
Stability of hydrogenated graphene: a first-principles study
Ding Yia,
Liu Yanga,
Shijie Xie*a and
Avadh Saxenab
aSchool of Physics, State Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, China. E-mail: xsj@sdu.edu.cn
bTheoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
First published on 10th February 2015
Abstract
In order to explain the disagreement between present theoretical and experimental investigations on the stability of hydrogenated graphene, we have systematically studied hydrogenated graphene with different configurations from the consideration of single-side and double-side adsorption using first-principles calculations. Both binding energy and formation energy are calculated to characterize the stability of the system. It is found that single-side hydrogenated graphene is always unstable. However, for double-side hydrogenation, some configurations are stable due to the increased carbon–carbon sp3 hybridization compared to single-side hydrogenation. Furthermore, it is found that the system is energetically favorable when an equal number of hydrogen atoms are adsorbed on each side of the graphene.
1 Introduction
Since the experimental discovery of graphene in 2004,1 this one-atom-thick material has attracted considerable attention due to its remarkable electronic and structural characteristics.2 Researchers have fruitfully investigated graphene to optimize its chemical and physical properties. For example, to open a sizable band gap in graphene for use in electronic and spintronic devices, several methods have been proposed, such as fabricating graphene nanoribbons,3 utilizing bilayer structures,4,5 and chemical modification.6,7 Among them, chemical adsorption of non-carbon atoms on graphene is a good way to open a band gap. When such atoms are adsorbed on a graphene surface, they form covalent bonds with the carbon (C) atoms. These C atoms change their hybridization from sp2 to sp3, which dramatically alters the electronic structure of graphene. In recent years, adsorbing hydrogen (H) atoms on the graphene surface has received the most consideration.8,9 On the one hand hydrogenation is the simplest adsorption, which is easily controlled and studied, whereas on the other hand hydrogenation is related to hydrogen storage,10–13 which is promising for application of graphene in energy storage. Since the adsorption of H atoms on graphene is so important, a systematic investigation of its stability is highly desirable.However, present investigations show that there is a disagreement about the stability of hydrogenated graphene. Some theoretical works predict that hydrogenated graphene is very stable.14–16 The calculated formation energy of hydrogenated graphene is negative and the absolute value is much higher than the heat that the room temperature can provide. For example, a widely studied fully hydrogenated graphene with C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H = 1:1, which is also called graphane, has a negative formation energy lower than that of benzene.14 Its binding energy appears to be rather similar to fluorinated graphene,15 which has been demonstrated to be stable both in theory15 and experiment.6,17 Nevertheless, experimental investigations have shown that hydrogenation of graphene is unstable, which rapidly loses H atoms at moderate temperature.18–21
:H = 1:1, which is also called graphane, has a negative formation energy lower than that of benzene.14 Its binding energy appears to be rather similar to fluorinated graphene,15 which has been demonstrated to be stable both in theory15 and experiment.6,17 Nevertheless, experimental investigations have shown that hydrogenation of graphene is unstable, which rapidly loses H atoms at moderate temperature.18–21
In experiments, graphene membranes are often set on a substrate, such as SiO2.18,19 In this situation, only one side of graphene is accessible for H adsorption, because the diffusion of atomic H along the graphene–SiO2 interface is negligible.19,22 In contrast, for free-standing graphene membranes, both sides can adsorb. Therefore, to answer the stability question of hydrogenation of graphene, we have to consider single-side as well as double-side adsorption.
In this paper, using first-principles calculations we systematically study the stability of single-side and double-side hydrogenated graphene. Both the binding energy and the formation energy are calculated to characterize the stability of the system. The remainder of the paper is organized as follows. The model and methods are described next; the results and discussion section is presented subsequently. Finally, the main conclusions are given.
2 Model and methods
The purple rhombus as sketched in Fig. 1 is used as the supercell, which is composed of 8C atoms marked as 1–8. For hydrogenated graphene, the number of adsorbed H atoms and the adsorbing positions result in a large number of configurations. In this work, we focus on a limited number of typical configurations. | ||
| Fig. 1 Top view structure of pristine graphene. The purple rhombus area is chosen as the supercell containing 8C atoms marked as 1–8. The yellow balls represent C atoms. | ||
Both the binding energy EB and the formation energy EF are introduced to characterize the stability of hydrogenated graphene. We adopt the definition of EB (EF) as the difference of the hydrogenated graphene with respect to pristine graphene per H atom for the corresponding atomic H (diatomic molecules H2).14,15 The corresponding expressions are given by:
| EB = [EHG − (EPG + nEH)]/n |
| EF = [EHG − (EPG + nEH2/2)]/n |
The first-principles calculations are performed using density functional theory (DFT) method implemented with SIESTA code,23 which was successfully applied previously to describe adsorption on graphene.24 The exchange–correlation potential is approximated by generalized gradient approximation (GGA) using Perdew–Burke–Ernzerhof (PBE) functional,25 which is usually invoked for electronic structure calculations of C–H systems. The double-ζ plus polarization function (DZP) basis set is used, together with a mesh cutoff of 400 Ry and the Brillouin zone is sampled by 20 × 20 × 1 k points. Since periodic boundary conditions are applied in all three dimensions, a vacuum layer of more than 20 Å is included to minimize the interaction between adjacent layers. All the atoms in the supercell are fully relaxed to fulfill the energy and force convergence of 10−5 eV and 10−3 eV Å−1, respectively. As hydrogenation may change the bond length and bond angle of graphene, both the size and the shape of supercell are allowed to relax in our calculations.
3 Results and discussion
First, we consider the single-side hydrogenation of graphene. Typical configurations of 1–7H atoms adsorbing on the supercell of 8C atoms are considered. As the fully single-side hydrogenated graphene (8H atoms) has a flat structure and the lengths of C–C covalent bonds are much longer than the standard sp3 C–C bond length of 1.54 Å, this configuration is not considered.26Table 1 shows all the configurations we chose for single-side adsorption in our calculations. For 1H or 7H adsorption, there is only one configuration, because every adsorbing site or vacant site is equivalent; for 2H and 6H adsorption, there are three configurations [ortho (o) meta (m) and para (p)] in all; for 4H adsorption, we only chose two typical configurations [chair (c) and boat (b)] that are widely studied;14,27 for 3H or 5H adsorption, we also chose two configurations which are obtained by subtracting or adding one equivalent atom from 4H configurations.
| n = 1 |  |
| n = 2 |  |
| n = 3 |  |
| n = 4 |  |
| n = 5 |  |
| n = 6 |  |
| n = 7 |  |
Fig. 2(a) shows the binding energy of single-side hydrogenation of graphene with different number of adatoms. The star point indicates the binding energy of one fluorine (F) atom adsorbing onto graphene as a reference. For the binding energy, it is found that all the values are negative, and have a similar order of magnitude as that of F adsorption. However, as shown in Fig. 2(b), the formation energies of hydrogenated graphene are always positive, which is quite different from the negative value of fluorinated graphene. The positive formation energy indicates that H atoms are easy to dissociate from the surface of graphene and form H2 molecules. It is also obtained that among all the configurations of single-side hydrogenation, 2H-p is the most stable one (C:H = 4:1), instead of the most widely studied 4H-c configuration (C:H = 2:1),28,29 which happens to be the most unstable one in our calculations.
 | ||
| Fig. 2 (a) Binding energy and (b) formation energy of single-side hydrogenation of graphene with different number of adatoms. The corresponding energy of each configuration is marked using its label. The dashed lines represent zero energy. The star points in (a) and (b) denote the binding energy and formation energy of one fluorine atom adsorbing onto graphene under the same condition, respectively. | ||
The processes of H atoms dissociating from graphene and forming H2 molecules have a barrier to overcome (breaking the C–H bond), and if two H atoms form a free H2 molecule far away from the graphene surface, the value of the barrier is related to the binding energy. However, if two adsorbed H atoms form a confined H2 molecule near the graphene surface, the barrier may be much lower than the calculated binding energy. The low barrier of H migration from one C atom to another can be used as an indirect evidence.30 The exothermic process of H2 formation also indicates the possibility, whereas desorption of F atoms is impossible, because the process is endothermic. Therefore, at moderate temperature, H atoms are easy to dissociate from graphene, when the adsorption only takes place on one side.
