
Electrochemical determination of phenanthrene based on anthraquinone sulfonate and poly diallyldimethylammonium chloride modified indium–tin oxide electrode
Maochao Weia,
Shuo Duanb,
Shan Liua,
Xiangli Zhenga,
Fangquan Xiaa and
Changli Zhou*a
aKey Laboratory of Chemical Sensing & Analysis in Universities of Shandong (University of Jinan), Jinan 250022, China. E-mail: chm_zhoucl@ujn.edu.cn; Fax: +86 531 82765969; Tel: +86 531 82765372
bCollege of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China
First published on 26th May 2015
Abstract
In this paper, specific electrochemical interaction between phenanthrene (Phe) and anthraquinone sulfonate (AQS) was first investigated by Cyclic Voltammetry (CV) and Differential Pulse Voltammetry (DPV). Poly diallyldimethylammonium chloride (PDDA) and AQS were modified onto indium–tin oxide (AQS/PDDA/ITO) by self-assembling. Owing to the specific interaction between AQS and Phe, the amount of Phe could be quantified by the electrochemical oxidation peak current difference of AQS at AQS/PDDA/ITO in the range from 1.0 × 10−12 to 1.0 × 10−9 mol L−1 with a linearity (r = 0.9942) and a low detection limit of 5.0 × 10−13 mol L−1 (S/N = 3). As an example of its practical application, the new sensor was used for quantitative determination of Phe in a standard sample and cloud-rain water samples of Mount Taishan with satisfactory results.
Introduction
Polycyclic aromatic hydrocarbons (PAHs), such as anthracene (Ant), phenanthrene (Phe), fluoranthene (Flu) and benzo[a]pyrene (BaP), are a category of compounds mainly formed by incomplete combustion and pyrolysis of organic substances.1 Due to their strong biotoxicity, carcinogenicity and low degradability, PAHs become important pollutants. This seriously threatens human life and the environment.2 Among the known 500 kinds of carcinogens, more than 200 kinds are related to PAHs.3 Moreover, 16 PAHs compounds are published in the list of priority pollutants in water by the US environmental protection agency (EPA).4 Therefore, it is imperative to establish a reliable method to detect and degrade PAHs. Presently, various analytical methods, including GC,5 HPLC,6,7 GC-MS,8 fluorescence,9 surface enhanced Raman spectroscopy,10 surface plasmon resonance,11 piezoelectric crystal,12 cytometric immunoassay,13 fiber optic sensor,14 and electrochemical biosensor15–17 had been developed to detect PAHs. GC, HPLC and GC-MS, as the most commonly used analytical methods; they usually need excessive time and energy on the previous separation or extraction. The separation or extraction processes always uses organic extraction agent and then causes secondary pollution. For that reason, the new determination method of PAHs, especially with characteristics of low pollution and energy consumption, was still expected.Electrochemical method has been widely applied into development of chemical and biological sensors18–21 due to its selectivity and sensitivity. Due to low pollution, low energy consumption, convenient operation and rapid analysis; electrochemical sensors, has been widely used for determining environmental and biologic species.22,23 In general, PAHs were not usually oxidized readily. The electrochemical detections of PAHs always focused on biosensor-platform or electrocatalytic oxidation, which included electrochemical immunosensor15–17 and electrochemical DNA sensor.24,25 Fahnrich reported an amperometric immunosensor for PAHs detection using screen-printed electrode.15 Wang reported an electrochemical immunosensor fabrication for benzo[a]pyrene detection using dendritic SiO2 nanoparticles for biomolecule immobilization.22 Pandey reported the application of electrochemical DNA biosensor for PAHs detection.24 However, enzyme inactivation could induce the decrease of the biosensor's stability and sensitivity. Electrocatalytic oxidation is another ways to detect PAHs. Electrocatalyst, including dendritic 7T-polythiophene,26 Ag/TiO2 nanotubes,27 TiO2,28 RuOx/TiO2,29 are used in electrochemical catalytic oxidation and degradation of PAHs. However, the main problems of electrocatalysis are the complex matrix effect and derivative reaction caused by high oxidation potential. Considering those conditions mentioned above, redox probes, with high stability and selectivity, are expected to play a significant role in designing PAHs electrochemical sensor.30,31
PAHs with specific aromatic rings can interact with corresponding substances through specific π–π conjugation, and the interactions varied with the number and special structural differences in the aromatic rings.32,33 The diversity of π–π conjugation interactions had been applied into PAHs sensors, such as quartz crystal microbalance12 and optical chemistry sensor.14 Via the π–π conjugation between PAHs and nitroaromatics, electrochemical TNT sensor with enhanced responses was developed.34 As a specific interaction, π–π conjugation might provide a potential method to absorb and detect PAHs. Particularly, phenanthrene (Phe) with three benzene rings has a specific open arched, planar structure. Phe might have special π–π conjugation interaction with corresponding substance.35,36 The π–π conjugation of Phe with humic subunits35 and monovalent ions36 were well discussed by theoretical calculation. More immunosensor for selective electrochemical determination of Phe has been reported.37 We believe the π–π conjugation of Phe might establish a unique interaction with the corresponding probe, and the probe redox could be involved in Phe detection.
In this study, specific electrochemical interaction between Phe and anthraquinone sulfonate (AQS) was firstly investigated by CVs and DPV. Due to this specific interaction, Phe could effectively promote the electrochemical redox of AQS. Thus, the interaction could help the researchers to establish a novel, facile method to determinate Phe concentration via detecting the electrochemical responses of AQS oxidation. AQS (specific redox probe) was modified onto indium–tin oxide (ITO, providing low-priced and modifiable electrode) through self-assembly method. A novel electrochemical sensor of Phe was firstly prepared based on the specific recognition towards Phe. This study showed the great significance in improving the selectivity of target analytes, and provided a potential method to develop novel environmental measurement techniques.
Experimental
Instruments and chemicals
Cyclic voltammetries (CVs) and differential pulse voltammetry (DPV) testings are performed on CHI842C electrochemical workstation (Chenhua Instrument Co., China). Electrochemical impedance spectroscopy (EIS) was recorded with IM6ex electrochemical workstation (ZAHNER Co., Germany). A traditional three-electrode system is employed, including an indium–tin oxide (ITO) electrode or a modified electrode as the working electrode, a platinum wire as the counter electrode, and an Ag/AgCl electrode as the reference electrode.The cloud-rain water samples of Mount Taishan were offered by school of environmental engineering, Shandong University. Phenanthrene (Phe) and other PAHs were obtained from Aladdin Reagent Co., Ltd. Anthraquinone-2-sulfonic acid sodium salt (providing water-solubility AQS) and poly diallyldimethylammonium chloride (PDDA) are purchased from J&K Chemical Reagent Co. All other chemicals are at least of analytical grade and used as received. Each PAH standard solution was diluted by anhydrous ethanol into a stock solution containing 1.0 mmol L−1 PAH. Ultra-pure water was used in all aqueous solutions. ITO (1 cm × 1 cm × 0.2 cm) was purchased from Thermo Electron Co.
Electrode preparation
In order to enhance the sensitivity and make it easy to use, AQS was immobilized onto the ITO to achieve a better and more stable analytical performance. The preparation process was similar to Liu's work,38 the ITO electrode was first immersed into PDDA solution (3 mg mL−1, containing 0.5 mol L−1 NaCl) for 20 minutes to adsorb positively charged PDDA as a precursor layer, and then the PDDA modified ITO electrode was immersed in a 1.0 mol L−1 anthraquinone-2-sulfonic acid sodium salt water solution for 3 h. After rinsing with ultra-pure water and air drying, AQS/PDDA/ITO was prepared.Results and discussion
EIS characterizations of electrodes
EIS experiment was a powerful tool to study the electrode interface properties and the result could provide the information of the electrode surface. The EIS results (Nyquist plots, −Z′′ vs. Z′) for different electrodes in a solution containing of 3.0 mmol L−1 [Fe(CN)6]3−/4− and 0.1 mol L−1 KCl with the frequencies swept from 105 to 0.1 Hz are shown in Fig. 1. The equivalent circuit (inset) is further chosen to fit the impedance data obtained in the experiments. As can be seen, the EIS results of both bare ITO electrode (unmodified) and the AQS/PDDA/ITO electrode showed semicircle in high frequency region and straight line in low frequency region. The Ret value in the low frequency region of bare ITO electrode (curve a) was estimated to be 768.2 Ω and AQS/PDDA/ITO (curve b) was estimated to be 5960.6 Ω, indicating that the electron transfer resistance increased greatly after the adsorption layer modified onto the electrode surface.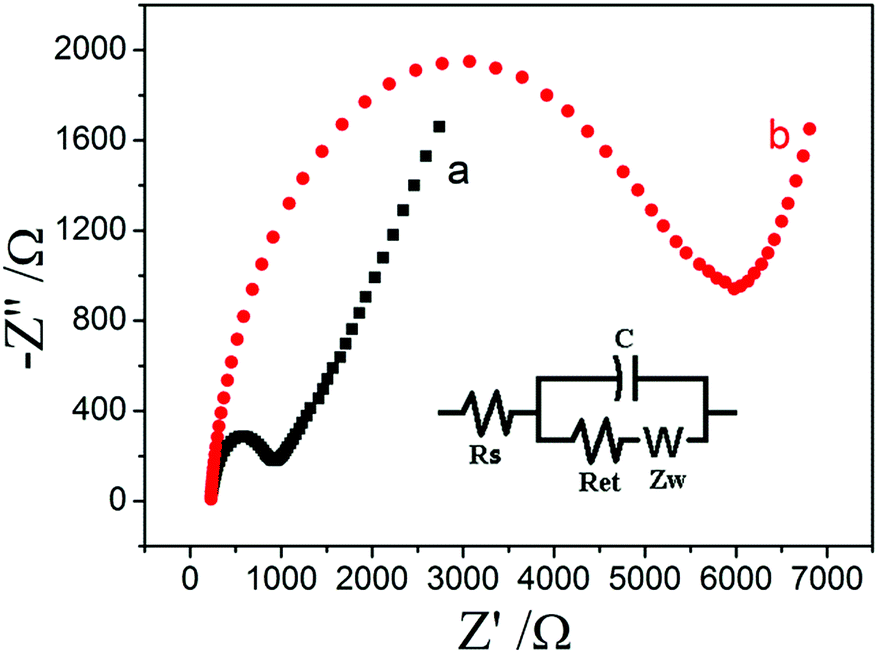 | ||
| Fig. 1 The Nyquist plots of bare ITO (a) and AQS/PDDA/ITO (b) in a solution containing of 3.0 mmol L−1 [Fe(CN)6]3−/4− and 0.1 mol L−1 KCl. | ||
Electrochemical properties of AQS/PDDA/ITO
CV is a most widely used technique for acquiring qualitative information in electrochemical reactions. The CV results of AQS at different electrodes in electrolyte solution (Acetic acid–sodium acetate (HAc–NaAc) buffer solution with 1.0 × 10−5 mol L−1 AQS, pH 3.5) were showed in Fig. 2. A slight obvious oxidation peak can be observed at about −0.28 V and almost not a reduction peak of AQS at bare ITO electrode (curve a). Compared with bare ITO electrode, AQS/PDDA/ITO (curve b) exhibited higher oxidation peak currents at about −0.22 V and a weak reduction peak at about −0.38 V. This phenomenon can be attributed to the immobilization of AQS and PDDA on the ITO electrode, which decrease the electrical conductivity and improve the reversibility of the system. However, the peak current responses of AQS redox get better at AQS/PDDA/ITO, which probably due to the increase load of AQS species adsorbed onto the PDDA layer. All the conditions above indicated that electrochemical responses of AQS could be greatly improved by immobilizing PDDA and AQS onto the surface of the ITO electrode.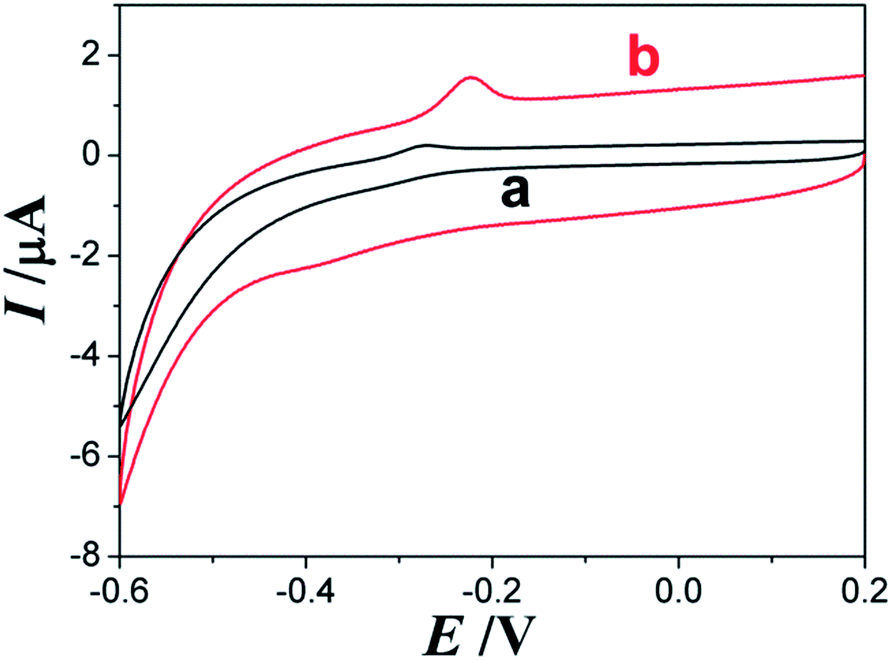 | ||
| Fig. 2 The CVs of AQS at bare ITO electrode (a) and AQS/PDDA/ITO (b) in HAc–NaAc buffer (pH 3.5). Scan rate: 100 mV s−1. | ||
In order to obtain a better electrochemical response, modified electrode at different buffer solutions and different pH were investigated by CV. And in HAc–NaAc solution, AQS/PDDA/ITO showed the best CV results. In addition, cyclic voltammograms of AQS/PDDA/ITO electrode in solutions of different pH values were recorded (Fig. 3). As pH increases, the anodic peak potential has shifted negatively. Considering the anodic peak currents and potentials, pH 3.5 HAc–NaAc solution was chosen as the best pH for the rest experiments.
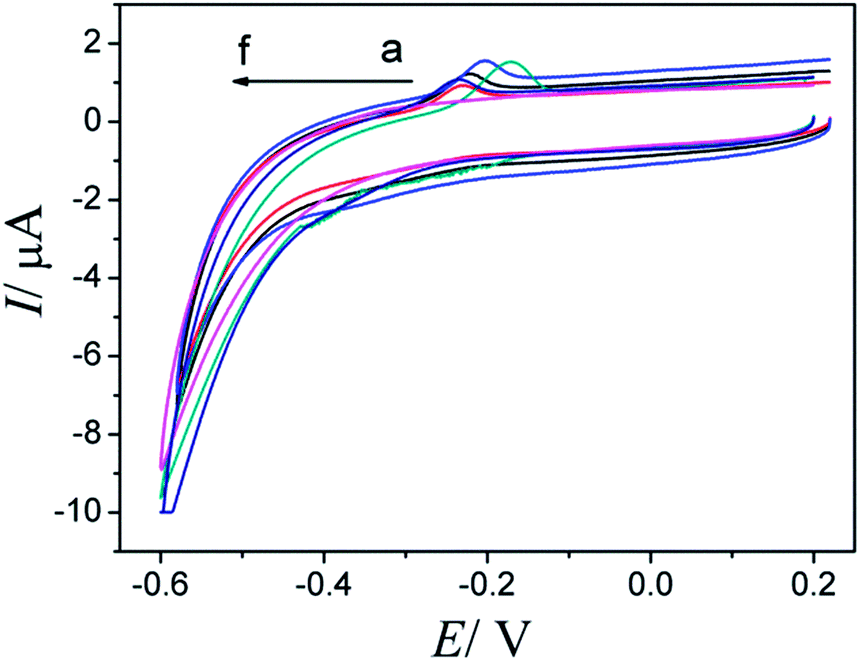 | ||
| Fig. 3 The CVs of AQS/PDDA/ITO in 0.1 mol L−1 NaAc–HAc buffer solution with different pH. From a to f: 3.0, 3.5, 4.0, 4.5, 5.0 and 5.5. | ||
The CVs behaviors of the AQS/PDDA/ITO electrode in NaAc–HAc buffer solution with a pH value of 3.5 at different scan rates were studied. It would be beneficial for us to understand the redox characters of AQS at the proposed modified electrode, and thus the following process for the construction of Phe sensor. As shown in Fig. 4, the anodic peak currents were linearly proportional to the scan rate in the range from 20 to 300 mV s−1 (Fig. 4 inset). The result indicated that the electro–oxidative reaction of AQS on the surface of electrode was a surface-controlled process.
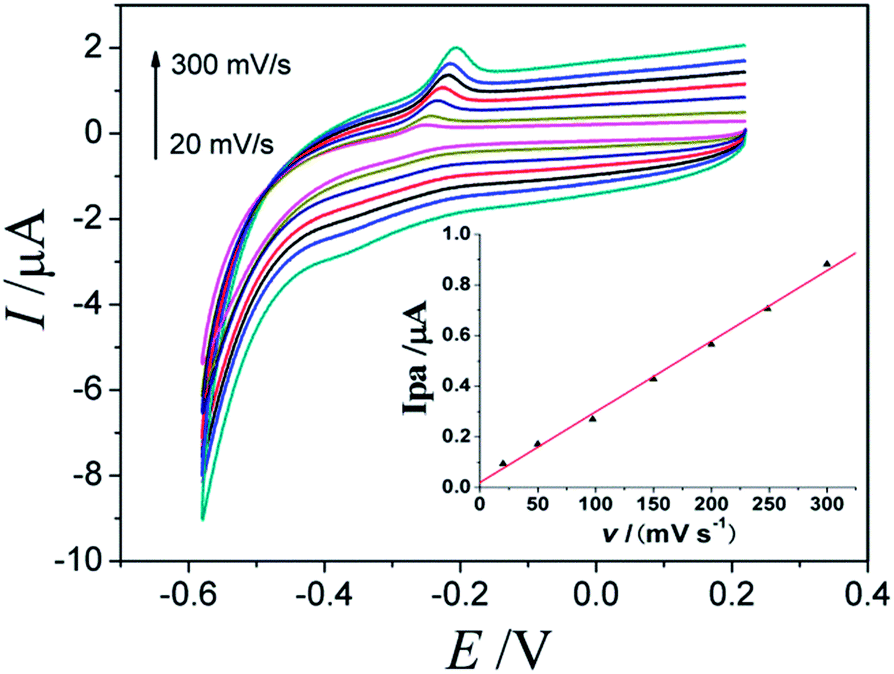 | ||
| Fig. 4 The CVs of AQS/PDDA/ITO electrode in pH 3.5 NaAc–HAc buffer solution with different scan rates from 20 to 300 mV s−1. Insert plot: the linear relationship between scanning rates and anodic peak currents. | ||
The specific interaction between AQS and Phe
DPV was a widely used electrochemical method which is more sensitive compared to CV. The anodic peak current of AQS on the proposed modified electrode at different concentrations of Phe were recorded by DPV. As shown in Fig. 5, The anodic peak current difference (ΔIp) is linear to the logarithm value of the concentrations of Phe (lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c) in the range of 1.0 × 10−12–1.0 × 10−9 mol L−1 with a regression equation of ΔIp = 0.3995lgc + 4.803 (μA, mol L−1, r = 0.9942) (Fig. 5, inset). The detection limit is evaluated to be 5.0 × 10−13 mol L−1 (S/N = 3). The obtained high sensitivity cannot be ascribed simply to the high sensitive DPV technology itself, it is reasonable that the weak interactions between the electroactive AQS and Phe contribute significantly to the excellent response behaviors of the proposed sensor. Even an extremely low concentration of Phe can perturb the redox behaviors of AQS obviously and thus large electrochemical signals can be recorded.
c) in the range of 1.0 × 10−12–1.0 × 10−9 mol L−1 with a regression equation of ΔIp = 0.3995lgc + 4.803 (μA, mol L−1, r = 0.9942) (Fig. 5, inset). The detection limit is evaluated to be 5.0 × 10−13 mol L−1 (S/N = 3). The obtained high sensitivity cannot be ascribed simply to the high sensitive DPV technology itself, it is reasonable that the weak interactions between the electroactive AQS and Phe contribute significantly to the excellent response behaviors of the proposed sensor. Even an extremely low concentration of Phe can perturb the redox behaviors of AQS obviously and thus large electrochemical signals can be recorded.
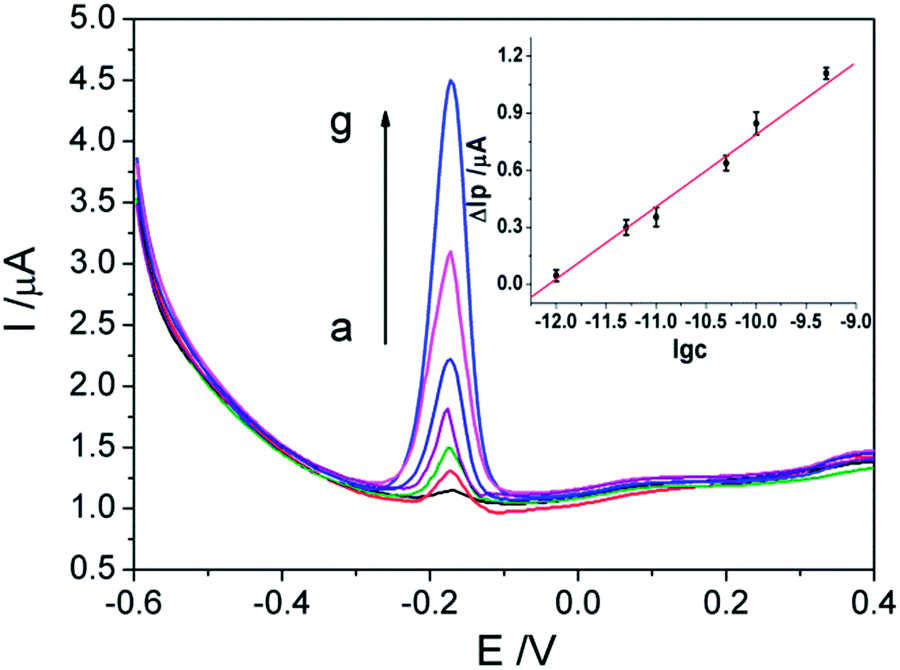 | ||
| Fig. 5 DPV curves of the AQS/PDDA/ITO in 0.1 mol L−1 NaAc–HAc solution (pH 3.5) containing various concentrations of Phe. From a to g: 0, 1.0 × 10−12, 5.0 × 10−12, 1.0 × 10−11, 5.0 × 10−11, 1.0 × 10−10 and 1.0 × 10−9 mol L−1. The inset shows the linear relationship between ΔIp and the logarithmic concentration of Phe. | ||
Stability, reproducibility and anti-interference performance of AQS/PDDA/ITO
Under the optimized conditions, the initial current response of AQS/PDDA/ITO was recorded by DPV. The electrode was kept at 4 °C for 30 days; the peak currents could maintain 96.0% (±0.5) of the initial value. The electrode regeneration was obtained by immersing the modified electrode into toluene overnight. The current response could recover 95.0% (±1.0) of the initial current response.To investigate the selectivity and anti-interference performance of AQS/PDDA/ITO, a series of PAHs, such as naphthalene (Nap), anthracene (Ant), benz[a]anthracene (BaA), benzo[a]pyrene (BaP) and pyrene (Pyr) are analyzed under the same condition (Fig. 6). The result showed that other PAHs had little influence on the AQS electrochemical oxidation. Considering their similar physical and chemical properties to Phe, the influence of Phe to AQS was verified as a specific π–π interaction, and its influence might derive from the particularly non-closed planar arched structure of Phe. We proposed that the structure could lock the benzene ring of AQS and form a planar interlocking structure, owing to the probable size-matched conjugation effect. The interlocked planar structure could effectively provide the larger effectively provide the larger electron transfer area to promote the AQS redox process.
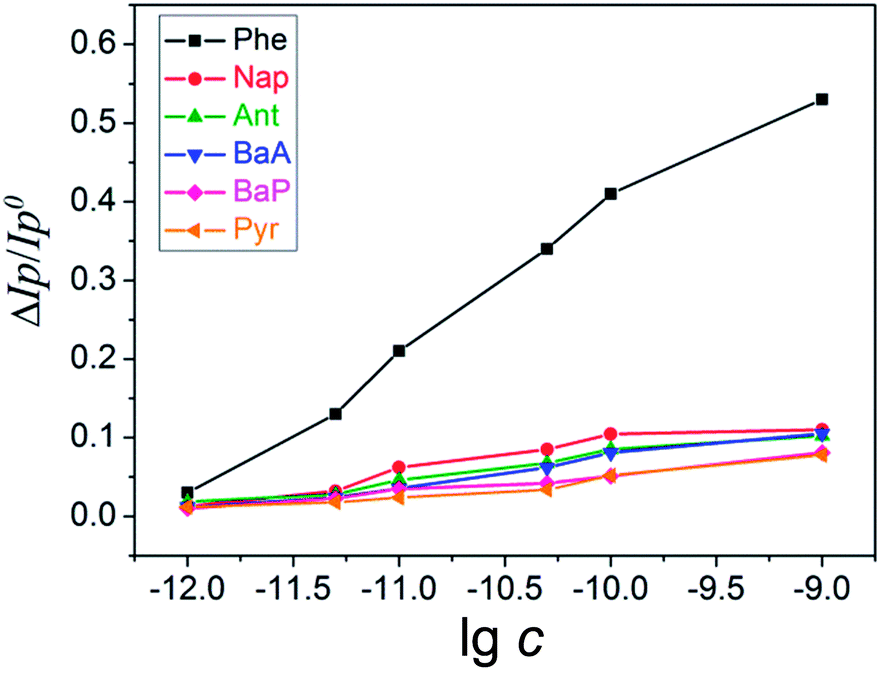 | ||
| Fig. 6 ΔIp/Ip0 of AQS/PDDA/ITO as functions of the concentrations of PAHs from top to bottom: Phe, Nap, Ant, BaA, BaP, Pyr. | ||
In contrast, a series of PAHs like Nap and Ant, they have no non-closed planar arched structure and they could not form the specific interaction with AQS (Scheme 1). Due to the specific π–π conjugation, Phe could be determined by detecting the enhancement of oxidation peak currents of AQS, and excluding the interference from other PAHs.
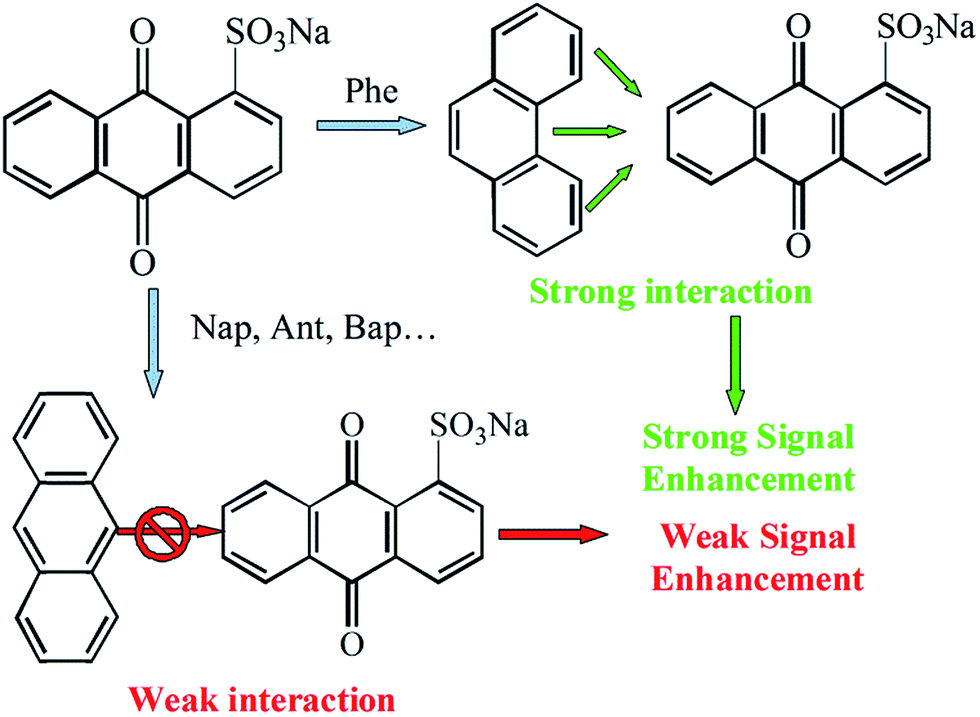 | ||
| Scheme 1 The scheme of conjugation effect towards AQS redox enhancement. | ||
Analytical applications
To evaluate the applicability of the proposed method, a standard liquid sample containing 16 PAHs compounds and cloud-rain water samples of Mount Taishan were tested with five parallel measurements. Before electrochemical assays, 1.0 mL sample was transferred to a 20 mL electrolytic cell, and then the sample pH was adjusted to 3.5 using 0.1 mol L−1 HAc and NaAc. Finally, 0.1 mol L−1 Hac–NaAC (pH 3.5) was added into the solution to constant volume to 10.0 mL. The electrochemical responses of blank sample and the samples were recorded by DPV respectively. The obtained ΔIp values were utilized to calculate the concentrations of Phe based on the linear equation as described above. The quantitative results were shown in Table 1. Noteworthy, this study showed satisfactory result compared to the GC coupled UV-vis monitor standard result. The result has substantiated the proposed method could be a great use in the practical detection of Phe.Conclusions
In this study, specific electrochemical interaction between Phe and AQS was firstly investigated by CVs and DPVs. Owing to a matching size conjugation, the resulting strong π–π interaction between Phe and AQS has effectively promoted the electrochemical redox of AQS. Thus, a novel and facile method to quantitative detection of Phe was established. Furthermore, the reliability and applicability of the proposed method have been validated by GC method. With more specific recognizing species be found, hopefully, this simple electrochemical method can be used in detecting more PHAs quantitatively.Acknowledgements
This study was supported by Shandong Provincial Natural Science Foundation, China (ZR2013BM003) and Science and Technology Development Plan Project of Shandong Province (2012G0022116).Notes and references
- A. L. Juhasz and R. Naidu, Int. Biodeterior. Biodegrad., 2000, 45, 57–88 CrossRef CAS.
- K.-H. Kim, S. A. Jahan, E. Kabir and R. J. Brown, Environ. Int., 2013, 60, 71–80 CrossRef CAS PubMed.
- K. Fretheim, J. Agric. Food Chem., 1976, 24, 976–979 CrossRef CAS.
- R.-A. Doong, S.-M. Chang and Y.-C. Sun, J. Chromatogr. A, 2000, 879, 177–188 CrossRef CAS.
- A. Darán and A. González, J. Environ. Sci. Technol., 2009, 6, 663–670 Search PubMed.
- L. Yang, Z. Wang and L. Xu, J. Chromatogr. A, 2006, 1104, 230–237 CrossRef CAS PubMed.
- W. Wei, R. Liang, Z. Wang and W. Qin, RSC Adv., 2015, 5, 2659–2662 RSC.
- S. Sporstol, N. Gjos, R. G. Lichtenthaler, K. O. Gustavsen, K. Urdal, F. Oreld and J. Skei, Environ. Sci. Technol., 1983, 17, 282–286 CrossRef CAS.
- T. Chetiyanukornkul, A. Toriba, T. Kameda, N. Tang and K. Hayakawa, Anal. Bioanal. Chem., 2006, 386, 712–718 CrossRef CAS PubMed.
- J. Du and C. Jing, J. Phys. Chem. C, 2011, 115, 17829–17835 CAS.
- N. Miura, M. Sasaki, K. V. Gobi, C. Kataoka and Y. Shoyama, Biosens. Bioelectron., 2003, 18, 953–959 CrossRef CAS.
- M. Liu, Q. X. Li and G. A. Rechnitz, Anal. Chim. Acta, 1999, 387, 29–38 CrossRef CAS.
- A. Meimaridou, W. Haasnoot, L. Noteboom, D. Mintzas, J. Pulkrabova, J. Hajslová and M. W. Nielen, Anal. Chim. Acta, 2010, 672, 9–14 CrossRef CAS PubMed.
- C.-Y. Yun, D. Dhital, J.-R. Lee, G. Park and I.-B. Kwon, Opt. Laser Technol., 2012, 44, 269–280 CrossRef CAS PubMed.
- K. Fähnrich, M. Pravda and G. Guilbault, Biosens. Bioelectron., 2003, 18, 73–82 CrossRef.
- G. Hanrahan, D. G. Patil and J. Wang, J. Environ. Monit., 2004, 6, 657–664 RSC.
- A. Ahmad and E. Moore, Analyst, 2012, 137, 5839–5844 RSC.
- B. J. Privett, J. H. Shin and M. H. Schoenfisch, Anal. Chem., 2010, 82, 4723–4741 CrossRef CAS PubMed.
- V. Suryanarayanan, C. T. Wu and K. C. Ho, Electroanalysis, 2010, 22, 1795–1811 CrossRef CAS PubMed.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473–4491 RSC.
- J. Liu, M. D. Morris, F. C. Macazo, L. R. Schoukroun-Barnes and R. J. White, J. Electrochem. Soc., 2014, 161, H301–H313 CrossRef CAS PubMed.
- C. Wang, M. Lin, Y. Liu and H. Lei, Electrochim. Acta, 2011, 56, 1988–1994 CrossRef CAS PubMed.
- J. Liu, S. Wagan, M. Dávila Morris, J. Taylor and R. J. White, Anal. Chem., 2014, 86, 11417–11424 CrossRef CAS PubMed.
- P. Pandey and H. Weetall, Anal. Chem., 1995, 67, 787–792 CrossRef CAS.
- R.-A. Doong, H.-M. Shih and S.-H. Lee, Sens. Actuators, B, 2005, 111, 323–330 CrossRef PubMed.
- C. Rassie, R. A. Olowu, T. T. Waryo, L. Wilson, A. Williams, P. G. Baker and E. I. Iwuoha, Int. J. Electrochem. Sci., 2011, 6, 1949–1967 CAS.
- J. Li, L. Yang, S. Luo, B. Chen, J. Li, H. Lin, Q. Cai and S. Yao, Anal. Chem., 2010, 82, 7357–7361 CrossRef CAS PubMed.
- M. García-Martínez, L. Canoira, G. Blázquez, I. Da Riva, R. Alcántara and J. Llamas, Chem. Eng. J., 2005, 110, 123–128 CrossRef PubMed.
- L.-H. Tran, P. Drogui, G. Mercier and J.-F. Blais, J. Hazard. Mater., 2009, 164, 1118–1129 CrossRef CAS PubMed.
- X. Zheng, D. Tian, S. Duan, M. Wei, S. Liu, C. Zhou, Q. Li and G. Wu, Electrochim. Acta, 2014, 130, 187–193 CrossRef CAS PubMed.
- S. Liu, M. Wei, X. Zheng, S. Xu and C. Zhou, Anal. Chim. Acta, 2014, 826, 21–27 CrossRef CAS PubMed.
- S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed.
- P. Samorí, N. Severin, C. D. Simpson, K. Müllen and J. P. Rabe, J. Am. Chem. Soc., 2002, 124, 9454–9457 CrossRef PubMed.
- H.-X. Zhang, Q. Chen, R. Wen, J.-S. Hu and L.-J. Wan, Anal. Chem., 2007, 79, 2179–2183 CrossRef CAS PubMed.
- H. Wijnja, J. J. Pignatello and K. Malekani, J. Environ. Qual., 2004, 33, 265–275 CrossRef CAS.
- S. Ikuta, J. Mol. Struct.: THEOCHEM, 2000, 530, 201–207 CrossRef CAS.
- E. J. Moore, M. P. Kreuzer, M. Pravda and G. G. Guilbault, Electroanalysis, 2004, 16, 1653–1659 CrossRef CAS PubMed.
- Y. Liu, Y. Yu, Q. Yang, Y. Qu, Y. Liu, G. Shi and L. Jin, Sens. Actuators, B, 2008, 131, 432–438 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |