
Spicarins A–D from acetylated extract of fungus Spicaria elegans KLA03†
Zhenjian Lin‡
,
Xinhua Ma,
Hongjuan Wei,
Dehai Li,
Qianqun Gu and
Tianjiao Zhu*
Key Laboratory of Marine Drugs, Chinese Ministry of Education, Ocean University of China, Qingdao 266003, PR China. E-mail: zhutj@ouc.edu.cn; Fax: +86-532-82033054; Tel: +86-532-82031632
First published on 9th April 2015
Abstract
Spicarins A (1) and B (2), two novel isobenzfuran-aspochalasin heterodimers, and spicarins C (3) and D (4), two isobenzofuran dimers, were isolated from the acetylated products of the marine-derived fungus Spicaria elegans KLA03. The structures of compounds 1–4 were elucidated by spectroscopic and/or X-ray diffraction methods. Compounds 3 and 4 inhibited lipopolysaccharide (LPS)-induced nitric oxide production in BV2 microglial cells.
Introduction
The successful preparation of structurally diverse compounds increases the chance of discovering novel skeleton compounds with interesting biological activity, and therefore, has been considered as one of the most important factors for drug discovery in the past decades. Natural products continue to be an excellent source of novel molecules of importance to drug discovery. Beside the traditional natural products discovery strategy, several strategies have been applied to extend the structure diversity. One approach, known as OSMAC (one strain many compounds), aims to activate silent genes in microorganisms by manipulating culture conditions.1 Another approach is combinatorial biosynthesis, such as engineering and designing artificial gene clusters to produce “unnatural” natural compounds.2 Alternatively, using the natural extract as an agent, newly developed strategies to extend the chemical space by yield semisynthetic compounds rather than mature ones have made a great progress. For instance, chemically engineered extracts (CEEs) has successfully enhanced the structural diversity and biological activities by chemical diversification of crude extracts of natural organisms.3During our exploration of the chemically diversity of the secondary metabolites produced by marine derived fungi, a series of cytochalasins with diverse amino acid component and difference PKS ring systems have been discovered from a fungal strain Spicaria elegans KLA03 by OSMA approaches.4–9 Very recently, we found a pyrogallol containing compound, eleganketal A, from S. elegans KLA03, which represented a new class of fungal polyketide compounds that possessed a novel spiro[isobenzofuran-1,3′-isochroman] ring system.10 Like eleganketal A, more related analogues existed in the fermentation products were not stable. We treated the crude extract of S. elegans KLA03 by acetic anhydride for stabilization of the potential phenol, alcohol or hemiacetal groups. The investigation of the acetylated products resulted in the identification of two previously undescribed polyketide heterodimers, composing partial eleganketal and aspochalasin unites, spicarins A (1) and B (2), together with two new related compounds spicarins C (3) and D (4) (Fig. 1). Herein, we report the discovery, structure elucidation and bioactivity of the acetylated derivatives.
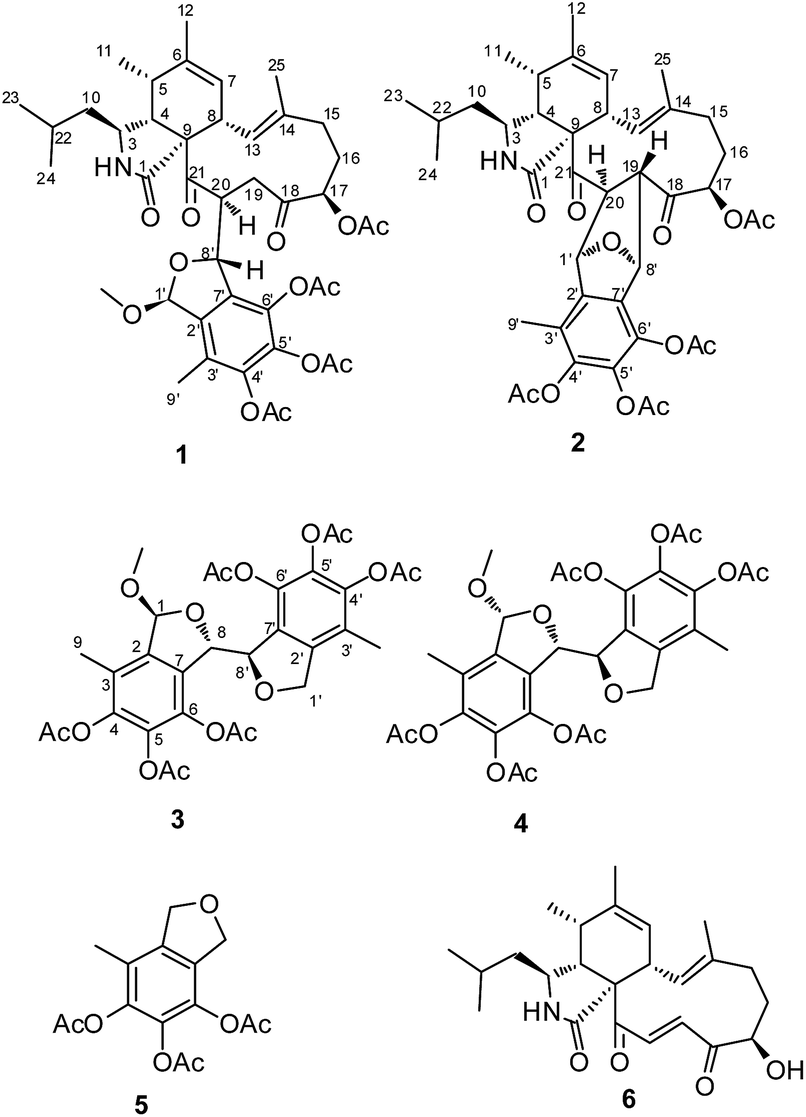 | ||
| Fig. 1 Structures of compounds 1–6. | ||
Results and discussion
By HRESIMS (m/z 780.3585 for [M + H]+ calcd C42H54NO13 for 780.3595 for 1 and 748.3355 for [M + H]+ calcd C41H50NO12 for 748.3333 for 2), the molecular formulas of 1 and 2 were determined as C42H53NO13 and C41H49NO12, respectively. Analysis of the 1D NMR data (Table 1) of 1 revealed 3 carbonyls, 9 quaternary carbons, 11 methines, 4 methylenes, 7 methyls (one of which is methoxyl) and four acetyls. First, comparison of the 1H and 13C NMR data with those of 4,5,6-trihydroxy-7-methyl-1,3-dihydroisobenzofuran (5)11 showed the presence of an isobenzofuran moiety, which was further confirmed by HMBC correlations (Fig. 2) from H-9′ to C-2′, C-3′ and C-4′, from H-8′ to C-1′ and C-2′, from H-1′ to C-7′, and from the protons of 1′-OCH3 to C-1′. Furthermore, comparison of the remaining 1D NMR data with those of the known aspochalasin B (6)12 indicated that 1 had the same 10-isopropyl-aspochalasin skeleton, except for the double bond of the α,β unsaturated ketone in 6 being replaced by a methine (δH 3.22 m, δC 54.3 CH) and a methylene (δH 3.75 dd, J = 18.7, 3.8 Hz, 2.60 dd, J = 18.7, 6.1 Hz, δC 38.4 (t)). As a result, the chemical shifts of the two carbonyls in 1 at 208.7 (C-21) and 201.7 (C-18) were shifted downfield by 3.5 ppm and 6.5 ppm, respectively, in comparison to that observed in 6 (205.2, C-21 and 195.6, C-18). This difference was confirmed by the HMBC correlations from H-19 to C-21 and C-18 and from H-20 to C-18. Finally, the HMBC correlation from H-20 to C-8′ and from H-8′ to C-21, as well as the 1H-1H COSY correlations between H-8′ and H-20, suggested that the 10-isopropyl-aspochalasins moiety and the methylisobenzofuran moiety were fused through C-8′ and C-20. The structure of 1 was further confirmed by single-crystal X-ray analysis (Fig. 3).| Position | 1 | 2 | ||
|---|---|---|---|---|
| 1H J = Hz | 13C | 1H J = Hz | 13C | |
| 1 | 175.5 (s) | 174.7 (s) | ||
| 2-NH | 5.93 brs | 6.07 brs | ||
| 3 | 3.03m | 51.1 (d) | 2.46m | 52.5 (d) |
| 4 | 2.53dd (4.8, 2.0) | 54.4 (d) | 3.00m | 51.3 (d) |
| 5 | 2.60m | 35.5 (d) | 2.47m | 35.4 (d) |
| 6 | 139.8 (s) | 140.6 (s) | ||
| 7 | 5.34 brs | 125.9 (d) | 5.37 brs | 125.9 (d) |
| 8 | 3.27 brd (10.4) | 42.6 (d) | 2.98m | 43.7 (d) |
| 9 | 68.3 (s) | 67.0 (s) | ||
| 10 | 1.34m; 1.24m | 48.2 (t) | 1.30m | 49.1 (t) |
| 11 | 1.19d (6.0) | 13.5 (q) | 1.16d (6.6) | 13.9 (q) |
| 12 | 1.74 brs | 20.1 (q) | 1.75 brs | 20.5 (q) |
| 13 | 6.18d (10.4) | 125.1 (d) | 6.26d (10.4) | 125.7 (d) |
| 14 | 137.5 (s) | 136.5 (s) | ||
| 15 | 2.32m; 1.87m | 28.7 (t) | 2.31m; 2.08m | 28.5 (t) |
| 16 | 2.25m; 2.04m | 35.5 (t) | 2.08m; 1.95m | 35.9 (q) |
| 17 | 5.28dd (8.8, 2.2) | 75.6 (d) | 5.60dd (4.4, 4.4) | 74.9 (d) |
| 17-COCH3 | 2.07s | 20.1 (q), 169.5 (s) | 2.10 s | 20.1 (q), 170.1 (s) |
| 18 | 201.7 (s) | 205.5qC | ||
| 19 | 3.75dd (18.7, 3.8); 2.60dd (18.7, 6.1) | 38.4 (t) | 3.11d (5.5) | 54.9 (d) |
| 20 | 3.22 m | 54.3 (d) | 5.02dd (5.0, 5.0) | 51.5 (d) |
| 21 | 208.7 (s) | 207.5 (s) | ||
| 22 | 1.54m | 25.2 (d) | 1.59m | 25.6 (q) |
| 23 | 0.92d (6.6) | 20.9 (q) | 0.94d (6.6) | 23.8 (q) |
| 24 | 0.91d (6.6) | 23.7 (q) | 0.93d (6.6) | 20.7 (q) |
| 25 | 1.45s | 16.1 (q) | 1.36s | 15.7 (q) |
| 1′ | 6.34d (2.2) | 104.5 (d) | 5.47d (4.9) | 81.4 (d) |
| 1′-OCH3 | 3.02s | 50.4 (q) | ||
| 2′ | 134.9 (s) | 133.6 (s) | ||
| 3′ | 126.3 (s) | 122.8 (s) | ||
| 4′ | 142.6 (s) | 140.5 (s) | ||
| 5′ | 135.9 (s) | 134.6 (s) | ||
| 6′ | 136.2 (s) | 140.1 (s) | ||
| 7′ | 132.3 (s) | 133.4 (s) | ||
| 8′ | 6.07dd (8.8, 2.2) | 82.9 (d) | 5.53 brs | 81.0 (d) |
| 9′ | 2.14s | 11.8 (q) | 1.95s | 21.0 (q) |
| 3 × COCH3 | 2.30, 2.23, 2.14s | 20.2, 20.2, 20.1 (q); 167.4, 167.2, 166.5 (s) | 2.30, 2.27, 2.24s | 20.2, 20.2, 20.1 (q); 167.4, 167.2, 167.0 (s) |
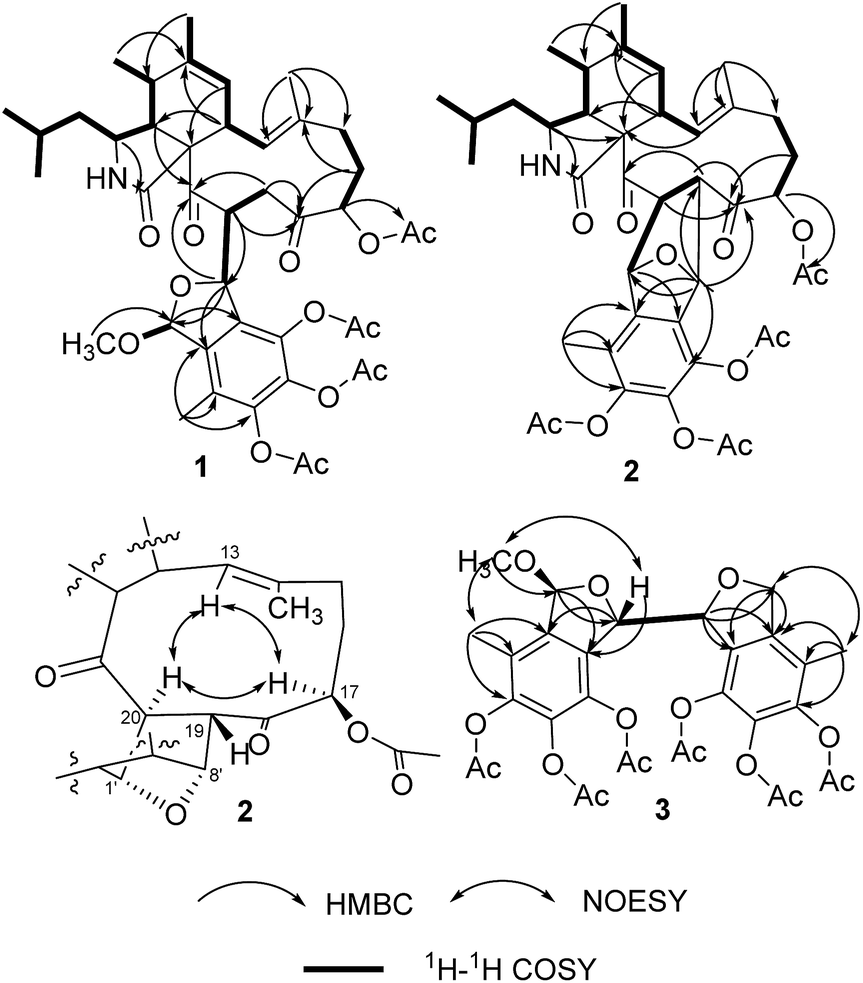 | ||
| Fig. 2 Key 1H-1H COSY, HMBC and NOESY correlations of 1, 2 and 3. | ||
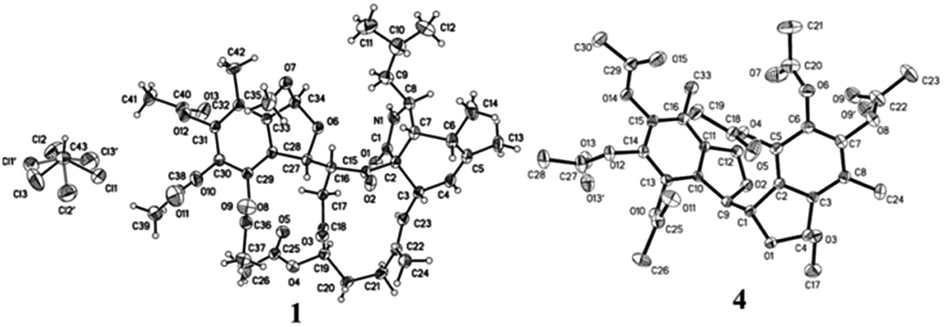 | ||
| Fig. 3 Final X-ray drawing of compounds 1 and 4. | ||
The 1H and 13C NMR data (Table 2) of 2 were similar to those of 1, except for the absence of a methoxyl group. In particular, the methyl acetal group at C-1′ was absent in the NMR spectra of 2, instead, an oxygenated methine group (δH 5.47, δC 81.4) presented (Table 2). Furthermore, a methine group (δH 3.11, δC 54.9) at the C-19 position of the aspochalasin moiety was observed instead of a methylene of that in 1. The 1H-1H COSY correlations between H-20 to H-1′ and H-19 and the HMBC correlation from H-8′ to C-19 and C-18 indicated there was a tetrahydrofuran ring between C-19/20 and C-1′/C-8′, which is also required by the unsaturation degree of the molecular formula of 2. Thus, the structure of compound 2 was deduced as an isobenzfuran-aspochalasin heterodimers, possessing a novel ‘7-oxabicyclo[2.2.1]heptane’ ring system (Fig. 4).
| Position | 3 | 4 | ||
|---|---|---|---|---|
| 1Ha J = Hz | 13Ca | 1H J = Hz | 13C | |
| a The chemical shifts in () were measured in DMSO-d6. | ||||
| 1 | 6.10 brs (6.08 brs) | 106.5 (106.8) (d) | 6.06 brs | 106.8 (d) |
| 1-OCH3 | 3.25 s (3.25 s) | 52.7 (54.5) (q) | 3.16 s | 55.5 (q) |
| 2 | 135.9 (136.8) (s) | 136.8 (s) | ||
| 3 | 125.9 (126.1) (s) | 126.0 (s) | ||
| 4 | 142.6 (142.8) (s) | 143.0 (s) | ||
| 5 | 135.9 (136.8) (s) | 136.8 (s) | ||
| 6 | 135.9 (136.7) (s) | 136.7 (s) | ||
| 7 | 129.2 (130.2) (s) | 130.2 (s) | ||
| 8 | 5.63 brs [5.45dd (2.6, 2.6)] | 85.3 (85.2) (d) | 5.37 brd (2.9) | 85.8 (d) |
| 9 | 2.13 s (2.07) | 11.7 (12.5) (q) | 2.04 s | 12.6 (q) |
| 1′ | 4.90d (12.4); 4.81 brd (12.4) (4.98d, 4.80d) | 73.3 (73.2) (t) | 5.02dd (11.5, 3.0); 4.99d (11.5) | 73.4 (t) |
| 2′ | 139.1 (139.5) (s) | 139.6 (s) | ||
| 3′ | 123.2 (123.6) (s) | 123.4 (s) | ||
| 4′ | 142.2 (143.2) (s) | 142.7 (s) | ||
| 5′ | 134.3 (135.0) (s) | 135.2 (s) | ||
| 6′ | 135.7 (136.5) (s) | 136.5 (s) | ||
| 7′ | 127.7 (128.6) (s) | 129.1 (s) | ||
| 8′ | 5.55 brs [5.36dd (2.3, 2.4)] | 86.0 (86.0) (d) | 5.27 brs | 86.3 (d) |
| 9′ | 2.00 s (1.98) | 12.9 (13.4) (q) | 2.00 s | 13.4 (q) |
| 6 × COCH3 | 2.29, 2.29, 2.22, 2.23, 2.13, 2.10 s, (2.10, 2.13, 2.25, 2.26, 2.32, 2.33 s) | 20.0, 20.0, 20.0, 20.0, 20.2, 20.2 (q), 167.5, 167.4, 167.3, 167.3, 166.9, 166.6 (s) (20.7, 20.7, 20.8, 20.8, 20.9, 20.9 (q), 168.0, 168.1, 168.3, 168.4, 168.7, 168.7 (s)) | 2.33, 2.32, 2.28, 2.22, 2.12, 2.10 s | 20.7, 20.8, 20.8, 20.9, 20.9, 21.0 (q), 168.8, 168.7, 168.6, 168.2, 168.1, 168.0 (s) |
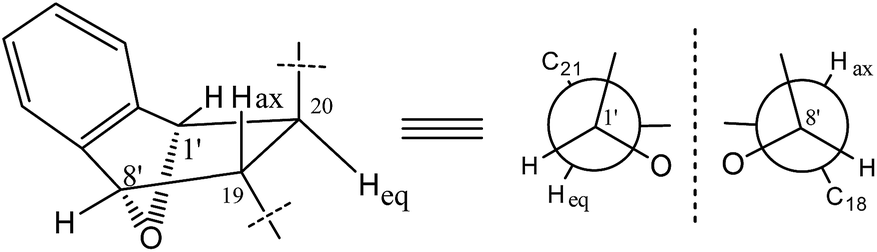 | ||
| Fig. 4 Partial conformational analysis of compound 2. | ||
The X-ray diffraction structure established the relative configuration of 1. The aspochalasin moiety has the same configuration as that in 6. The furan ring adopted a trans form, and the bond between C-20 and C-8′ was in a threo form. Since the absolute configuration of the perhydroisoindol-1-one moiety of aspochalasin has been presumed to be conserved, which was biosynthetically derived from a Diels–Alder-type reaction.7 Thus the absolute configuration of 1 was proposed as 3S, 4R, 5S, 8S, 9S, 17R, 20S, 1′S, 8′S. The relative configuration of the ‘7-oxabicyclo[2.2.1]heptane’ moiety in 2 was determined by the J-based conformational analysis. Based on the analysis of the model molecule rac-7-oxabicyclo-[2.2.1]heptane-2,3-dicarboxylic acid,13 the dihedral angle from the proton at the bridgehead carbon to the axial proton is almost 90° (J ≈ 0), and almost 40° (J ≈ 5.0) to the equatorial proton. In the 1H NMR data of 2, the observed JH-1′,H-20 = 4.9 and JH-8′,H-19 ≈ 0 suggested that the H-20 was in a equatorial bond and the H-19 was in a axial bond. The current NMR data did not support the relative configuration between the ‘7-oxabicyclo[2.2.1]heptane’ moiety and the perhydroisoindol-1-one moiety.
Compounds 3 and 4 were obtained as pale yellow powder. The molecular formula of both 3 and 4 were determined as C31H32O15 by HRESIMS m/z 667.1613 for [M + Na]+ for 3 and 667.1630 for [M + Na]+ for 4, calcd C31H32O15Na for 667.1639. Compounds 3 and 4 showed very similar 1D NMR signals (Table 1), suggesting they are diastereoisomers. Analysis of the 1D NMR data (Table 1) revealed 3 had 12 aromatic quaternary carbons, 2 oxygenated methines, an oxygenated methylene, an acetalic methine, 3 methyls and 6 acetyls. In comparison to the 1D NMR data of 5, 3 showed the presence of two fully substituted benzene rings, indicating that 3 has structure of dimer of 5. This hypothesis was confirmed by analysis of the HMBC and 1H-1H COSY spectra (Fig. 2). The HMBC correlations from H-1 to C-7, from H-8 to C-2, and from H-9 to C-2, C-3 and C-4 indicated a methylisobenzofuran moiety, of which a methyl acetal group at C-1 was assigned by HMBC correlation from the protons of 1-OCH3 to C-1. The other methylisobenzofuran moiety was confirmed by the HMBC correlations from H-8′ to C-2′ and C-1′, from H-1′ to C-7′, from H-9′ to C-2′, C-3′ and C-4′. The 1H-1H COSY correlation between H-8 and H-8′ established the fusion of the two methylisobenzofuran moieties through C-8 and C-8′.
The relative configurations of 3 and 4 were determined by the NOESY correlations and/or X-ray diffraction. The X-ray diffraction of 4 (Fig. 3) determined the relative configuration as 1R*,8S*,8′R*. The dihedral angle between H-8 and H-8′ is −79°, which is consistent with the small coupling constant (2.9 Hz) between H-8 and H-8′ in 4. The methyl acetal group and H-8 was in the opposite face of the furan ring. In contrast, the NOESY correlation between the protons of 1-OCH3 and H-8 (Fig. 2) indicated the methyl acetal group at C-1 and H-8 was in the same face of the furan ring in 3. Since the coupling constant between H-8 and H-8′ in 3 is also very small (2.6 Hz), the relative configuration of 3 was tentatively assigned as 1S*,8S*,8′R*.
Compounds 1–4 were tested for their cytotoxicities against the MOLT-4, BEL-7402, HL-60 and A-549 cell lines. None of them showed any activity. In vitro anti-inflammatory assay, compounds 3 and 4 inhibited lipopolysaccharide (LPS)-induced nitric oxide production in BV2 microglial cells, with IC50 values of 30 and 75 μM, respectively.
Experimental section
General experimental procedures
1H, 13C NMR and DEPT and 2D-NMR spectra were recorded on a JEOL JNM-ECP 600 spectrometer using TMS as internal standard and chemical shifts were recorded as δ values. NOESY experiments were carried out using a mixing time of 0.5 s. ESI-MS was measured on a Q-TOF ULTIMA GLOBAL GAA076 LC mass spectrometer. Semipreparative HPLC was performed using an ODS column [Shin-pak ODS (H), 10 × 250 mm, 5 μm, 4 mL min−1].X-ray structure determination
X-ray diffraction intensity data were collected on a MAC DIP-2030K diffractometer with graphite-monochromater Mo Kα radiation (λ = 0.71073 Å) by the ω scan technique [scan width 0–180°, 2θ ≤ 50°]. Hydrogen positions were found from difference Fourier maps and geometric calculations. All calculations were carried out on a personal computer using the SHELX-97 program system. Compound 1: (ESI†); altogether 8079 reflections were collected, of which 3626 with |F|2 ≥ 2σ|F|2 equation were observed. The structure was solved by direct methods and refined by block-matrix least-squares procedure to R1 = 0.0715, wR2 = 0.1901. Compound 4: (ESI†); altogether 5599 reflections were collected, of which 2599 with |F|2 ≥ 2σ|F|2 equation were observed. The structure was solved by direct methods and refined by block-matrix least-squares procedure to R1 = 0.0635, wR2 = 0.2171.Fungal material
The fungus Spicaria elegans was isolated from the marine sediments collected in Jiaozhou Bay, China and was preserved in the China Center for Type Culture Collection (patent depositary number: KLA03 CCTCC M 205049). Working stocks were prepared on potato dextrose agar slants stored at 4 °C.Fermentation and extraction
The fungus was incubated at 28 °C under shaking condition for 8 days in 400–500 mL conical flasks containing the liquid medium (200 mL per flask) composed of glucose (20 g L−1), peptone (5 g L−1), maltose (3 g L−1) and yeast extract (3 g L−1) and sea-water after adjusting its pH to 7.0. The fermented whole broth (30 L) was extracted three times with EtOAc to give an EtOAc solution, which was concentrated under reduced pressure to give a crude extract (15.5 g).Extract diversification and purification
The dry crude extract (15.5 g) was passed through a silica column to remove the fatty acids, polar sugar and salts. The pre-purified extract was dried and dissolved in 500 mL dry pyridine and 200 mL acetic anhydride, the mixture was stirring at room temperature for 72 h. after that, the solvent was removed by vacuum and the residue was fractionationed on a silica gel column using gradient elution of CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH. The fractions that contains major compounds were further purified by extensive HPLC (70% MeOH, 4.0 mL min−1) gave compounds 1 (20 mg), 2 (5.0 mg), 3 (15.6 mg), 4 (12.3 mg) and 5 (30.0 mg). Compound 6 was purified previously from underivatized extract of KLA03.
:MeOH. The fractions that contains major compounds were further purified by extensive HPLC (70% MeOH, 4.0 mL min−1) gave compounds 1 (20 mg), 2 (5.0 mg), 3 (15.6 mg), 4 (12.3 mg) and 5 (30.0 mg). Compound 6 was purified previously from underivatized extract of KLA03.
Cytotoxicity assay
Cytotoxic activity was evaluated by the MTT method using MOLT4, A-549, HL-60 and BEL-7402 cell lines. The cell lines were grown in RPMI-1640 supplemented with 10% FBS under a humidified atmosphere of 5% CO2 and 95% air at 37 °C. An aliquot (200 μL) of those cell suspensions at a density of 5 × 104 cell mL−1 was plated in 96 well microtiter plates and incubated for 24 h at the above condition. Then 2 μL of the test compound solutions (in DMSO) at different concentrations was added to each well and further incubated for 72 h in the same condition. MTT solution (20 μL of 5 mg mL−1 in IPMI-1640 medium) was added to each well and incubated for 4 h. Old medium containing MTT (150 μL) was then gently replaced by DMSO and pipetted to dissolve any formazan crystals which had formed. Absorbance was then determined on a SPECTRA MAX PLUS plate reader at 570 nm. Vp-16 (etoposide) was used as the positive control with IC50 values of 0.28, 0.63, 0.04 and 1.03 μM against MOLT4, A-549, HL-60 and BEL-7402 cells, respectively.Lipopolysaccharide (LPS)-induced inflammation inhibition assay
Nitrite in culture supernatants, a stable by-product of NO, was measured by Griess reagent (iNtRON) according to the instructions provided by the manufacturer. Briefly, cells (1 × 105 cells per well) were seeded in 96 well microtiter plates and incubated for 24 h in growth medium under a humidified atmosphere of 5% CO2 and 95% air at 37 °C. Cells were then pretreated with compounds at various concentrations in serum-free medium for 45 min. After pretreatment, LPS (0.1 mg mL−1) was added, and the cells were incubated for 16 h. After LPS exposure, duplicates of supernatants (100 μL) were collected and mixed with 50 μL of Griess reagent. The absorbance was measured with a 595 nm filter by a microplate spectrophotometer. The concentration of nitrite released in the supernatants was calculated with the linear equation derived from the standard curve generated by known concentrations. Pyrrolidine dithiocarbamate (PDTC) was used as positive control, which has an IC50 value of 0.058 μM.Conclusions
Spicarins A (1) and B (2) are members of a large family of leucine-derived cytochalasans. In natural occurring cytochalasans, the α,β-unsaturated ketone moiety at C-19–C-21 in the macrocyclic ring of this type cytochalasans has been found to be substituted by different functional groups, such as a adenine unit in alachalsins and a anthranilate containing peptide moiety in aspochalamins.7 Spicarin A (1) and B (2) is the first example of carbon–carbon bonds substitution at C-19 and C-20 of aspochalasins class, and represented a type of unprecedented skeleton compounds. Although it is uncertain whether the carbon–carbon bonds substitution at C-19 and C-20 of aspochalasins formed enzymatically or during the acylation reaction, this study suggested that simple derivatization can be very useful to generate/identify novel skeleton compounds.Acknowledgements
This work was financially supported by the Chinese National Natural Science Fund (no. 21372208, 41376147, 41176120, 30772640); the NSFC-Shandong Joint Fund for Marine Science Research Centers (U1406402), the National High Technology Research and Development Program of China (no. 2013AA092901).Notes and references
- H. B. Bode, B. Bethe, R. Hoefs and A. Zeeck, ChemBioChem, 2002, 3, 619 CrossRef CAS.
- S. Horinouchi, J. Antibiot., 2008, 61, 709 CrossRef CAS PubMed.
- I. A. Ramallo, M. O. Salazar, L. Mendez and R. L. E. Furlan, Acc. Chem. Res., 2011, 44, 241 CrossRef CAS PubMed.
- R. Liu, Q. Gu, W. Zhu, C. Cui, G. Fan, Y. Fang, T. Zhu and H. Liu, J. Nat. Prod., 2006, 69, 871 CrossRef CAS PubMed.
- R. Liu, Z. Lin, T. Zhu, Y. Fang, Q. Gu and W. Zhu, J. Nat. Prod., 2008, 71, 1127 CrossRef CAS PubMed.
- Z. Lin, T. Zhu, G. Zhang, H. Wei and Q. Gu, Can. J. Chem., 2009, 87, 486 CrossRef CAS.
- Z. Lin, T. Zhu, H. Wei, G. Zhang, H. Wang and Q. Gu, Eur. J. Org. Chem., 2009, 3045 CrossRef CAS PubMed.
- Z. Lin, T. Zhu, L. Chen and Q. Gu, Chin. Chem. Lett., 2010, 21, 824 CrossRef CAS PubMed.
- F. Wang, H. Wei, T. Zhu, D. Li, Z. Lin and Q. Gu, Chem. Biodiversity, 2011, 8, 887 CAS.
- Y. Luan, H. Wei, Z. Zhang, Q. Che, Y. Liu, T. Zhu, A. Mandi, T. Kurtan, Q. Gu and D. Li, J. Nat. Prod., 2014, 77, 1718 CrossRef CAS PubMed.
- Y. Ishikawa, T. Ito and K. H. Lee, Nihon Yukagakkaishi, 1996, 45, 1321 CAS.
- W. Keller-Schierlein and E. Kupfer, Helv. Chim. Acta, 1979, 62, 1501 CrossRef CAS PubMed.
- Y. Wang, R. Hu and Y. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1442 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 709723 and 709724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra01923k |
| ‡ Current address: Department of Medicinal Chemistry, L. S. Skaggs Pharmacy Institute, University of Utah, Salt Lake City, UT, USA. |
| This journal is © The Royal Society of Chemistry 2015 |