Next, we consider the double-side hydrogenation of graphene. On the basis of single-side hydrogenation we investigated, we adsorb H atoms on all the vacant sites at the other side of graphene, forming double-side adsorption. The number of H atoms adsorbed on each side is denoted as Hu and Hd, respectively. The ratio Hu:Hd is used to distinguish between different configurations. For example, for configuration 7H of single-side adsorption, the vacant site is position 2 as marked in Fig. 1, and one H atom is adsorbed on this site at the other side of graphene to form double-side adsorption. All the configurations we calculated for double-side hydrogenation are listed in Table 2. Because of the symmetry, some of the configurations are equivalent, such as 1:7 and 7:1. For convenience, the same labels are used as in single-side hydrogenation.
| Hu:Hd = 4:4 |
 |
| Hu:Hd = 5:3 |
 |
| Hu:Hd = 6:2 |
 |
| Hu:Hd = 7:1 |
 |
Comparing the corresponding configurations with the same labels in Tables 1 and 2, we find that adding H atoms changes the C backbone a little. The structures of double-side adsorption are similar to their counterparts, so the comparison of corresponding configurations makes sense, which can reveal the changes when more H atoms are adsorbed on the other side of graphene. Of course, double-side adsorption of Hu:Hd = 1:7 has similar structure to single-side adsorption with 7H atoms (but not to the 1H atom).
The binding energy and formation energy of double-side hydrogenation are shown in Fig. 3, including corresponding data of single-side hydrogenation for comparison. It is found that both the binding energies and the formation energies of the double-side adsorption are lower than those of the single-side adsorption, which means that the system becomes more stable when hydrogen atoms are adsorbed on graphene from both sides. Much more important is the formation energy; as shown in Fig. 3(b), with the increasing ratio of Hu:Hd, the formation energies change from negative to a positive value. As indicated above, a negative (positive) formation energy indicates the system of hydrogenated graphene is stable (unstable). Therefore, our calculation shows that 4H and 5H adsorption are stable, but 7H adsorption is still unstable. Comparing to the normal room temperature (∼25 meV), the calculated formation energies of 4H and 5H adsorption are an order of magnitude higher, so these configurations are still stable. This phenomenon indicates that adding H atoms on the other side strengthens the binding of C and H atoms.
 | ||
| Fig. 3 (a) Binding energy and (b) formation energy of single-side and double-side hydrogenation of graphene with different configurations. The dashed lines represent zero energy. | ||
The configurations of 4H-c for two kinds of adsorption have the most obvious change as shown in Fig. 3, and they also have been given the most consideration among hydrogenated graphene studies, for example, in the works of Wang and Ding et al., where foreign atoms are doped in the two configurations and half-metallic behavior is obtained.31,32 Therefore, we select the two configurations for further analysis of their electronic structure. Fig. 4 shows the contour plots based on electron density of the two configurations. For single-side adsorption, more electrons distribute around their own atoms as shown in Fig. 4(a), while for the double-side case, more electrons distribute between C and H atoms and become shared electrons, corresponding to stronger C–H covalent bonds.
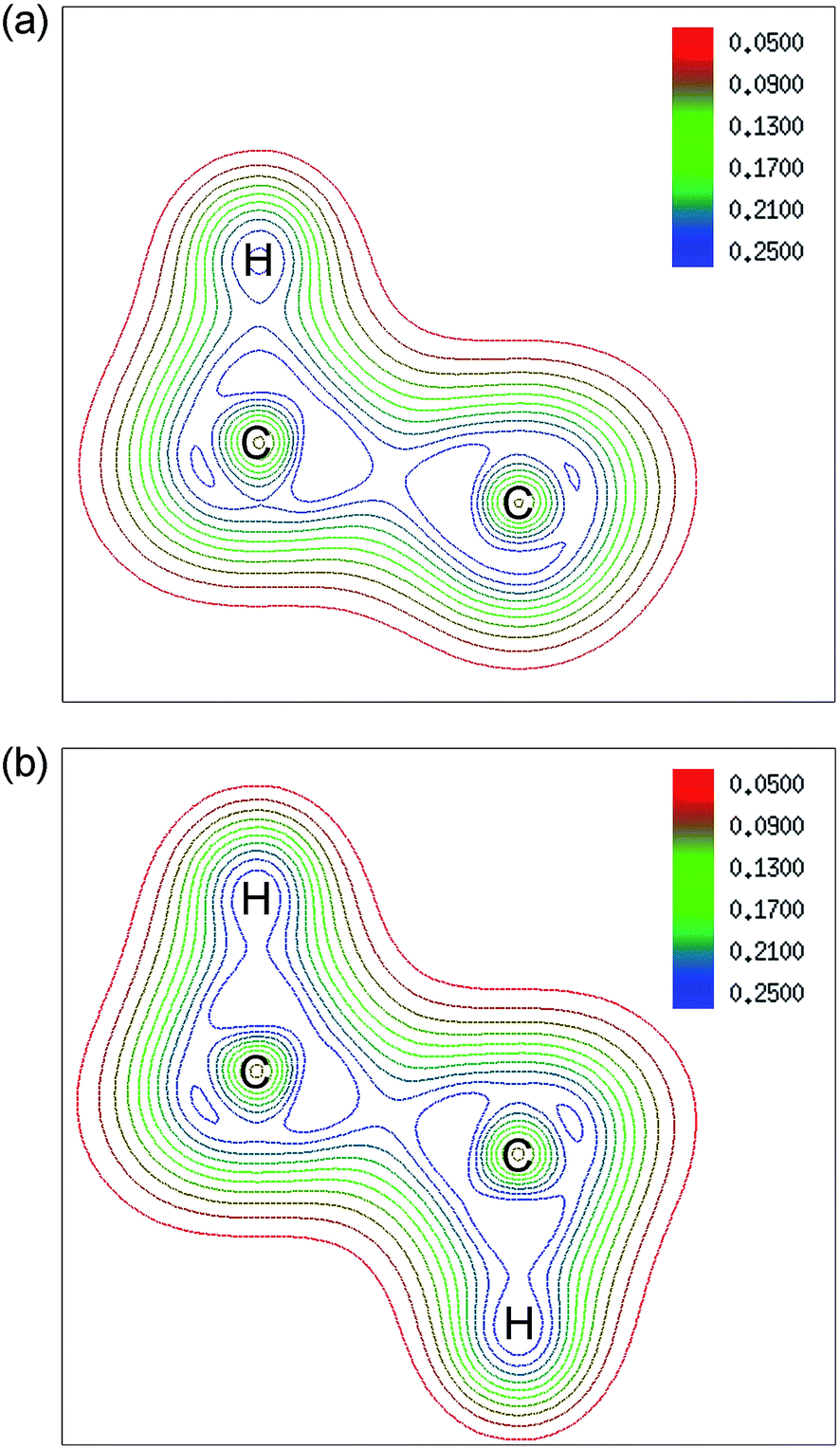 | ||
| Fig. 4 Contour plots based on electron density of 4H-c for (a) single-side and (b) double-side adsorption. The plane which contains two C atoms (position 1 and 2 marked in Fig. 1) and their adsorbed H atoms is selected. Contours of the valence electron density are plotted from 0.05 a.u. to 0.25 a.u. with colors changing from red to blue. | ||
Considering the influence on the geometric structures by adding H atoms on the other side, we provide a comparison of C–C bond lengths and C–C–C bond angles for all the configurations in Table 3. As is well known, adsorption on graphene is a process of breaking π bonds and producing additional σ bonds, and transforming C–C sp2 hybridization to sp3. Compared to single-side adsorption, the calculated C–C–C bond angles of double-side are closer to 109.5°, which is the bond angle in diamond and results in enhanced system stability. The configuration of 4H-c of double-side adsorption has the most standard sp3 hybridization, where both the C–C bond length and the C–C–C bond angle are closest to those in diamond, leading to the most stable hydrogenated graphene. Furthermore, from left to right in Table 3, the C–C bond length and C–C–C bond angle of double-side hydrogenation increase gradually, and the same tendency is found in Fig. 3. In the work of adsorption of H2 molecules on graphene studied by Zhou et al.,13 it was found that higher the symmetry, the stronger the binding energy, which is consistent with our findings. In summary, when the ratio of adsorbing H atom of the two sides tends to 1:1, the hybridization of C–C bond is close to sp3 and the C–H covalent bonds are strengthened. From our calculations, it is found that not all the configurations of double-side hydrogenated graphene are stable. Previous theoretical studies focused on the configurations with the same number of H atoms on each side and obtained stable results. However, in experiment, even though the graphene membranes can be set as free-standing and both sides are accessible to atomic H, the randomness of adsorption and a higher probability of adsorption on upper surface quite likely lead to a metastable state, and even the uncorrelated H frustrated domains would prevent the formation of configurations with Hu:Hd = 4:4.9,16 Therefore, double-side hydrogenation of graphene in experiments may lead to unstable states.
| 4H-c | 4H-b | 5H-c | 5H-b | 6H-p | 6H-m | 6H-o | 7H | ||
|---|---|---|---|---|---|---|---|---|---|
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) CC CC |
Single-side | 1.505 | 1.524 | 1.520 | 1.536 | 1.548 | 1.549 | 1.550 | 1.567 |
| Double-side | 1.539 | 1.548 | 1.551 | 1.554 | 1.562 | 1.562 | 1.564 | 1.573 | |
![[small theta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c9.gif) CCC CCC |
Single-side | 115.1 | 116.5 | 117.7 | 117.7 | 118.6 | 118.6 | 118.9 | 119.2 |
| Double-side | 111.4 | 111.8 | 113.3 | 113.3 | 114.5 | 114.9 | 115.7 | 116.7 | |
4 Conclusions
In summary, we have systematically studied the stability of hydrogenated graphene considering different number of adatoms and positions, and provided a reasonable explanation for the disagreement between present theoretical and experimental investigations on the stability of hydrogenated graphene. When adsorption takes place only on one side of graphene, the system is unstable, even though the binding energy of C–H bond has the same order of magnitude as that of C–F bond. However, double-side hydrogenation of graphene is more stable, as the C–C sp3 hybridization is enhanced, leading to the stronger combination of C and H atoms. Therefore, if we intend to get more stable hydrogenated graphene in experiments, double-side adsorption seems necessary, and a better control of the number of H atoms adsorbing on the two sides of the graphene membrane.Acknowledgements
The authors thank Prof. Nujiang Tang of Nanjing University and Dr Qian Feng of Fujian Normal University for the useful discussions and suggestions. This work was financially supported by the National Natural Science Foundation of the People's Republic of China (Grant no. 11174181 and no. 21161160445) and 111 project B13029. This work was also supported in part by the U.S. Department of Energy.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- D. Yi, D. Hou, S. Li and S. Xie, Nanoscale, 2013, 5, 9118–9122 RSC.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951–954 CrossRef CAS PubMed.
- Y. Zhang, T. T. Tang, C. Girit, Z. Hao, M. C. Martin, A. Zettl, M. F. Crommie, Y. R. Shen and F. Wang, Nature, 2009, 459, 820–823 CrossRef CAS PubMed.
- Q. Feng, N. Tang, F. Liu, Q. Cao, W. Zheng, W. Ren, X. Wan and Y. Du, ACS Nano, 2013, 7, 6729–6734 CrossRef CAS PubMed.
- D. Hou, J. Wei and S. Xie, Phys. Chem. Chem. Phys., 2011, 13, 13202–13206 RSC.
- A. Savchenko, Science, 2009, 323, 589 CrossRef CAS PubMed.
- M. Pumera and C. H. A. Wong, Chem. Soc. Rev., 2013, 42, 5987–5995 RSC.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668 CAS.
- S. S. Han, H. Jung, D. H. Jung, S.-H. Choi and N. Park, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155408 CrossRef.
- V. Tozzini and V. Pellegrini, Phys. Chem. Chem. Phys., 2013, 15, 80–89 RSC.
- W. Zhou, J. Zhou, J. Shen, C. Ouyang and S. Shi, J. Phys. Chem. Solids, 2012, 73, 245–251 CrossRef CAS PubMed.
- J. O. Sofo, A. S. Chaudhari and G. D. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153401 CrossRef.
- O. Leenaerts, H. Peelaers, A. D. Hernández-Nieves, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195436 CrossRef.
- M. Z. S. Flores, P. A. S. Autreto, S. B. Legoas and D. S. Galvao, Nanotechnology, 2009, 20, 465704 CrossRef CAS PubMed.
- R. R. Nair, W. Ren, R. Jalil, I. Riaz, V. G. Kravets, L. Britnell, P. Blake, F. Schedin, A. S. Mayorov, S. Yuan, M. I. Katsnelson, H. M. Cheng, W. Strupinski, L. G. Bulusheva, A. V. Okotrub, I. V. Grigorieva, A. N. Grigorenko, K. S. Novoselov and A. K. Geim, Small, 2010, 6, 2877–2884 CrossRef CAS PubMed.
- S. Ryu, M. Y. Han, J. Maultzsch, T. F. Heinz, P. Kim, M. L. Steigerwald and L. E. Brus, Nano Lett., 2008, 8, 4597–4602 CrossRef CAS PubMed.
- D. C. Elias, R. R. Nair, T. M. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610–613 CrossRef CAS PubMed.
- R. Balog, B. Jørgensen, J. Wells, E. Lægsgaard, P. Hofmann, F. Besenbacher and L. Hornekær, J. Am. Chem. Soc., 2009, 131, 8744–8745 CrossRef CAS PubMed.
- Z. Luo, T. Yu, K. J. Kim, Z. Ni, Y. You, S. Lim, Z. Shen, S. Wang and J. Lin, ACS Nano, 2009, 3, 1781–1788 CrossRef CAS PubMed.
- H. J. Xiang, E. J. Kan, S.-H. Wei, X. G. Gong and M. H. Whangbo, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 165425 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordeón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- D. W. Boukhvalov, M. I. Katsnelson and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035427 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- B. S. Pujari, S. Gusarov, M. Brett and A. Kovalenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 041402 CrossRef.
- D. K. Samarakoon and X. Q. Wang, ACS Nano, 2009, 3, 4017–4022 CrossRef CAS PubMed.
- J. Zhou, Q. Wang, Q. Sun and P. Jena, Appl. Phys. Lett., 2011, 98, 063108 CrossRef PubMed.
- A. N. Rudenko, F. J. Keil, M. I. Katsnelson and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 081405 CrossRef.
- D. W. Boukhvalov, Phys. E, 2010, 43, 199–201 CrossRef CAS PubMed.
- Y. Wang, Y. Ding, S. Shi and W. Tang, Appl. Phys. Lett., 2011, 98, 163104 CrossRef PubMed.
- Y. Ding, Y. Wang, J. Ni, L. Shi, S. Shi, C. Li and W. Tang, Nanoscale Res. Lett., 2011, 6, 190 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |