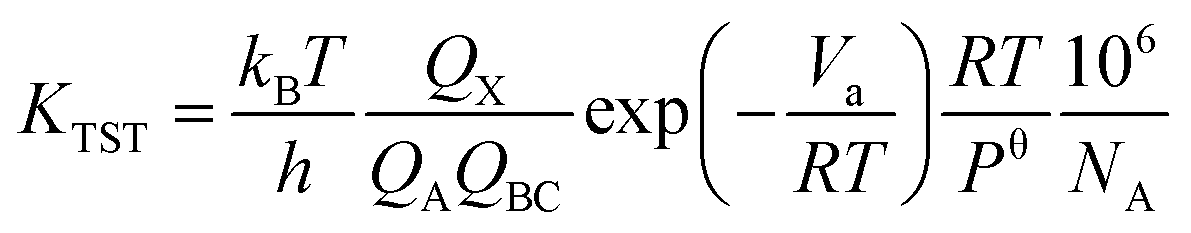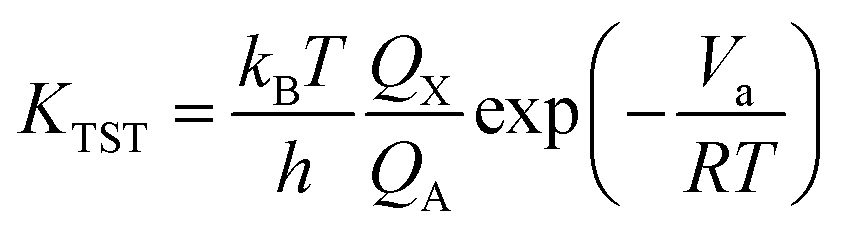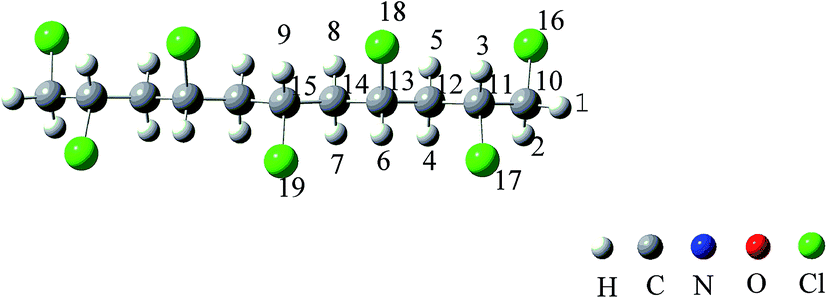DOI:
10.1039/C5RA00612K
(Paper)
RSC Adv., 2015,
5, 37988-37994
The OH-initiated chemical transformation of 1,2,4,6,8,10,11-heptachloroundecane in the atmosphere†
Received
12th January 2015
, Accepted 7th April 2015
First published on 9th April 2015
Abstract
Short chain chlorinated paraffins (SCCPs) have recently drawn public attention because they have properties similar to persistent organic pollutants. In this study, 1,2,4,6,8,10,11-heptachloroundecane (HCU) is selected as a model molecule to investigate the chemical transformation of SCCPs using density functional theory (DFT) methods. After hydroxyl (OH) radicals initiate hydrogen (H) atom abstraction reactions, the produced intermediates could be further oxidized in the presence of O2/NO. The main products were found to be chloro-aldehydes and chloro-ketones after dechlorination reactions, or chloro-hydrins after reaction with H2O. These products have strong water solubility and polarity. Cl and OH radicals, which have strong reactivity, are also generated in the degradation process. Rate constants were calculated using transition state theory and the Arrhenius formulas were fitted. The total rate constant for the reaction of HCU with OH radicals is about 1.67 × 10−12 cm3 per molecule per s at room temperature. The atmospheric lifetime of HCU relative OH radicals is about 7.1 days.
Introduction
Chlorinated paraffins (CPs) are complex mixtures of polychlorinated n-alkanes, which include many congeners and isomers.1 Generally, CPs can be classified into short chain CPs (SCCPs, C10–C13), medium chain CPs (MCCPs, C14–C17) and long chain CPs (LCCP, C18–C30) according to their carbon chain length (molecular formula: CxH(2x−y+2)Cly).2
Since the 1930s, hundreds of CPs have been used as additives in sealants, paints, rubbers, metal working fluids, flame retardants and plasticizers.3 Recently, more and more attention has been paid to their harmful properties, especially to SCCPs.
As by-products in the production of MCCPs and LCCPs, SCCPs have properties such as potential toxicity, persistence, bioaccumulation and long-range transport, which are similar to persistent organic pollutants (POPs).4,5 At the Conference of the Stockholm Convention (SC) for persistent organic pollutants, SCCPs have been reviewed five times from 2007 to 2012 (except in 2011) for their draft risk profile. However, the review is postponed because studies have been insufficient.6,7 However, several countries including the United States and Canada and the European Union still restrict the use of SCCPs.8 It can be expected that once SCCPs are listed in the SC, the manufacturing activities of CPs and the relevant economy will be deeply influenced.7
SCCPs are ubiquitous in both natural and human living environments in air, water, sediment, aquatic organisms, and terrestrial wildlife.9–15 The partition coefficients for n-octanol–water vary from 5.06 to 8.12,3,16 whereas the coefficients for n-octanol–air are about 8.2–9.8, indicating bioaccumulation and biomagnification through the food chain.17 Exposure experiments prove that SCCPs are carcinogenic to rats and can also cause the chronic toxicosis of aquatic organisms.18 in addition, a series of reports about their toxicological effects indicate that there is potential harmful impact on human health.19,20
Usually SCCPs can be discharged from or deposited into the municipal wastewater network and finally be accumulated in the wastewater treatment plants (WWTPs).21 From 14 WWTPs in the UK, Steven et al. observed that the concentration of SCCPs in sludge varied from 7 to 200 mg per kg dry weight.22 A nationwide survey of sewage sludge samples from 52 WWTPs was carried out in China. It was found that concentrations of SCCPs ranged between 0.80 and 52.7 mg per kg dry weight and most of the samples shared the similar congener distribution patterns.23 It could be speculated that the improper disposal of sludge may cause secondary pollution due to the stability of SCCPs.
Owing to the increased public awareness, advanced equipment and improved evaluation methods, SCCPs have been studied for many years and important scientific data have been obtained.7 Recently, Jiang's group studied the concentration and distribution of SCCPs in farm soils,10 the distribution patterns in 52 WWTPs nationwide,23 the environmental fate of SCCPs, their behavior and possible removal in sewage treatment plants,24 their migration behavior in the coastal East China Sea,25 and their spatial and temporal trends in Bohai and Yellow Sea.26 In China, Wang also investigated the air, soil and atmospheric deposition processes of SCCPs in the Pearl River Delta.27 However, SCCPs are very complex mixtures and are difficult to separate using current experimental conditions. In addition, there are few standard analytical methods.7 Moreover, the reported results are different in different laboratories, sometimes even with 300% deviations.28 Therefore, most of the studies are still at the monitoring stage, and a few reports on experiments about dechlorination or degradation of SCCPs could be found. Zhang synthesized nanoscale zero-valent iron particles to test the reductive dehalogenation of SCCPs.29 In Lu's experiment, the degradation efficacy was tested by a bacterial strain in pure culture, in which SCCPs were utilized to supplement carbon and energy.22
Recent studies showed that the density functional theory (DFT) could offer accurate prediction of kinetics, pathways and products by calculating the reaction mechanism in many fields.30–32 To the best of our knowledge, quantum chemical studies on SCCPs are very rare. In order to make up for the inadequacy of degradation experiments,7 a specific model molecule is selected to explore theoretical possibilities and completed mechanisms of the dechlorination or degradation of SCCPs for the first time in a computational approach with quantum chemistry. Some useful data for related experiments will be provided.
In atmospheric chemistry, reactions initiated by OH radicals are of paramount importance. They can be produced and recycled by solar ultraviolet radiation spontaneously.33 Thus, we assume that the reactions of SCCPs with OH radicals may be the dominant removal process. In this study, the OH-initiated degradation reactions of the model molecule, in the presence of O2, NO and H2O, are selected to be calculated with density functional theory (DFT). The rate constants are also computed by transition state theory (TST).
Computational methods
Quantum chemistry calculation
To select a proper model molecule, the optimization is calculated at the b3lyp/6-31+g(d) level. In reaction processes, the optimization of reactants, transition states, intermediates and products are calculated at the M062x/6-31+g(d,p) level. Based on previous reports, the M062x function performs consistently for computing energies and enthalpies of small and large organic molecules with lower computational costs.34–38 It takes weak interactions into account and appears to provide energy predictions, performance, and reliable and reasonable descriptions comparable to the best performing function available currently.39 The vibrational frequencies are also calculated at the same level to confirm the nature of stationary points.
Kinetic calculation
Transition state theory (TST) is used to determine rate constants with a statistical representation. The model is based on the potential interaction between reactants and products.40 Taking a bimolecular reaction as example:
The rate constant is calculated by applying the following formula.
| |
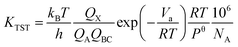 | (2) |
For unimolecular reactions,
| |
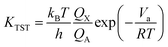 | (3) |
where
kB is the Boltzmann constant,
T is the temperature,
h is the Planck constant,
R is universal gas constant,
Va is the barrier,
Pθ is standard pressure and
NA is Avogadro constant.
Qx is the partition function of the transition state, whereas
QA and
QBC are those of the reactants. The reaction rates were calculated with temperature ranging from 200 K to 400 K in 10k steps.
Results and discussion
Selection of target molecule
Because the quantum chemical calculation is based on the analysis of specific molecules, the concrete structure must be known. As we know, SCCPs are complex mixtures with thousands of unseparated homologs and isomers,1 and there is no particular representative in published reports. Based on our experiences, we would like to select one molecule derived from SCCPs, but with relatively stronger abundance and lower energy. Firstly, the general formula (CxH(2x−y+2)Cly, x = 10–13, y = 1–13) is confirmed. From various published reports, we found that many SCCPs samples in different environment media displayed very similar congener distribution patterns and relative abundance,10,23,25–27 with Cx (x = 10, 11) and Cly (y = 6, 7, 8) as the dominant congener group. After initial comparison on relative abundance, C11H17Cl7 was chosen as the target formula.
Secondly, we identified the structure with lowest energy, which is stable and abundant among the isomers. One hundred structures were drawn randomly, in which chlorine atoms were substituted for different numbers of hydrogen atoms on one carbon atom. Then their energies were compared. Results showed that those with only one chlorine atom on one carbon had lower energies. Therefore, the scope was narrowed down naturally.
According to permutation and combination theory, for mono-substituted C11H17Cl7 on each carbon atom, there are 170 isomers altogether in this category. All of them were drawn and optimized. The structure with the lowest energy was chosen as the model molecule to be analyzed afterwards. It is 1,2,4,6,8,10,11-heptachloroundecane (HCU) which is symmetrical (Fig. 1).
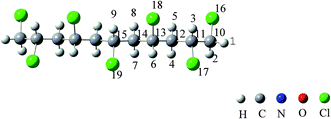 |
| | Fig. 1 The atom numbers on the structure of 1,2,4,6,8,10,11-heptachloroundecane (HCU). | |
OH-initiated abstraction reactions
As shown in Fig. 1, there are 9 different hydrogen atoms in the HCU structure and they can be initiated by OH radical in atmospheric oxidations. Hydrogen atoms are labeled from no. 1 to no. 9 for convenience. Under atmospheric conditions, OH radicals can easily initiate chemical reactions by attacking hydrogen atoms because it is strongly nucleophilic. Therefore, H atom abstraction from HCU should be the first step. It is evident that the 9 hydrogen atoms can be divided into 4 types according to the relative location: H1(H2), H3, H4(H5, H7, H8) and H6(H9), which will be discussed separately.
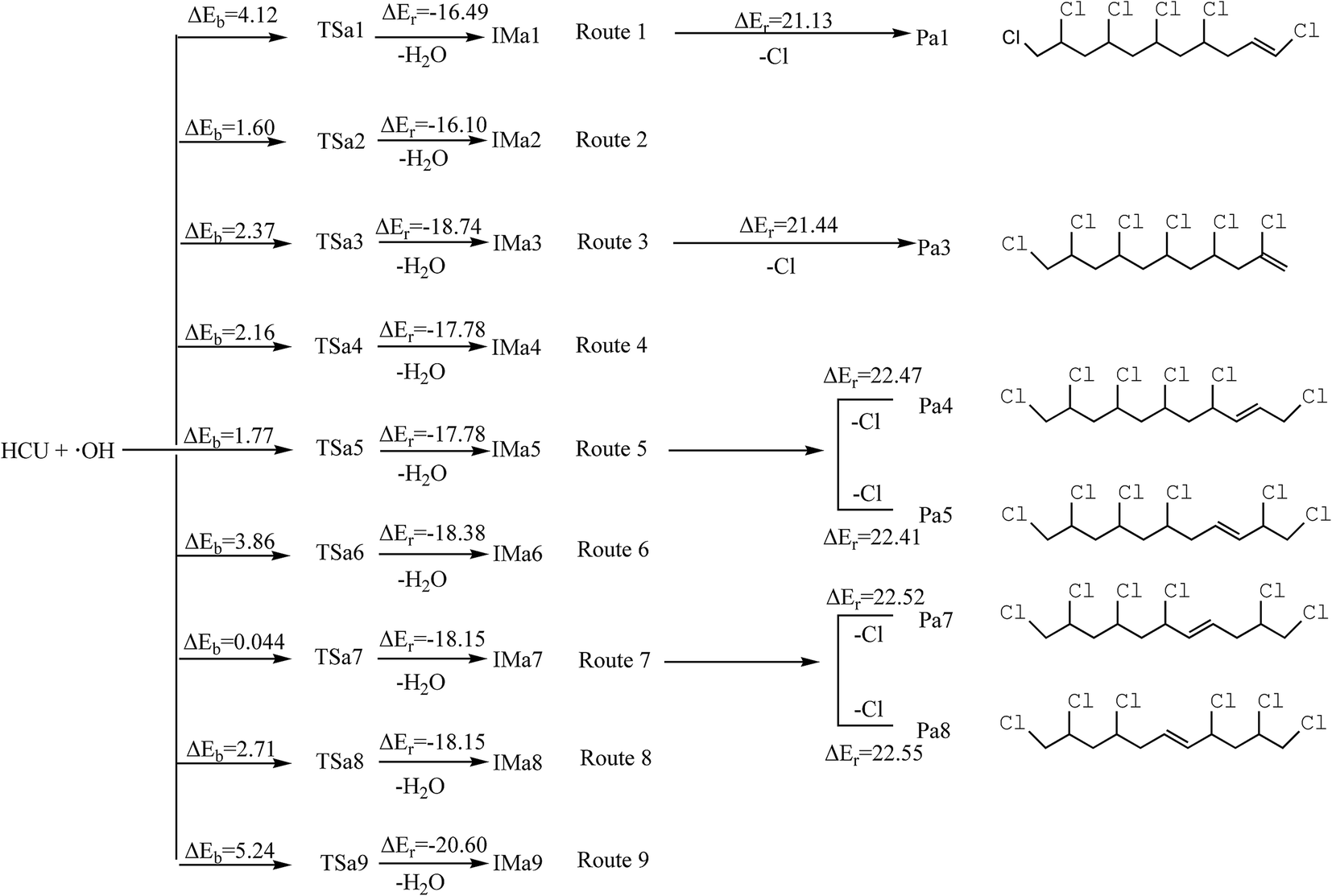
H1 and H2 can be abstracted by the OH radicals. The transition states (TSa1 and TSa2) can be found in this process, in which the transition vectors display the motion of the H1(H2) atom between the C10 atom and the O atom in the OH radical. The abstraction pathways can generate H2O and IMa1 via a potential barrier of 4.12 kcal mol−1 and IMa2 with the barrier of 1.60 kcal mol−1, releasing 16.49 and 16.10 kcal mol−1 of energy. The reaction mechanisms of route 3 to 9 are similar to those in route 1 and 2, and the potential energy profiles are shown in Scheme 1. Generally, the potential barriers of TSa3 to TSa9 are 0.044–5.24 kcal mol−1, and all the other abstraction reactions are strongly exothermic, releasing 17.78–20.60 kcal mol−1 energy (Scheme 1). ΔEb is the potential barrier and ΔEr is the reaction heat in the elementary reaction.
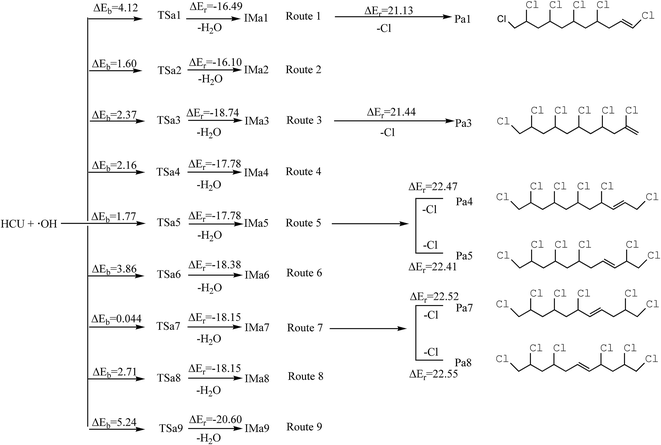 |
| | Scheme 1 Schematic diagram of the reaction pathways in the H abstraction and dechlorination reaction mechanisms of HCU with the barriers ΔEb (kcal mol−1) and reaction heats ΔEr (kcal mol−1). | |
The data indicate that all types of H abstraction reactions have low barriers and give out considerable energy. The above mentioned reactions are expected to occur easily and may play an important role in the degradation of HCU in the atmosphere. Because the intermediates are still active, they will be further oxidized by O2/NO.
It should be noted that H1 and H2 are connected to the same C atom. After abstraction and geometrical optimization, IMa1 and IMa2 are identical, and so are IMa4 and IMa5, IMa7 and IMa8. Therefore only IMa1, IMa3, IMa5, IMa6, IMa7, IMa9 are selected for further discussion.
Subsequent reactions
The intermediates from IMa1 to IMa9 are rich in energy, and they may carry out further reactions through two pathways: dechlorination and oxidation by O2/NO.
Dechlorination to alkene
As shown in Fig. 1, the numbers of carbon atoms from 10 to 15 and chlorine atoms from 16 to 19 are also labeled. After abstraction reactions, the bond length between intermediate atoms changes a lot, which may naturally lead to further dechlorination.
For IMa1, the bond length of C10–Cl16 and C10–C11 becomes shorter, varying from 1.80 Å to 1.72 Å and from 1.52 Å to 1.49 Å, respectively, and the C10 atom becomes unsaturated. It is assumed that the structure tends to eliminate Cl17 to form a double bond between C10 and C11. As for IMa5, the distance between C12 and C11 or C12 and C13 becomes shorter too (from 1.50 Å to 1.47 Å). Moreover, the neighboring C–Cl bond gets elongated (from 1.80 Å to 1.84 Å). Hence, the structure could be transformed into two types of alkenes (Pa4 and Pa5) by dechlorination on either side. The dechlorination of IMa3 is similar to that of IMa1, whereas that of IMa7 is similar to IMa5. For IMa6 or IMa9, there is no chlorine atom connected to the C atom next to C13 or C15. As a result, the intermediates could not generate alkenes after dechlorination (Scheme 1). However, no transition state could be found in this process and the reactions are all endothermic, with the energy of 21.132–2.55 kcal mol−1. Therefore, the pathways of dechlorination need to absorb heat from the environment.
Reactions in the presence of O2/NO
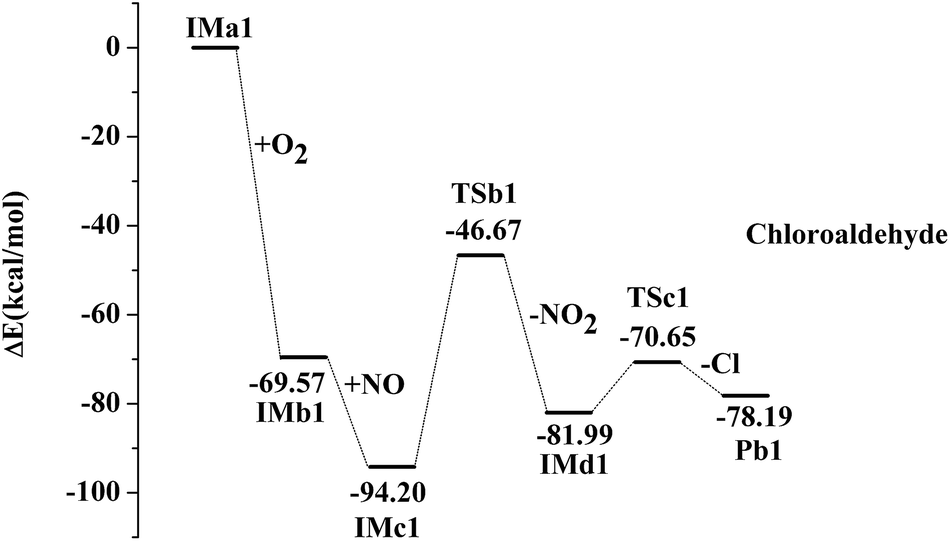
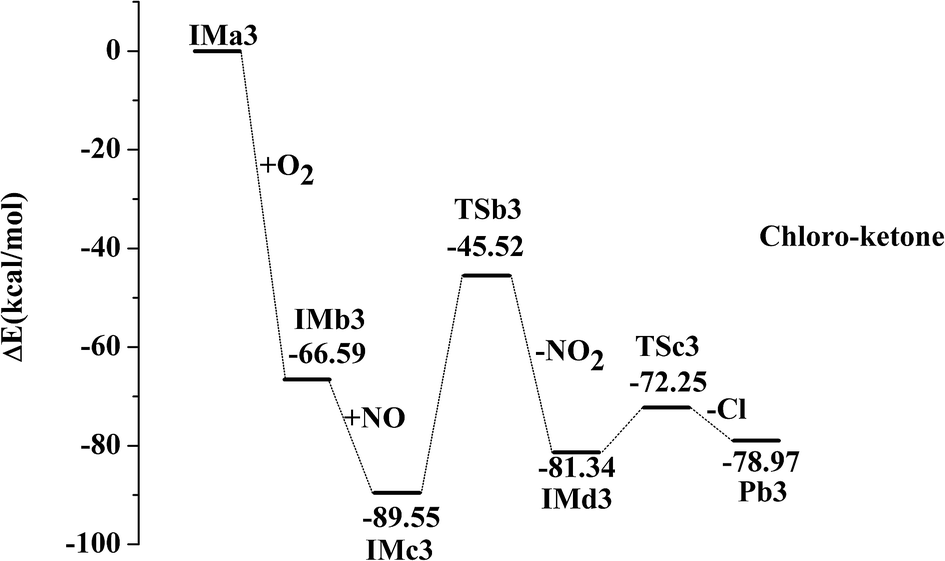
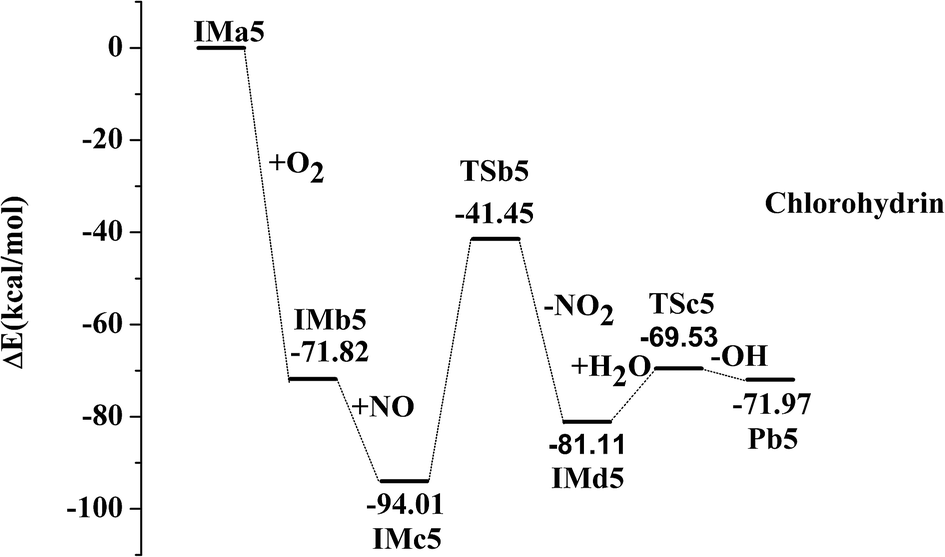
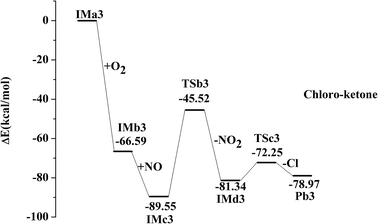
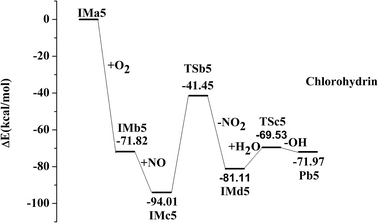
IMa1 is taken as an example for the second pathway. It could be further oxidized by ubiquitous O2 in the atmosphere, which is a barrierless process. The oxidation reaction is strongly exothermic, and up to 69.57 kcal mol−1 of energy is released. The generated IMb1 will react with NO immediately to form IMc1 and the reaction releases 24.63 kcal mol−1 of energy as a barrierless process. Promptly, IMc1 reacts in a directly unimolecular decomposition via cleavage of the O–O bond to produce IMd1 and NO2. The elimination has to cross a transition state with an energy barrier of 47.53 kcal mol−1 and absorbs a little energy of 12.21 kcal mol−1. However, the energy in need is far lower than that released from the previous reactions (IMa1 → IMb1 → IMc1). Therefore, this entire process could occur easily. The mechanisms in those processes from IMa1 to IMa9 are similar, with slight differences in their energy. The profiles of potential energy surfaces of IMa1, IMa3, IMa5 are drawn in Fig. 2–4 as representatives, and IMa6, IMa7 and IMa9 are shown as follows.
| IMa6 + O2 → IMb6, ΔEr = −66.51 kcal mol−1 |
| IMb6 + NO → IMc6, ΔEr = −23.80 kcal mol−1 |
| IMc6 → TSb6 → IMd6 + NO2, ΔEb = 48.53 kcal mol−1, ΔEr = 10.06 kcal mol−1 |
| IMa7 + O2 → IMb7, ΔEr = −70.00 kcal mol−1 |
| IMb7 + NO → IMc7, ΔEr = −22.62 kcal mol−1 |
| IMc7 → TSb7 → IMd7 + NO2, ΔEb = 52.73 kcal mol−1, ΔEr = 13.05 kcal mol−1 |
| IMa9 + O2 → IMb9, ΔEr = −65.58 kcal mol−1 |
| IMb9 + NO → IMc9, ΔEr = −21.67 kcal mol−1 |
| IMc9 → TSb9 → IMd9 + NO2, ΔEb = 45.06 kcal mol−1, ΔEr = 7.08 kcal mol−1 |
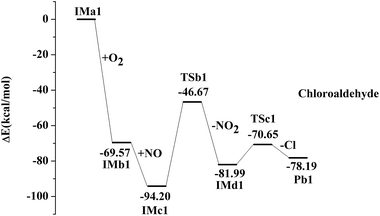 |
| | Fig. 2 Profile of the potential energy surface for the oxidation and further dechlorination of IMa1. | |
 |
| | Fig. 3 Profile of the potential energy surface for the oxidation and further dechlorination of IMa3. | |
 |
| | Fig. 4 Profile of the potential energy surface for the oxidation and further dechlorination of IMa5. | |
Formation of products
After the NO2 elimination reaction, intermediates from IMd1 to IMd9 are active, which could further undergo decomposition or addition reactions to form alcohol, aldehyde or ketone by different routes. For IMd1, IMd3, IMd6 and IMd9, the fracture of the C–Cl bond and the formation of the carbon–oxygen double bonds may occur to generate the products (Pb1, Pb3, Pb6 and Pb9). The Cl atoms are also separated in the processes through transition states. Pb1 is chloro-aldehyde but Pb3, Pb6 and Pb9 are chloro-ketones.
| IMd1 → TSc1 → Pb1 + Cl, ΔEb = 11.34 kcal mol−1, ΔEr = 3.81 kcal mol−1 |
| IMd3 → TSc3 → Pb3 + Cl, ΔEb = 9.09 kcal mol−1, ΔEr = 2.36 kcal mol−1 |
| IMd6 → TSc6 → Pb6 + Cl, ΔEb = 7.76 kcal mol−1, ΔEr = −0.95 kcal mol−1 |
| IMd9 → TSc9 → Pb9 + Cl, ΔEb = 8.84 kcal mol−1, ΔEr = 1.50 kcal mol−1 |
For IMd5 and IMd7, no Cl atoms are attached to the reactive C atoms and carbon–oxygen double bonds are not generated. However, the reaction between unsaturated O atoms and H2O should not be ignored.
| IMd5 + H2O → TSc5 → Pb5 + OH, ΔEb = 11.59 kcal mol−1, ΔEr = 9.15 kcal mol−1 |
| IMd7 + H2O → TSc7 → Pb7 + OH, ΔEb = 13.28 kcal mol−1, ΔEr = 11.98 kcal mol−1 |
It is known that H2O is the third most prevalent component in the atmosphere and accounts for about 1.5% by mass as vapor and about 2% if clouds and ice crystals are included.41 The active IMd5 and IMd7 could seize the H atom from H2O to form chlorohydrins (Pb5 and Pb7), with OH radicals regenerated simultaneously. It is well recognized that OH radicals contribute a lot in atmospheric chemistry as the dominant reactive species, which can initiate a new round of reactions of HCU, or to degrade other organic compounds in the troposphere.42 To date, the entire mechanism of general degradation is discussed in detail. The above degradation mechanism is similar to that of other SCCPs compounds.
Kinetics property
The kinetic calculations are carried out with the TST method as well. The barrierless O2/NO oxidation reactions are not included. A suitable range of 200–400 K is chosen to calculate rate constants, and they are fitted in the Arrhenius formula k = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT). The reaction rates at the temperature of 298 K and the formula are shown in Table 1. They can provide helpful information for the fate assessment of pollutants and the atmospheric model study.43 In the multichannel reaction of HCU with OH radical, the total constant is about 1.67 × 10−12 cm3 per molecule per s at 298 K. Recently, Li has calculated the OH radical reaction rate constant with different SCCPs, with the results that kOH of C11H16Cl8 and C10H15Cl7 are 1.6 and 2.0 × 10−12 cm3 per molecule per s at 298 K.43 It was reported that the average OH radical concentration is 9.7 × 105 molecule per cm3 (ref. 44) and the atmospheric lifetime of HCU can be approximated by τ = 1/(kOH[OH]),45–47 which is determined to be 7.1 days. OH radicals seem to play an important role in the elimination process of HCU.
exp(−Ea/RT). The reaction rates at the temperature of 298 K and the formula are shown in Table 1. They can provide helpful information for the fate assessment of pollutants and the atmospheric model study.43 In the multichannel reaction of HCU with OH radical, the total constant is about 1.67 × 10−12 cm3 per molecule per s at 298 K. Recently, Li has calculated the OH radical reaction rate constant with different SCCPs, with the results that kOH of C11H16Cl8 and C10H15Cl7 are 1.6 and 2.0 × 10−12 cm3 per molecule per s at 298 K.43 It was reported that the average OH radical concentration is 9.7 × 105 molecule per cm3 (ref. 44) and the atmospheric lifetime of HCU can be approximated by τ = 1/(kOH[OH]),45–47 which is determined to be 7.1 days. OH radicals seem to play an important role in the elimination process of HCU.
Table 1 Arrhenius formulas of rate constants k for elementary reactions at 200–400 K
| Reactions |
k298 K |
Arrhenius formulas |
R2 |
| cm3 per molecule per s. s−1. |
| HCU + OH → TSa1 → IMa1 + H2O |
7.28 × 10−15 |
k(T) = 2.00 × 10−11exp(−2353.2/T)a |
0.9999 |
| HCU + OH → TSa2 → IMa2 + H2O |
3.29 × 10−13 |
k(T) = 1.28 × 10−11exp(−1083.4/T)a |
0.9994 |
| HCU + OH → TSa3 → IMa3 + H2O |
1.44 × 10−13 |
k(T) = 2.05 × 10−11exp(−1471.5/T)a |
0.9997 |
| HCU + OH → TSa4 → IMa4 + H2O |
1.21 × 10−13 |
k(T) = 1.20 × 10−11exp(−1364.9/T)a |
0.9996 |
| HCU + OH → TSa5 → IMa5 + H2O |
5.72 × 10−14 |
k(T) = 2.96 × 10−12exp(−1169.5/T)a |
0.9995 |
| HCU + OH → TSa6 → IMa6 + H2O |
8.20 × 10−16 |
k(T) = 1.46 × 10−12exp(−2222.9/T)a |
0.9998 |
| HCU + OH → TSa7 → IMa7 + H2O |
6.75 × 10−13 |
k(T) = 1.89 × 10−12exp(−300.29/T)a |
0.9918 |
| HCU + OH → TSa8 → IMa8 + H2O |
3.39 × 10−13 |
k(T) = 8.59 × 10−11exp(−1642.5/T)a |
0.9997 |
| HCU + OH → TSa9 → IMa9 + H2O |
1.94 × 10−15 |
k(T) = 3.51 × 10−11exp(−2915.4/T)a |
0.9999 |
| IMc1 → TSb1 → IMd1 + NO2 |
1.00 × 10−19 |
k(T) = 1.98 × 1016exp(−24213/T)b |
0.9999 |
| IMc3 → TSb3 → IMd3 + NO2 |
9.76 × 10−21 |
k(T) = 5.18 × 1012exp(−22448/T)b |
0.9999 |
| IMc5 → TSb5 → IMd5 + NO2 |
1.64 × 10−24 |
k(T) = 1.57 × 1015exp(−26741/T)b |
0.9999 |
| IMc6 → TSb6 → IMd6 + NO2 |
7.88 × 10−22 |
k(T) = 8.41 × 1014exp(−24715/T)b |
0.9999 |
| IMc7 → TSb7 → IMd7 + NO2 |
2.28 × 10−24 |
k(T) = 2.93 × 1015exp(−26829/T)b |
0.9999 |
| IMc9 → TSb9 → IMd9 + NO2 |
4.41 × 10−20 |
k(T) = 1.33 × 1014exp(−22967/T)b |
0.9999 |
| IMd1 → TSc1 → Pb1 + Cl |
7.37 × 105 |
k(T) = 4.02 × 1014exp(−5988.5/T)b |
0.9999 |
| IMd3 → TSc3 → Pb3 + Cl |
2.29 × 105 |
k(T) = 2.76 × 1012exp(−4853.0/T)b |
0.9999 |
| IMd5 + H2O → TSc5 → Pb5 + OH |
8.58 × 10−23 |
k(T) = 7.11 × 10−14exp(−6113.1/T)a |
0.9999 |
| IMd6 → TSc6 → Pb6 + Cl |
1.26 × 109 |
k(T) = 1.62 × 1015exp(−4184.0/T)b |
0.9999 |
| IMd7 + H2O → TSc7 → Pb7 + OH |
8.24 × 10−23 |
k(T) = 1.19 × 10−13exp(−6964.9/T)a |
0.9999 |
| IMd9 → TSc9 → Pb9 + Cl |
4.23 × 107 |
k(T) = 3.36 × 1014exp(−4728.5/T)b |
0.9999 |
The experimental information for the degradation reaction system of SCCPs is absent. There is no comparison between experimental results and the calculated data. It is expected that the data and mechanism in this study could provide a reference for future experiments; thus, more studies on SCCPs are needed.
Conclusions
Though SCCPs have the properties of long-range transport and persistence, they could be degraded in the atmosphere to some extent, especially in the presence of OH radical. Other mechanisms initiated by NO3 or O3 would be further studied.
Based on our study, we predict that the SCCPs can be transformed to chlorinated alcohols, aldehydes or ketones in the degradation processes, which have higher reactivity and water solubility than those of SCCPs. Then the subsequent atmospheric reactions will occur easily and Cl atoms are released. As a highly active radical, Cl atoms contribute to oxidative chemistry. Compared to OH radicals, Cl reacts up to 100 times faster in the analogous oxidation of organic compounds, as several experimental investigations have indicated.48–51 In addition, Cl atoms intensify ozone depletion as well, which exerts a negative effect on ecological and environmental conditions. Alternatively, with sufficient oxides of nitrogen, Cl reacts quickly with organic molecules, leading to ozone formation in turn.52 Evidently, chlorinated alcohols, aldehydes, ketones and Cl atoms will produce profound impact on environmental quality and human health.
Acknowledgements
This work is supported by Marie Curie International Research Staff Exchange Scheme Fellowship within the 7th European Community Framework Program (no. 295132), the National Natural Science Foundation of China (no. 21277082, 21337001), Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province (no. BS2012HZ009), and Program for New Century Excellent Talents in University (NCET-13-0349), Project for science and technology development of Shandong province (2014GSF117028) and Beijing National Laboratory for Molecular Science (no. 20140160).
Notes and references
- F. J. Santos, J. Parera and M. T. Galceran, Anal. Bioanal. Chem., 2006, 386, 837–857 CrossRef CAS PubMed.
- G. T. Tomy, A. T. Fisk, J. B. Westmore and D. C. G. Muir, Rev. Environ. Contam. Toxicol., 1998, 158, 53–128 CAS.
- H. Bettina, F. Hermann, V. Wolfgang and C. Mehmet, Environ. Sci. Technol., 2011, 45, 2842–2849 CrossRef CAS PubMed.
- H. J. Poremski, S. Wiandt and T. Knacker, Organohalogen Compd., 2001, 52, 397–400 CAS.
- Q. Li, J. Li, Y. Wang, Y. Xu, X. Pan, G. Zhang, C. Luo, Y. Kobara, J. Nam and K. C. Jones, Environ. Sci. Technol., 2012, 46, 11948–11954 CrossRef CAS PubMed.
- Eighth meeting of the Persistent Organic Pollutants Review Committee (POPRC.8), Geneva, Switzerland, Oct. 15–19, 2012.
- T. Wang, Y. Wang and G. Jiang, Environ. Sci. Technol., 2013, 47, 11924–11925 CrossRef CAS PubMed.
- E. Sverko, G. T. Tomy, C. H. Märvin and D. C. G. Muir, Environ. Sci. Technol., 2012, 46, 4697–4698 CrossRef CAS PubMed.
- M. Houde, D. C. G. Muir, G. T. Tomy, D. M. Whittle, C. Teixeira and S. Moore, Environ. Sci. Technol., 2008, 42, 3893–3899 CrossRef CAS.
- L. Zeng, T. Wang, W. Han, B. Yuan, Q. Liu, Y. Wang and G. Jiang, Environ. Sci. Technol., 2011, 45, 2100–2106 CrossRef CAS PubMed.
- P. Castells, J. Parera, F. J. Santos and M. T. Galceran, Chemosphere, 2008, 70, 1552–1562 CrossRef CAS PubMed.
- M. Chen, X. Luo, X. Zhang, M. He, S. Chen and B. Mai, Environ. Sci. Technol., 2011, 45, 9936–9943 CrossRef CAS PubMed.
- I. O. Koh, W. Rotard and W. H. P. Thiemann, Chemosphere, 2002, 47, 219–227 CrossRef CAS.
- U. E. Fridén, M. S. McLachlan and U. Berger, Environ. Int., 2011, 37, 1169–1174 CrossRef PubMed.
- K. H. Harada, T. Takasuga, T. Hitomi, P. Wang, H. Matsukami and A. Koizumi, Environ. Sci. Technol., 2011, 45, 7019–7027 CrossRef CAS PubMed.
- D. T. H. M. Sijm and T. L. Sinnige, Chemosphere, 1995, 31, 4427–4435 CrossRef CAS.
- B. C. Kelly, M. G. Ikonomou, J. D. Blair, A. E. Morin and F. A. P. C. Gobas, Science, 2007, 317, 236–239 CrossRef CAS PubMed.
- H. M. Cooley, A. T. Fisk, S. C. Wiens, G. T. Tomy, R. E. Evans and D. C. G. Muir, Aquat. Toxicol., 2001, 54, 81–99 CrossRef CAS.
- A. T. Fisk, G. T. Tomy, C. D. Cymbalisty and D. C. G. Muir, Environ. Toxicol. Chem., 2000, 19, 1508–1516 CrossRef CAS PubMed.
- B. Burýšková, L. Bláha, D. Vršková, K. Šimková and B. Maršálek, Acta Vet. Brno, 2006, 75, 115–122 CrossRef.
- M. Lu, J. Chem. Technol. Biotechnol., 2014, 89, 1118 CrossRef CAS PubMed.
- J. L. Stevens, G. L. Northcott, G. A. Stern, G. T. Tomy and K. C. Jones, Environ. Sci. Technol., 2003, 37, 462–467 CrossRef CAS.
- L. Zeng, T. Wang, T. Ruan, Q. Liu, Y. Wang and G. Jiang, Environ. Pollut., 2012, 160, 88–94 CrossRef CAS PubMed.
- L. Zeng, H. Li, T. Wang, Y. Gao, K. Xiao, Y. Du, Y. Wang and G. Jiang, Environ. Sci. Technol., 2013, 47, 732–740 CrossRef CAS PubMed.
- Z. Zhao, H. Li, Y. Wang, G. Li, Y. Cao, L. Zeng, J. Lan, T. Wang and G. Jiang, Environ. Sci. Technol., 2013, 47, 5013–5022 CrossRef CAS PubMed.
- L. Zeng, R. Chen, Z. Zhao, T. Wang, Y. Gao, A. Li, Y. Wang, G. Jiang and L. Sun, Environ. Sci. Technol., 2013, 47, 11449–11456 CrossRef CAS PubMed.
- Y. Wang, J. Li, Z. Cheng, Q. Li, X. Pan, R. Zhang, D. Liu, C. Luo, X. Liu, A. Katsoyiannis and G. Zhang, Environ. Sci. Technol., 2013, 47, 2679–2687 CrossRef CAS PubMed.
- F. Pellizzato, M. Ricci, A. Held, H. Emons, W. Bohmer, S. Geiss, S. Iozza, S. Mais, M. Petersen and P. Lepom, Trends Anal. Chem., 2009, 28, 1029–1035 CrossRef CAS PubMed.
- Z. Zhang, M. Lu, Z. Zhang, M. Xiao and M. Zhang, J. Hazard. Mater., 2012, 243, 105–111 CrossRef CAS PubMed.
- M. Altarawneh, B. Z. Dlugogorski, E. M. Kennedy and J. C. Mackie, J. Phys. Chem. A, 2006, 110, 13560–13567 CrossRef CAS PubMed.
- Q. Zhang, X. Qu and W. Wang, Environ. Sci. Technol., 2007, 41, 6109–6116 CrossRef CAS.
- J. Zhou, J. Chen, C. Liang, Q. Xie, Y. Wang, S. Zhang, X. Qiao and X. Li, Environ. Sci. Technol., 2011, 45, 4839–4845 CrossRef CAS PubMed.
- C. Zhang, T. Sun and X. Sun, Environ. Sci. Technol., 2011, 45, 4756–4762 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Org. Lett., 2006, 8, 5753–5755 CrossRef CAS PubMed.
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2010, 6, 395–404 CrossRef CAS PubMed.
- S. Rayne and K. Forest, J. Mol. Struct.: THEOCHEM, 2010, 948, 102–107 CrossRef CAS PubMed.
- S. Rayne and K. Forest, Nat. Preced., 2010 DOI:10.1038/npre.2010.5183.1.
- S. Rayne and K. Forest, Nat. Preced., 2010 DOI:10.1038/npre.2010.4865.1.
- B. Boekfa, S. Choomwattana, P. Khongpracha and J. Limtrakul, Langmuir, 2009, 25, 12990–12999 CrossRef CAS PubMed.
- P. R. P. Barreto, A. F. A. Vilela and R. Gargano, J. Mol. Struct.: THEOCHEM, 2003, 639, 167–176 CrossRef CAS PubMed.
- X. Sun, C. Zhang, Y. Zhao, J. Bai, Q. Zhang and W. Wang, Environ. Sci. Technol., 2012, 46, 8148–8155 CrossRef CAS PubMed.
- R. Atkinson, Chem. Rev., 1986, 86, 69–201 CrossRef CAS.
- C. Li, H. Xie, J. Chen, X. Yang, Y. Zhang and X. Qiao, Environ. Sci. Technol., 2014, 48, 13808–13816 CrossRef CAS PubMed.
- R. G. Prinn, R. F. Weiss, B. R. Miller, J. Huang, F. N. Alyea, D. M. Cunnold, P. J. Fraser, D. E. Hartley and P. G. Simmonds, Science, 1995, 269, 187–192 CAS.
- G. P. Brasseur, J. J. Orlando and G. S. Tyndall, in Atmospheric Chemistry and Global Change, Oxford University Press, USA, 1999 Search PubMed.
- X. Tang, Y. Zhang and M. Shao, in Atmospheric Environmental Chemistry, Higher Education Press, Beijing, China, 2006 Search PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, in Upper and Lower Atmosphere, Academic press, New York, 1999 Search PubMed.
- H. B. Singh and J. F. Kasting, J. Atmos. Chem., 1988, 7, 261–285 CrossRef CAS.
- B. J. Finlayson-Pitts, Res. Chem. Intermed., 1993, 19, 235–249 CrossRef CAS PubMed.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS PubMed.
- K. W. Oum, M. J. Lakin, D. O. DeHaan, T. Brauers and B. J. FinlaysonPitts, Science., 1998, 279, 74–76 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Atmospheric Chemistry: Fundamentals and Experimental Techniques, Wiley, New York, 1986 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The structures and energies of all the 170 mono-substituted C11H17Cl7 isomers and structures of intermediates, transition states and products. See DOI: 10.1039/c5ra00612k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.