DOI:
10.1039/C5RA02417J
(Communication)
RSC Adv., 2015,
5, 30216-30221
Solvothermal synthesis, structure, and fluorescence properties of three d10 polymers assembled from semi-rigid V-shaped aza-bridged multicarboxylate†
Received
11th February 2015
, Accepted 18th March 2015
First published on 19th March 2015
Abstract
Hydro(solvo)thermal reactions between a new flexible multicarboxylate ligand, 2,2′-azanediyldibenzoic acid (H2L), and M(OH)2 (M = Cd, Zn) in the presence of different N-donor ancillary ligands, 4,4′-bipyridine (bpy) and pyridine (py), afford three novel coordination polymers: [Cd2(L)2(bpy)1.5(H2O)]n (1), [Cd2(L)(Py)2]n (2), and [Zn2(L)2]n (3). These polymers show great differences in regard to their structures and properties due to the variation in the ancillary ligands and in the coordination geometry of the metal ions. Compound 1 exhibits a 3D supramolecular network assembled from 2D layered architecture composed of dinucelar cadmium subunits via intermolecular π⋯π and C–H⋯π interactions. The supramolecular structure of compound 2 is built from the packing of two homochiral helical chain. Compound 3 possesses two crystallographically nonequivalent metal atoms with Zn1 in a four-coordinated tetrahedron geometry and Zn2 in a five-coordinated trigonal bipyramid geometry. Five different coordination modes, ranging from μ2, μ3 and μ4, have been observed in the H2L ligands. The solid-state fluorescence spectra show that complexes 1 and 2 exhibit small shifts to the blue, while 3 is red shifted with respect to the free ligand.
1. Introduction
In the field of supramolecular chemistry and crystal engineering, the design and synthesis of coordination polymers is an emerging field owing to their structural aesthetics and topologies as well as diverse functional properties.1–3 Among the various kinds of complexes fabricated, those with intriguing fluorescent properties attract great attention due to their potential applications in light-emitting diodes (LEDs).4,5 The specific ligands used are very important in the construction of these coordination polymers.
As an important family of multidentate O-donor ligands, organic aromatic polycarboxylate ligands have been extensively employed in the preparation of such metal–organic complexes for their versatile coordination modes. Furthermore, in comparison with the rigid aromatic multicarboxylate ligands with one benzene ring as the central molecular framework, the so-called semi-rigid V-shaped multicarboxylate ligands with two benzene rings bridged by an nonmetallic atom (C, O, S, or N atom) as the central molecular framework exhibit increased flexibility and are therefore able to lead to metal complexes with diverse structures because of the free rotation of two benzene rings around the bridged non-metallic atom.1,6
Current research is concerned with semi-rigid V-shaped multicarboxylate ligands with the two aromatic motifs bridged by ‘–CH2–’ or ‘–O–’;2,6,7 conversely, reports on V-shaped multicarboxylate ligands where the two benzene ring arms are connected by ‘–NH–’ are relatively rare.5–8 With the aim of understanding the coordination chemistry of V-shaped aza-bridged multicarboxylate ligands, we synthesized 2,2′-azanediyldibenzoic acid and implemented solvothermal reactions on it.
Pleasingly, three structurally different coordination polymers were obtained with different ancillary ligands and d10 metal salts (Cd, Zn). Here we report the syntheses, crystal structures, and fluorescent properties of these title intermediates.
2. Experimental section
2.1. Materials and synthesis
All reagents and solvents employed in the present work were obtained from commercial sources and used directly without further purification. 2,2′-Azanediyldibenzoic acid (H2L) was synthesized according to the literature as detailed below:9 2-chlorobenzoic acid (3.130 g, 20 mmol), 2-aminobenzoic acid (5.486 g, 40 mmol), anhydrous K2CO3 (1.382 g, 10 mmol) and Cu (0.1 g) were combined in 15 mL N,N-dimethylformamide (DMF) and refluxed at 150 °C for 2 h. After cooling down to room temperature, 50 mL aqueous HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were added to the reaction slowly, and a light-yellow precipitate was produced. The mixture was filtered after stewing for 12 h; the crude product was washed with water (20 mL) and then dried in air. Recrystallization in tetrahydrofuran yielded the final product (45.5%). Anal. calcd for C14H11NO4: C 65.37, H 4.31, N 5.45; found: C 65.12, H 4.28, N 5.50. IR/cm−1 (KBr): 3331(m), 1667 (s), 1515 (s), 1447 (s), 1411 (s), 1322 (s), 1165 (s), 1044 (m), 989 (m), 924 (m), 751(s).
:1) were added to the reaction slowly, and a light-yellow precipitate was produced. The mixture was filtered after stewing for 12 h; the crude product was washed with water (20 mL) and then dried in air. Recrystallization in tetrahydrofuran yielded the final product (45.5%). Anal. calcd for C14H11NO4: C 65.37, H 4.31, N 5.45; found: C 65.12, H 4.28, N 5.50. IR/cm−1 (KBr): 3331(m), 1667 (s), 1515 (s), 1447 (s), 1411 (s), 1322 (s), 1165 (s), 1044 (m), 989 (m), 924 (m), 751(s).
Crystals of compounds 1, 2 and 3 were isolated by hydrothermal reaction. The synthesis route is shown in Scheme 1. A heavy walled Pyrex tube containing a mixture of H2L (0.0516 g, 0.2 mmol), 4,4′-bipyridine (0.0312 g, 0.2 mmol), Cd(OH)2 (0.0292 g, 0.2 mmol), water (2 mL) and ethanol (1 mL) was frozen and sealed under vacuum, then placed inside an oven at 140 °C for 72 h and then cooled to room temperature at a rate of 5 °C h−1. The yield obtained on yellow block crystals for 1 was 35% (0.035 g) (based on ligand). Calc. for l: C 51.32, H 3.38, N 6.96; found for l: C 51.30, H 3.39, N 6.97. IR (KBr, cm−1) 3256 (m), 1604 (m), 1549 (s), 1496 (s), 1392 (s), 1275 (m), 1159 (m), 682 (m). Complex 2 was synthesized by a similar procedure as for complex 1 except pyridine (0.2 mL) was used instead of 4,4′-bipyridine, colorless needle-like crystals for 2 were obtained with a yield of 53% (0.056 g) (based on ligand). Calc. for 2: C 54.77, H 3.61, N 7.99; found for 2: C 54.75, H 3.62, N 8.01. IR (KBr, cm−1): 3251 (m), 1580 (s), 1501 (s), 1394 (s), 1281 (m), 756 (m). Complex 3 was prepared by solvothermal reaction between H2L (0.0516 g, 0.2 mmo1) and Zn(OH)2 (0.0198 g, 0.2 mmol) in a mixture of water (2 mL) and ethanol (1 mL) as the solvent without any ancillary ligands. The isolated yield of the yellow crystals of 3 was 86% (0.055 g) (based on ligand). Calc. for 3: C 52.40, H 2.81, N 4.37; found for 3: C 52.35, H 2.80, N 4.38. IR (KBr, cm−1) 3332 (m), 1667 (s), 1575 (s), 1516 (s), 1448 (s), 1410 (m), 1321 (m), 1248 (s), 750 (s).
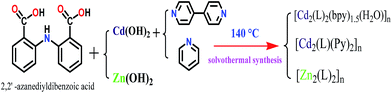 |
| | Scheme 1 The preparation of coordination complexes 1–3. | |
2.2. Characterization
The FT-IR spectra were recorded on a Nicolet Magna 750 FT-IR spectrometer using KBr pellets in the range of 4000–400 cm−1. Elemental analysis was performed on a Perkin-Elmer 240C elemental analyzer. The room-temperature X-ray powder diffraction spectra (XRPD) were recorded on a Rigaku D/Max-2500 diffractometer at 40 kV, 100 mA with a Cu-target tube and a graphite monochromator. Simulation of the XRPD pattern was carried out by the single-crystal data and diffraction-crystal module of the mercury (Hg) program available free of charge via the internet at http://www.iucr.org. Absorption spectra were carried out on a Jasco-1500 CD spectrometer with DRCD-574 integrating sphere at ambient temperature. The solid absorption spectra were measured on the resulting complexes as crystals (ca. 0.5 mg) in 100 mg of oven-dried KBr. The baseline correction was performed with the spectrum of a pure KBr disk. Spectra were recorded for the wavelength range 250–700 nm for all the disks and the data pitch was 0.1 nm. PL emission spectra were measured on the resulting ground crystal powder at room temperature using a spectrofluorophotometer (JASCO, FP-6500) with a xenon lamp (150 W) as a light source and a PMT voltage of 500 V.
2.3. Single-crystal structure determination
X-ray single crystal structures were determined with a Bruker Smart APEX II CCD diffractometer. The diffraction data were collected using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at room temperature. Empirical absorption corrections were applied by using the SADABS program. The structures were solved by direct methods and refined by the full-matrix least-squares based on F2 using SHELXTL-97 program. All non-hydrogen atoms were refined anisotropically and the hydrogen atoms of organic ligands were located from the successive difference Fourier syntheses. Detailed information about the crystal data and structure determination for the title compounds is summarized in Table SI.† The selected intra-atomic distances and bond angles are given in Table SII.†
3. Results and discussion
3.1. Description of the structure
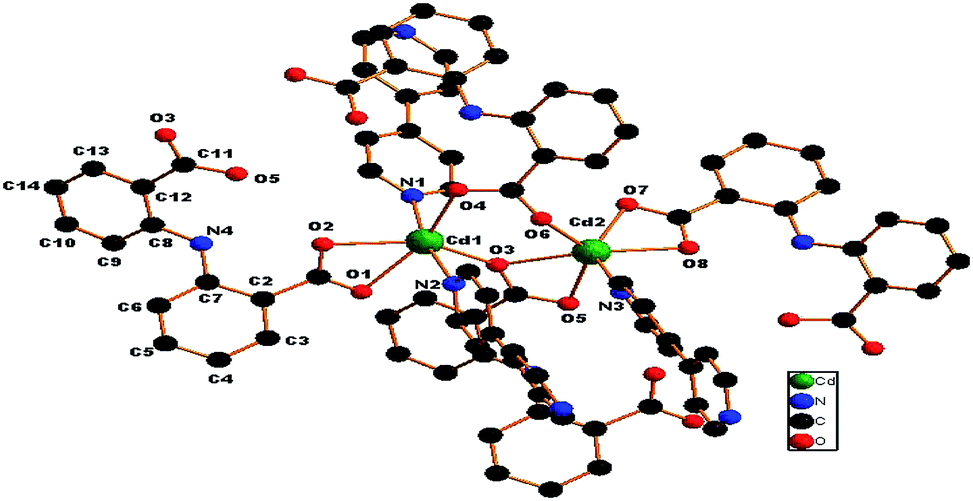
[Cd2(L)2(bpy)1.5(H2O)]n (1). Compound 1 crystallized in a triclinic crystal system and a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. As shown in Fig. 1, the asymmetric unit contains two independent Cd(II) ions, two 2,2′-azanediyldibenzoic acid molecules, one and a half bpy, and two solvated water molecules. Compound 1 features an unusual 2D two-layered structure constructed by a binuclear cadmium unit with a Cd1⋯Cd2 distance of 3.8367 Å; both Cd1 and Cd2 atoms can be described as displaying a distorted octahedral geometry, and each Cd1 atom is coordinated to four oxygen atoms of three ligands (by two monodentate carboxylic groups and one chelating carboxylic group) in the equatorial plane and two nitrogen atoms from bpy molecules along the axial position. The Cd2 atom is coordinated to five oxygen atoms of three L ligands (by two chelating carboxylic groups and one monodentate carboxylic group) and one nitrogen atom from bpy. The Cd–O and Cd–N distances range from 2.222(4) to 2.389(4) Å and 2.267(4) to 2.355(4) Å respectively, which are similar to those found in other Cd(II) complexes.10–12 The Cd1–O6 distance is 2.9172 Å, which suggests a nonnegligible interaction.13 It is worth noting that H2L in 1 adopts two different coordination modes (μ3-kO:kO,O′:kO′′,O′′′; μ3-kO:kO′:kO′′,O′′′) to bridge three Cd(II) atoms in O,O′-chelating and monodentate fashions (Fig. 4A and B).
space group. As shown in Fig. 1, the asymmetric unit contains two independent Cd(II) ions, two 2,2′-azanediyldibenzoic acid molecules, one and a half bpy, and two solvated water molecules. Compound 1 features an unusual 2D two-layered structure constructed by a binuclear cadmium unit with a Cd1⋯Cd2 distance of 3.8367 Å; both Cd1 and Cd2 atoms can be described as displaying a distorted octahedral geometry, and each Cd1 atom is coordinated to four oxygen atoms of three ligands (by two monodentate carboxylic groups and one chelating carboxylic group) in the equatorial plane and two nitrogen atoms from bpy molecules along the axial position. The Cd2 atom is coordinated to five oxygen atoms of three L ligands (by two chelating carboxylic groups and one monodentate carboxylic group) and one nitrogen atom from bpy. The Cd–O and Cd–N distances range from 2.222(4) to 2.389(4) Å and 2.267(4) to 2.355(4) Å respectively, which are similar to those found in other Cd(II) complexes.10–12 The Cd1–O6 distance is 2.9172 Å, which suggests a nonnegligible interaction.13 It is worth noting that H2L in 1 adopts two different coordination modes (μ3-kO:kO,O′:kO′′,O′′′; μ3-kO:kO′:kO′′,O′′′) to bridge three Cd(II) atoms in O,O′-chelating and monodentate fashions (Fig. 4A and B).
 |
| | Fig. 1 Asymmetric unit of 1 (H atoms of ligand are omitted for clarity). | |
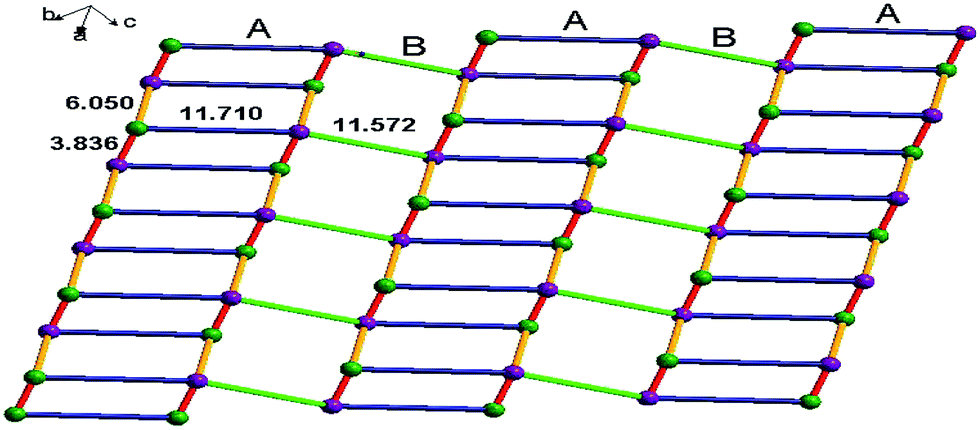
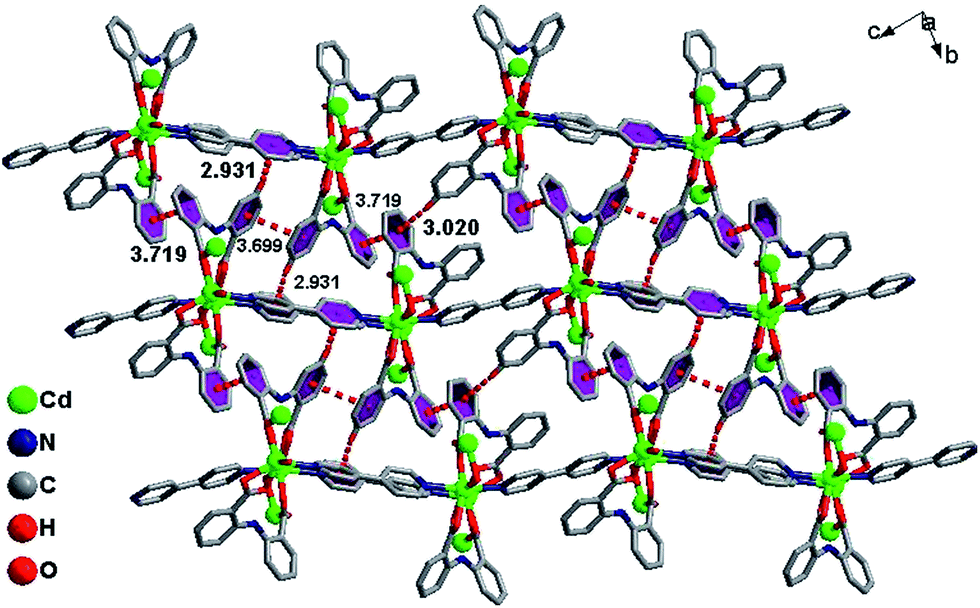
The infinite 2D wave-like layer structure of 1 is assembled from two kinds of ladder-like chains (A and B). The ligands connect adjacent binuclear cadmium units with a distance of 6.050 Å (orange line; the red line means binuclear cadmium unit) in mode A and B as in a vertical ladder armrest chain. The bpy molecules connecting Cd1 and Cd2 with a distance of 11.710 Å (blue line) constitute the ladder cleats of ladder A, the other bpy molecules connecting adjacent Cd1 atoms with a distance of 11.572 Å (light green) form the ladder cleats of ladder B (Fig. 2). Abundant π⋯π (3.699 Å, 3.719 Å) and C–H⋯π (2.931 Å, 3.020 Å) interactions further assemble the 2D layer to a 3D supermolecular structure in slipped AAAA stacking fashion (Fig. 3).14
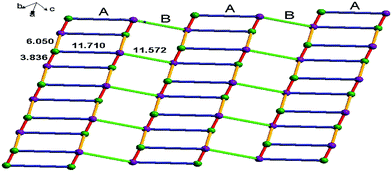 |
| | Fig. 2 The 2D wave-like layer structure assembled from two kinds of ladder-like structure of 1, the violet dots indicate Cd1 atoms, and green dots indicate Cd2 atoms. | |
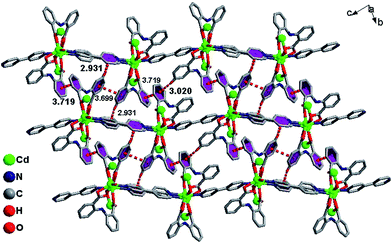 |
| | Fig. 3 π⋯π, C–H⋯π interactions and AAAA stacking fashion for 1. | |
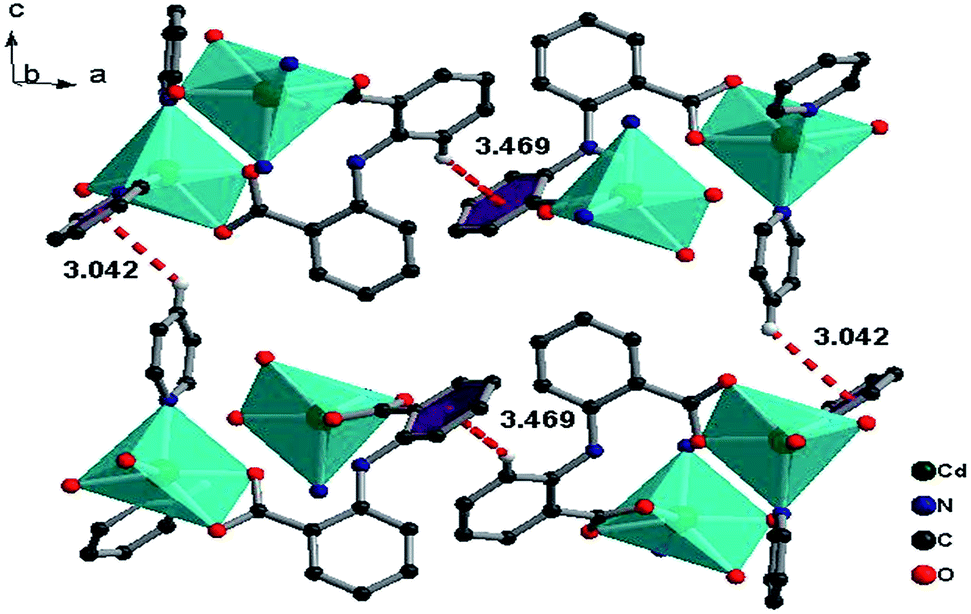
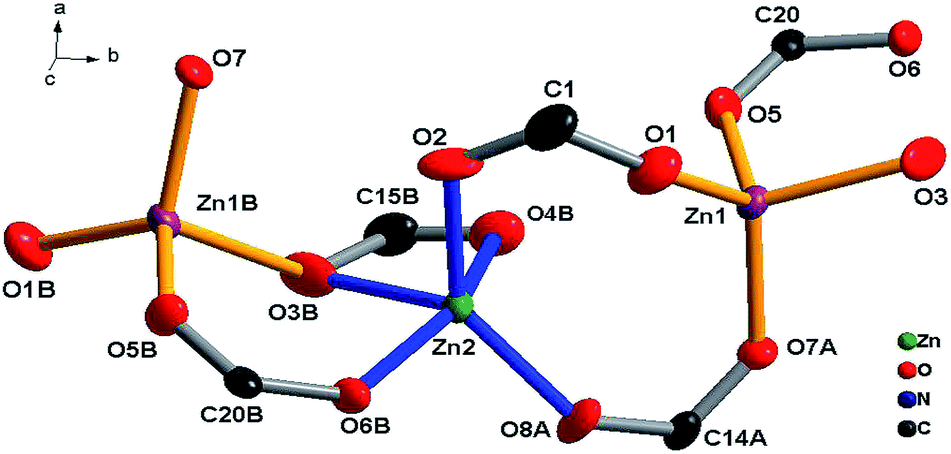
[Zn2(L)2]n (3). The X-ray diffraction study for 3 reveals that it crystallizes in a space group P21/c, and each molecular unit consists of two L ligands and two zinc atoms. Zinc usually prefers tetrahedral coordination, however, 3 exhibits a four-coordinated tetrahedral mode as well as a five-coordinated rectangular-pyramidal mode with Zn–O bonds ranging from 1.900 to 2.245 Å.16–18 Two carboxylate groups with different L connecting Zn1 and Zn2 atoms form a dinucelar unit with an eight-member ring Zn1–O1–C1–O2–Zn2–O8A–C14A–O7A (Fig. 8). A 1D ribbon structure is formed along the b-axis by connecting the zinc dinuclear units through the joint atom O3B and two carboxylate groups in a μ2 chelating mode (Fig. 9).
 |
| | Fig. 8 Coordination environment of Zn center, only the Zn, O and alpha-carbon atoms are shown for clarity (30% thermal ellipsoids). | |
 |
| | Fig. 9 A view of 1D ribbon chain structure of 3 along the c-axis. | |
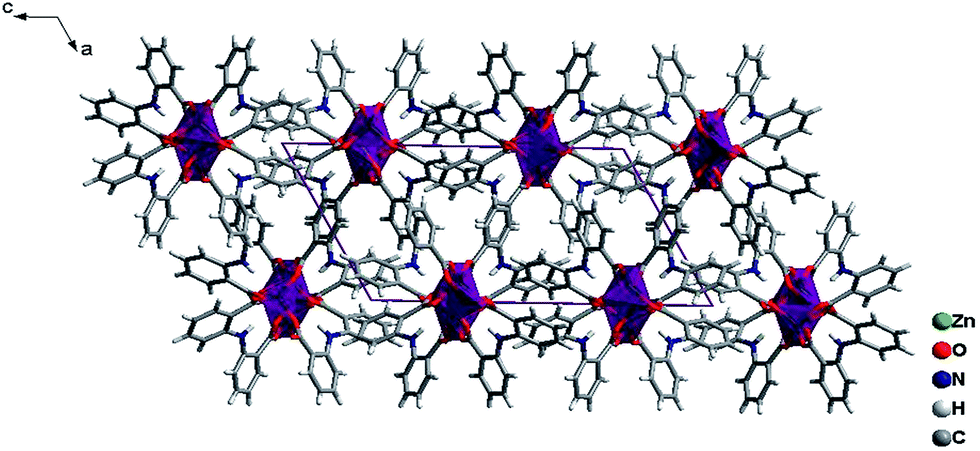
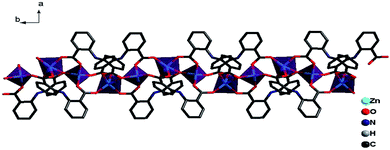
The ligands L adopt two kinds of μ4-coordination mode (μ4-kO:kO,O′:kO′′:kO′′′; μ4-kO:kO′:kO′′:kO′′′) to bridge four Zn(II) ions in O,O′-chelating and monodentate fashions. The dihedral angle between the two phenyl rings of the two L ligands is 41.4° and 39.3° respectively, and the dihedral angles between the carboxylate groups and the corresponding phenyl rings are 19.6° and 28.2° for L of mode D, and 22.5° and 23.9° for mode E (Fig. 4). Finally, The weak C–H⋯π and π⋯π interactions and other noncovalent interaction-static attracting forces, like Coulombic and van der Waals forces among the ribbon chains, link the chain structure into a 3D-dimensional network (Fig. 10).15,19,20
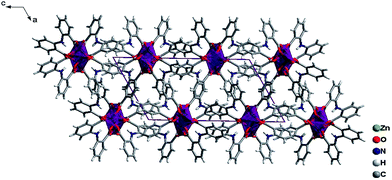 |
| | Fig. 10 A view of the packing diagrams for 3 along b-axis, the 1D chain extending along the b-axis and arraying in ac plane. | |
3.2. Powder X-ray data
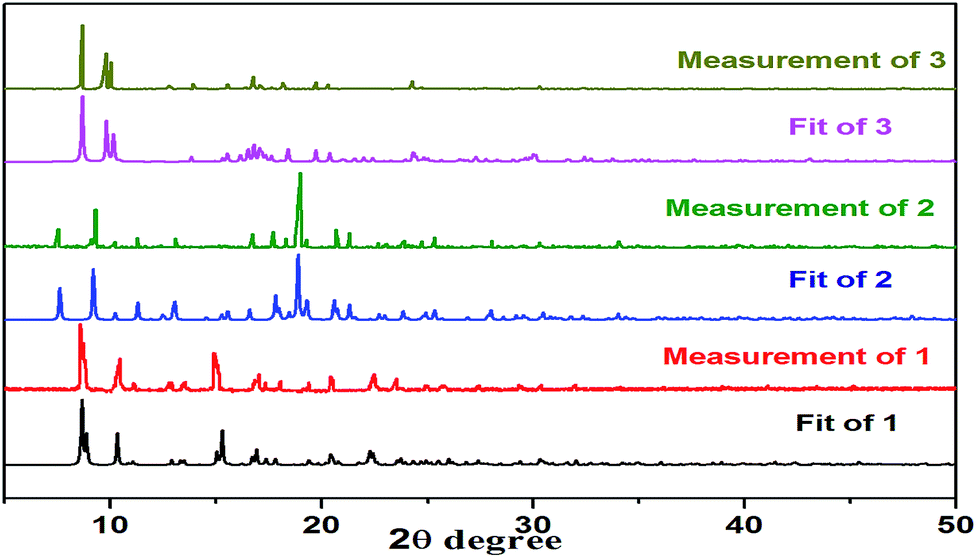
XRD was used to check the purity of 1–3. The results show that all the peaks displayed in the measured patterns at room temperature closely match those in the simulated patterns generated from single-crystal diffraction data, indicating that single phases of 1–3 were formed (Fig. 11).
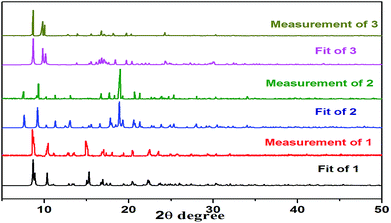 |
| | Fig. 11 XRD powder patterns of complexes 1–3. | |
3.3. Photophysical and electrochemical properties
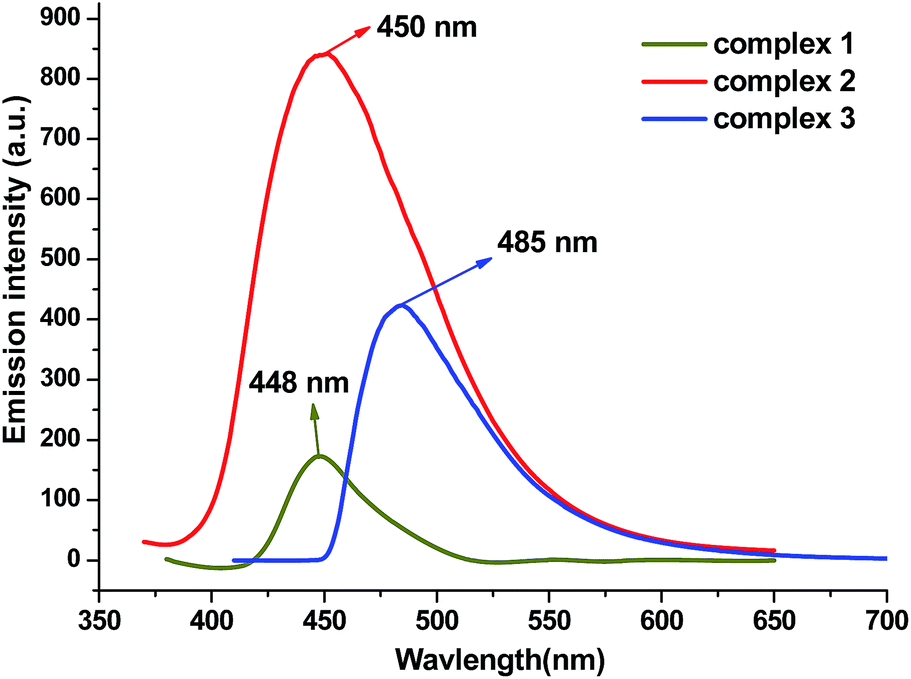
Metal–organic coordination complexes constructed from d10 metal atoms (or d10 metal clusters) and conjugated organic ligands are promising candidates for hybrid photoactive materials with potential applications such as light-emitting diodes (LEDs).21 The solid-state absorption spectra and luminescent emission spectra of 1, 2, 3 and free H2L ligand were studied at room temperature (Fig. 12 and ESI Fig. S1 and S2†). Both complexes and ligand exhibit absorption values in the 220–400 nm region. One robust emission peak at 457 nm with the excitation at 350 nm was observed from free H2L ligand, which is attributable to the π–π* transitions; similar emissions were also found from complex 1 and 2 (448 nm for 1 and 450 nm for 2), the closeness in the emission wavelength among 1, 2 and H2L suggests that the emissions of 1 and 2 are mainly ligand-centered electronic excitation.22,23 However the emission peak of 3 possesses a 28 nm red shift from the free ligand, and we tentatively assign this to the intra ligand (π–π*) fluorescence. The weakened luminescence emissions of the three complexes compared to the free ligand may be attributed to the flexibility of the H2L free ligand to the metal cluster, which effectively increases the flexibility of the metal–organic framework and the loss of energy by radiationless decay.24
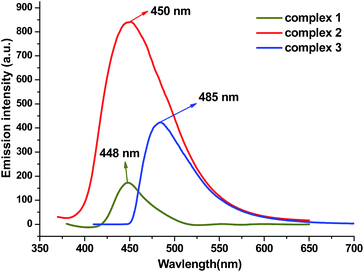 |
| | Fig. 12 Fluorescent emission spectra of 1, 2, 3 in the solid state at room temperature. | |
4. Conclusion
In conclusion, a versatile flexible semi-rigid V-shaped aza-bridged multicarboxylate ligand has been introduced to construct three new d10 metal complexes. Fine control over the synthetic conditions such as the choice of auxiliary ligand, solvent, and metal salt generated three metal complexes of 2,2′-azanediyldibenzoic acid which show structures ranging from one-dimensional to two dimensional framework. Five coordination modes of H2L ranging from μ2, μ3 to μ4 have been observed. The study reveals that the H2L ligand coordinates with the metal in a chelate mode and monodentate fashions to link three different metal centers resulting in two-dimensional structures in complex 1. Conversely, the ligand coordinates in a chelating fashion to link two different metal centers to generate one-dimensional structures in complex 2, and coordinates with the metal in chelate and monodentate modes to link four metal centers to offer one-dimensional structures in complex 3 (Fig. 4). This reveals that introduction of different ancillary ligands can mediate the coordination model of carboxylate groups and therefore afford structures with dimensional diversity. Moreover, complexes 1–3 exhibit solid-state fluorescence properties at room temperature.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant no. 21471070, 21461010, 21261009 and 21206062), Young Scientist Foundation of Jiangxi Province and Natural Science Foundation of Jiangxi Province (GJJ11453), Jiangxi Province Science and Technology Support Program (20131102040132).
References
- K. K. Bisht and E. Suresh, Inorg. Chem., 2012, 51, 9577 CrossRef CAS PubMed.
- H. L. Wang, K. Wang, D. F. Sun, Z.-H. Ni and J. Z. Jiang, CrystEngComm, 2011, 13, 273 Search PubMed.
- R. P. Davies, R. Less, P. D. Lickiss, K. Robertson and A. J. P. White, Cryst. Growth Des., 2010, 10, 4571 CAS.
- K. H. He, Y. W. Li, Y. Q. Chen, W. C. Song and X. H. Bu, Cryst. Growth Des., 2012, 12, 2730 CAS.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissogloua and G. Psomas, Dalton Trans., 2010, 4517 RSC.
- H. L. Wang, D. P. Zhang, D. F. Sun, Y. T. Chen, L. F. Zhang, L. J. Tian, J. Z. Jiang and Z. H. Ni, Cryst. Growth Des., 2009, 9, 5273 CAS.
- X. L. Zhang, M. F. Jin, Y. Qiu and F. Guo, J. Inorg. Organomet. Polym., 2011, 21, 498 CrossRef CAS.
- E. J. Gao, Y. Huang, M. C. Zhu, L. Wang, F. C. Liu, H. Y. Liu, S. Ma, Q. Z. Shi, N. Wang and C. Y. Shi, Inorg. Chem. Commun., 2009, 872 CrossRef CAS PubMed.
- J. E. Field and D. Venkataraman, Chem. Commun., 2002, 306 RSC.
- G. A. Farnum, A. L. Pochodylo and R. L. L. Duca, Cryst. Growth Des., 2011, 11, 678 CAS.
- X. L. Zhao, L. L. Zhang, H. Q. Ma, D. Sun, D. X. Wang, S. Y. Feng and D. F. Sun, RSC Adv., 2012, 2, 5543 RSC.
- L. F. Ma, J. H. Qin, L. Y. Wang and D. S. Li, RSC Adv., 2011, 1, 180 RSC.
- Y. Z. Tang, J. B. Xiong, Y. H. Tan, Y. Wang, Y. P. Deng, Q. Xu and H. R. Wen, Inorg. Chim. Acta, 2014, 410, 82 CrossRef CAS PubMed.
- S. Hazra, B. Sarkar, S. Naiya, M. G. B. Drew, A. Frontera, D. Escudero and A. Ghosh, Cryst. Growth Des., 2010, 10, 1677 CAS.
- S. Q. Zang, Y. Su, J. G. Lin, Y. Z. Li, S. Gao and Q. J. Meng, Inorg. Chim. Acta, 2009, 362, 2440 CrossRef CAS PubMed.
- Q. B. Bo, H. Y. Wang, J. L. Miao and D. Q. Wang, RSC Adv., 2012, 2, 11650 RSC.
- J. Sahu, M. Ahmad and P. K. Bharadwaj, Cryst. Growth Des., 2013, 13, 2618 CAS.
- A. Karmakar, H. M. Titi and I. Goldberg, Cryst. Growth Des., 2011, 11, 2621 CAS.
- A. K. Ghosh, D. Ghoshal, T. H. Lu, G. Mostafa and N. R. Chaudhuri, Cryst. Growth Des., 2004, 4, 851 CAS.
- P. V. Dau, M. Kim, S. J. Garibay, F. H. L. Munch, C. E. Moore and S. M. Cohen, Inorg. Chem., 2012, 51, 5671 CrossRef CAS PubMed.
- K. H. He, Y. W. Li, Y. Q. Chen, W. C. Song and X. H. Bu, Cryst. Growth Des., 2012, 12, 2730 CAS.
- D. Tian, Y. Li, R. Y. Chen, Z. Chang, G. Y. Wang and X. H. Bu, J. Mater. Chem. A, 2014, 2, 1465 CAS.
- K. K. Bisht and E. Suresh, Inorg. Chem., 2012, 51, 9577 CrossRef CAS PubMed.
- K. H. He, W. C. Song, Y. W. Li, Y. Q. Chen and X. H. Bu, Cryst. Growth Des., 2013, 12, 1064 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CIF files of the structures. CCDC 1048011–1048013. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra02417j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.