate buffer solution was added to adjust the sample pH (6.0) using diluted NaOH and HCl solutions. Then, 0.5 mg of ammonium pyrrolidinedithiocarbamate was added. After waiting for approximately 5 minutes for complex formation, an extractor solvent consisting of tetrahydrofuran (600 μL) and 1-decanol (150 μL) was injected rapidly into the sample. The mixture was sonicated for 5 minutes, and a cloudy solution was obtained. During this time, the supramolecular solvent spontaneously formed in the bulk solution. This solution was centrifuged at 4000 rpm for 10 minutes. Then, the supramolecular solvent phases that had a low density separated fully from the aqueous phase and formed a solvent solution in the upper phase. The lower water phase was taken up with a pipette and discarded. The volume of the supramolecular solvent (about 120–150 μL) was completed to 500 μL with methanol. Then, 100 μl of the solution was injected into the micro-sampling introduction system connected to the FAAS nebulizer. The measurements made using the microinjection system were performed in a continuous aspiration mode. The same procedure was applied for the blank solutions. | ||
| Fig. 1 Schematic presentation of the presented procedure (SM-DLLME). | ||
2.4. Applications
Tap, well, sea and underground water samples were collected from various regions of Turkey. These samples were filtered through membrane filters with 0.45 μm pores. Then, the developed microextraction technique was applied to all water samples.A human hair sample was taken from a man using stainless steel scissors. All glassware and plastic equipment were immersed in hydrochloric acid to avoid contamination. Then all equipment was washed with deionised water and dried. The hair sample was washed with acetone and deionised water.33 This procedure was performed again in order to thoroughly remove adhered contaminants. Then, the hair sample was dried and was ready to be analyzed by the methods developed. First, 10 mg of the human hair sample was carefully weighted in a beaker. Then, 5 mL concentrated HNO3 (65%) and 10 mL H2O2 (30%) were added to the hair sample. A watch glass was placed over the beaker and the sample digested at 100 °C. Mixtures were evaporated on the hot plate to dryness, and 5 mL concentrated HNO3/10 mL H2O2 was added again and evaporated to dryness. Distilled water was added to the residue using a micropipette, and the sample was transferred to a 50 mL conical-bottom glass centrifuge tube. The final volume of the solution was completed to 5 mL. After the pH of the sample was adjusted to 6.0 using diluted sodium hydroxide and hydrochloric acid, the developed microextraction method was applied to the human hair sample.
3. Results and discussion
3.1. Effects of pH
In the extraction operation, the optimum pH of the sample solution is very important to form complexes of trace metal ions with a suitable complexing agent and for their passage into the extraction phase from the water phase.34 Thus, the effect of pH on the recovery of Cu(II) was studied in the range of 2.0–8.0. The results in Fig. 2 show that the recovery % increased beyond pH 4. The results were obtained quantitatively in the range of pH 4–7, and the recovery % decreased after the pH 8. Therefore, pH 6 was selected as a suitable pH and used in subsequent experiments. | ||
| Fig. 2 Effect of the pH on the recovery of Cu(II) by SM-DLLME (n = 3). | ||
3.2. Influence of the amount of APDC
SM-DLLME depends on the formation of a hydrophobic complex between the analyte and the chelating agent.35,36 To increase the extraction efficiency, the effects of the recovery of the amount of ligand that was added to the solution at the studied pH was examined by adding chelating agent in the range 0.1 to 1.5 mg APDC (0.1%). The results obtained are depicted in Fig. 3. The results reveal that 0.5 mg and greater than 0.5 mg of chelating agent was suitable to obtain a quantitative recovery %. When the amount of chelating agent added was less than 0.5 mg, the recoveries observed were not quantifiable. Thus, 0.5 mg ligand was enough to convert all copper to metal complex. Therefore, the amount of ligand was chosen to be 0.5 mg to achieve quantitative recoveries (>95%), and this amount of ligand was used in later studies.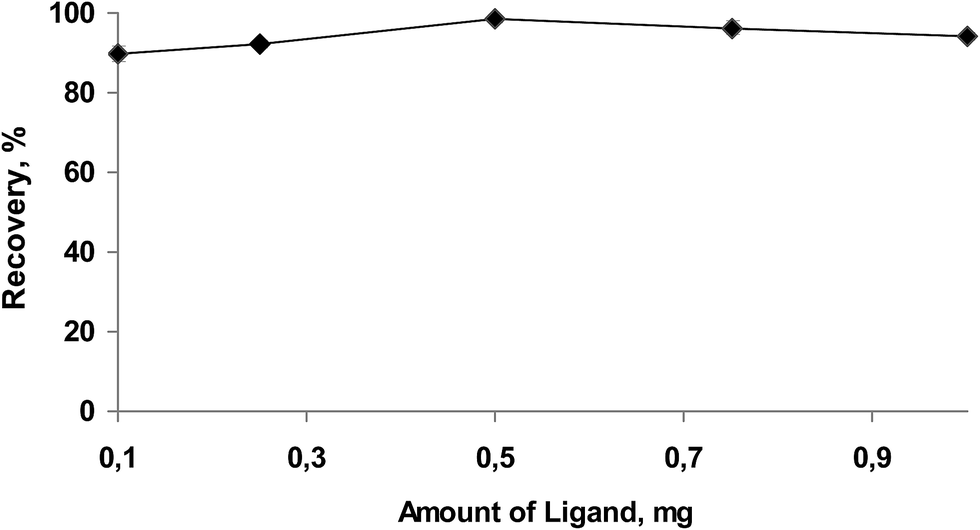 | ||
| Fig. 3 Effect of amount of APDC on the recovery of Cu(II) obtained from SM-DLLME (n = 3). | ||
3.3. Type and amount of supramolecular solvent
The influence of the supramolecular extraction solvent composition on the microextraction of copper was examined. To obtain the formation of a suitably cloudy solution, different extraction solvents including 1-decanol, undecanol and decanoic acid were tested with THF. THF acts as a dispersing solvent and causes self-assembly of decanol, which is composed of reversed micelles.21 The recoveries of copper with decanoic acid–THF, undecanol–THF and 1-decanol–THF were 78% ± 3%, 85% ± 3% and 98% ± 2%, respectively. A quantitative recovery (>95%) was obtained with 1-decanol–THF. Therefore, 1-decanol was used to form the supramolecular solvent together with THF.The amount of 1-decanol–THF greatly influences both the volume of extractant yielded and the extraction efficiency in the cloudy solution. Hence, first the volume of 1-decanol was studied in the range of 50–250 μL. The results are depicted in Fig. 4. The recovery % of Cu(II) was found to be quantitative when the volume of 1-decanol was above 100 μL. Above 200 μL, the recoveries decreased. Hence, 150 μL 1-decanol was determined to be the optimum extraction solvent volume to achieve quantitative extraction of Cu–APDC complex.
 | ||
| Fig. 4 Effect of volume of 1-decanol on the recovery of Cu(II) by SM-DLLME (n = 3). | ||
To examine the effect of the volume of THF, a series of microextraction solvents that had different volumes of THF (in the range of 200–1000 μL) and a fixed amount of 1-decanol (150 μL) were prepared. The optimum volume of THF was determined to ensure better micelle formation, better dispersion and quantitative recovery of Cu(II). The recovery of Cu(II) was not quantitative with up to 600 μL of THF (Fig. 5). Above this volume, the recoveries were sufficient to obtain quantitative results. Therefore, 600 μl of THF was selected with 1-decanol as the supramolecular solvent, and this volume was used in subsequent studies.
 | ||
| Fig. 5 Effect of volume of THF on the recovery of Cu(II) obtained from SM-DLLME (n = 3). | ||
Efficient quantitative extraction for copper(II) with the presented microextraction system was obtained when the 1-decanol–THF volume ratio was 0.250. The recoveries of copper(II) were not quantitative at other volume ratios of decanol–THF. This result is consistent with the literature.22–24
3.4. Effects of ultrasonic mixing time and centrifugation time
After 1-decanol–THF was injected into the model solution medium, model solutions were kept in an ultrasonic bath from 2–10 minutes, and the recoveries were investigated. Depending on the ultrasonication time, the recovery of analytes increased linearly up to 5 min. This ultrasonic bath time (5 min) was adequate for obtaining the maximum recovery %. Thus, this time was selected as optimal for subsequent studies.The centrifugation time used for extraction is an important experimental parameter in terms of separating the extraction phase exactly from the water phase. For this reason, the influence of centrifugation time was studied with model solutions at 4000 rpm (3500 rcf) from 2–10 minutes. Because the best separation was achieved in 10 minutes, the sample solutions were centrifuged for 10 minutes in subsequent microextraction studies.
3.5. Sample volume
Various experiments were carried out using different sample volumes between 10–50 mL to investigate the effect of sample volume on the recovery of analyte. Other experimental conditions were kept constant. Copper recovery was found to increase quantitatively (≥95%) up to 30 mL, and with a sample volume greater than 30 ml, the recovery decreased. Subsequently, we used a sample volume up to 30 mL with this developed method. As a result, the preconcentration factor was calculated to be 60 (the first volume: 30, the final volume: 0.5 mL).3.6. Effects of matrix components
Because of the matrix effect, trace heavy metals cannot be determined directly with FAAS.37–43 Therefore, model solutions containing interference ions (alkali, alkaline earth and other ion and metals) that can be found in real samples were prepared to examine the influence of the matrix effect on the microextraction of Cu(II). The general procedure was applied to these solutions containing 0.5 μg of copper in 10 ml and different amounts of foreign ions. The tolerance values were defined according to the amounts of interference ions. Table 1 shows the results for the interference ions. As can be seen in Table 1, matrix ions had no important interference effect on the determination of Cu(II) up to the amounts used to obtained maximum tolerance values.| Foreign ions | Added as | Amount of foreign ions (mg L−1) | Recovery, % |
|---|---|---|---|
| Na+ | NaNO3 | 3000 | 101 ± 4 |
| K+ | KCl | 3000 | 100 ± 2 |
| Mg2+ | Mg(NO3)2·6H2O | 2000 | 98 ± 4 |
| Ca2+ | Ca(NO3)2·4H2O | 2000 | 106 ± 6 |
| Cl− | KCl | 3000 | 98 ± 3 |
| SO42− | Na2SO4 | 1000 | 95 ± 3 |
| Mn2+ | Mn(NO3)2·4H2O | 10 | 97 ± 2 |
| Cr3+ | Cr(NO3)3·9H2O | 10 | 101 ± 3 |
| Cd2+ | Cd(NO3)2·4H2O | 10 | 101 ± 4 |
| Zn2+ | Zn(NO3)2·6H2O | 10 | 101 ± 3 |
| Co2+ | Co(NO3)2·6H2O | 10 | 102 ± 2 |
| Pb2+ | Pb(NO3)2 | 5 | 96 ± 1 |
| Fe3+ | Fe(NO3)3·9H2O | 2.5 | 98 ± 4 |
| Ni2+ | Ni(NO3)3·6H2O | 2.5 | 96 ± 4 |
3.7. Analytical performance
To obtain the calibration curves, five standard solutions were used as the supramolecular solvent. Several analytical parameters such as enhancement factor (EF), limit of detection (LOD), limit of quantification (LOQ) and repeatability were obtained upon applying the developed method. The calibration curve equation was found to be A = 0.00394 + 0.054C (A: absorbance of the solution, C: determined Cu concentration of the solution). The correlation coefficient (R2) was 0.995. The limit of detection (LOD) and limit of quantification (LOD) were determined by using blank solutions. The LOD was calculated by using of the ratio of three times the standard deviation of the absorbance of ten blank samples to the slope of the calibration curve (3 s m−1) and was 0.11 μg L−1. The LOQ was calculated by using of the ratio of ten times the standard deviation of the absorbance of ten blank samples to the slope of the calibration curve (10 s m−1) and was 0.34 μg L−1. The relative standard deviation (RSD)% was calculated to determine the precision of the method by using ten model solutions that contained 25 μg L−1 of copper(II) and was found to be 2.2%. The enhancement factor (EF) calculated as the ratio of the slope of the calibration curve of copper(II) after preconcentration to that prior to preconcentration was found to be 60.3.3.8. Accuracy and application of the method
In order to test the reliability of this method, addition/recovery studies were conducted on samples of water and hair. Different amounts of copper were added to water and hair samples. The recoveries obtained after the addition of known amounts of copper compared to those from the real samples can be seen in Tables 2 and 3. The recoveries ranged between 95–102% for water and hair samples. According to the results, the copper recoveries were consistent with the amounts of copper added.| Tap water from Canakkale | Well water from Sivas | Marmara sea water from Istanbul | ||||||
|---|---|---|---|---|---|---|---|---|
| Added (μg) | Founda (μg) | % Recovery | Added (μg) | Founda (μg) | % Recovery | Added (μg) | Founda (μg) | % Recovery |
| a Mean ± standard deviation. | ||||||||
| 0.00 | 0.023 ± 0.005 | — | 0.00 | 0.028 ± 0.004 | — | 0.00 | 0.039 ± 0.009 | — |
| 0.25 | 0.268 ± 0.030 | 98 | 0.25 | 0.281 ± 0.007 | 101 | 0.25 | 0.282 ± 0.007 | 97 |
| 0.50 | 0.532 ± 0.030 | 102 | 0.50 | 0.516 ± 0.007 | 98 | 0.50 | 0.539 ± 0.009 | 100 |
In order to evaluate the validity and applicability of the developed method, the method based on SM-DLLME method was applied under optimum conditions with TMDA-51.3 and TMDA-64.2 water – trace element and NCS ZC 8100 2b human hair certified reference materials (CRMs). The results are shown in Table 4. The recovery percentages ranged between 95% and 100%. The copper recoveries obtained were close to the certified values.
| TMDA-51.3 water – trace elements (μg L−1) | TMDA-64.2 water – trace elements (μg L−1) | NCS ZC 8100 2b human hair (μg g−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| Certified value | Found | Recovery (%) | Certified value | Found | Recovery (%) | Certified value | Found | Recovery (%) |
| a Mean ± standard deviation. | ||||||||
| 89.2 | 88.8 ± 8a | 100 | 274 | 263 ± 10 | 96 | 33.6 ± 2.3 | 31.9 ± 1.2 | 95 |
The results from the application of the developed microextraction method to the determination of copper in hair and water samples are given in Table 5.
| Concentration of copper, μg L−1 | |
|---|---|
| Well water from Kayseri | 126 ± 5 |
| Sea water from Marmara sea from Çanakkale | 158 ± 5 |
| Underground water from Kayseri | 353 ± 2 |
3.9. Comparison between the proposed method and other extraction methods
A comparison of the SM-DLLME method and some of the published extraction methods for preconcentration and determination of Cu(II) was made, and the results are presented in Table 6. As can be seen, the proposed method was comparable to other reported methods in which flame atomic spectrometry was applied as the detection method. The values of LOD, relative standard deviation and enhancement factor are generally good except for a few exceptions.44–51 Thus, the presented method can be used for the determination of trace levels of copper in real samples.| Method | Sample | EFa | LODb (μg L−1) | RSDc (%) | Ref. |
|---|---|---|---|---|---|
| a Enrichment factor.b Limit of detection.c Relative standard deviation; DLLME: Dispersive liquid–liquid microextraction, UA-IL-DLLME: ultrasonic assisted-ionic liquid based-liquid–liquid microextraction, SFODME: floating organic drop microextraction, MWNTs: solid phase extraction on multi-walled carbon nanotubes, CPE: cloud-point extraction, SM-DLLME: supramolecular solvent liquid–liquid microextraction. | |||||
| DLLME | Water | 42 | 3.00 | 5.1 | 43 |
| UA-IL-DLLME | Food | 100 | 0.17 | 3.0 | 44 |
| DLLME | Cereals, vegetables | 55 | 0.16 | 1.5 | 45 |
| UA-IL-DLLME | Water | 50 | 1.90 | 3.8 | 46 |
| SFODME | Water | 333 | 0.40 | 0.9 | 47 |
| MWNTs | Water | 60 | 1.46 | — | 48 |
| CPE | Food | 29 | 1.50 | 6.4 | 49 |
| SM-DLLME | Water, human hair | 60.3 | 0.11 | 2.2 | This work |
4. Conclusions
The supramolecular-solvent based dispersive liquid–liquid microextraction method based on the formation of reverse micelles of decanol dispersed in THF/water has many great advantages for the accurate determination of copper using microsampling FAAS. The application of the SUPRAS method is quite simple and rapid. A single extraction is sufficient, and other extraction procedures are not needed. The separation and preconcentration of copper and preparation for sample analysis procedures takes approximately 35–40 minutes. The method is an environmentally friendly method, because it does not require too much organic solvent. The amounts of THF (600 μL) and 1-decanol (150 μL) used to create the SUPRAS phase are very low. Because the extraction solvent does not need to be evaporated, direct extraction solvent analysis (at the microliter level) can be done to analyze the copper. In addition, a large amount of sample volume is not required, and a very small sample volume (500 μL) can be analyzed with FAAS using this method. In addition, the selectivity of the method is good, and no interference effect in the presence of a matrix component was observed. Based on these advantages, the developed method is a practical and reliable method for the determination of copper in environmental water samples and human hair.References
- A. Duran, M. Soylak and S. A. Tuncel, J. Hazard. Mater., 2008, 155, 114–120 CrossRef CAS PubMed.
- S. L. Belli and A. Zirino, Anal. Chem., 1993, 65, 2583–2589 CrossRef CAS.
- V. K. Gupta, M. L. Yola, N. Atar, Z. Ustundag and A. O. Solak, Electrochim. Acta, 2013, 112, 541–548 CrossRef CAS PubMed.
- M. Ghaedi, K. Niknam, K. Taheri, H. Hosseinian and M. Soylak, Food Chem. Toxicol., 2010, 48, 891–897 CrossRef CAS PubMed.
- World Health Organization, (2008), Drinking Water Quality: Third Edition incorporating the First and Second Addenda, Volume 1: Recommendations, Geneva.
- Council of the European Union, (1998), Council Directive 98/83/EC on the Quality of Water Intended for Human Consumption.
- F. A. Aydin and M. Soylak, Talanta, 2007, 72, 187–192 CrossRef CAS PubMed.
- M. Soylak and Y. Akkaya, Trace Elem. Electrolytes, 2002, 19, 100–104 CAS.
- M. Soylak, A. Uzun and L. Elci, Trace Elem. Electrolytes, 2002, 19, 15–19 CAS.
- M. Soylak, S. Saracoglu and L. Elci, Trace Elem. Electrolytes, 2003, 20, 160–165 CrossRef CAS.
- O. Yalcinkaya, O. M. Kalfa and A. R. Turker, Turk. J. Chem., 2010, 34, 207–217 CAS.
- D. Nasser, J. Nasrin and A. Y. Kumar, Turk. J. Chem., 2008, 32, 561–570 Search PubMed.
- A. Jain and K. K. Verma, Anal. Chim. Acta, 2011, 706, 37–65 CrossRef CAS PubMed.
- X. Wen, Q. Deng, J. Guo and S. Yang, Spectrochim. Acta, Part A, 2011, 79, 508–512 CrossRef CAS PubMed.
- N. Philiswa, J. Nomngongo and N. Catherine, Spectrochim. Acta, Part B, 2014, 98, 54–59 CrossRef PubMed.
- M. Soylak and E. Yılmaz, Desalination, 2011, 275, 297–301 CrossRef CAS PubMed.
- H. Jiang, B. Hu, B. Chen and W. Zu, Spectrochim. Acta, Part B, 2008, 63, 770–776 CrossRef PubMed.
- M. R. Moghadam, S. Dadfarnia and A. M. H. Shabani, J. Hazard. Mater., 2011, 186, 169–174 CrossRef CAS PubMed.
- S. Dadfarnia and A. M. H. Shabani, Anal. Chim. Acta, 2010, 658, 107–119 CrossRef CAS PubMed.
- S. Jafarvand and F. Shemirani, Sep. Sci., 2011, 34, 455–661 CrossRef CAS PubMed.
- A. Ballesteros-Gomez, S. Rubio and D. Perez-Bendito, J. Chromatogr. A, 2009, 1216, 530–539 CrossRef CAS PubMed.
- S. Jafarvand and F. Shemirani, Anal. Methods, 2011, 3, 1552–1559 RSC.
- A. Moral, M. D. Sicilia and S. Rubio, J. Chromatogr. A, 2009, 1216, 3740–3745 CrossRef CAS PubMed.
- S. Jafarvand and F. Shemirani, J. Sep. Sci., 2011, 34, 455–461 CrossRef CAS PubMed.
- F. Rezaei, Y. Yaminia, M. Moradi and B. Daraei, Anal. Chim. Acta, 2013, 804, 135–142 CrossRef CAS PubMed.
- E. Yilmaz and M. Soylak, Talanta, 2014, 126, 191–195 CrossRef CAS PubMed.
- F. J. López-Jiménez, S. Rubio and D. Pérez-Bendito, Food Chem., 2010, 121, 763–769 CrossRef PubMed.
- C. Caballo, M. D. Sicilia and S. Rubio, Anal. Chim. Acta, 2013, 761, 102–108 CrossRef CAS PubMed.
- E. M. Costi, M. D. Sicilia and S. Rubio, J. Chromatogr. A, 2010, 1217, 6250–6257 CrossRef CAS PubMed.
- S. Jafarvand and F. Shemirani, J. Sep. Sci., 2011, 34, 455–461 CrossRef CAS PubMed.
- A. Ballesteros-Gomez and S. Rubio, Anal. Chem., 2012, 84, 342–349 CrossRef CAS PubMed.
- M. Soylak and E. Yilmaz, Atom Spectrosc., 2013, 34, 15–19 CAS.
- P. Bermejo-Barrera, O. Muniz-Naveiro, A. Moreda-Pineiro and A. Bermejo-Barrera, Forensic Sci. Int., 2000, 107, 105–120 CrossRef CAS.
- Naeemullah, M. Tuzen, T. G. Kazi, D. Citak and M. Soylak, J. Anal. At. Spectrom., 2013, 28, 1441–1445 RSC.
- M. Salahinejad and F. Aflaki, J. Chem. Health Risks, 2013, 3, 55–64 Search PubMed.
- A. Garcia-Prieto, L. Lunar, S. Rubio and D. Pérez-Bendi, Anal. Chim. Acta, 2008, 617, 51–58 CrossRef CAS PubMed.
- A. Gundogdu, D. Ozdes, C. Duran, V. N. Bulut, M. Soylak and H. B. Senturk, Chem. Eng. J., 2009, 153, 62–69 CrossRef CAS PubMed.
- A. A. Ensafi, M. Jokar and M. Ghiaci, J. Iran. Chem. Soc., 2015, 12, 457–467 CrossRef CAS.
- H. B. Senturk, A. Gundogdu, V. N. Bulut, C. Duran, M. Soylak, L. Elci and M. Tufekci, J. Hazard. Mater., 2007, 149, 317–323 CrossRef CAS PubMed.
- M. Soylak and N. D. Erdogan, J. Hazard. Mater., 2006, 137, 1035–1041 CrossRef CAS PubMed.
- M. A. Farajzadeh, M. Bahram, B. G. Mehr and J. A. Jöhnsson, Talanta, 2008, 75, 832–840 CrossRef CAS PubMed.
- M. Soylak and L. Elci, J. Trace Microprobe Tech., 2000, 18, 397–403 CAS.
- M. Tuzen and M. Soylak, J. Hazard. Mater., 2009, 162, 724–729 CrossRef CAS PubMed.
- N. Jalbani and M. Soylak, Food Chem., 2015, 167, 433–437 CrossRef CAS PubMed.
- K. Shrivas and N. K. Jaiswal, Food Chem., 2013, 141, 2263–2268 CrossRef CAS PubMed.
- M. Rajabi, S. Asemipour, B. Barfi, M. R. Jamali and M. Behzad, J. Mol. Liq., 2014, 194, 166–171 CrossRef CAS PubMed.
- C. A. Sahin and I. Tokgöz, Anal. Chim. Acta, 2010, 667, 83–87 CrossRef CAS PubMed.
- M. Soylak and O. Ercan, J. Hazard. Mater., 2009, 168, 1527–1531 CrossRef CAS PubMed.
- V. A. Lemos, M. S. Santos, G. T. David, M. V. Maciel and M. A. Bezerra, J. Hazard. Mater., 2008, 159, 245–251 CrossRef CAS PubMed.
- V. N. Bulut, A. Gundogdu, C. Duran, H. B. Senturk, M. Soylak, L. Elci and M. Tufekci, J. Hazard. Mater., 2007, 146, 155–163 CrossRef CAS PubMed.
- M. Ghaedi, F. Ahmadi, Z. Tavakoli, M. Montazerozohori, A. Khanmohammadi and M. Soylak, J. Hazard. Mater., 2008, 152, 1248–1255 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |