
Photocatalytic oxidative desulfurization of dibenzothiophene by C/TiO2@MCM-41 nanoparticles under visible light and mild conditions
Mahshid Zarrabia,
Mohammad H. Entezari*ab and
Elaheh K. Goharshadic
aSonochemical Research Center, Department of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, 91775, Mashhad, Iran. E-mail: moh_entezari@yahoo.com
bEnvironmental Research Center, Department of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, 91775, Mashhad, Iran
cNano Research Center, Department of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, 91775, Mashhad, Iran
First published on 9th April 2015
Abstract
Today, due to the environmental pressures on the sulfur content of gasoline and fuel cell applications, refineries need a very deep desulfurization process to reach the ultra-low sulfur diesel (ULSD, 1 ppm). In this research, dibenzothiophene (DBT) in n-octane solution (as a diesel fuel model) was completely converted into biphenyl by visible light irradiation in the presence of C/TiO2@MCM-41 (CTM-41) as a photocatalyst under mild conditions. For the first time, the conversion was carried out through oxidation in one phase without using oxidant. For synthesizing CTM-41, tetra-butylorthotitanate was used as a precursor of Ti. Carbon species were identified in CTM-41 by X-ray photoelectron spectroscopy and UV-Vis spectroscopy. The self-doped carbon in CTM-41 was attributed to the tetra-butylorthotitanate used as a precursor of Ti. In the synthesis process, there was no carbon precursor addition. The formation of two types of carbon (carbonate species and coke-like structure in lattice of TiO2) is responsible for the visible photocatalytic activity of the catalyst. The samples were characterized by X-ray diffraction, FTIR and UV-Vis spectroscopy. The specific surface areas of the prepared catalysts determined by the nitrogen adsorption–desorption isotherm at 77 K were 922.42 m2 g−1 and 767.54 m2 g−1. The GC-MS analysis confirmed the conversion of DBT to biphenyl. Based on the experimental results, the hole and hydroxyl radical as reactive species in the proposed mechanism played the key roles in the photocatalytic desulfurization process.
1. Introduction
Ultra deep desulfurization of fuels has attracted a great deal of attention. This is not only for legislation enforcing environmentally friendly transportation fuel but also the severe requirement of liquid hydrocarbon fuels with sulfur content of less than 1 ppmw for fuel cell applications.1,2 Currently, the sulfur removal from various liquid hydrocarbon streams is conducted by the catalytic hydrodesulfurization (HDS) process at 300–400 °C and 3–6 MPa hydrogen pressure with high hydrogen consumption in refineries. Although HDS process is a conventional technology for desulfurization of liquid fuels, it cannot efficiently eliminate refractory compounds such as alkylthiophenes, benzothiophene (BT), dibenzothiophene (DBT), and its derivatives.1,3–5 Oxidative desulfurization (ODS) is considered to be one of the promising new methods for deep desulfurization of fuels. The photocatalytic ODS is attracting interest, because of its low cost and mild operation of conditions.6–10Photocatalysis has emerged as a promising technology for the degradation of organic pollutants. TiO2 is the most widely used photocatalyst in various applications due to its high photocatalytic activity, lower cost, nontoxic nature, chemical inertness, and simplicity but it has some disadvantages.1,11–13 However, TiO2 can agglomerate very easily during the synthesis process which has adverse effects on the catalytic efficiency. In addition, in the bare TiO2 nanoparticles a rapid recombination can be occurred for the photogenerated electron–hole pairs. These disadvantages limit wide spread use of the TiO2 for various applications. Uniform ordered channels of mesoporous materials such as MCM-41 can control the particle size of TiO2 and efficiently prevent particles from agglomeration.
In general, the photocatalytic activity of TiO2@MCM-41 is higher than TiO2. This is due to long-lived photo induced charge separation, large pore size, and high specific surface of MCM-41 which has the ability to adsorb some bulky reactants. The introduction of transition metals into MCM-41 increases the active sites of the photocatalyst.14–16
Another disadvantage of TiO2 is exhibiting low efficiency under visible light because it has large band gap energy of 3.2 eV, corresponding to a threshold wavelength of 388 nm. Much study was performed by UV light irradiation because TiO2 can only absorb UV light. In general the photocatalysts with narrow band gaps are suitable for desulfurization under visible light irradiation.
The doping of nonmetallic elements has been regarded as a promising strategy for improving visible light activity of TiO2-based photocatalysts.17,18 Among such elements, carbon, sulfur, and nitrogen atoms have been shown to effectively enhance the visible light activity of TiO2. These nonmetallic atoms can substitute in the lattice structure of TiO2 causing substantial improvements in the visible light activity as compared to that of pure TiO2.19–21
In this work, dibenzothiophene was successfully degraded at low temperature and atmospheric pressure by CTM-41 under visible light irradiation. For the first time, the photocatalytic degradation was carried out without adding an oxidant. It was confirmed that the presence of carbon in the synthesized photocatalyst has a major role in the degradation. In addition, a mechanism was proposed for the degradation of DBT.
2. Experimental
2.1. Materials
Tetra-butylorthotitanate (98%), ethanol (99.5%), dimethyl sulfoxide (DMSO, 99%), ammonium oxalate (AO, 99.5%) and hexane were supplied by Merck. Dibenzothiophene (DBT), titanium tetrachloride (TiCl4, 99.9%) and p-benzoquinone (BQ) from Sigma-Aldrich, n-octane (95%) from Samchun, MCM-41 from Gas Chemical Company of China, and de-ionized water were used for washing and sample preparation.2.2. Preparation of the catalyst
The mixture was stirred for 30 min at room temperature. Then, a certain amount of TBOT was added. The mixture was stirred and refluxed at 343 K for 20 h under nitrogen atmosphere. The solid was separated from the mixture using centrifugation at rate of 5000 rpm. The product was washed four times with dry ethanol. The solid was then hydrolyzed in a beaker with 150 mL of deionized water. The mixture was stirred for another 4 h. Then, the product was washed with deionized water, centrifuged at rate of 5000 rpm, and finally air dried at 373 K. The white powder as modified sample was obtained after calcination at 773 K for 2 h.22
2.3. Characterization techniques
The X-ray diffraction (XRD) patterns (2θ = 10–80°) were recorded by Bruker-AXS, D8 Advance model at a scanning rate of 0.04° s−1. The wide angle and small angle X-ray diffraction were performed with monochromatized Cu-Kα radiation (λ = 1.5406 Å) and Co-Kα radiation (λ = 1.7890 Å), respectively. The Fourier-transformed infrared spectra (FT-IR, Avatar 370) using the KBr pellet method was recorded on a Thermo-Nicolet spectrometer. The optical absorption spectrum of the DBT was measured using UV-Vis spectroscopy (Unico 2800). The X-ray photoelectron spectroscopy (XPS) was performed using an ESCA PHI 5000 instrument and Al-Kα irradiation (1486.6 eV) to determine the chemical states of Ti, S, O, and C atoms in the prepared samples. The N2 adsorption isotherms of samples were measured at 77 K on a Bel Japan apparatus (Belsorp mini II model) to calculate BET specific surface areas, pore volumes, and pore sizes of the powder samples. The structural of the samples were determined with transmission electron microscope (TEM, Philips CM-120). The GC (Agilent 6890N) and MS (Agilent 5975B) were also used for the determination of the by products produced during the degradation process.2.4. Photocatalytic degradation of DBT
The degradation was performed under constant stirring in a 50 mL glass vessel containing 10 mL of n-octane solution of DBT (300 ppm) as a fuel model and 0.015 g CTM-41 as a catalyst. Before illumination, the suspension was magnetically stirred for 45 min to attain adsorption–desorption equilibrium between photocatalyst and DBT in the dark. It was then illuminated with two 300 W tungsten lamps (visible irradiation) at 333 K for 5 h. The absorption of DBT was monitored with a UV-Vis spectrophotometer at λmax = 325 nm. The strong absorbance at 325 nm suggests an n–π* excitation similar to that for the sulfur atom in DBT.23 The absorption was converted to the concentration through the standard curve. The degradation of DBT was calculated according to the initial and final concentration of DBT in the solution:where x is the percent of degradation, C0 and C (mg L−1) stand for the initial and final concentration of DBT, respectively. For approving the degradation of DBT, the liquid phase was separated from the catalyst by centrifuging. Then, the organic phase and catalyst were extracted for GC-MS analyses.

3. Results and discussion
3.1. Characterization of the catalyst
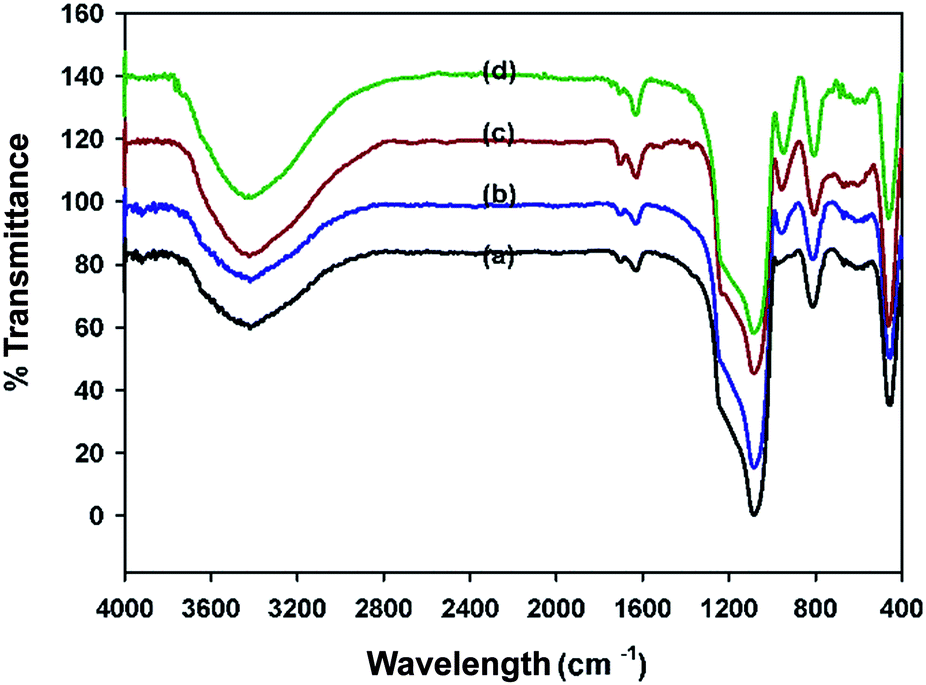 | ||
| Fig. 1 FT-IR spectra: (a) MCM-41, (b) CTM-41 (Si/Ti = 50, TBOT), (c) CTM-41 (Si/Ti = 5, TBOT), (d) CTM-41 (Si/Ti = 5, TiCl4). | ||
 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of (a) MCM-41, (b) CTM-41 (Si/Ti = 50), (c) CTM-41 (Si/Ti = 5). | ||
The first adsorption at low pressure (P/P° < 0.01) was caused by monolayer adsorption of N2 on the walls of the mesopores. Upon monolayer adsorption, multilayer of nitrogen starts to develop at higher relative pressure of nitrogen (P/P° = 0.01–0.34). At relative pressure between 0.34 and 0.40, a sharp inflection due to capillary condensation with mesopores is observed. The sharpness of the step indicates a uniform pore size distribution. The sharpness of the step diminishes with titania loading which implies that the highly mesoporous structure was decreased but the structural regularity is not significantly affected. A summary of results obtained from N2 adsorption–desorption isotherms of MCM-41 and CTM-41 with two different Si/Ti molar ratios are given in Table 1. The increase of the metal content leads to a gradual reduction in pore volume and total surface area. It may be due to the formation of large TiO2 nanoparticles but decreasing of titanium incorporated in the pore wall (Si–O–Ti) from CTM-41 (Si/Ti = 50) to CTM-41 (Si/Ti = 5) causes the increasing pore diameter from CTM-41 (Si/Ti = 50) to CTM-41 (Si/Ti = 5).
| D (nm) | SBET (m2 g−1) | VT (cm3 g−1) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a D (nm): average pore diameter, SBET (m2 g−1): surface area, V (cm3 g−1): total pore volume. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MCM-41 | 3.63 | 1076.50 | 0.98 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTM-41 (Si/Ti = 50) | 3.42 | 922.42 | 0.79 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTM-41 (Si/Ti = 5) | 3.55 | 767.54 | 0.68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.1.3.1. Small angles. The small angle XRD (SAXRD) profiles of MCM-41 and CTM-41 with two different Ti loadings are presented in Fig. 3. It is shown that MCM-41 pattern has a strong peak at 2θ = 2.55° and two weak peaks at 2θ = 4.43°, 5.13° assignable to the crystallographic planes of (100), (110), and (200), respectively. This pattern proves the high order of the pores in the MCM-41 mesoporous structure.
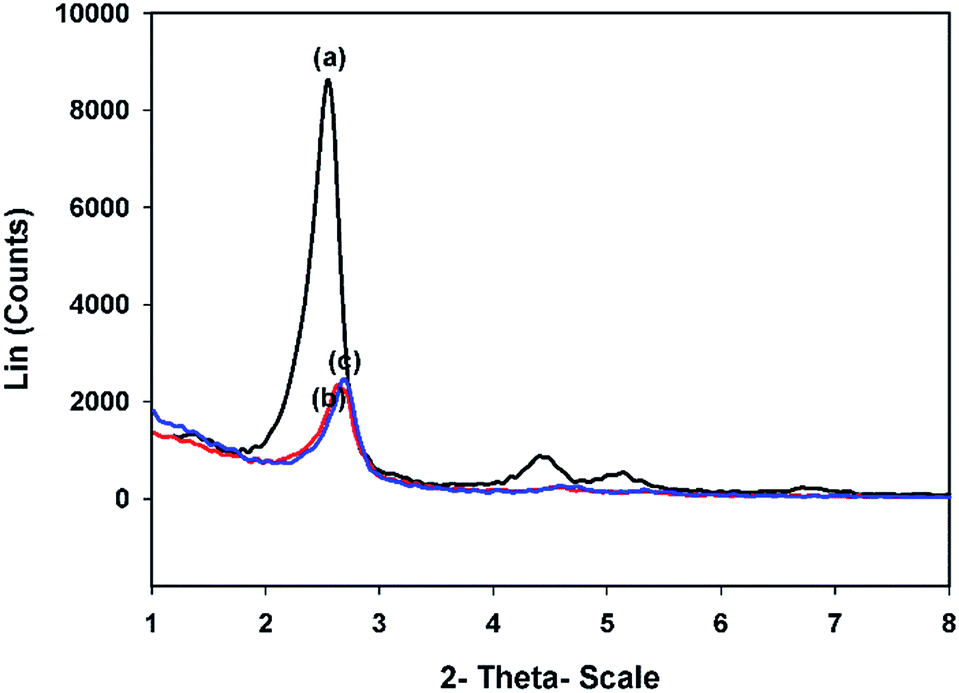 | ||
| Fig. 3 SAXRD patterns of (a) MCM-41, (b) CTM-41 (Si/Ti = 50), (c) CTM-41 (Si/Ti = 5). | ||
The SAXRD data, wall thickness (W), unit cell parameter (a0), and micro-strain (ε) are listed in Table 2. When the Ti content increased, a decrease in the intensity of the main diffraction peak (100 peak) was observed. This is due to the scattering contrast decreases between the pores and pore walls. The CTM-41 samples show strong d100 reflection peak which suggests a well order nanostructure of MCM-41 which was retained even after the loading of titania. As the Ti content increased, a small shift in the position of main peak to the higher angles was observed. This is due to the contraction of mesoporous framework which may be correlated with the calcination of samples. Therefore, the interplanar distance (d100) and the hexagonal unit cell parameter (a0) of the samples decreased with increasing of Ti content in the samples. The wall thickness (W) of the hexagonal arranged pore was calculated from W = a0 − D. The results showed that the increase of wall thickness in CTM-41 (0.02) can be attributed to the Ti incorporation in the wall of the pore but the decrease of wall thickness in CTM-41 (0.2) may be correlated to the crystalline titania which was not incorporated in the wall. The broadening of XRD peaks (Δβ = 0.03°) are observed for Ti doped CTM-41 (0.02). The small changes in the broadening of the peak can be due to the increase of micro-strain. The small changes are attributed to the incorporation of Ti into the Si–OH bands on the surface of the pore. The FWHM (β) value of CTM-41 (0.2) shows the inverse trend which indicates formation of TiO2 (anatase) phase. The micro-strain (ε) was calculated according to the formula of micro-strain which is: ε = (β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ)/4.
cosθ)/4.
| 2θ (°) | d100 (nm) | a0 (nm) | W (nm) | FWHM (β) | ε (10−3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a d100 (nm): interplanar distance, a0 (nm): hexagonal unit cell parameter, calculated from a0 = 2d100/31/2, W (nm): wall thickness, calculated using W = a0 − D. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MCM-41 | 2.55 | 4.110 | 4.640 | 1.007 | 0.287 | 1.249 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTM-41 (50) | 2.63 | 3.949 | 4.508 | 1.090 | 0.290 | 1.265 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTM-41 (5) | 2.67 | 3.877 | 4.434 | 0.886 | 0.272 | 1.181 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.1.3.2. Large angles. Fig. 4 shows the wide angle X-ray diffraction patterns. The XRD pattern of the pure MCM-41 shows a broad peak between 2θ = 15° and 30° which confirms the amorphous silica structure.25 After incorporating of TiO2 into the MCM-41, a decrease in the intensity of the maximum peak was observed. The existence of broad peak indicates that the composites are still amorphous. There are no XRD peaks for the TiO2 in the CTM-41 (Si/Ti = 50) composite which are related to the condensation of Ti with pendant Si–OH groups on the pore surface and generating tetrahedral Ti species instead of TiO2 as a secondary phase segregation. In addition, the sizes of formed TiO2 may be very small to detect. Meanwhile, for CTM-41 (Si/Ti = 5), additional peaks were appeared at 2θ = 25.3240, 37.9860, 48.0520, 54.7630, and 62.6760. These peaks are attributed to the presence of anatase phase of TiO2 as secondary phase segregation. The average crystal size of the anatase phase for CTM-41 (Si/Ti = 5) was determined by the Scherrer formula and it was 6.89 nm. The emergence of new peaks in the wide angle and the inverse trend of the FWHM value for CTM-41 (Si/Ti = 5) are reasons for entity anatase phase. The shift of position of the maximum intensity to a higher value is probably resulted from the slight structural change as the micro-strain increases.
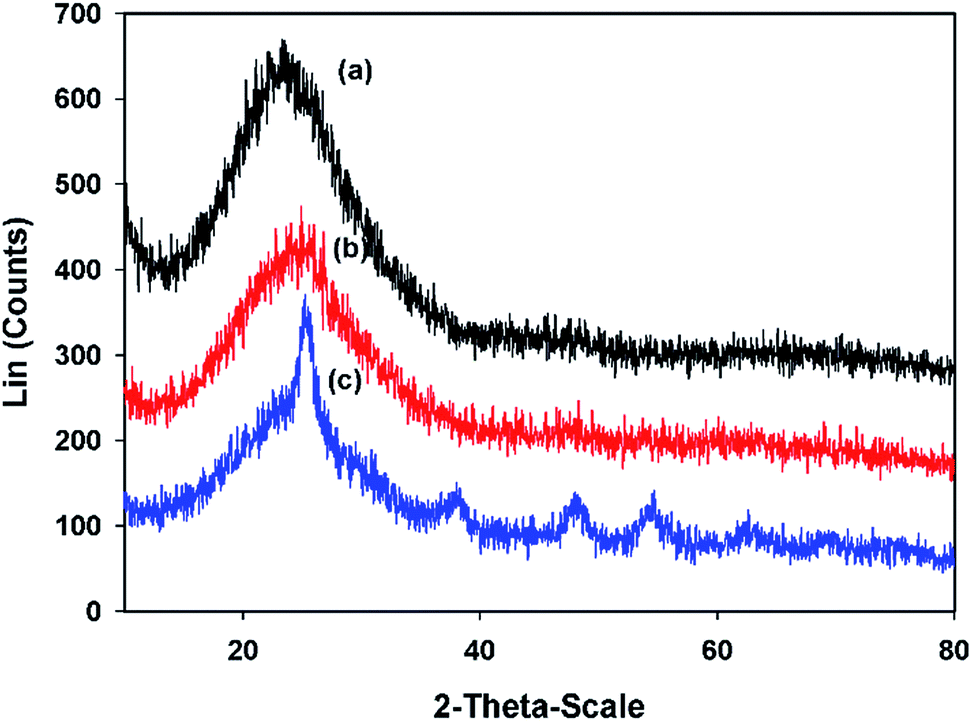 | ||
| Fig. 4 XRD wide-angle patterns of (a) MCM-41, (b) CTM-41 (Si/Ti = 5), (c) CTM-41 (Si/Ti = 50). | ||
 | ||
| Fig. 5 TEM image and the selected-area diffraction patterns of CTM-41 (Si/Ti = 5). | ||
The long and straight mesopores of MCM-41 support is conserved and unaffected by the presence of titania.
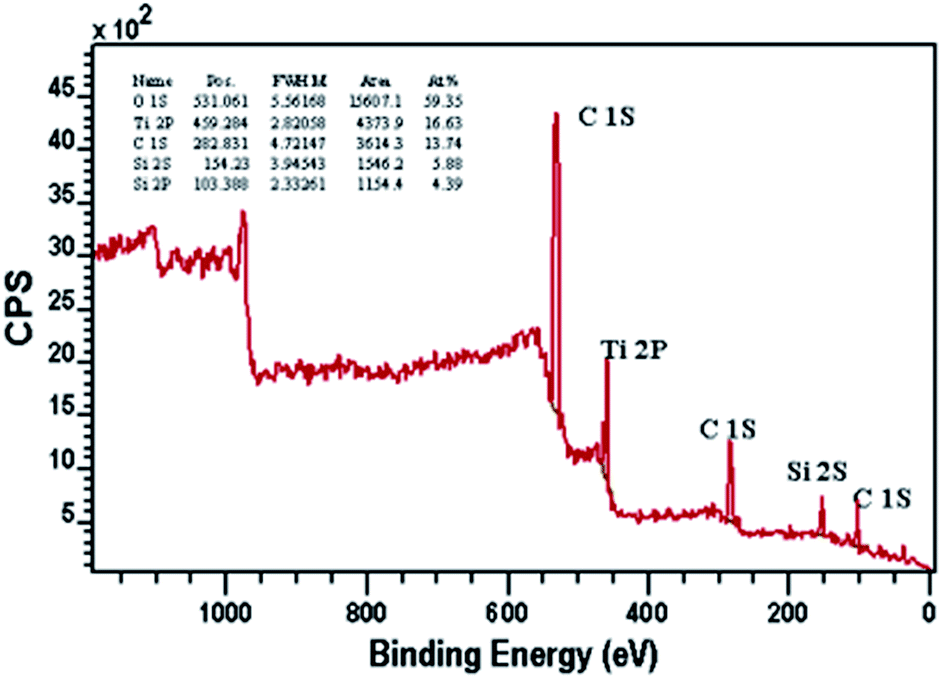 | ||
| Fig. 6 The XPS survey spectrum of CTM-41 (Si/Ti = 5). | ||
Fig. 7 shows the XPS peaks of the carbon-doped in the composite. This is due to the diffusion of the carbon atoms during the synthesis process. Alkoxide residues after hydrolysis may result in carbon entering. Therefore, it was suggested that the peak at 283.7 eV is originated from Ti–alkoxide derived C–C species which is different from adventitious carbon species. These carbon residues may form highly condense and coke-like structure around TiO2 nanoparticles and play the role of a sensitizer like organic dye molecules.26
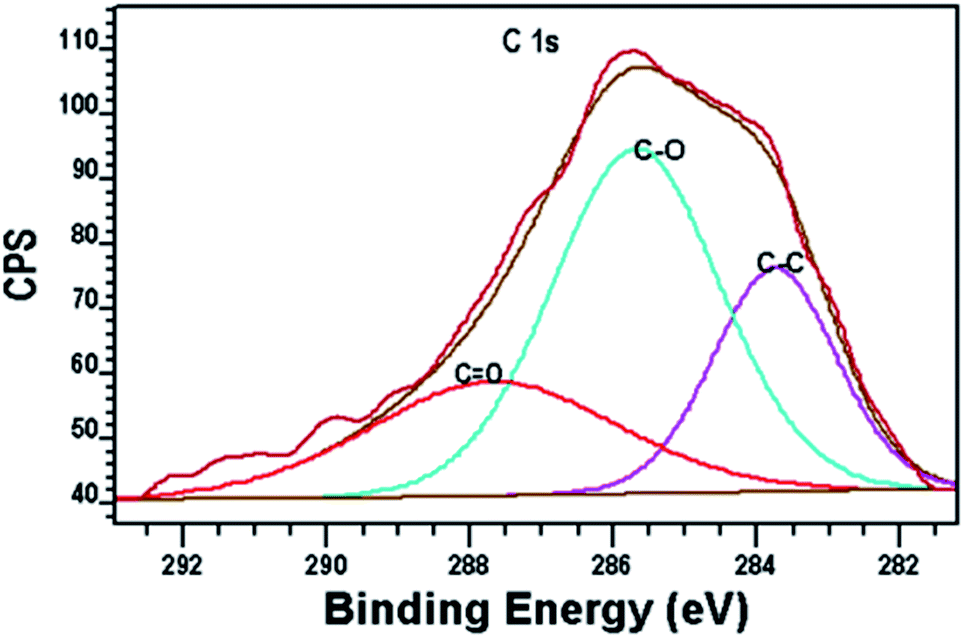 | ||
| Fig. 7 The XPS spectrum of the C. | ||
A peak around 281 eV resulting from Ti–C bond is not observed, so carbon is not substituted by oxygen atom in the anatase lattice.31
Other two peaks around 286 and 288 eV are attributed to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively which show the incorporation of carbon atoms into the interstitial positions of the TiO2 lattice. The formation of carbonate ions or carbon atoms into the lattice of TiO2 can be occurred by replacing titanium atoms in the form of Ti–O–C.27–30 A peak around 281 eV resulting from Ti–C bond is not observed, so carbon is not substituted by oxygen atom in the anatase lattice.31
O, respectively which show the incorporation of carbon atoms into the interstitial positions of the TiO2 lattice. The formation of carbonate ions or carbon atoms into the lattice of TiO2 can be occurred by replacing titanium atoms in the form of Ti–O–C.27–30 A peak around 281 eV resulting from Ti–C bond is not observed, so carbon is not substituted by oxygen atom in the anatase lattice.31
Fig. 8 shows the XPS spectrum of Ti 2p region. The Ti 2p signals split into two peaks, which attributed to the Ti 2p3/2/Ti 2p1/2 binding energy region. The peak Ti p3/2/Ti 2p1/2 for TiO2 in the CTM-41 sample appears at 460.3/465.5 eV. In comparison to the binding energy of TiO2 anatase (458.8/464.5 eV),25,32 there is a slight red-shift about (1.5/1 eV) for TiO2 in the CTM-41 sample. This shows that Ti3+ species are formed in the carbon-doped titania which indicates presence of carbonate ions because of interstitial doping of carbon in lattice.16,27,33 The peak at 460.7/466.7 eV is consistent with silica, where cross-linking Ti–O–Si bonds. The O 1s spectra are shown in Fig. 9. The low binding energy near 530 eV is ascribed to anatase (Ti–O–Ti). The band near 532.2 eV is due to Si–O–Ti and terminal TiOH. The peak at 534.9 eV is attributed to SiO2, H2O and bridged TiOH. The binding energy at 536.2 eV is assigned to be Si–OH. The XPS results for the grafted sample CTM-41 (Si/Ti = 5) are summarized in Table 3.
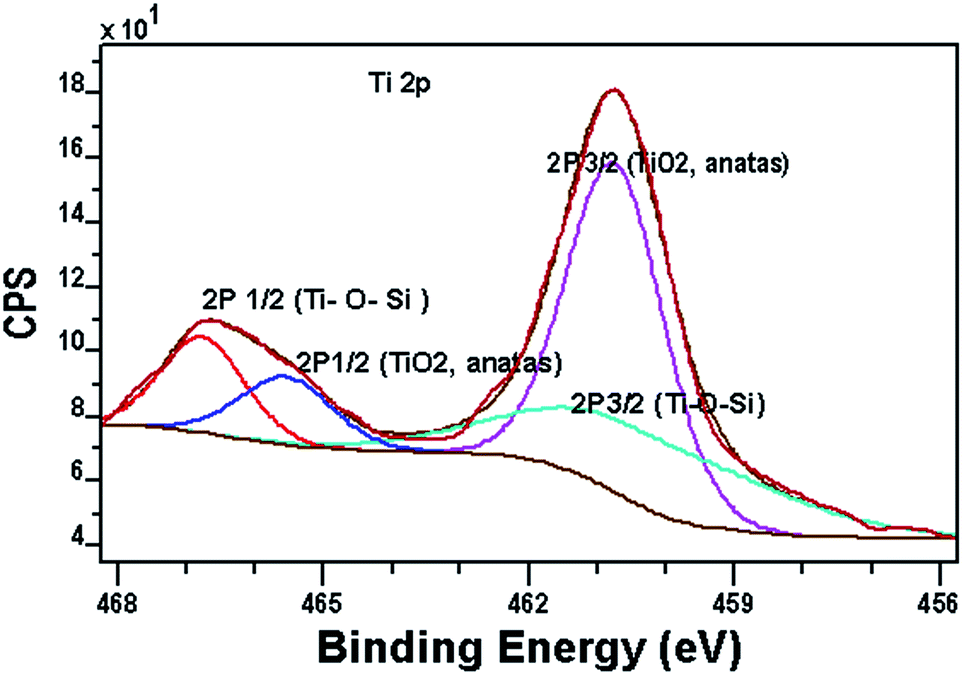 | ||
| Fig. 8 The XPS spectrum of the Ti 2p. | ||
 | ||
| Fig. 9 The XPS spectrum of the O 1s. | ||
| Element/orbital | Binding energy (eV) | Atomic content (%) | Assignment |
|---|---|---|---|
| Si 2p | 105.4 | 9.73 | SiO2 |
| Si 2s | 156.2 | 13.1 | |
| O 1s | 530.0 | 0.83 | TiO2 |
| 532.2 | 17.43 | Si–O–Ti | |
| 534.9 | 13.76 | SiO2, (TiOH) | |
| 536.2 | 1.48 | Si–OH | |
| Ti 2p3/2/Ti 2p1/2 | 460.7/466.7 | 12.02 | Ti–O–Si |
| 460.3/465.5 | 7.64 | TiO2 (anatase) | |
| C 1s | 283.7 | 5.83 | O–Ti–C |
| 285.7 | 12.2 | (CC, C–C) |
|
| 287.6 | 5.97 | CO32− |
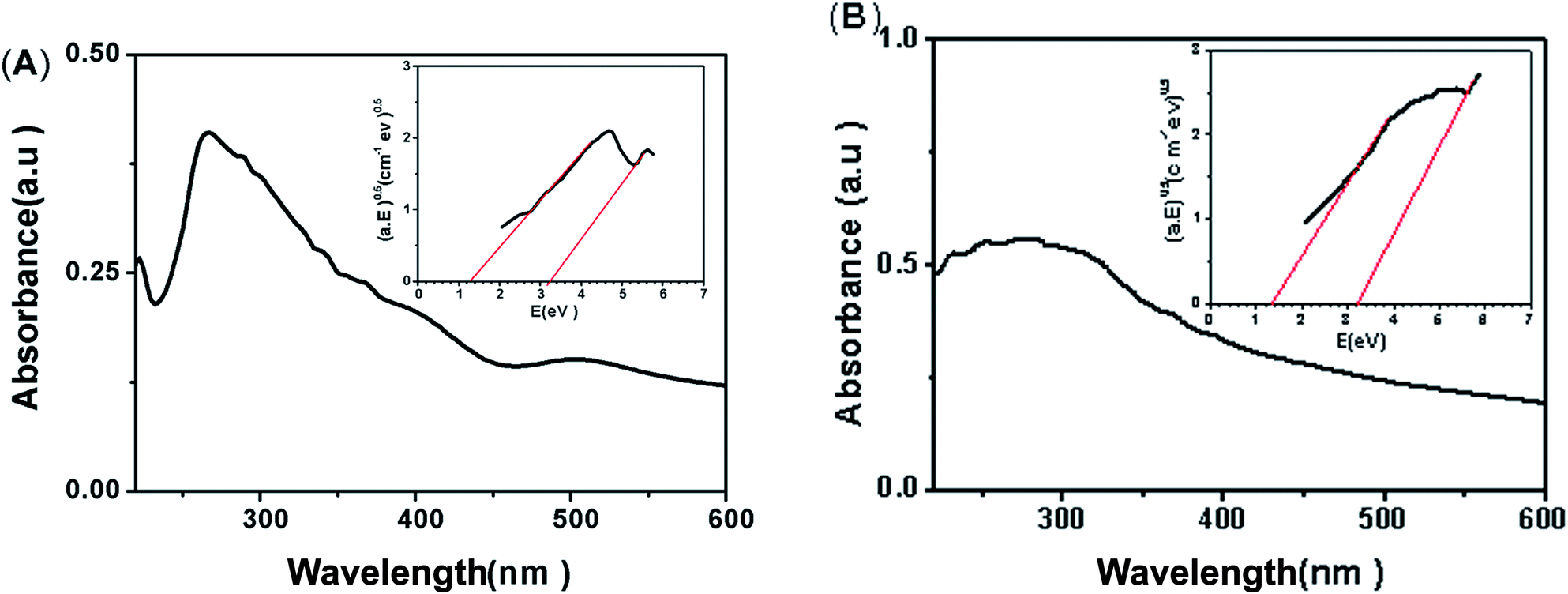 | ||
| Fig. 10 Absorption spectra and Tauc's plots for obtaining the band gap (A) CTM-41 (Si/Ti = 50), (B) CTM-41 (Si/Ti = 5). | ||
Palanivelu et al. reported that the band gap narrowing is due to the carbon impurity states which are attributed to substitutional C atoms replacing O atoms (Ti–C bonds) or interstitial C atoms (carbonate ions).27,35–38 As the XPS spectra show, carbon is not substituted by oxygen atom in the lattice of anatase (absent of Ti–C bonds at 281 eV); therefore, the band gap narrowing for the CTM-41 can be explained by the isolated C 2p states of ions in the band gap of TiO2.12,37 Then, the carbonate values of 1.3 eV and 3.2 eV represent the band gap energies of the C/TiO2 between valence and interbands and between the valence and conduction bands, respectively.39–41 Previous reports show that the formation of carbonate species could cause an obvious long-tail absorption in the visible region.42,43 Therefore, existence of long-tail absorption in UV-Vis spectrum of the samples also confirms the presence of carbonate ions.43
Further analysis of the UV-Vis absorption spectrum indicates that CTM-41 (Si/Ti = 5) has a stronger visible light absorption from 410 to 600 nm than that of observed for CTM-41 (Si/Ti = 50). On the other hand, the visible light absorption is improved with the increase of carbon content (see Table 4).
| Sample | % C | % H |
|---|---|---|
| CTM-41 with TiCl4 precursor (5) | 0.08 | 0.60 |
| CTM-41 with TBOT precursor (5) | 1.80 | 0.88 |
| CTM-41 with TBOT precursor (50) | 0.53 | 0.65 |
3.2. Mechanism of carbon formation in CTM-41
Based on the XPS analysis of CTM-41, the peak at 283.7 eV shows the alkoxide residues after hydrolysis. Therefore, it is suggested that Ti(OC4H9)4 is hydrolyzed by H2O. Butyl alcohol (BuOH) and Ti(OH)4 were formed. Then, dibutyltitanate was decomposed by calcination at 500 °C (Fig. 11).44 Furthermore, in order to confirm that TBOT acts as a precursor of carbon, the CHN elemental analysis on CTM-41 samples with TiCl4 and TBOT as precursors were performed. The CTM-41 samples were synthesized with TiCl4 and TBOT as precursors by the similar procedure (see Section 2.2.1 and 2.2.2).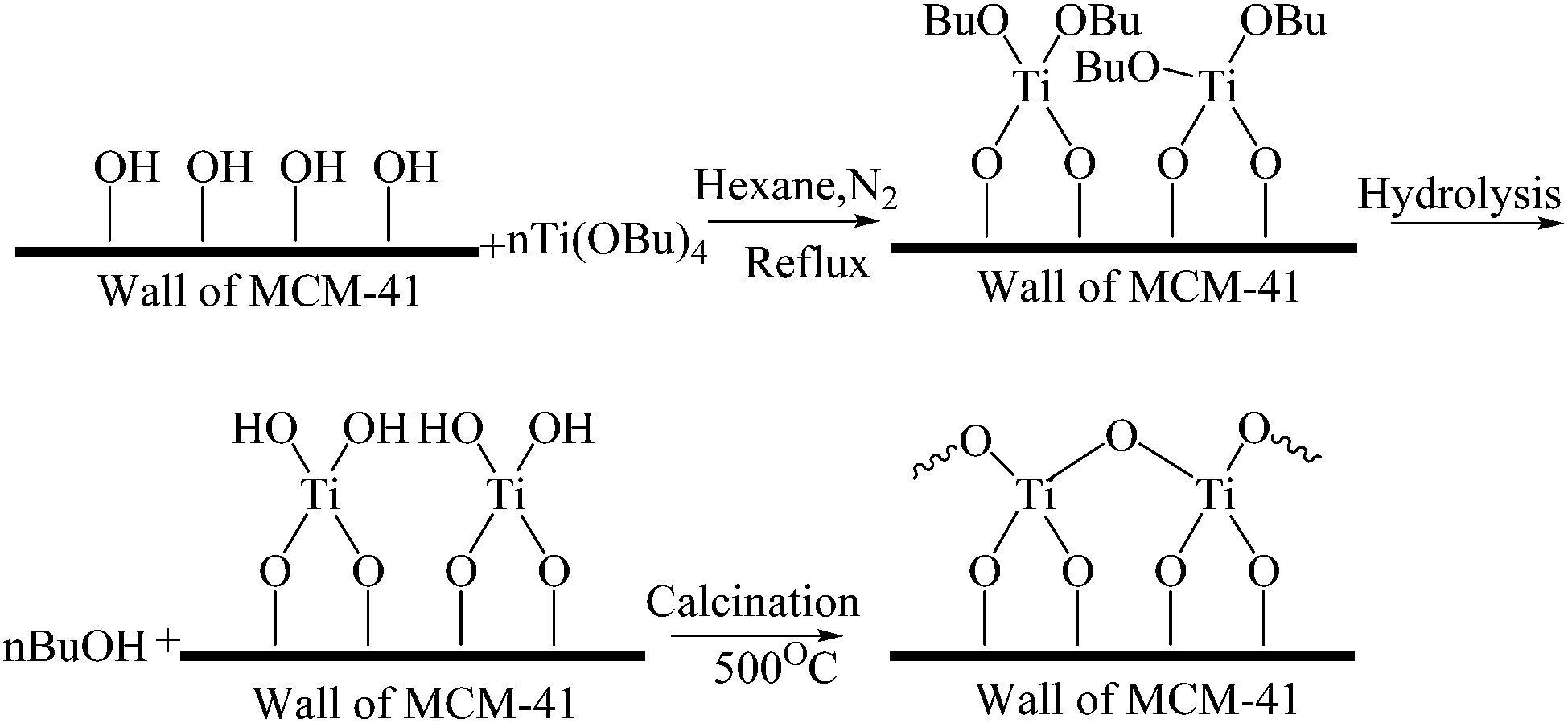 | ||
| Fig. 11 Schematic reaction between MCM-41 and TBOT. | ||
Table 4 shows that the catalyst prepared by TiCl4 contains 0.08% carbon, while the sample prepared by TBOT contains 0.53% and 1.8% carbon in the catalyst with the ratio of Si/Ti = 50 and Si/Ti = 5, respectively.
The results indicate that carbon can be self-doped when butyl alcohol (BuOH) is formed by the hydrolysis of titanium dibutyltitanate and decomposition by calcination.
3.3. Photocatalytic degradation of DBT by CTM-41
The catalytic performance of CTM-41 was investigated in dark and under visible irradiation with two ratios of Ti loading (Fig. 12).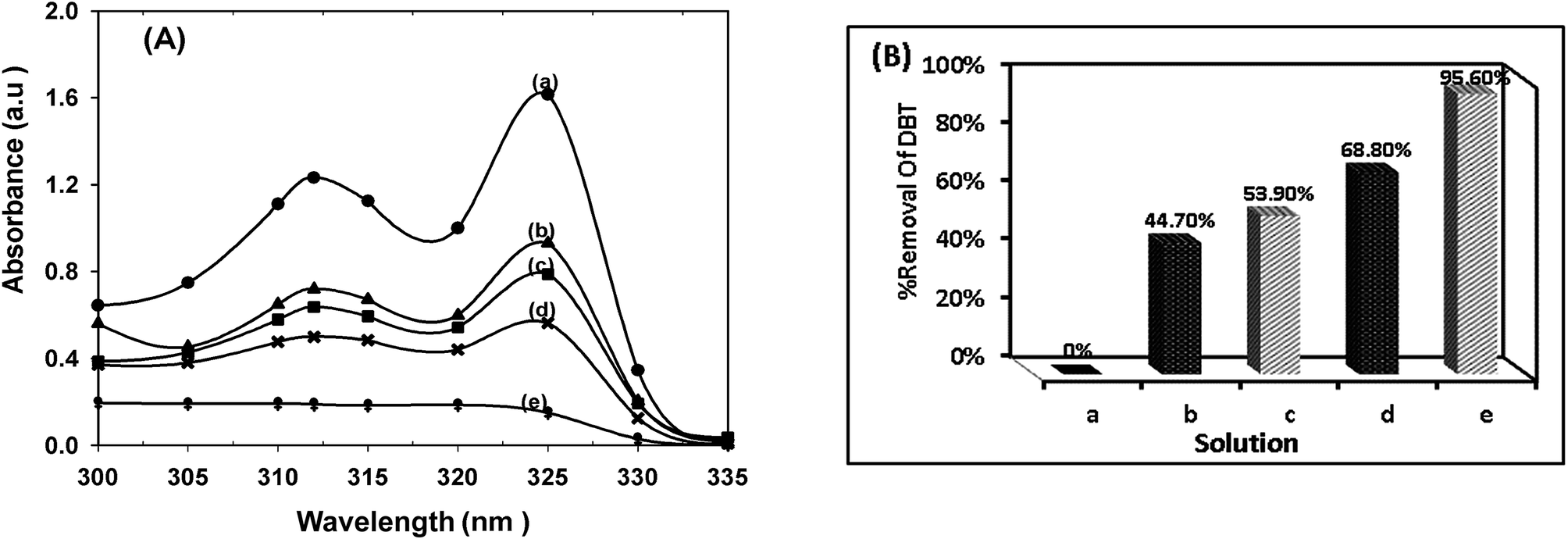 | ||
| Fig. 12 (A) UV-Vis absorption spectra. (B) Percentage of DBT removal (a) standard solution of DBT (300 ppm), (b) DBT solution in the presence of CTM-41 (Si/Ti = 50) in dark for 45 min, (c) DBT solution in the presence of CTM-41 (Si/Ti = 5) in dark for 45 min, (d) DBT solution in the presence of CTM-41 (Si/Ti = 50) in dark for 45 min and light for 5 h, (e) DBT solution in the presence of CTM-41 (Si/Ti = 5) in dark for 45 min and light for 5 h, condition: under two lamps (300 W) irradiation, 10 mL DBT (300 ppm), 0.015 g catalyst, 60 °C. | ||
With increasing Ti loading, the removal of DBT was increased in dark and light too. The removal of DBT in dark was proceeded via adsorption which is attributed to the mesoporosity of CTM-41 and Lewis acid sites of tetrahedral Ti species in the framework of the catalyst. Increasing Ti loading leads to an increase in acid sites.15,37,45 As shown in Fig. 12, the removal of DBT was 53.9% by the catalyst with lower ratio (Si/Ti = 5) and 44.7% with higher ratio (Si/Ti = 50) in dark. These results represent that the Lewis acid sites of tetrahedral Ti species for the adsorption are more important than pore volume and surface area of the catalyst. The removal of DBT under visible light can be proceeded in the presence of the carbon.
In other words, the degradation of dibenzothiophene depends on the amount of carbon and Si/Ti ratio. The amount of carbon was also increased with the ratio of Si/Ti (see Table 4).
3.4. Photocatalytic desulfurization mechanism
Some experiments were designed to elucidate the degradation mechanism. At first, one experiment was carried out in DBT solution without photocatalyst. The results showed that the absorption does not change with increasing the irradiation time. It means that DBT is stable in the presence of light in the absence of photocatalyst.Also, some experiments were performed using different radical scavengers39
As shown in Fig. 13, when the dimethyl sulfoxide (DMSO) was added as a scavenger for hydroxyl radicals, desulfurization of DBT was remarkably inhibited (reduced from 95.6% to 12.7%).
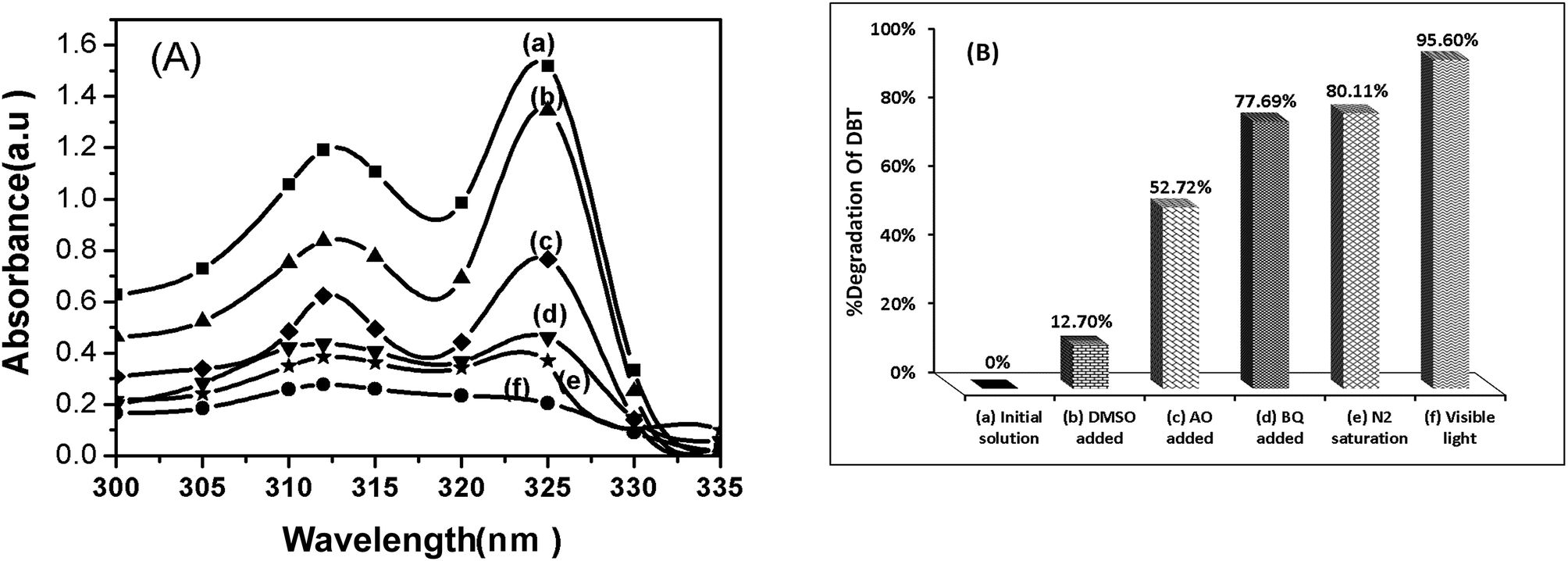 | ||
| Fig. 13 The effect of radical scavengers on the photocatalytic degradation of DBT (A) UV-Vis absorption spectra. (B) Percentage of DBT degradation (a) standard solution of DBT (300 ppm), (b) with DMSO, (c) with AO, (d) with BQ, (f) without scavenger, condition: 0.015 g CTM-41 (Si/Ti = 5) under visible light irradiation for 5 h. | ||
This behaviour was attributed to the reaction of DMSO with hydroxyl radicals and reducing its concentration. When p-benzoquinone (BQ) was added for the superoxide anion radicals, desulfurization of DBT was reduced from 95.6% to 77.6%. This implies that the superoxide anion radicals (O2−˙) play minor roles in the degradation of DBT.
Also, the usage of inert N2-saturated atmosphere causes a low desulfurization. Addition of ammonium oxalate (AO) as a scavenger for holes reduced desulfurization from 95.6% to 52.7%. Therefore, hydroxyl radicals (OH˙) and holes (h+) are the primary oxidants in our photocatalytic oxidation. The energy diagram for CTM-41 presents the proposed mechanism. This is based on the formation of impurity level (C 2p level of isolated carbonate ions) above the valence band of TiO2 and the role of photosensitizer of coke-like species.
Under visible light irradiation, the electrons excitation from the valence to impurity level (C 2p level) and from impurity level to conduction band led to the generation of electron–hole pairs. The coke-like species also play the role of photosensitizer (P) like organic dyes which can be excited and injected electrons into the conduction band of TiO2. Then, the rate of electron transfer to the adsorbed oxygen onto surface of TiO2 nanoparticles increased. The electrons can be trapped by the isolated tetrahedrally coordinated Ti-oxide species (Ti4+) dispersed in MCM-41 to generate Ti3+ ion. In the presence of O2, the Ti3+ sites easily react with O2 and form O2−˙.36
| e− + Ti4+OH → Ti3+OH |
| Ti3+OH + O2 → Ti4+OH + O2−˙ |
The O2−˙ can generate hydroxyl radicals (OH˙) via reaction with H+.
| O2−˙ + H+ → HO˙2 |
| HO˙2 + H+ + e− → H2O2 |
| H2O2 + e− → OH˙ + OH− |
The suggested sources of H+ are:
(1) The surface's hydroxyl groups of catalyst can act as an electron donor for photo-generated H+.
(2) The hydroxyl groups of absorbed water vapor in the air on the surface of TiO2@MCM-41.36
The photo-generated holes in C 2p states could not thermodynamically oxidize water or hydroxyl on catalyst surface. This is due to the lower potential of holes in the C 2p states (1.3 eV) than the standard redox potential of OH˙/H2O (2.7 eV) as shown in Scheme 1. Therefore, the holes generated in the C 2p states acted as an oxidizing sites only for the adsorbed DBT,46 not for the water or hydroxyl on the catalyst surface.
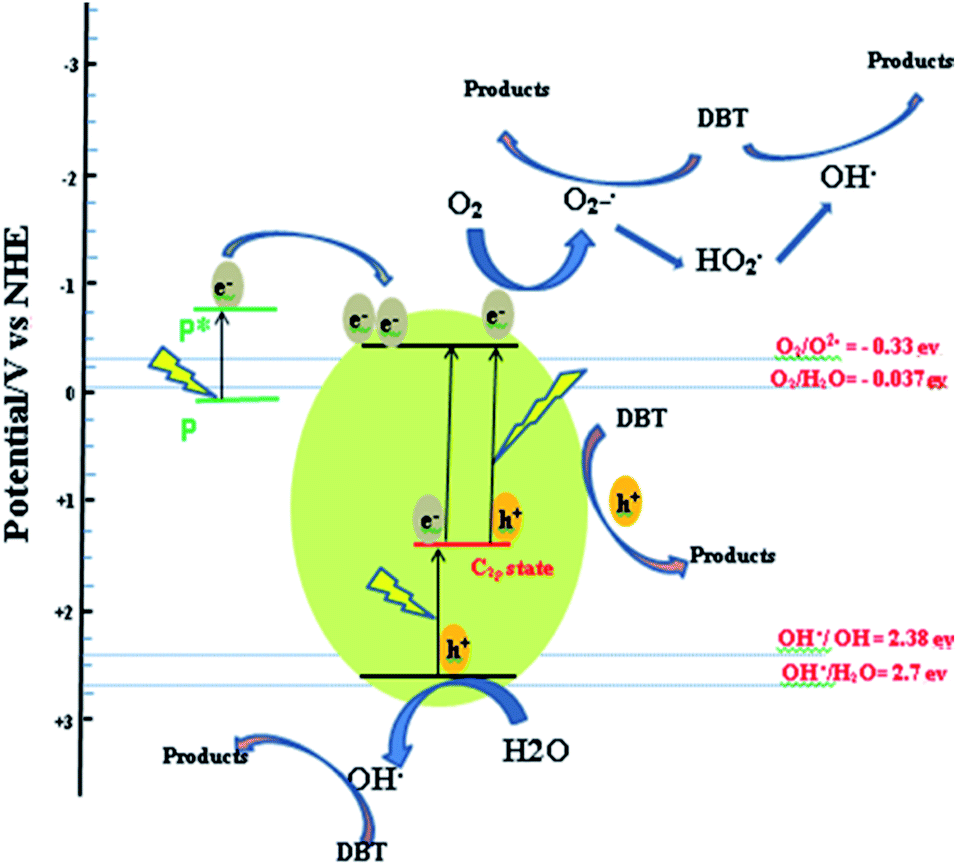 | ||
| Scheme 1 Proposed mechanism for DBT degradation. | ||
Another experiment was conducted to understand the effect of UV light irradiation. As shown in Fig. 14, the photo degradation efficiency of DBT decreased under UV light irradiation. To examine the cause for decreasing degradation of DBT, some experiments using radical scavengers were performed under UV irradiation. It can be seen that, under UV irradiation, the addition of AO as a scavenger for holes did not change the degradation of DBT. This result clearly suggests that holes (h+) are not formed in C 2p states. This observation is reasonable because UV irradiation can only excite the electrons from valance band to the conduction band of TiO2 but UV irradiation cannot excite the electrons from interband (C 2p level) to conduction band of TiO2. The generated holes in valance band of TiO2 can thermodynamically oxidize water and hydroxyl ions to produce hydroxyl radicals. The oxidation power of hydroxyl radicals (relative oxidation power = 2.05) is weaker than that of holes (relative oxidation power = 2.35).47 Therefore, the degradation of DBT decreases under UV light. As shown in Fig. 14, it is found that hydroxyl radicals (OH˙) and O2−˙ are the primary oxidants in the photocatalytic oxidation of DBT under UV light irradiation.
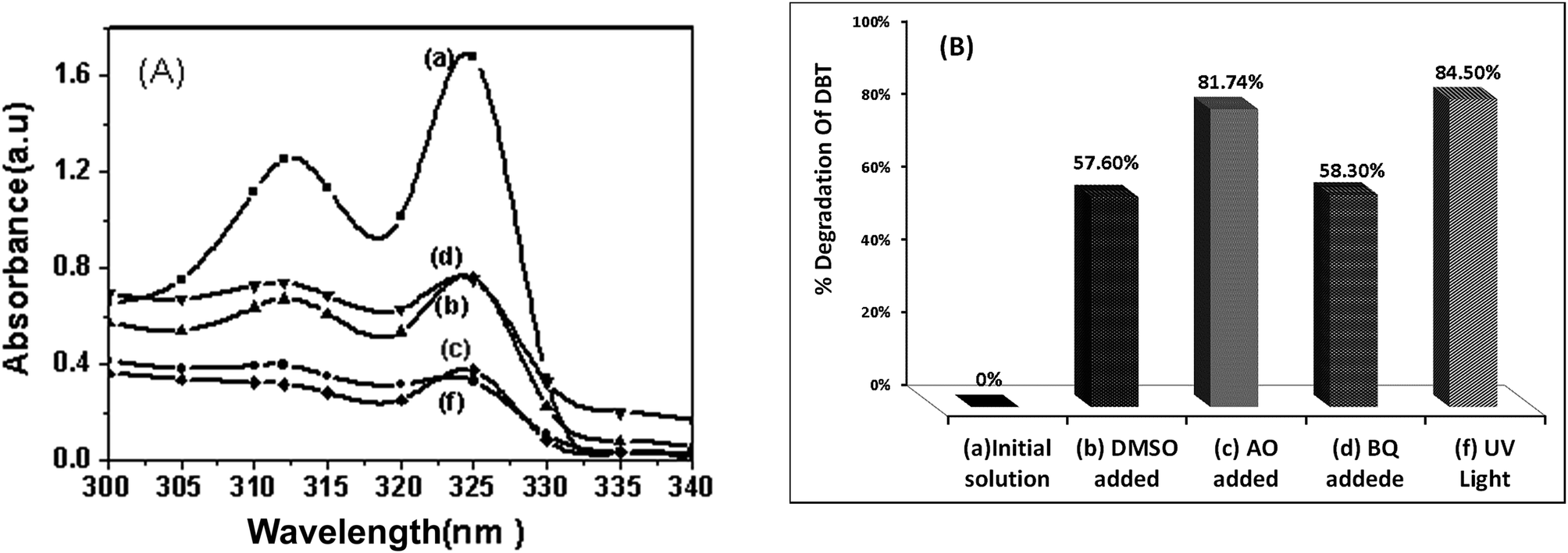 | ||
| Fig. 14 The effect of radical scavengers on the photocatalytic degradation of DBT (A) UV-Vis absorption spectrum. (B) Percentage of DBT degradation (a) standard solution of DBT (300 ppm), (b) with DMSO, (c) with AO, (d) with BQ, (f) without scavenger, condition: 0.015 g CTM-41 (Si/Ti = 5) under UV light irradiation for 5 h. | ||
3.5 Possible DBT photocatalyzed degradation path
In order to describe the photodegradation pathway, the GC-MS analysis was performed on three solutions:(1) Before photocatalytic degradation, the DBT solution (300 ppm) was analyzed by GC-MS. The GC chromatogram showed a peak at retention time (tr) of 34.78 min. This peak was identified by GC-MS analysis and compared with standard samples. The retention time was corresponded to DBT.
(2) After photocatalytic degradation and catalyst separation by centrifuge, the GC-MS detected a large peak with the retention time of 24.96 min. This retention time was related to the production of biphenyl. Any peak containing a sulfur compound in the solution was not detected.
(3) To examine the adsorption of sulfur compounds on the surface, the catalyst separated from previous step was added to alcohol and stirred at about 30 °C. After desorption, the solution was collected and analyzed by GC-MS analysis. The sulfur species were not observed on the GC chromatogram.
Since the formation of biphenyl was observed from DBT solution after photocatalytic degradation and sulfur compounds were not detected, it is plausible to assume that the sulfur compounds were escaped as gas from the solution.
Based on the literature,48,49 the DBT (1) can be converted to DBT sulfone (2) in the presence of OH˙ and it is shown in Scheme 2. Shiraishi et al.50 reported that when DBT sulfone was dissolved in 2-propanol and photo irradiated at λ > 280 nm by a high-pressure mercury lamp under air, the concentration of DBT sulfone decreases with time and the biphenyl concentration increased. In this work, as is shown in Scheme 2, first DBT (1) can convert to DBT sulfone (2) in the presence of OH˙ and then DBT sulfone (2) is photoexcited by photo irradiation to form highly reactive 1,5-biradical (3) intermediate and then it is hydrogenated by OH groups of CTM-41 and further decomposed to form finally biphenyl (5).
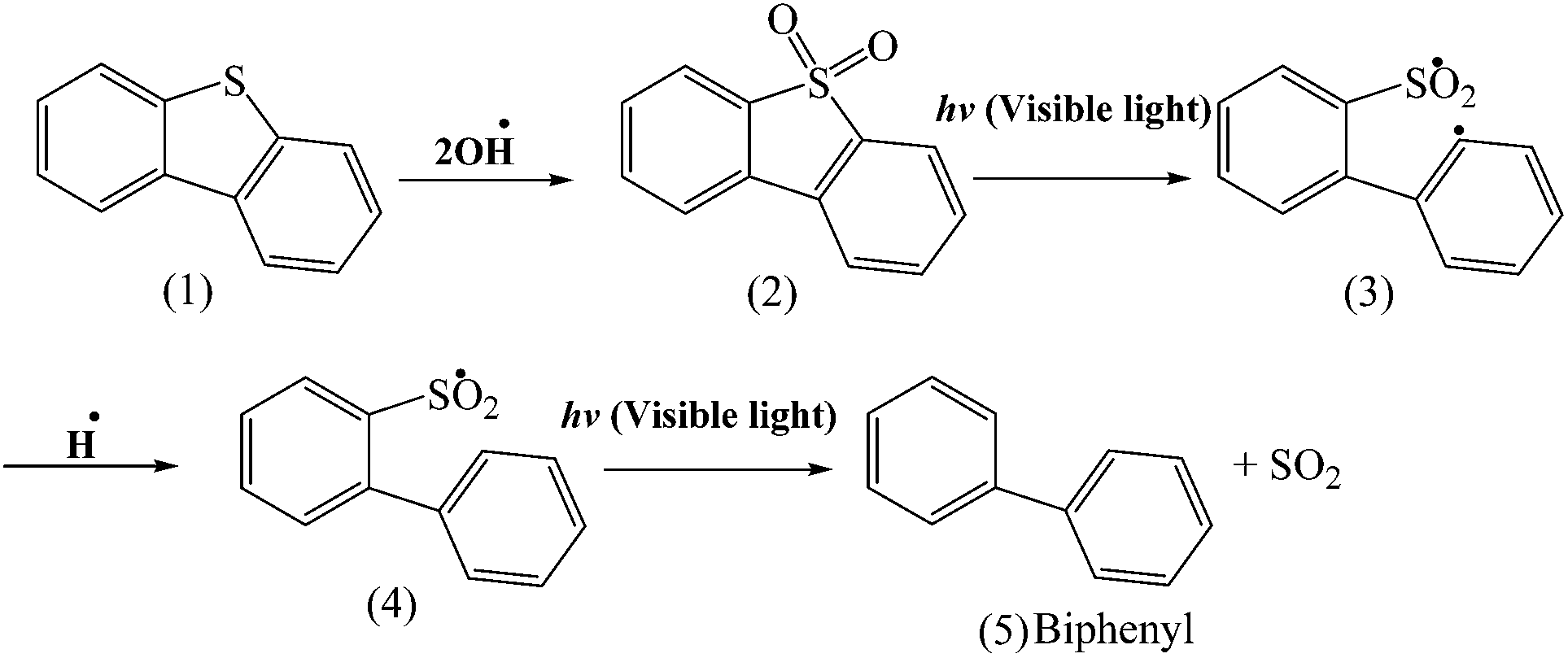 | ||
| Scheme 2 Possible DBT photocatalyzed degradation path. | ||
3.6 Recycling of the catalyst
The recyclability of the CTM-41 (Si/Ti = 5) was performed by testing consecutive cycles of the photocatalytic process. Fig. 15 shows the percentage of desulfurization for this compound. It was observed that the catalytic system could be recycled at least two times with little decrease in DBT removal. After using, CTM-41 nanoparticles were separated from the solution with centrifuging and then used in another cycle.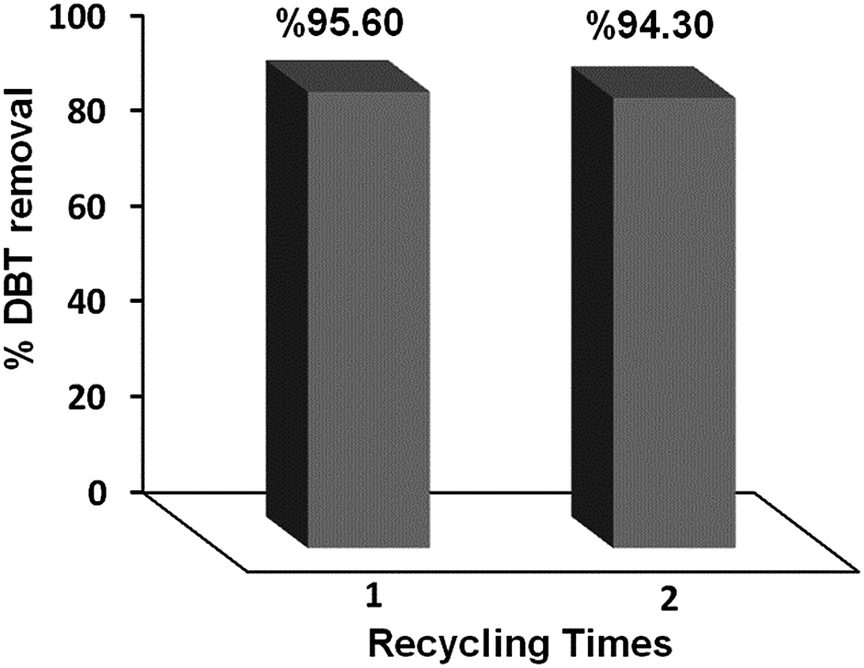 | ||
| Fig. 15 Reusability of the photocatalyst. | ||
4. Conclusions
In this work, carbon self-doped TiO2@MCM-41 (CTM-41) nanoparticles were prepared. The CTM-41 nanoparticles with measured band gaps of 1.3 eV and 3.2 eV exhibited high photocatalytic activity. These band gaps were assigned to the isolated C 2p states of carbonate ions. According to the XPS and UV analyses, there are two forms of C in CTM-41: interstitial carbons (carbonate species) and coke-like structure which have both been considered as the source of the extended optical absorption in the visible range. The C-doped TM-41 samples exhibited an enhanced absorption in the visible light region. The results suggested that the photocatalytic degradation of DBT is 95.6% when CTM-41 (Si/Ti = 5) was used as a catalyst. The results of GC-MS analysis showed that DBT converts to biphenyl under visible light irradiation and mild conditions within 5 h. The photocatalytic degradation of DBT by CTM-41 could be done via direct reactions of DBT with holes and OH˙ trapped on the surface.Acknowledgements
The support of Ferdowsi University of Mashhad (Research and Technology) is appreciated for the Project (3/19276, 17/10/2011). This work has also been supported by the “Iranian National Science Foundation: INSF” (no. 90001893).References
- C. Su, B. Y. Hong and C. M. Tseng, Sol–gel preparation and photocatalysis of titanium dioxide, Catal. Today, 2004, 96(3), 119–126 CrossRef CAS PubMed.
- H. Tao, T. Nakazato and S. Sato, Energy-efficient ultra-deep desulfurization of kerosene based on selective photooxidation and adsorption, Fuel, 2009, 88(10), 1961–1969 CrossRef CAS PubMed.
- A. Samokhvalov, et al., Adsorption and desorption of dibenzothiophene on Ag-titania studied by the complementary temperature-programmed XPS and ESR, Appl. Surf. Sci., 2011, 257(8), 3226–3232 CrossRef CAS PubMed.
- A. H. M. Shahadat Hussain and B. J. Tatarchuk, Mechanism of hydrocarbon fuel desulfurization using Ag/TiO2–Al2O3 adsorbent, Fuel Process. Technol., 2014, 126, 233–242 CrossRef CAS PubMed.
- A. Samokhvalov, et al., Surface characterization of Ag/Titania adsorbents, Appl. Surf. Sci., 2010, 256(11), 3647–3652 CrossRef CAS PubMed.
- Y. Jia, G. Li and G. Ning, Efficient oxidative desulfurization (ODS) of model fuel with H2O2 catalyzed by MoO3/γ-Al2O3 under mild and solvent free conditions, Fuel Process. Technol., 2011, 92(1), 106–111 CrossRef CAS PubMed.
- X. Zhou, et al., Oxidative desulfurization of dibenzothiophene based on molecular oxygen and iron phthalocyanine, Fuel Process. Technol., 2009, 90(2), 317–323 CrossRef CAS PubMed.
- W. Guo, et al., Oxidative desulfurization of diesel with TBHP/isobutyl aldehyde/air oxidation system, Appl. Energy, 2011, 88(1), 175–179 CrossRef CAS PubMed.
- K. Castillo, et al., Oxidation of dibenzothiophene to dibenzothiophene-sulfone using silica gel, J. Catal., 2009, 268(2), 329–334 CrossRef CAS PubMed.
- A. Al-Abduly and V. K. Sharma, Oxidation of benzothiophene, dibenzothiophene, and methyl-dibenzothiophene by ferrate(vi), J. Hazard. Mater., 2014, 279, 296–301 CrossRef CAS PubMed.
- A. Fujishima, T. N. Rao and D. A. Tryk, Titanium dioxide photocatalysis, J. Photochem. Photobiol., C, 2000, 1(1), 1–21 CrossRef CAS.
- A. Fujishima and X. Zhang, Titanium dioxide photocatalysis: present situation and future approaches, C. R. Chim., 2006, 9(5–6), 750–760 CrossRef CAS PubMed.
- Y. Lan, Y. Lu and Z. Ren, Mini review on photocatalysis of titanium dioxide nanoparticles and their solar applications, Nano Energy, 2013, 2(5), 1031–1045 CrossRef CAS PubMed.
- A. A. Belhekar, S. V. Awate and R. Anand, Photocatalytic activity of titania modified mesoporous silica for pollution control, Catal. Commun., 2002, 3(10), 453–458 CrossRef CAS.
- D. Zhao, et al., Rapid and facile synthesis of Ti-MCM-48 mesoporous material and the photocatalytic performance for hydrogen evolution, Int. J. Hydrogen Energy, 2010, 35(11), 5276–5283 CrossRef CAS PubMed.
- X. Yan, et al., Preparation, Characterization and Photocatalytic Activity of TiO2 Formed from a Mesoporous Precursor, J. Porous Mater., 2004, 11(3), 131–139 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, Nitrogen-Concentration Dependence on Photocatalytic Activity of TiO2−xNx Powders, J. Phys. Chem. B, 2003, 107(23), 5483–5486 CrossRef CAS.
- J. H. Park, S. Kim and A. J. Bard, Novel Carbon-Doped TiO2 Nanotube Arrays with High Aspect Ratios for Efficient Solar Water Splitting, Nano Lett., 2005, 6(1), 24–28 CrossRef PubMed.
- C. Lettmann, et al., Visible light photodegradation of 4-chlorophenol with a coke-containing titanium dioxide photocatalyst, Appl. Catal., B, 2001, 32(4), 215–227 CrossRef CAS.
- R. Leary and A. Westwood, Carbonaceous nanomaterials for the enhancement of TiO2 photocatalysis, Carbon, 2011, 49(3), 741–772 CrossRef CAS PubMed.
- H. J. Yun, et al., Facile preparation of high performance visible light sensitive photo-catalysts, Appl. Catal., B, 2010, 94(3–4), 241–247 CrossRef CAS PubMed.
- S. Zheng and L. Gao, Synthesis and characterization of Pt, Au or Pd clusters deposited titania-modified mesoporous silicate MCM-41, Mater. Chem. Phys., 2003, 78(2), 512–517 CrossRef CAS.
- J. Robertson and T. J. Bandosz, Photooxidation of dibenzothiophene on TiO2/hectorite thin films layered catalyst, J. Colloid Interface Sci., 2006, 299(1), 125–135 CrossRef CAS PubMed.
- S. Zheng, et al., Preparation, characterization and photocatalytic properties of singly and doubly titania-modified mesoporous silicate MCM-41 by varying titanium precursors, J. Mater. Chem., 2001, 11(2), 578–583 RSC.
- H. Yang, Y. Deng and C. Du, Synthesis and optical properties of mesoporous MCM-41 containing doped TiO2 nanoparticles, Colloids Surf., A, 2009, 339(1–3), 111–117 CrossRef CAS PubMed.
- K. Nishizawa, M. Okada and E. Watanabe, New preparation method of visible light responsive titanium dioxide photocatalytic films, Mater. Sci. Appl., 2014, 5, 112–123 CAS.
- K. J. S. I. a. Y.-S. L. Palanivelu, Carbon Doping of TiO2 for Visible Light Photo Catalysis – A review, Carbon: Sci. Technol., 2007, 8(3), 214–224 Search PubMed.
- S. Sakthivel and H. Kisch, Daylight Photocatalysis by Carbon-Modified Titanium Dioxide, Angew. Chem., Int. Ed., 2003, 42(40), 4908–4911 CrossRef CAS PubMed.
- C. Di Valentin and G. Pacchioni, Trends in non-metal doping of anatase TiO2: B, C, N and F, Catal. Today, 2013, 206, 12–18 CrossRef CAS PubMed.
- T.-H. Xu, et al., Band structures of TiO(2) doped with N, C and B, J. Zhejiang Univ., Sci., B, 2006, 7(4), 299–303 CrossRef CAS PubMed.
- Q. Chen, et al., Enhanced visible photocatalytic activity of titania-silica photocatalysts: effect of carbon and silver doping, Catal. Sci. Technol., 2012, 2(6), 1213–1220 CAS.
- B. J. Aronson, C. F. Blanford and A. Stein, Solution-Phase Grafting of Titanium Dioxide onto the Pore Surface of Mesoporous Silicates: Synthesis and Structural Characterization, Chem. Mater., 1997, 9(12), 2842–2851 CrossRef CAS.
- L. Guanglong, et al., Synthesis, characterization and photocatalytic evaluation of visible light activated C-doped TiO2 nanoparticles, Nanotechnology, 2012, 23(29), 294003 CrossRef PubMed.
- K. Madhusudan Reddy, S. V. Manorama and A. Ramachandra Reddy, Bandgap studies on anatase titanium dioxide nanoparticles, Mater. Chem. Phys., 2003, 78(1), 239–245 CrossRef.
- V. Etacheri, et al., A Highly Efficient TiO2−xCx Nano-heterojunction Photocatalyst for Visible Light Induced Antibacterial Applications, ACS Appl. Mater. Interfaces, 2013, 5(5), 1663–1672 CAS.
- L.-B. Xiong, et al., Ti3+ in the Surface of Titanium Dioxide: Generation, Properties and Photocatalytic Application, J. Nanomater., 2012, 2012, 13 Search PubMed.
- Y.-H. Hsien, et al., Photodegradation of aromatic pollutants in water over TiO2 supported on molecular sieves, Appl. Catal., B, 2001, 31(4), 241–249 CrossRef CAS.
- X. Yang, et al., Highly visible-light active C- and V-doped TiO2 for degradation of acetaldehyde, J. Catal., 2007, 252(2), 296–302 CrossRef CAS PubMed.
- P. Wang, et al., Carbon-sensitized and nitrogen-doped TiO2 for photocatalytic degradation of sulfanilamide under visible-light irradiation, Water Res., 2011, 45(16), 5015–5026 CrossRef CAS PubMed.
- J. Yang, et al., Visible-light photocatalysis in nitrogen–carbon-doped TiO2 films obtained by heating TiO2 gel–film in an ionized N2 gas, Thin Solid Films, 2008, 516(8), 1736–1742 CrossRef CAS PubMed.
- C. Di Valentin, G. Pacchioni and A. Selloni, Theory of Carbon Doping of Titanium Dioxide, Chem. Mater., 2005, 17(26), 6656–6665 CrossRef CAS.
- D. Chen, et al., Carbon and Nitrogen Co-doped TiO2 with Enhanced Visible-Light Photocatalytic Activity, Ind. Eng. Chem. Res., 2007, 46(9), 2741–2746 CrossRef CAS.
- G. Dai, et al., A simple preparation of carbon and nitrogen co-doped nanoscaled TiO2 with exposed {001} facets for enhanced visible-light photocatalytic activity, J. Mol. Catal. A: Chem., 2013, 368–369, 38–42 CrossRef CAS PubMed.
- S. Zheng, et al., Synthesis, characterization and photocatalytic properties of titania-modified mesoporous silicate MCM-41, J. Mater. Chem., 2000, 10(3), 723–727 RSC.
- G. A. Eimer, et al., The influence of Ti-loading on the acid behavior and on the catalytic efficiency of mesoporous Ti-MCM-41 molecular sieves, Catal. Today, 2008, 133–135, 639–646 CrossRef CAS PubMed.
- S. Banerjee, et al., New Insights into the Mechanism of Visible Light Photocatalysis, J. Phys. Chem. Lett., 2014, 5(15), 2543–2554 CrossRef CAS.
- R. Munter, Advanced oxidation processes – current status and prospects, Proc. Est. Acad. Sci., Chem., 2001, 50(2), 59–80 CAS.
- Z. Juan, et al., Photocatalytic oxidation dibenzothiophene using TS-1, Chem. Eng. J., 2010, 156(3), 528–531 CrossRef PubMed.
- R. Vargas and O. Núñez, The photocatalytic oxidation of dibenzothiophene (DBT), J. Mol. Catal. A: Chem., 2008, 294(1–2), 74–81 CrossRef CAS PubMed.
- Y. Shiraishi, et al., Photochemical Production of Biphenyls from Oxidized Sulfur Compounds Obtained by Oxidative Desulfurization of Light Oils, Energy Fuels, 2002, 17(1), 95–100 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |