
Structural and magnetic properties of GaN/Mn nanopowders prepared by an anaerobic synthesis route†
Mariusz Drygasa,
Jerzy F. Janik*a,
Miroslaw M. Buckob,
Jacek Goskcd and
Andrzej Twardowski*c
aAGH University of Science and Technology, Faculty of Energy and Fuels, al. Mickiewicza 30, 30-059 Krakow, Poland. E-mail: janikj@agh.edu.pl
bAGH University of Science and Technology, Faculty of Materials Science and Ceramics, al. Mickiewicza 30, 30-059 Krakow, Poland
cUniversity of Warsaw, Faculty of Physics, ul. Pasteura 5, 02-093 Warszawa, Poland. E-mail: andrzej.twardowski@fuw.edu.pl
dWarsaw University of Technology, Faculty of Physics, ul. Koszykowa 75, 00-662 Warszawa, Poland
First published on 16th April 2015
Abstract
A new oxygen-free molecular precursor system based on (i) ammonolysis in refluxing/liquid NH3 of selected mixtures of gallium tris(dimethyl)amide Ga(NMe2)3 and manganese bis(trimethylsilyl)amide Mn[N(SiMe3)2]2 (Me = CH3, initial Mn-contents = 0.1, 5, 20, 50 at.%) followed by (ii) pyrolysis under flowing ammonia gas at 500, 700, and 900 °C afforded a range of nanocrystalline powders in the GaN/Mn system. The nanopowders were characterized mainly by powder XRD diffraction, FT-IR spectroscopy, Raman spectroscopy, SEM/EDX morphology examination, and XRF elemental analysis. Magnetization measurements as a function of magnetic field and temperature were carried out with a SQUID magnetometer. Structurally, the materials were shown to be single-phases based on the gallium nitride lattice. The presence of small quantities of residual amorphous Mn/N/Si/C species due to an incomplete transamination/removal of the trimethylsilylamide groups during ammonolysis was deduced from the XRF, FT-IR, Raman, and magnetization data. Magnetic properties for all nanopowders consistently pointed to a paramagnetic GaMnN phase with antiferromagnetic interactions among Mn-centers that under favorable circumstances reached the level of 3.8 at.% Mn. The paramagnetic phase was accompanied by a residual antiferromagnetic phase due to a facile oxidation in air of excessive Mn-containing by-products.
1. Introduction
Gallium nitride GaN suitably doped with a magnetic transition metal (TM = Mn, Fe, Cr, etc.), which can be abbreviated as GaN/TM or GaTMN, has been anticipated for many promising applications as a dilute magnetic semiconductor DMS. The DMS materials are crucial in spintronics – spin based electronics which utilize both the semiconducting and magnetic properties of one material and some recent contributions and comprehensive reviews of the subject are available.1 The known DMS materials are usually produced either as kinetically-stabilized TM-doped thin films or monocrystals. In this regard, sintered GaN-based DMS ceramics prepared from nanopowders can, potentially, be a new convenient form of such materials for many challenging applications.2 The reproducible preparation and characterization of GaN-based DMS nanopowders are, therefore, indispensable for further progress in this area.The preparation of GaN-based thin films and monocrystals doped with selected transition metals including Mn was the subject of studies carried out by some of the authors of this report.3 The attention was also given to dope the nitride microcrystalline powders via the ammonothermal synthesis.4 GaN/Mn nanocrystalline powders were previously prepared by some of us using both the aerosol-assisted and anaerobic synthesis methods. The aerosol-assisted synthesis used the readily available oxygen-bearing nitrates of gallium and manganese and yielded some Mn-centers incorporated in the GaN lattice but it was plagued by the formation of a residual antiferromagnetic manganese oxide MnO by-product.5 The anaerobic synthesis dwelled on the anticipated incorporation of Mn-centers into the gallium imide framework, the latter formed by transamination/deamination chemistry in the oxygen-free gallium(III) tris(dimethyl)amide/ammonia reaction system, Ga(NMe2)3/NH3 (Me = CH3).6 Based on magnetization measurements, up to 3.5% of Mn-centers were incorporated in the hexagonal GaN nanopowders. However, the use of the oxygen-bearing manganese(III) acetylacetonate dopant resulted again in some antiferromagnetic MnO impurity.
There is only a handful of other reports concerned with magnetic doping of GaN powders. In one report, Mn-doped GaN microcrystalline powders containing ca. 0.1% of Mn were made by solid-state metathesis using Mn, Li3N, and Ga2O3 as precursors.7 In another study, similar powders, claimed with up to 7 at.% of incorporated Mn, were prepared in the gallium(III) nitrate/manganese(II) nitrate precursor system via conversion to oxides and, finally, reactions with ammonia at elevated temperatures.8 In a different approach, magnetic GaN/GaN:Mn core/shell nanowires were synthesized in a multistep process that included GaN nanowire preparation by vapor phase MOCVD and the nanowire surface modification by chemical vapor transport with the use of the Ga and MnCl2 precursor mixture.9 Modifications of a route to GaN nanowires, based on the use of GaN powders and MnCl2, resulted in hexagonal GaN-based nanowires modified in bulk with Mn up to 10%.10
Looking back at the transamination/deamination chemistry explored earlier by us, one can envision that the application of an oxygen-free bimetallic precursor system such as made of gallium(III) tris(dimethyl)amide/manganese(II) bis(dimethyl)amide, Ga(NMe2)3/Mn(NMe2)2, would create advantageous conditions for mixed organometallic species and, after reactions with ammonia, it would favor the evolution of Ga–N–Mn–N linkages whereas circumvent the formation of manganese oxygen by-products. This kind of chemistry was earlier confirmed by us in the system gallium(III) tris(dimethyl)amide/aluminum(III) tris(dimethyl)amide which upon reactions with ammonia afforded solid solutions of GaAlN at high temperatures.11 However, manganese(II) bis(dimethyl)amide is unknown and our initial attempts to make it via metathesis of MnX2 (X = Cl, Br) and LiNMe2 have missed the target. On the other hand, a related silylated amine derivative, i.e., manganese(II) bis(trimethylsilyl)amide Mn[N(SiMe3)2]2, is available from the metathetical reaction in the system MnX2 (X = Cl, Br)/LiN(SiMe3)2.12 In general, both the metal silylamide and metal regular amide derivatives are thought to undergo similar ammonolysis/nitridation reactions with the evolution of volatile HN(SiMe3)2 or HNMe2, respectively, and NH3, and the formation of a nitride with increasing temperatures. This will be true if the crucial ammonolysis step results in complete transamination and efficient removal of the amine groups from the system. Otherwise, the final materials can be contaminated with silylamine group decomposition by-products, mainly, TM–N–Si–C species.
First, for the Mn-silylamide derivative, one can expect under favorable conditions a temperature-driven idealized reaction sequence shown below.13
| Mn[N(SiMe3)2]2 + 2NH3 → Mn(NH2)2 (amide) + ↑2HN(SiMe3)2 transamination |
| Mn(NH2)2 → Mn(NH) (imide) + ↑NH3 deamination |
| 3Mn(NH) → {–Mn–N–Mn–N–Mn–} (nitride) + ↑NH3 deamination |
Second, gallium tris(dimethyl)amide is known to react with NH3 paralleling the chemistry above and forming the polymeric gallium imide at room temperature.11,14 The imide upon nitriding pyrolysis at elevated temperatures is then converted to pure GaN nanopowders. The overall pathway constitutes a reference precursor system in the current study.
| Ga(NMe2)3 + 3NH3 → Ga(NH2)3 (amide) + ↑3HNMe2 transamination |
| 2Ga(NH2)3 → 2/n{Ga(NH)3/2}n (imide) + ↑3NH3 deamination |
| 2/n{Ga(NH)3/2}n → {–Ga–N–Ga–N–} (nitride) + ↑NH3 deamination |
Therefore, the combination of managanese(II) bis(trimethylsilyl)amide and gallium(III) tris(dimethyl)amide provides a viable precursor system for the formation of mixed metal-nitride linkages already at the bimetallic precursor level, which creates advantageous conditions for incorporating Mn-centers in the evolving GaN lattice at higher temperatures.
In this study, we utilized the bimetallic system Ga(NMe2)3/Mn[N(SiMe3)2]2 with various initial Ga![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn atomic ratios and conversion temperatures to promote the GaN/Mn powder formation under a broad range of conditions. The resulting nanopowders were thoroughly characterized including detailed measurements of their magnetic properties.
:Mn atomic ratios and conversion temperatures to promote the GaN/Mn powder formation under a broad range of conditions. The resulting nanopowders were thoroughly characterized including detailed measurements of their magnetic properties.
2. Experimental
Synthesis
The synthesis method used to make GaN/Mn powders was based on the reported anaerobic preparation of pure nanocrystalline gallium nitride GaN from polymeric gallium imide {Ga(NH)3/2}n.14 To achieve the synthesis of various Mn-doped materials while conforming to the basic procedure, two major mixtures of Ga(NMe2)3 and Mn[N(SiMe3)2]2 with the initial Mn-contents of 5 and 20 at.% were employed. Also, one precursor mixture was made with 0.1 at.% Mn and another one with 50 at.% Mn to check the extreme metals compositions. Additionally, a sample with 10 at.% Mn was prepared to investigate a modification of the crucial transamination stage. For the 0.1, 20, and 50 at.% Mn-contents, a two-stage pyrolysis of the respective room temperature precursor under a NH3 flow, 0.5 L min−1, was performed: first stage, 18 h at 150 °C and second stage, 4 h at 500, 700 or 900 °C to afford gray-beige to gray-brownish final powders. For the 5 at.% Mn-content bimetallic precursor, one-stage pyrolysis under a NH3 flow, 0.5 L min−1, 4 h at 500, 700 or 900 °C was carried out to yield similarly colored powders. For this Mn-content, one additional sample was prepared at 500 °C via the two-stage pyrolysis. The resulting samples reflected both the various initial proportions of Mn and the range of pyrolysis temperatures as the predominant experimental factors. The synthesis routes are shown in Fig. 1.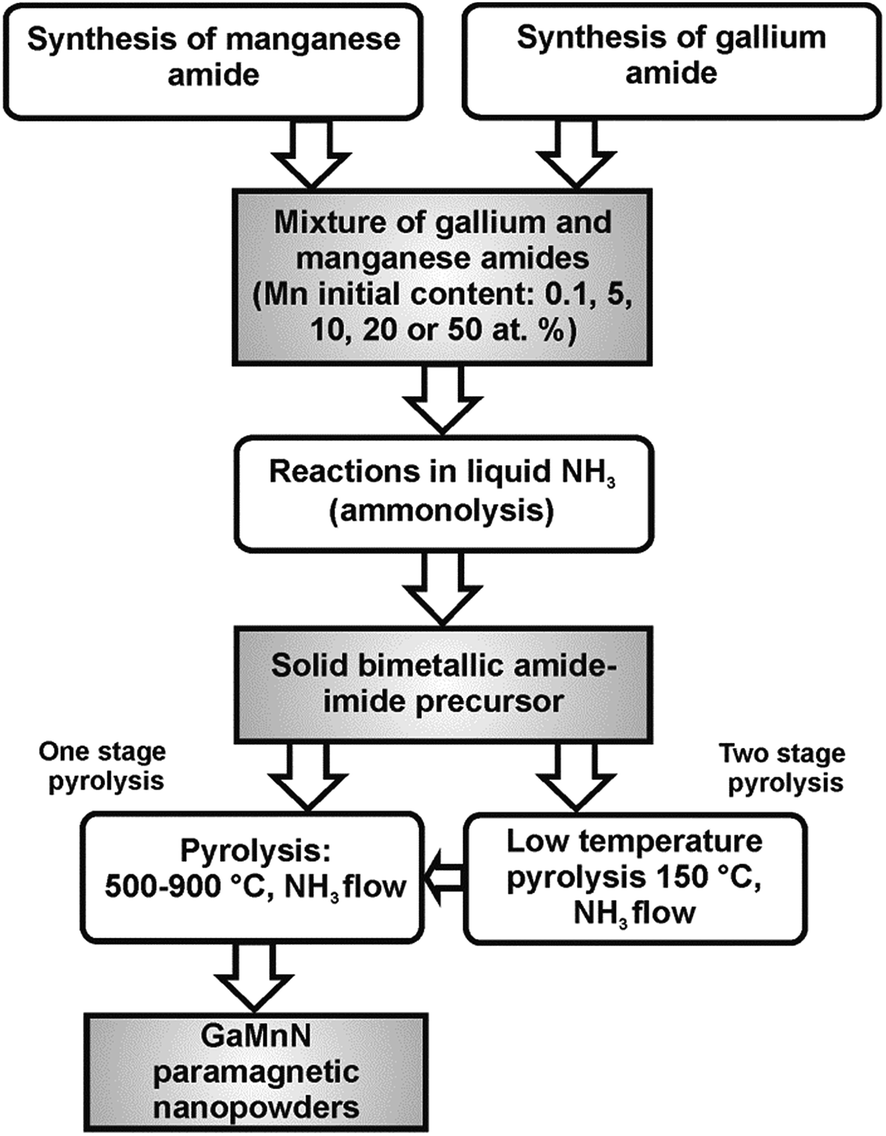 | ||
| Fig. 1 Synthesis of GaN/Mn nanopowders in the system Ga(NMe2)3/Mn[N(SiMe3)2]2/NH3. | ||
Sample labeling
The samples were labeled to show the basic parameters of their preparation, i.e., the initial Mn-content and the temperature pyrolysis levels. For example, the label 20Mn-150/700 refers to the powder from the bimetallic precursor containing 20 at.% Mn and pyrolyzed, first, at 150 °C followed by pyrolysis at 700 °C whereas 5Mn-900 describes the powder from the relevant precursor with 5 at.% Mn which was pyrolyzed only at 900 °C.Characterization
All product powders were characterized by the standard powder XRD technique with a X'Pert Pro PANalytical diffractometer (Cu Kα source; 2Θ = 20–80°). Average crystallite sizes were evaluated from the Scherrer's equation applying the Rietveld refinement method. For the evaluation, changes of the line profile parameters compared to a standard sample were utilized. Our standard was a polycrystalline alumina sintered body with an average grain size over 5 μm subjected to stress relief annealing. The profile parameters depend on the instrument settings used for data collection and on the profile function used for the refinement. In our analysis the full Voigt function was used to describe the profile of the measured diffraction lines. The total profile width is a convolution of the Gaussian profile part and of the Lorentzian profile part and these parts are combined numerically. In such a method, the full width at half-maximum (fwhm) is only one of several fitted parameters. X-ray fluorescence XRF determinations were performed on PANalytical WDXRF Axios mAX spectrometer equipped with a 4 kW Rh lamp. FT-IR spectra for solids (KBr pellets) were collected on a Nicolet 380 spectrometer. Micro-Raman analysis was done by HORIBA LabRAM HR spectrometer with spectral resolution 0.5 cm−1. SEM/EDX study was performed using a Hitachi Model S-4700 scanning electron microscope. Magnetization of the samples was measured as a function of magnetic field (up to 7 Tesla) and temperature (2–400 K) using a superconducting quantum interference device (SQUID) magnetometer. The powder samples were placed in capsules showing controlled diamagnetic signal. To reduce the ratio of holder signal to signal of the measured sample, the sample mass was maximized with respect to the upper limit of the SQUID operation range.3. Results and discussion
The idea to use the bimetallic system Ga(NMe2)3/Mn[N(SiMe3)2]2/NH3 (Me = CH3) to make GaN/Mn powders via thermally-driven transamination/deamination chemistry has a firm support from the available data on the related conversions as already discussed.11,13,14 The crucial transamination step in this precursor system is usually incomplete and the presence of some contaminant amorphous N–Si–C species from the thermal decomposition of residual N(SiMe3)2 groups cannot be excluded. Namely, at sufficiently high temperatures, the competing decomposition pathways start to prevail and lead, first, to polymeric N–Si–C species and, eventually, to silicon nitride and/or silicon carbide by-products.15,16 Al this means that the application of Mn[N(SiMe3)2]2 in the transamination/deamination reactions can be, potentially, a source of this kind of contaminations. It can especially be true in this study where excess quantities of the manganese precursor are purposely applied.In order to evaluate the suitability of pure Mn[N(SiMe3)2]2 to form nitrides, a study of its transamination/deamination chemistry was carried out and the results will be published elsewhere. In summary of this research, the reactions of the compound with liquid NH3 appear to result in an incomplete ammonolysis and yield at room temperature only partially transaminated precursor containing mostly the –NH2 and –N(SiMe3)2 functionalities. The reactions of this precursor with ammonia gas already at the temperature as low as 150 °C yield pure nanocrystalline manganese nitride η-Mn3N2. One could conclude, therefore, that upon suitable adjustments the manganese precursor would undergo equally efficient reactions with ammonia in the bimetallic system Ga(NMe2)3/Mn[N(SiMe3)2]2.
FT-IR spectroscopy of precursors
In Fig. 2, the FT-IR spectra for the reference monometallic reaction systems, Ga(NMe2)3/liquid NH3 and Mn[N(SiMe3)2]2/liquid NH3, and for the 5, 10, 20, and 50 at.% Mn-content/liquid NH3 bimetallic reaction systems are shown. The FT-IR spectrum for product from the system Ga(NMe2)3/liquid NH3 (upper left) is typical for polymeric gallium imide with residual NMe2 groups.14 The medium intensity broad bands at 3136 cm−1 and 1506 cm−1 are for N–H vibrations in the imide groups Ga–N(H)–Ga, and a strong broad peak at 557 cm−1 is for Ga–N stretches in the Ga–N lattice. The remaining peaks reflect the residual NMe2 groups.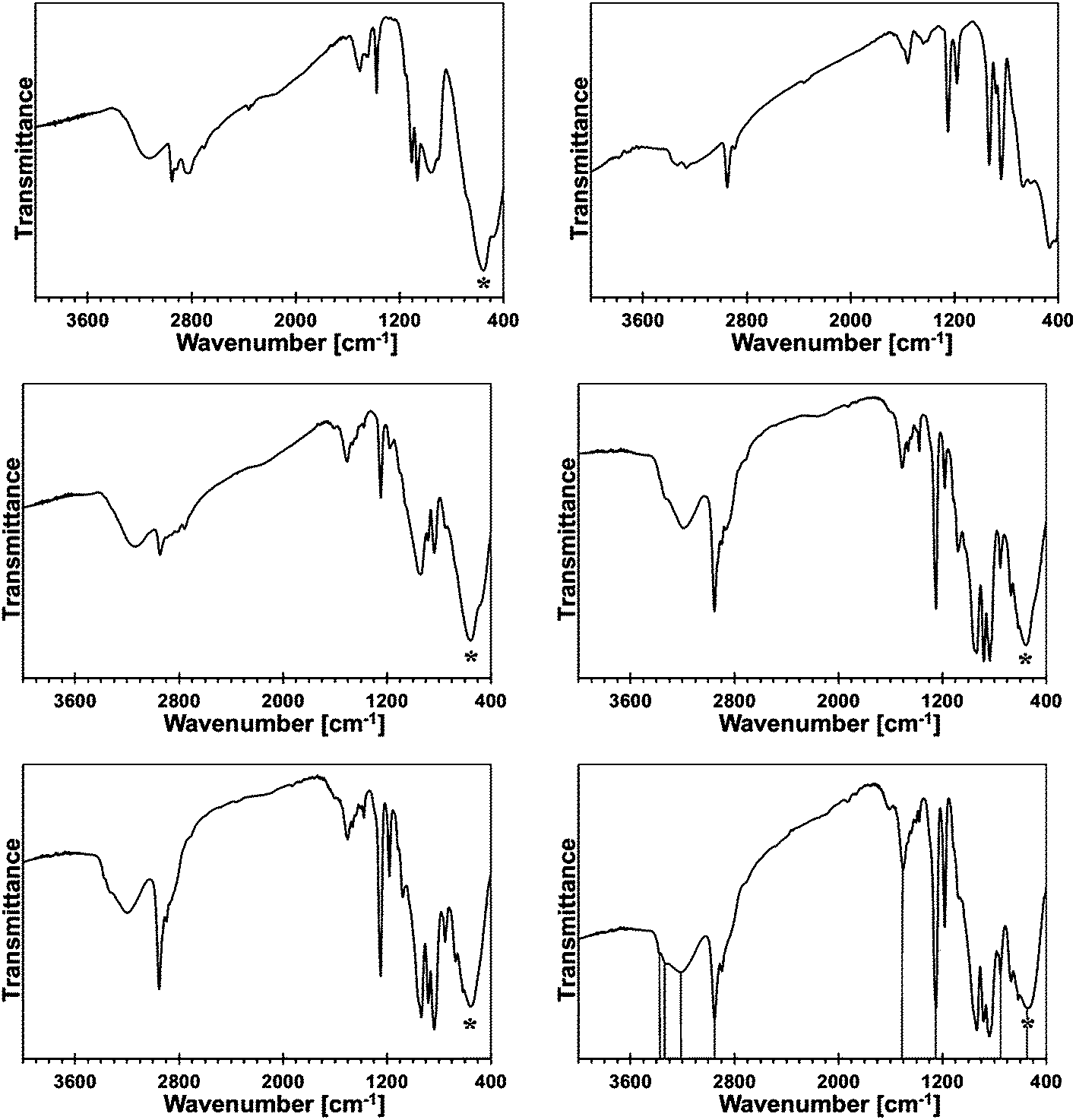 | ||
| Fig. 2 FT-IR spectra of mono and bimetallic precursors after reactions with liquid NH3: upper left – Ga(NMe2)3/liq. NH3, upper right –Mn[N(SiMe3)2]2/liq. NH3, middle left – 5 at.% Mn, middle right – 10 at.% Mn, lower left – 20 at.% Mn, lower right – 50 at.% Mn. Asterisks show the Ga–N stretch, vertical lines in the lower right are guides to the eye for important bands. | ||
The FT-IR spectrum for an insoluble product (upper right), which is isolated at room temperature from the reaction of pure Mn[N(SiMe3)2]2 in liquid NH3, confirms its incomplete transamination as discussed earlier. There are two peaks at 3332 and 3267 cm−1 that are assigned to N–H stretches and one peak at 1564 cm−1 assigned to N–H deformation in the Mn–NH2 amide groups. The remaining peaks are assigned to unconverted Mn–N(SiMe3)2 groups. Especially, a relatively strong peak at ca. 1250 cm−1 is diagnostic for SiMe3 groups. In conclusion, the product appears to be best approximated as {Mn[N(SiMe3)2]x(NH2)2−x}.
The FT-IR spectra for the 5 (middle left), 10 (middle right), 20 (lower left), and 50 at.% Mn (lower right) are all consistent in that the relative intensities of the peaks assigned to residual N(SiMe3)2 groups increase in a systematic way with the higher and higher proportion of initial Mn. This is best seen when comparing the peaks at ca. 2950/2900 and 1250 cm−1 for the C–H stretches and Si–CH3 deformation in the Mn–N(SiMe3)2 groups, respectively, to the strong peak at ca. 550–560 cm−1 for the Ga–N stretches. The predominant peaks for N–H species become now characteristic of the imide type groups, i.e., at ca. 3190–3220 and 1500–1510 cm−1. This suggests that a significant proportion of the Mn–NH2 groups reacts with in statu nascendi Ga–NH2 to form Ga–N(H)–Mn linkages according to Ga–NH2 + H2N–Mn → {Ga–N(H)–Mn} + ↑NH3. In this regard, a new peak of weak to medium intensity at 754 cm−1 is detected for all the bimetallic precursors. The peak is absent in the infrared spectra of both pure metal-derived precursors. We assign this peak tentatively to Mn–N stretches in the bimetallic {Ga–N(H)–Mn}-type of species.
Some of the bimetallic precursors were pyrolyzed under ammonia at 150 °C for 18 h before the pyrolysis at higher temperatures. This was done to promote an advantageous removal of the residual N(SiMe3)2 groups via transamination. This temperature is known to be safely below a range of SiMe3 thermal decomposition starting at 250–300 °C.15,16 The FT-IR spectra for the 10, 20, and 50 at.% Mn precursors of this type are shown in Fig. 3.
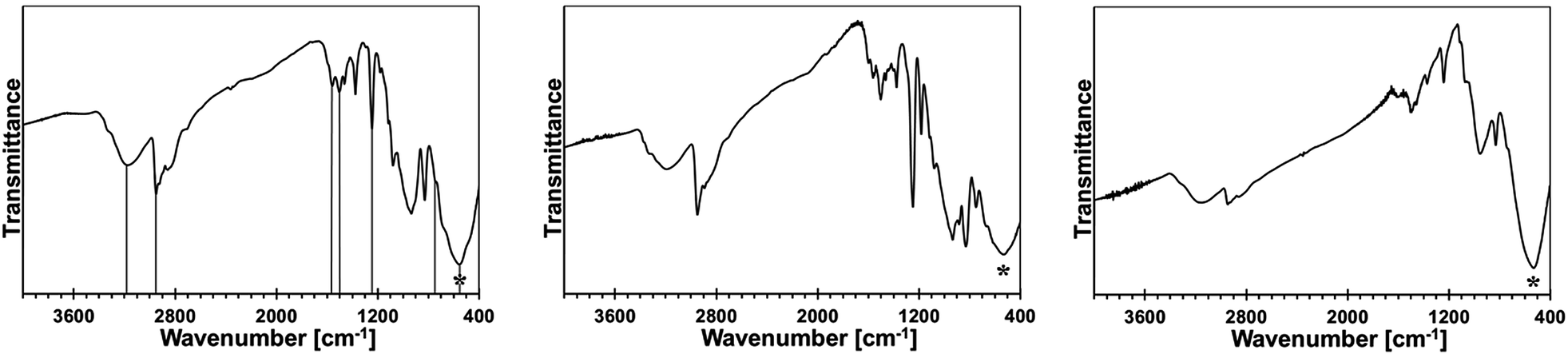 | ||
| Fig. 3 FT-IR spectra of bimetallic precursors after reactions in liquid NH3 that were additionally pyrolyzed under ammonia at 150 °C, 18 h: left – 10 at.% Mn, middle – 20 at.% Mn, right – 50 at.% Mn. Asterisks show the Ga–N stretch, vertical lines in the left are guides to the eye for important bands. | ||
The major conclusion that is drawn from comparison of these spectra with the respective spectra for their starting counterparts in Fig. 2 is that the pyrolysis under ammonia at 150 °C results in further transamination/removal of the N(SiMe3)2 groups but still some of them are retained. This is clearly seen by comparing the relative intensities of the C–H stretch in Si–CH3 at 2950 cm−1 to the imide N–H stretch at 3170–3200 cm−1 or the Si–CH3 deformation at 1245–1250 cm−1 to the Ga–N stretch at 560 cm−1 in the related spectral pairs. Also, a detailed inspection of the spectra supports the notion that the progressing transamination of Mn–N(SiMe3)2 groups during the 150 °C-pyrolysis results in increasing quantities of the Mn–NH2 groups but does not have any significant impact on an additional Ga–N(H)–Mn linkage formation as judged by the relative intensities of the diagnostic peak at ca. 750 cm−1.
SEM/EDX examination
The morphology of the powders was examined by SEM/EDX and typical images are shown in Fig. 4–6. In Fig. 4, the pictures for the initially 5 at.% Mn-content materials pyrolyzed at 500, 700, and 900 °C are included. In all these cases, a similar grain-type morphology of aggregates in confirmed. The element mapping confirms in all cases that both Ga and Mn are homogenously dispersed. An attempted mapping for Si was within a level of background noise. | ||
| Fig. 4 SEM images of powders with initial 5 at.% Mn-content: left – 5Mn-500, middle – 5Mn-700, right – 5Mn-900. | ||
Fig. 5 shows the SEM pictures for all pyrolyzed powders with the initial 20 at.% Mn-content. The overall appearance of the large pieces seen at the relatively low magnification suggests a transient gluey state of the precursor before full solidification to blocky chunks, especially, at 500 and 700 °C. The evolution of regularly shaped crystallites with increasing pyrolysis temperatures is clearly evident. This is already visible at 700 °C (middle picture) and takes a form of heterogenous areas with distinct morphologies at 900 °C (right picture). In the latter, a columnar growth of elongated sharp-ended microcrystallites can be compared with a grainy texture of the large bulk pieces. EDX spot analyses, labeled with the numbers in white, were carried out for distinct morphological features in the samples. Due to superposition of neighboring particles and varying sample thicknesses, only semi-quantitative results were obtained. For 20Mn-150/500 (Fig. 5, left), smooth and solid area 1 is characteristic of the largest Mn-content among the analyzed spots whereas area 3 in the morphologically different piece has this content lower by one order of magnitude relative to Ga. For 20Mn-150/700 (Fig. 5, middle), spot 1 in the grainy aggregate shows a similar Ga/Mn ratio compared to spot 1 in the previously discussed sample, and spot 2 in an apparently blocky crystallite is significantly enriched in Mn, which indicated specific phase evolution and separation after pyrolysis at 700 °C. This phenomenon is clearly seen in sample 20Mn-150/900 from yet higher temperature of pyrolysis at 900 °C (Fig. 5, right). The sharp-ended microcrystallites analyzed in spot 1 show a lower Mn-content than the blocky aggregate in spot 2. It is also found that the increased proportions of Mn were frequently associated with higher proportions of Si, the latter element seen at the relatively low levels in all samples.
 | ||
| Fig. 5 SEM images of powders with initial 20 at.% Mn-content: left – 20Mn-150/500, middle – 20Mn-150/700, right – 20Mn-150/900. The numbers in white show morphologically different areas analyzed by EDX. | ||
Two bimetallic precursors with the extreme Mn-contents of 0.1 and 50 at.% were pyrolyzed under ammonia at 900 °C. The SEM images of the resulting powders (not shown) confirmed for 0.1Mn-150/900 the homogenous grainy morphology well compared with the product from the respective 5 at.% Mn-content conversion (Fig. 4, right). For 50Mn-150/900, large microcrystallites were embedded in the agglomerated matrix of much smaller particles. For this material, the EDX analyses showed increased proportions of Mn and Si in the large crystallites compared with the bulky matrix.
The EDX mapping was done for a large inhomogeneous piece in 50Mn-150/900 and the results are shown in Fig. 6. Generally, there are regions associated mostly with a solid crust with well correlated Si and Mn element distributions but there are also small areas where Mn does not correlate either with Si or Ga as, for example, shown with the ellipse. There are also features that can be attributed to some combination of the Mn and Si elements with no Ga contribution, which are enclosed in the black and white rectangle. These results point out to a complex composite make-up of the powder.
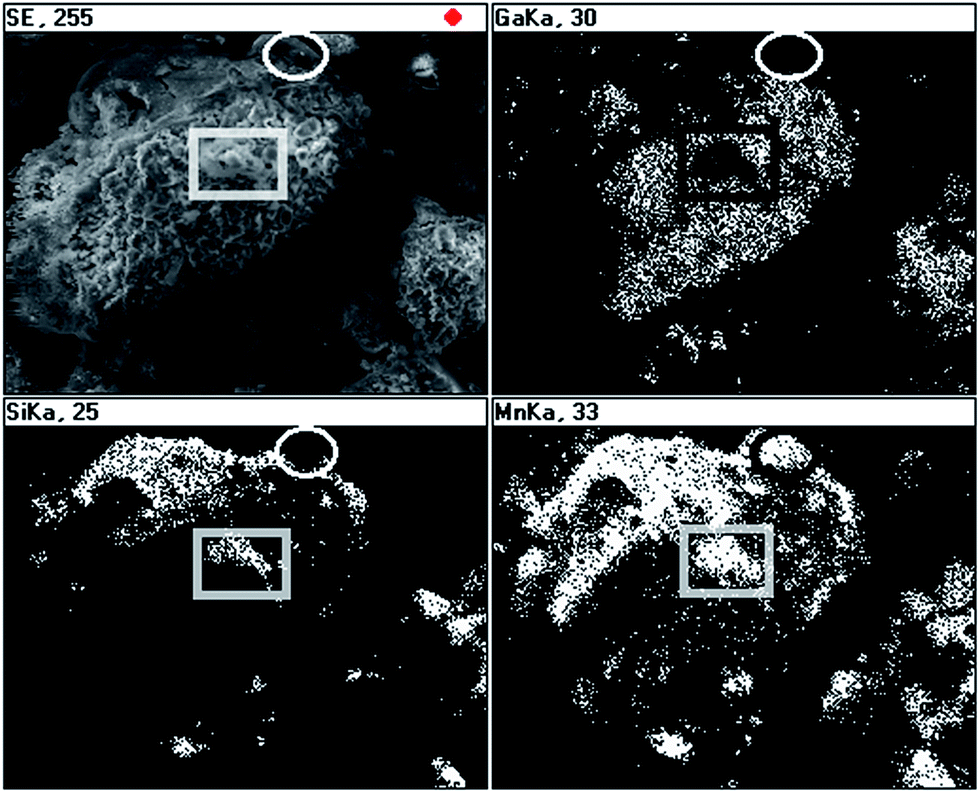 | ||
| Fig. 6 SEM/EDX element mapping for powder with initial 50 at.% Mn-content: upper left – low-magnification image for 50Mn-150/900, upper right – Ga-distribution, lower left – Si-distribution, lower right – Mn-distribution. | ||
XRF analysis
The SEM/EDX analyses were consistent with incomplete transamination chemistry during ammonolysis at ambient, retention of some N(SiMe3)2 groups, and their thermal degradation under reactive conditions of the pyrolysis under an ammonia flow. The residual N–Si–C moieties became the source of Si that could engage a part of Mn, especially if excess of Mn was available, to form by-products in the system Mn–N–Si–C in addition to the target materials in the system Ga–Mn–N. In order to estimate the elemental composition of the powders, X-ray fluorescence XRF determinations were done and the results were normalized for the Ga, Mn, and Si contents. Table 1 contains the results for the 5 and 20 at.% Mn-contents and Table 2 for the 0.1 and 50 at.% Mn-contents.| Element X | 500 °C | 700 °C | 900 °C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Amount [at.%] | Ga:X |
Mn:Si |
Amount [at.%] | Ga:X |
Mn:Si |
Amount [at.%] | Ga:X |
Mn:Si |
|
Initial Mn-content 5 at.% (one-stage pyrolysis, Ga![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :Mn = 19:1) :Mn = 19:1) |
|||||||||
| Ga | 94.0 | 1 | — | 94.3 | 1 | — | 91.8 | 1 | — |
| Mn | 3.9 | 19.0 | — | 4.1 | 18.0 | — | 4.3 | 16.9 | — |
| Si | 2.1 | 17.8 | 0.9 | 1.6 | 23.7 | 1.3 | 3.9 | 9.5 | 0.6 |
|
|||||||||
| Initial Mn-content 20 at.% (two-stage pyrolysis, Ga:Mn = 4:1) |
|||||||||
| Ga | 84.4 | 1 | — | 84.8 | 1 | — | 81.6 | 1 | — |
| Mn | 12.7 | 5.2 | — | 12.5 | 5.3 | — | 13.1 | 4.9 | — |
| Si | 2.9 | 11.6 | 2.2 | 2.7 | 12.7 | 2.4 | 5.3 | 6.2 | 1.3 |
| Element X | 900 °C | ||||
|---|---|---|---|---|---|
| Initial Mn-content 0.1 at.% (Ga:Mn = 999:1) |
Initial Mn-content 50 at.% (Ga:Mn = 1:1) |
||||
| Amount [at.%] | Ga:X |
Amount [at.%] | Ga:X |
Mn:Si |
|
| Ga | 99.8 | 1 | 73.7 | 1 | — |
| Mn | 0.2 | 351 | 21.6 | 2.7 | — |
| Si | — | — | 4.7 | 6.3 | 2.3 |
For the samples with the initial 5 and 20 at.% Mn-contents, the major conclusion is that a majority of the initial Mn, if not all, remains in the powders regardless of the pyrolysis temperature (Table 1). The second conclusion is that quite significant quantities of Si are retained as evaluated from the Mn:Si element ratios in the product powders. The initial Mn:Si ratio in Mn[N(SiMe3)2]2 is 1:4 or 0.25. If one of the two N(SiMe3) groups is removed via transamination, this ratio will be 0.5. The latter value is close to only one case of 5Mn-900 with Mn:Si equal to 0.6. In all other powders, it is larger than that reaching 2.4 for 20Mn-150/700. This ratio depends to some extent on the applied temperature – it peaks up after pyrolysis at 700 °C and is the lowest after pyrolysis at 900 °C. It is a result of a fine interplay between the competing transamination of MnN(SiMe3)2 with removal of volatile silazanes and the thermal decomposition of silazanes with Si retention at increased temperatures.
Somewhat different properties are displayed by the system with the extremely large initial proportion of the manganese precursor, i.e., 50 at.% or Ga:Mn = 1 (Table 2). The product 50Mn-150/900 has the Ga:Mn ratio equal to 2.7 which is consistent with significant losses of manganese. Apparently, such a large starting quantity of Mn[N(SiMe3)2]2 causes some unreacted excess of it to sublime out during initial heating stages. Interestingly, the Mn:Si ratio of 2.3 is in the range of 1.3–2.4 seen for the powders from the initial 20 at.% Mn-content indicating, again, similar factors that lead to retention of Si in the products.
The use of the extremely small proportion of Mn[N(SiMe3)2]2 of 0.1 at.% (Ga:Mn = 999:1) resulted in the 900 °C-powder that was analyzed with 0.2 at.% of Mn at the background noise level of the method as applied. This could be the major reason for the relatively large discrepancy between the initial and analyzed Mn-contents.
Powder XRD determinations
Some XRD patterns consisted of extremely broad peaks (“halos”) that are typical for nanocrystalline GaN in the low nanosized range. They can be indexed either as the hexagonal h-GaN or cubic c-GaN polytype. In fact, they are characteristic of phase inhomogeneous GaN which can be best described as nanocrystallites made of hexagonal and cubic closed-packed layers.14,17 In several cases, the deconvolution of patterns by best fitting failed to accommodate for real peak intensities and this was assumed to result mainly from complex texturing/preferential crystallite growth that was often confirmed by SEM examinations. An unusual broadening of peak bases, that was seen in some cases, was attributed to bimodal size distributions, i.e., the presence of larger crystallites and much smaller ones that belonged to two different distribution modes in the nanosized range. However, it is quite possible that this phenomenon is due to specific crystallite texturing, instead. There is also one case (vide infra) where very characteristic broadenings of the reflections indicate an extrinsic (double deformation) type of stacking faults present in the 2H GaN lattice as reported earlier by one of the co-authors.18The XRD patterns for the 5 at.% Mn-derived powders (500, 700, and 900 °C) are presented in Fig. 7 and for the 20 at.% Mn-derived powders (500, 700, and 900 °C) in Fig. 8. In Fig. 9, the XRD patterns for the extreme compositions are shown, i.e., for the powders prepared at 900 °C from the initial 0.1 and 50 at.% Mn-contents. Fig. 9 includes also the bar charts for hexagonal h-GaN and cubic c-GaN. Table 3 contains the basic structural parameters for the identified phases which were calculated from the XRD data.
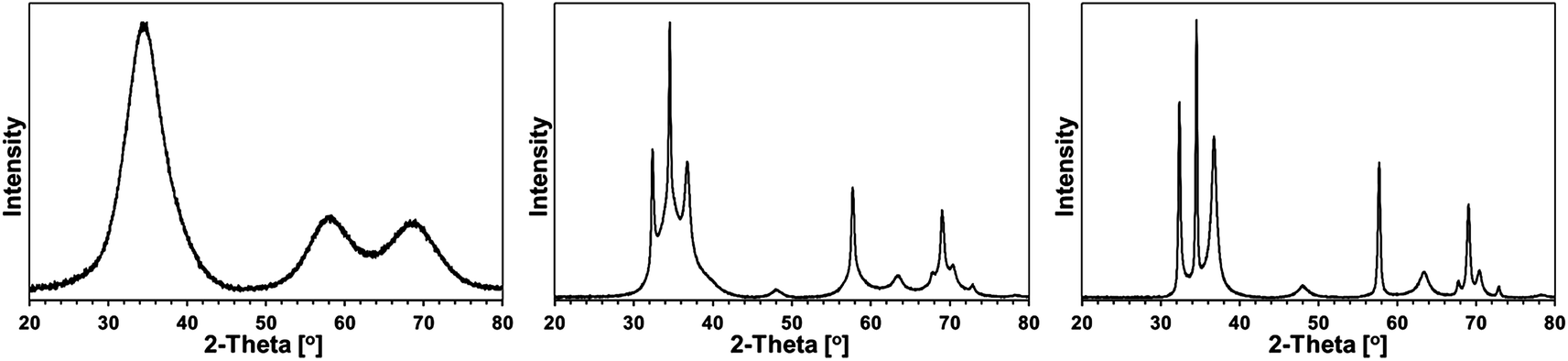 | ||
| Fig. 7 XRD patterns for powders with initial 5 at.% Mn-content: left – 5Mn-500, middle – 5Mn-700, right – 5Mn-900. | ||
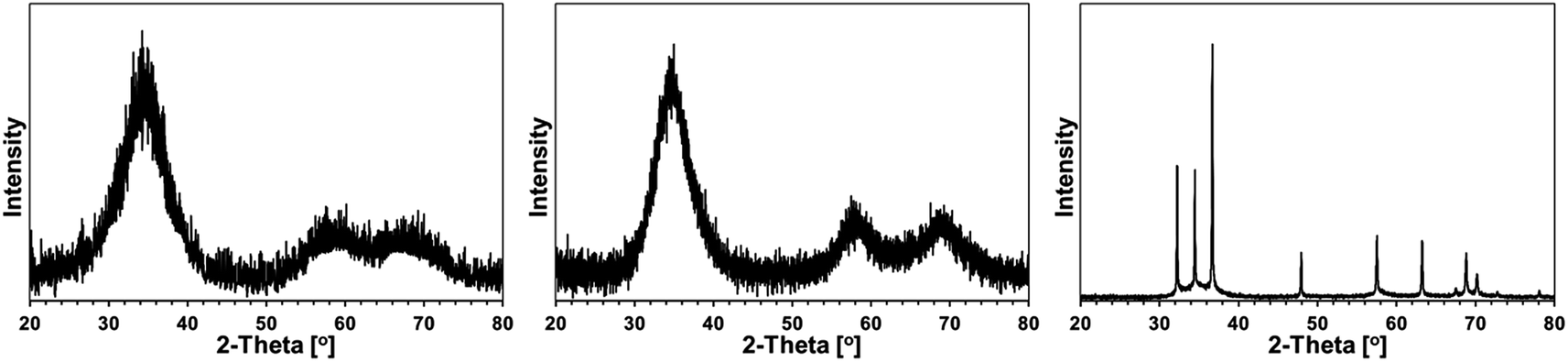 | ||
| Fig. 8 XRD patterns for powders with initial 20 at.% Mn-content: left – 20Mn-150/500, middle – 20Mn-150/700, right – 20Mn-150/900. | ||
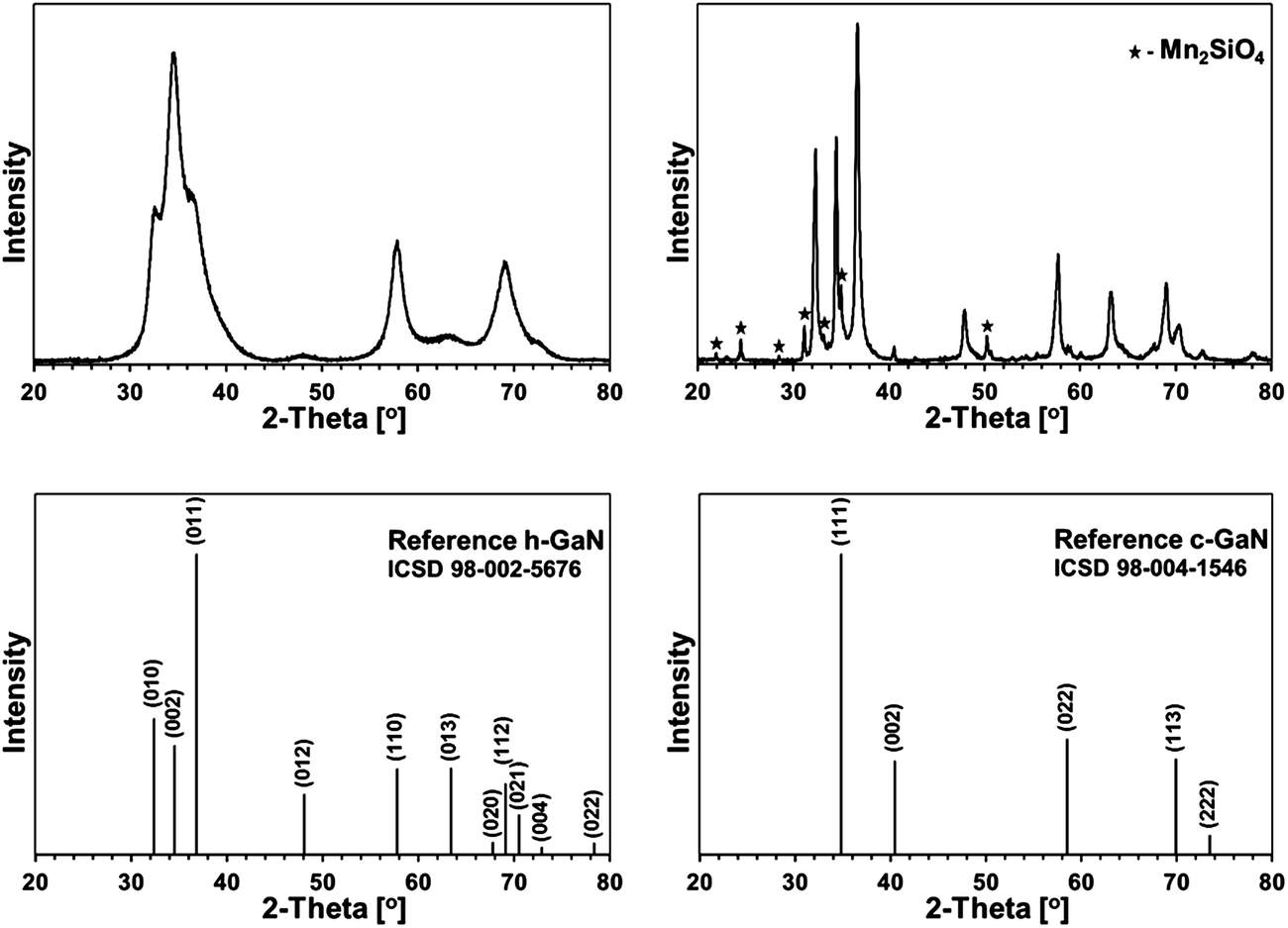 | ||
| Fig. 9 XRD patterns for powders with the extreme Ga:Mn compositions: upper left – 0.1Mn-150/900, upper right – 50Mn-150/900. The peaks assigned to orthorombic Mn2SiO4 are indicated with asterisks. The bar charts in the lower side are for hexagonal h-GaN (ICDD 98-002-5676) – lower left and cubic c-GaN (ICDD 98-004-1546) – lower right. | ||
| Sample | Phase: content [%] | Lattice parameters a [Å], c [Å] | Average crystallite size D [nm] |
|---|---|---|---|
| 5Mn-500 | c-GaN (phase inhomogeneous): 100 | a = 4.45 | 1.5 |
| 5Mn-700 | c-GaN: 29 | a = 4.51 | 4 |
| h-GaN (large nano): 18 | a = 3.20 | 22 | |
| c = 5.19 | |||
| h-GaN (small nano): 53 | a = 3.19 | 3 | |
| c = 5.27 | |||
| 5Mn-900 | h-GaN (large nano): 39 | a = 3.20 | 36 |
| c = 5.19 | |||
| h-GaN (small nano): 61 | a = 3.19 | 5 | |
| c = 5.19 | |||
| 20Mn-150/500 | c-GaN (phase inhomogeneous): 100 | a = 4.69 | 1.1 |
| 20Mn-150/700 | c-GaN (phase inhomogeneous): 100 | a = 4.57 | 1.6 |
| 20Mn-150/900 | h-GaN (large nano): 44 | a = 3.20 | >100 |
| c = 5.19 | |||
| h-GaN (small nano): 56 | a = 3.19 | 3 | |
| c = 5.22 | |||
| 0.1Mn-150/900 | c-GaN: 54 | a = 4.52 | 5 |
| h-GaN: 46 | a = 3.20 | 4 | |
| c = 5.29 | |||
| 50Mn-150/900 | c-GaN: 2 | a = 4.44 | 45 |
| h-GaN: 79 | a = 3.20 | 22 | |
| c = 5.19 | |||
| Mn2SiO4: 19 | — | — |
The patterns for all three 5 at.% Mn-derived powders (Fig. 7) can be satisfactorily indexed as belonging to the GaN polytypes with bimodal size distributions. No other crystalline phases are discernible. Apparently, the small quantity of Si detected by XRF in these materials (Table 1) is either below detection limits of X-ray diffraction or it is due to amorphous characteristics of the likely N–Si–C-bearing species.
In particular for 5 at.% Mn-contents, powder 5Mn-500 shows a typical “halo” pattern for phase-inhomogeneous, low temperature prepared GaN powders on the verge of crystallinity with an estimated average crystallite size D of 1.5 nm. The pyrolysis at the higher temperature of 700 °C (5Mn-700) results in some c-GaN, D = 4 nm, and the major h-GaN, the latter best fitted with two modes of size distribution, D = 3 and D = 22 nm. After the pyrolysis at 900 °C (5Mn-900), no c-GaN is found and the exclusive h-GaN is characteristic of the larger average crystallite sizes in the bimodal distribution, D = 5 and D = 35. These results differ somewhat from the comparable synthesis of pure GaN via the same anaerobic route by the appearance of c-GaN in 5Mn-700 and the bimodal size distribution of h-GaN in the powders. This implies specific impact of the Mn-bearing species on GaN crystallization.
The powders with the initial 20 at.% Mn-contents are characteristic of retarded GaN crystallization up to 700 °C (Fig. 8 and Table 1). The pattern for 20Mn-150/500 confirms the beginning of crystallization and phase-inhomogeneous GaN, which is typical for the pyrolysis at 500 °C. However, a very similar pattern and the crystallographic features are obtained also for 20Mn-150/700, which is rather surprising and uncommon for GaN powders prepared in this temperature range. Apparently, this is due to a thinning effect that is likely caused by the available residues from decomposition of some N(SiMe3)2 groups. Pyrolysis at 900 °C results in the exclusive h-GaN polytype with a bimodal size distribution that includes a fraction of very well crystallized h-GaN, D > 100 nm, in addition to a fine fraction with D = 3 nm. The SEM images support the formation of both the large elongated microcrystals and the much smaller size homogeneous nanocrystallites, the latter forming blocky agglomerates (Fig. 6, right). In this regard, such small GaN nanocrystallites have not yet been observed by us at this relatively high pyrolysis temperature. Similarly as for the 5 at.% Mn-content, the only phase is that of GaN and the Mn-additive plays a significant role in its crystallization.
Fig. 9, left, shows a XRD pattern for the powder prepared with the extremely small proportion of initial Mn of 0.1 at.% upon pyrolysis at 900 °C. It is worth to note that it is the least crystallized GaN obtained by us in this temperature range over several years of studies on this and related precursor routes to GaN powders. And for the first time, the cubic polytype c-GaN is observed after pyrolysis at 900 °C in such a high proportion of 54% (Table 3). The average crystallite sizes for c-GaN, D = 5 nm, and for h-GaN, D = 4 nm, are remarkably low and, certainly, are to be traced to the Mn-additive that even in small quantities has a pronounced impact on GaN crystallization, possibly, through Mn doping.
The initial 50 at.% Mn-content precursor mixture upon pyrolysis at 900 °C yielded a powder reactive during sample handling in air. The presence of significant quantities of manganese orthosilicate Mn2SiO4 (19%) can be explained by adventitious oxidation of some nanosized or amorphous by-products, which resulted from decomposition of excessive amounts of the manganese precursor. It is worth underlining that this is the only case in the entire study that a crystalline component other than GaN-based polytype(s) is detected by XRD. In addition to Mn2SiO4, the product is composed of the major h-GaN in 79%, D = 22 nm, and the minor c-GaN, D = 45 nm, the latter in the relatively low amount of 2%.
Specifically, it can reasonably be argued that Mn2SiO4 could have formed by oxidation from such plausible by-products as many of manganese silicides MnxSiy19 or the binary manganese silicon nitride MnSiN2,20 not to mention very possible transient Mn–N–Si–C species from decomposition of the excess of Mn[N(SiMe3)2]. In this regard, the oxidation of Mn11Si19 in dry oxygen at 800 °C was found to produce amorphous silica SiO2 layers on the silicide's surface rather than forming manganese silicate.21a As a matter of fact, the so-called silicon monoxide “SiO” was used in reaction with MnCl2 to make Mn4Si7 nanowires that were covered with amorphous silica, which supported the propensity of manganese silicides to be oxidized with the preferential formation of amorphous silica.21b This picture can be, however, more complex if one considers a prior incorporating by manganese silicide some available carbon to form Mn–Si–C by-products.22 On the other hand, the presence of N-containing by-products with the Mn–N–Si or Mn–N–Si–C make-up seems to be better substantiated in the reaction system with ammonia. In particular, manganese silicon nitride MnSiN2 was found to be stable up to 1000 °C and when oxidized with an Ar/O2 mixture it formed various manganese silicates (rhodonite and braunite) and amorphous silica.20a
FT-IR spectroscopy of products
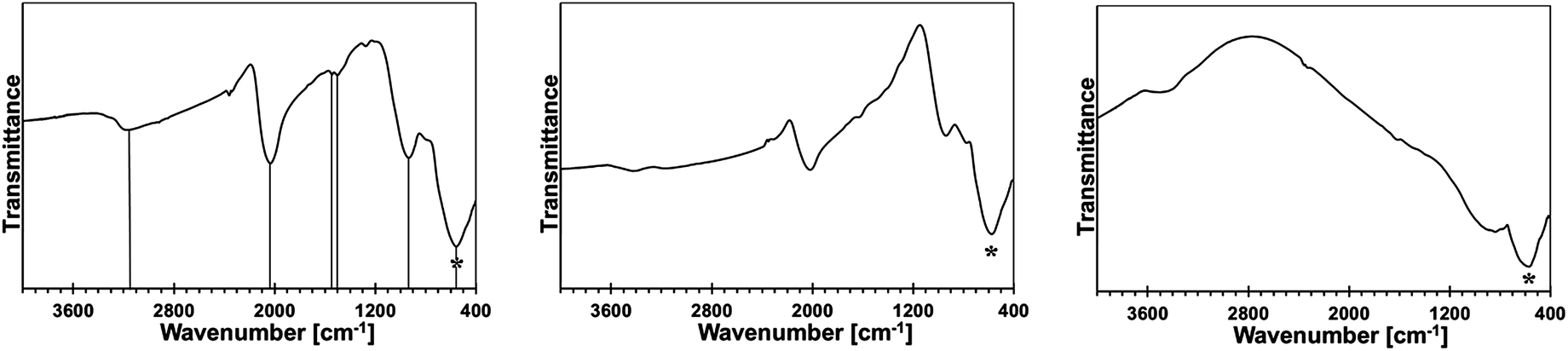
An additional insight is gained from infrared spectra that provide bonding information irrespective of crystallinity, therefore, complementing the powder XRD determinations. The FT-IR spectra for the 5 at.% Mn-derived powders (500, 700, and 900 °C) are presented in Fig. 10 and for the 20 at.% Mn-derived powders (500, 700, and 900 °C) in Fig. 11. The opaque characteristics of the KBr pellets caused occasional deformations of the baseline. The broad bands seen at 3430 and 1630 cm−1 are for adventitious water adsorbed during sample handling and KBr pellet preparation.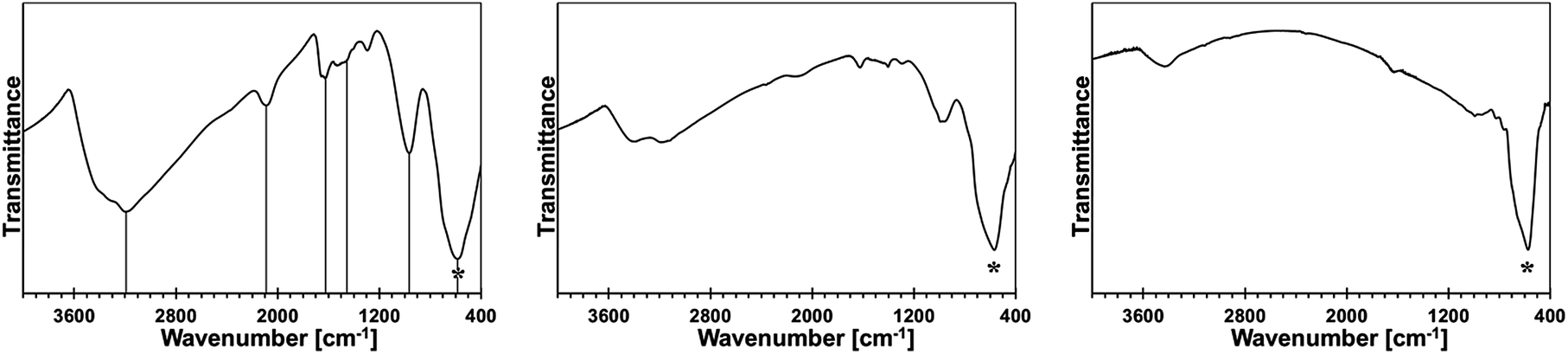 | ||
| Fig. 10 FT-IR spectra for powders with initial 5 at.% Mn-content: left – 5Mn-500, middle – 5Mn-700, right – 5Mn-900. Asterisks show the Ga–N stretch, vertical lines in the left are guides to the eye for important bands. | ||
 | ||
| Fig. 11 FT-IR spectra for powders with initial 20 at.% Mn-content: left – 20Mn-150/500, middle – 20Mn-150/700, right – 20Mn-150/900. Asterisks show the Ga–N stretch, vertical lines in the left are guides to the eye for important bands. | ||
The common feature in all spectra of the powders with the initial 5 wt% Mn-content is a strong band at 560–580 cm−1 for the Ga–N stretches in GaN [Fig. 10]. Variations in this peak's position in the range ca. 550–600 cm−1 and its symmetry and broadness for GaN nanopowders are a function of combined experimental and materials factors, among them particle size and size distribution, surface properties, oxygen content, crystallinity, present polytypes, and KBr sample turbidity. Interestingly, the spectra for 5Mn-500 and 5Mn-700 also show, respectively, a weak and a very weak band at ca. 2070–2090 cm−1 that is typical for Si–H stretches and the similar intensity bands at 1400–1500 cm−1 for CH2 deformations.23 This is accompanied by a medium to a weak intensity band at 950–980 cm−1 which is in the range of Si–H deformation mode and Si–N stretches and by medium intensity bands at ca. 3150–3400 cm−1 and 1600–1620 cm−1 assigned to N–H vibrations.24 These features are reduced in intensity with increasing pyrolysis temperature and they are absent in the 900 °C-powder, the latter having a dominant band at 570 cm−1 for GaN.
The spectral features are consistent with the incomplete transamination chemistry and retention of residual N(SiMe3)2 groups followed by their decomposition under a reactive ammonia atmosphere. It is known that SiMe3 groups and related polycarbosilanes start to decompose at temperatures as low as 250–300 °C with the formation of Si–H functionalities, which can persist up to 700–800 °C.23 Based on the FT-IR spectra, there are thus indications that the polymeric/amorphous {Mn–N(H)–Si(H)–CH2–} species are formed at lower pyrolysis temperatures. This agrees well with the XRD/EDX and XRF analyses that confirmed the presence of Si in the powders.
The FT-IR spectra for the powders with the initial 20 at.% Mn-content (Fig. 11) confirm the findings discussed earlier for the 5 at.% Mn-content. It is worth to note that the manganese nitride η-Mn2N3, which is otherwise formed in the reaction of pure Mn[N(SiMe3)2] and NH3, is reported to show no active vibrational mode in the mid-infrared range. The relevant bands are now more intense due to the higher proportion of the manganese precursor used in excess and, therefore, higher quantities of the by-products. The dominant band is at 560–570 cm−1 for GaN. The Si–H stretch at 2030 cm−1 is very intense for 20Mn-150/500, reduced in intensity for 20Mn-150/700, and absent for 20Mn-150/900 after pyrolysis at 900 °C. There are rather weak bands for the N–H vibrations at 3150–3170 and 1550 cm−1. The spectrum for 20Mn-150/900 is relatively simple and contains the major GaN-related band at 570 cm−1 and a weak and broad band extending from ca. 800 to 950 cm−1 that reflects some amorphous by-products. These qualitative results can be compared with the XRF analyses in Table 1. The analyzed ratio Mn:Si is approximately twice as high for the 20 at.% Mn-derived powders compared with the 5 at.% Mn-derived powders.
The FT-IR spectra for the 0.1 at.% Mn-150/900 and 50 at.% Mn-150/900 powders (not shown) generally display features supporting the presence of the major GaN-based product and some by-products. In this regard, the former powder shows the predominant Ga–N stretch at 580 cm−1 whereas the latter one specifically indicates that the by-product can be one of manganese silicates – a set of three medium intensity bands at 818, 857, and 945 cm−1 match well the strongest bands at 820, 867, and 950 cm−1 recorded for mineral tephroite which is orthorhombic Mn2SiO4.25 Based on this evidence and the relevant XRD data (Fig. 9) the 50 at.% Mn-150/900 powder appears to be composed of two crystalline components, i.e., GaN-type phase and Mn2SiO4. The latter could result from post-reaction adventitious oxidation of the initially formed decomposition by-products {Mn–N(H)–Si(H)–CH2}. This is further supported by the XRF data that show for this powder the ratio Mn:Si equal to 2.3 to be compared with such a ratio of 2 in Mn2SiO4. In conclusion, the majority of Mn seems to be associated with the air sensitive by-products whereas only part of it may have entered doping reactions with the gallium nitride precursor towards GaMnN.
Raman spectroscopy of products
The Raman spectra for the 700 and 900 °C-derived materials are shown in Fig. 12 and the best-fitted bands are listed in Table 4.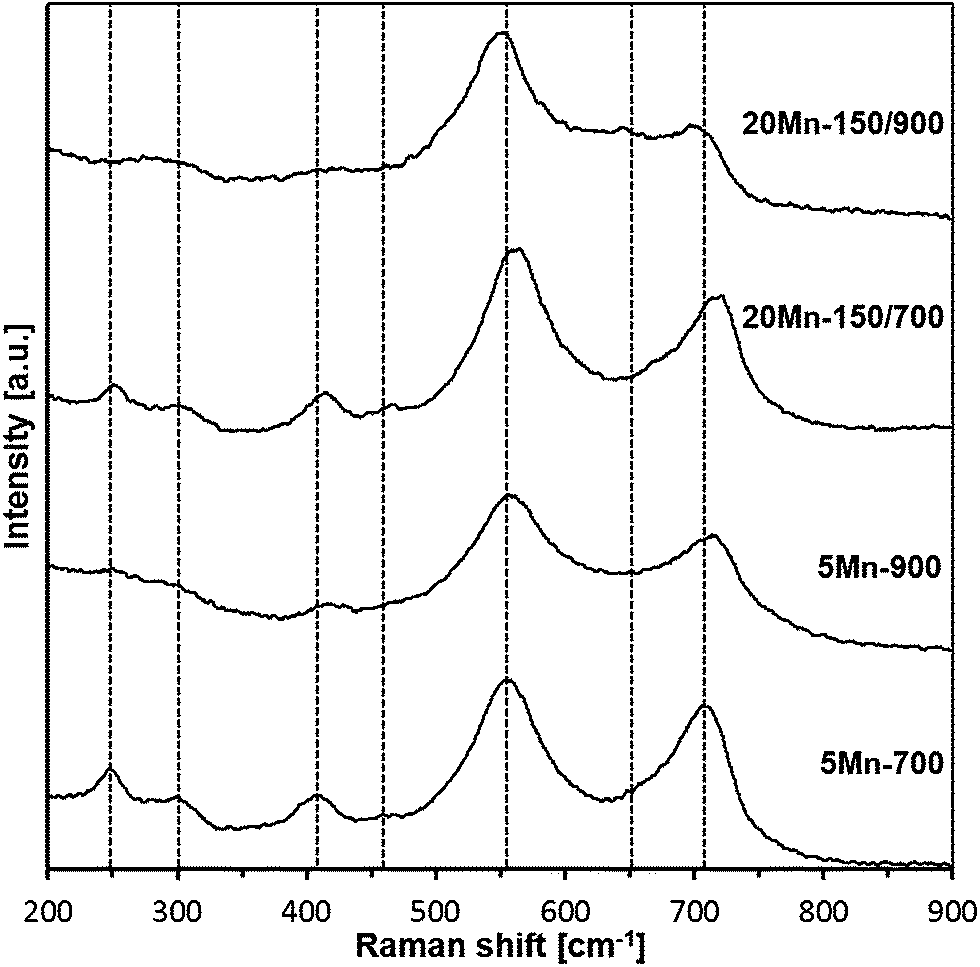 | ||
| Fig. 12 Raman spectra for 700 and 900 °C-derived nanopowders. | ||
| Assignment | 5Mn-700 | 20Mn-150/700 | 5Mn-900 | 20Mn-150/900 |
|---|---|---|---|---|
| Raman shifts [cm−1] (sh – shoulder; vw – very weak) | ||||
| LO (hexagonal and/or cubic GaN lattices) | 709 | 718 | 716 | 701 |
| N-vacancy in doped GaN | 660 (sh) | 670 (sh) | 660 (sh) | 649 (vw) |
| E2 (h-GaN lattice) | 555 | 561 | 558 | 548 |
| TO (c-GaN lattice) | ||||
| By-product | 466 (vw) | 460 | 475 (sh) | — |
| 2-Phonon difference mode | 406 | 413 | 417 | 416 |
| Disorder-activated/acoustic phonon at zone boundary | 300 | 301 | 300 (vw) | 278 |
| By-product | 247 | 252 | 244 (vw) | — |
Generally, each of four spectra contains a similar set of bands, alas, of varying intensities. The two strongest bands are typical for GaN lattices, i.e., the band in the range 701–718 cm−1 is the LO mode in h-GaN and/or in c-GaN whereas the band at 548–561 cm−1 is assigned to the E2 mode in h-GaN and/or TO mode in c-GaN.26 The rather weak bands at 649–670, 406–417, and 278–301 cm−1 are frequently observed in GaN with extensive lattice defects or doped GaN lattices including Mn-doping.26d Finally, the weak to very weak features at 400–475 and 244–252 cm−1 could not be identified in any of the available reports on Raman studies in related systems and are thus assigned to by-product contaminants. It is instructive to relate the major bands to the reported Raman shifts for the pure/undoped GaN powders prepared with the same method and at the same temperatures although measured under different laser irradiation conditions.27 For the 700 °C-derived pure GaN nanopowder, bands at 332, 410, 550, 637, and 744 cm−1 are observed implying lattice defects in GaN. In the 900°C-derived powder, there are mostly bands at 410, 570, and 730 cm−1 consistent with much better crystallized GaN. The latter property is also reflected by an enhanced relative intensity of the LO phonon mode26c agreeing well with the previously discussed XRD data. Of special interest is also an observation of the shift to lower frequencies for the E2 and LO modes compared with pure GaN, which has been seen on several occasions for Mn-doped GaN/GaMnN.26e,f In conclusion, the Raman study is consistent with the synthesis of Mn-doped GaN nanopowders that show improved crystallinity and smaller quantities of contaminants with increased synthesis temperature.
Magnetization measurements
Magnetization of the samples was measured at magnetic fields B up to 7 Tesla in the temperature range 2 K < T < 400 K. For all the samples, a diamagnetic contribution of the host GaN material was taken into account. The representative data of magnetization M as a function of magnetic field B at different temperatures are shown for sample 5Mn-150/500 in Fig. 13 and 14, the latter being an expanded high temperature part of the former.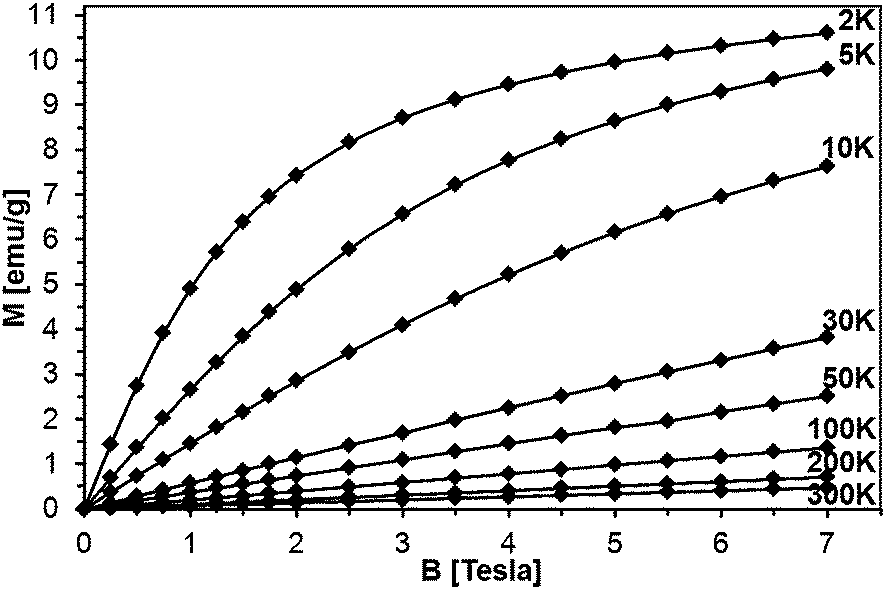 | ||
| Fig. 13 Magnetization of sample 5Mn-150/500 as a function of magnetic field B at different temperatures. The lines connect experimental points and are guides to the eye, only. | ||
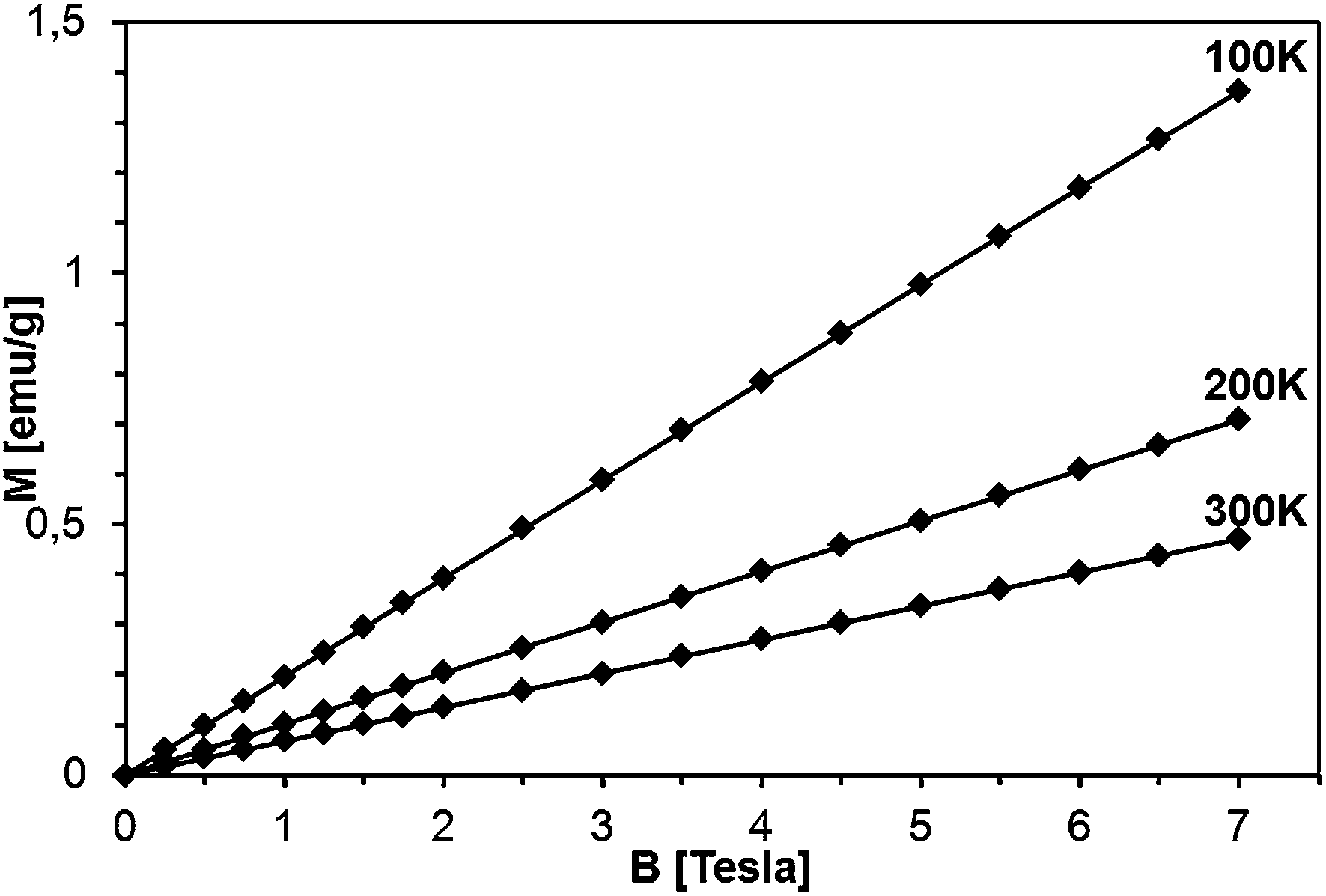 | ||
| Fig. 14 Magnetization of sample 5Mn-150/500 as a function of magnetic field at T = 100, 200, and 300 K. The lines are guides to the eye, only. | ||
The magnetization reveals rather typical paramagnetic, Brillouin-like behavior: it tends to saturate with increasing B at the lowest temperatures, whereas at higher temperatures (T > 100 K) is practically linear with B. Similar behavior is observed for all the samples, as displayed in Fig. 15, where curves M vs. B at T = 2 K are shown. It should be noted that the measured magnetization data do not correlate with the initial Mn-contents used for synthesis. This suggests that only fraction of the used manganese enters the paramagnetic Mn-doped GaN lattice since the observed M(B,T) dependence is typical for GaN doped with Mn.4,6,28 In this regard, GaMnN grown previously by other techniques usually suffered from ferromagnetic precipitations of MnxNy (or GaxMny) formed during the growth process.29 These precipitations manifest themselves by a rapid increase of magnetization at low magnetic fields (B < 0.5 Tesla) and are clearly visible at the temperatures T > 50 K. A detailed examination of the low field data (Fig. 13–15) shows that our samples are free of such effects. Therefore, one may conclude that the applied synthesis method, in contrast to many previously reported methods for GaMnN growth, produces powders free of ferromagnetic precipitations.
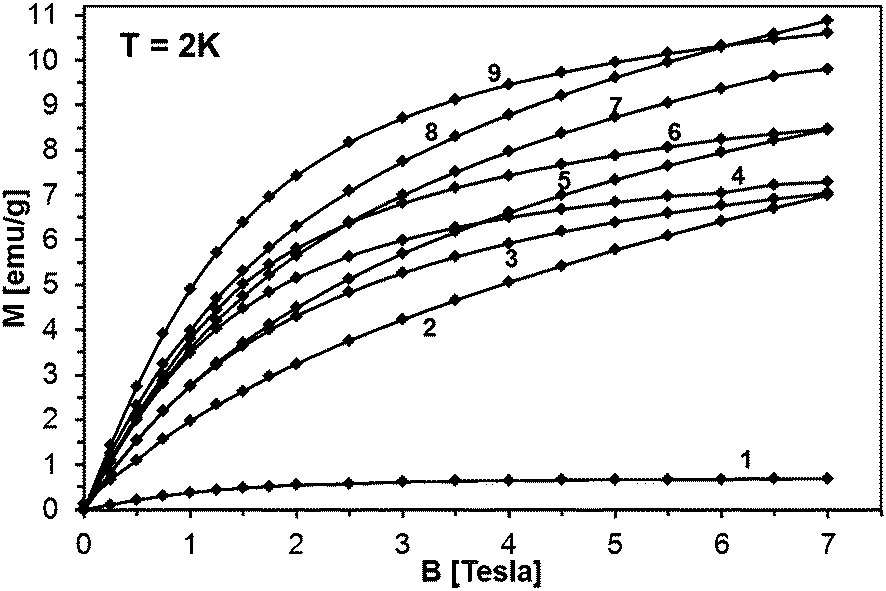 | ||
| Fig. 15 Magnetization M as a function of magnetic field strength B at T = 2 K for selected powders: 1 – 0.1Mn-150/900, 2 – 50Mn-150/900, 3 – 5Mn-900, 4 – 5Mn-500, 5 – 20Mn-150/900, 6 – 5Mn-700, 7 – 20Mn-150/500, 8 – 20Mn-150/700, 9 – 5Mn-150/500. The lines are guides to the eye, only. | ||
It should be noted that magnetization decays with temperature rather slowly, much slower than expected for a typical Brillouin-type paramagnet. This is exemplified in Fig. 16, where magnetization of sample 50Mn-150/900 is shown as a function of temperature, together with function 1/T, pinned to the data at the lowest temperature. The observed behavior suggests that the excess contribution to the magnetization results from yet another magnetic phase neither paramagnetic GaMnN nor a ferromagnetic one.
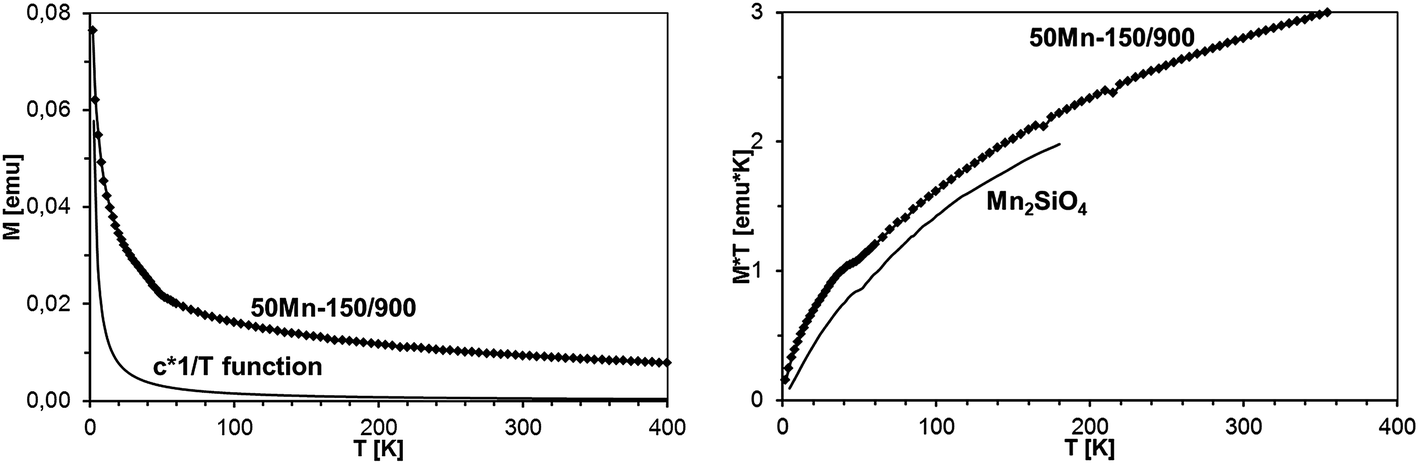 | ||
| Fig. 16 Left – magnetization of sample 50Mn-150/900 as a function of temperature at B = 1 Tesla. The solid line shows c × 1/T function, where c was chosen to match the experimental data at T = 2 K. Right – M × T vs. T plots for sample 50Mn-150/900 and for Mn2SiO4.32a | ||
This observation is farther confirmed if one tries to describe the measured magnetization using a standard method of effective Brillouin function,30 very effectively used in modeling the systems of interacting magnetic ions randomly distributed in nonmagnetic host lattice.29,31,32 We recall that this formalism assumes magnetization in the form that is shown below in eqn (1).
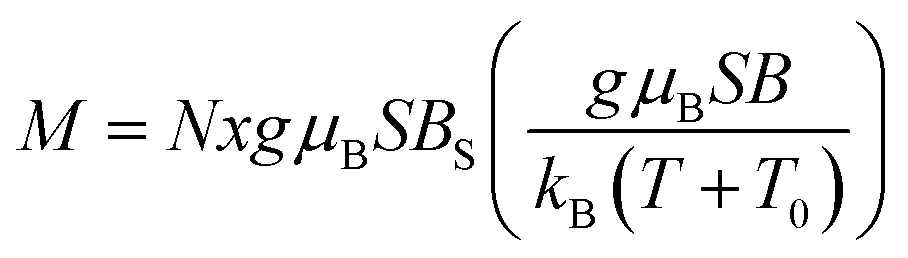 | (1) |
where BS(z) is Brillouin function, N is number of GaN unit cells in 1 g, x is Mn molar concentration, S is the spin of Mn-ion (5/2 in the case of GaMnN), and T0 is effective temperature reflecting interaction between Mn-ions (T0 < 0 for FM coupling and T0 > 0 for AFM interaction).30 Typically, N and T0 are adjustable parameters which should provide proper description in the entire temperature range.
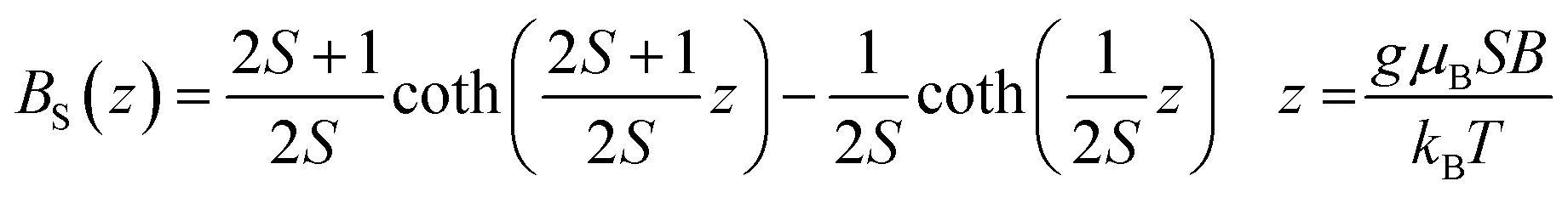
The sample fit is depicted in Fig. 17 for powder 50Mn-150/900, at T = 2 K and 300 K. The fit at 2 K is acceptable although not perfect (x = 0.023 and T0 = 3.9 K). However, there is no way to describe the 300 K data with parameters x and T0 obtained for T = 2 K. The calculated magnetization according to eqn (1) recovers only a small fraction of the measured magnetization.
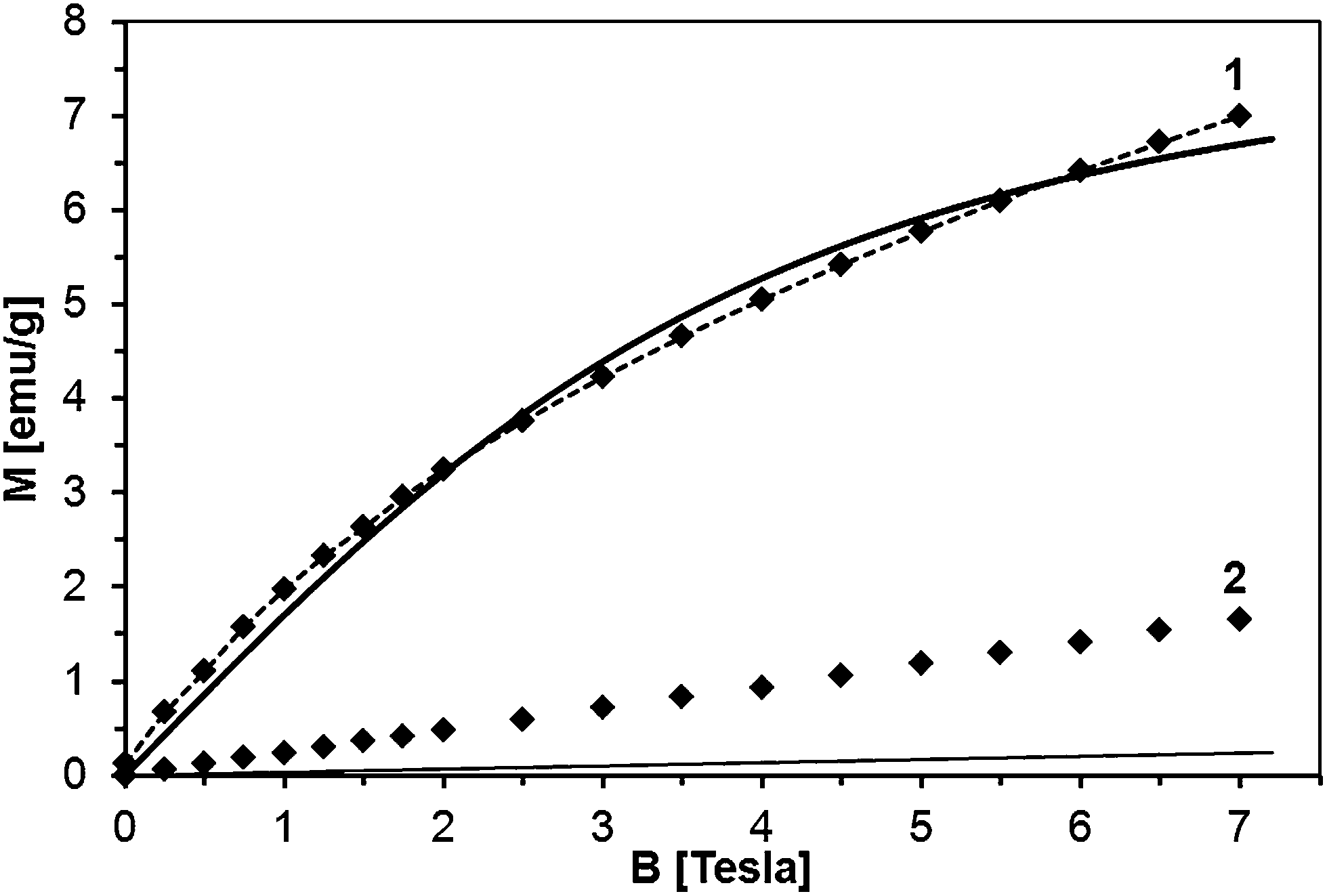 | ||
| Fig. 17 Magnetization of sample 50Mn-150/900 as a function of magnetic field strength at T = 2 K (1) and T = 300 K (2). Solid lines depict effective Brillouin function model described in the text: thick solid line – T = 2 K, T0 = 3.9 K, x = 0.023; thin solid line – T = 300 K, T0 = 3.9 K, x = 0.023. Dashed line – Brillouin function complemented by the term b × B. | ||
Apparently, the model of the Mn-ion system incorporated in the GaN lattice cannot satisfactorily describe the measured magnetization only by itself. A part of the measured magnetization must result from a distinct magnetic phase other than homogenous GaMnN. Since this additional magnetic contribution seems to be linear with magnetic field (Fig. 17), does not follow the 1/T decay characteristic for paramagnets (Fig. 16), and shows no effects of fast saturation at low fields typical for ferromagnetic phases (Fig. 14), we ascribe it to an antiferromagnetic (AFM) phase containing Mn. This phase (or phases) must have been formed during the synthesis. A possible candidate for this phase is Mn2SiO4, which can be a likely oxidation by-product in our synthesis and is known to be an antiferromagnet.32 Moreover magnetization of Mn2SiO4 reveals a characteristic bump around 40 K which is also visible in our data (Fig. 16, right). Actually, our XRD study directly confirmed the presence of the orthorhombic Mn2SiO4 in powder 50Mn-150/900 (Fig. 9). Having this in mind, the measured magnetization should be the sum of two contributions, i.e., the Brillouin-type contribution described by eqn (1) and the antiferromagnetic contribution, which can be modeled by a linear function of magnetic field B:
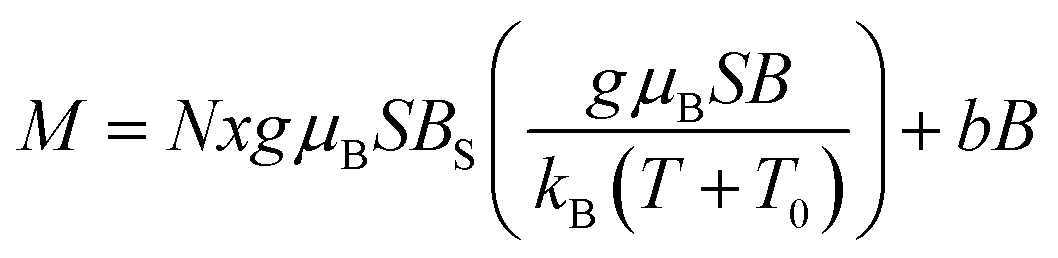 | (2) |
Fitting magnetization data with eqn (2) provides an excellent description of the experimental data as shown in Fig. 17. We note that in the case of powder 50Mn-150/900, with the highest initial Mn-content, the antiferromagnetic contribution constitutes more than half of the measured magnetization. The Mn concentration estimated from the Brillouin-like contribution in GaMnN is about 3 at.%. Similar fits for the other powders show that regardless the initial Mn-contents the actual Mn concentration in GaMnN is found at around 3 at.% with the exception of powder 5Mn-150/500 for which it reaches 3.8 at.%. The rest of Mn that stays in the products seems to form the AFM phase/phases.
For all the samples, T0 is rather small (not larger than 5 K) and positive indicating AFM interactions among Mn-ions in the GaMnN phase. These observations are in agreement with the previous reports on GaMnN for which the nearest neighbor exchange integral was estimated to be J = −1.9 K.28
The standard way to demonstrate interactions in the Brillouin-type paramagnetic system is plotting M × T vs. T. We recall that for a non-interacting magnetic system one gets a straight horizontal line whereas for an interacting system the line bends at low temperatures (where thermal energy is comparable to exchange coupling): upward for ferromagnetic exchange interactions and downward for antiferromagnetic ones. In the present case, the measured magnetization is the sum of those for GaMnN and for the AFM phase (most probably Mn2SiO4), so the M (measured) × T vs. T curve is the sum of the M (GaMnN) × T vs. T and M (AFM) × T vs. T curves as depicted in Fig. 18. The plots of M (measured) × T vs. T are shown in Fig. 19 for all four powders prepared from the initial 5 at.% Mn-content precursor. Since at sufficiently high temperatures (T several times larger than T0, so approx. T = T + T0) the M (GaMnN) × T vs. T is a straight horizontal line, the observed increase of M (measured) × T vs. T with increasing temperature (Fig. 18 and 19) is due to the AFM contribution M (AFM) × T vs. T. Direct subtraction of M (AFM) × T vs. T is rather difficult because the actual content of the AFM phase is not known. However, if the functional dependence of M (AFM) × T vs. T is known,32c one can suitably scale it and subtract it in the way that M × T(T) for T > 200 K yields a straight horizontal line (Fig. 19, right). For a sufficiently small/residual mass of by-product the resulting constant value of M × T (T > 200 K) should be proportional to the number of magnetic ions Nx (eqn (3)),
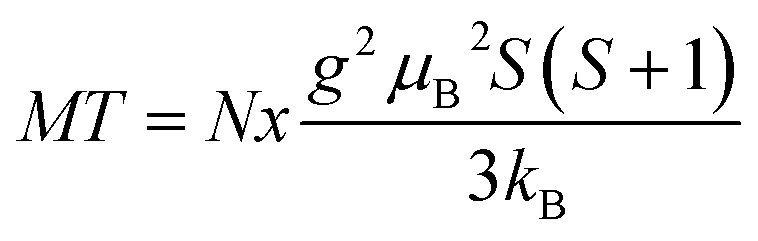 | (3) |
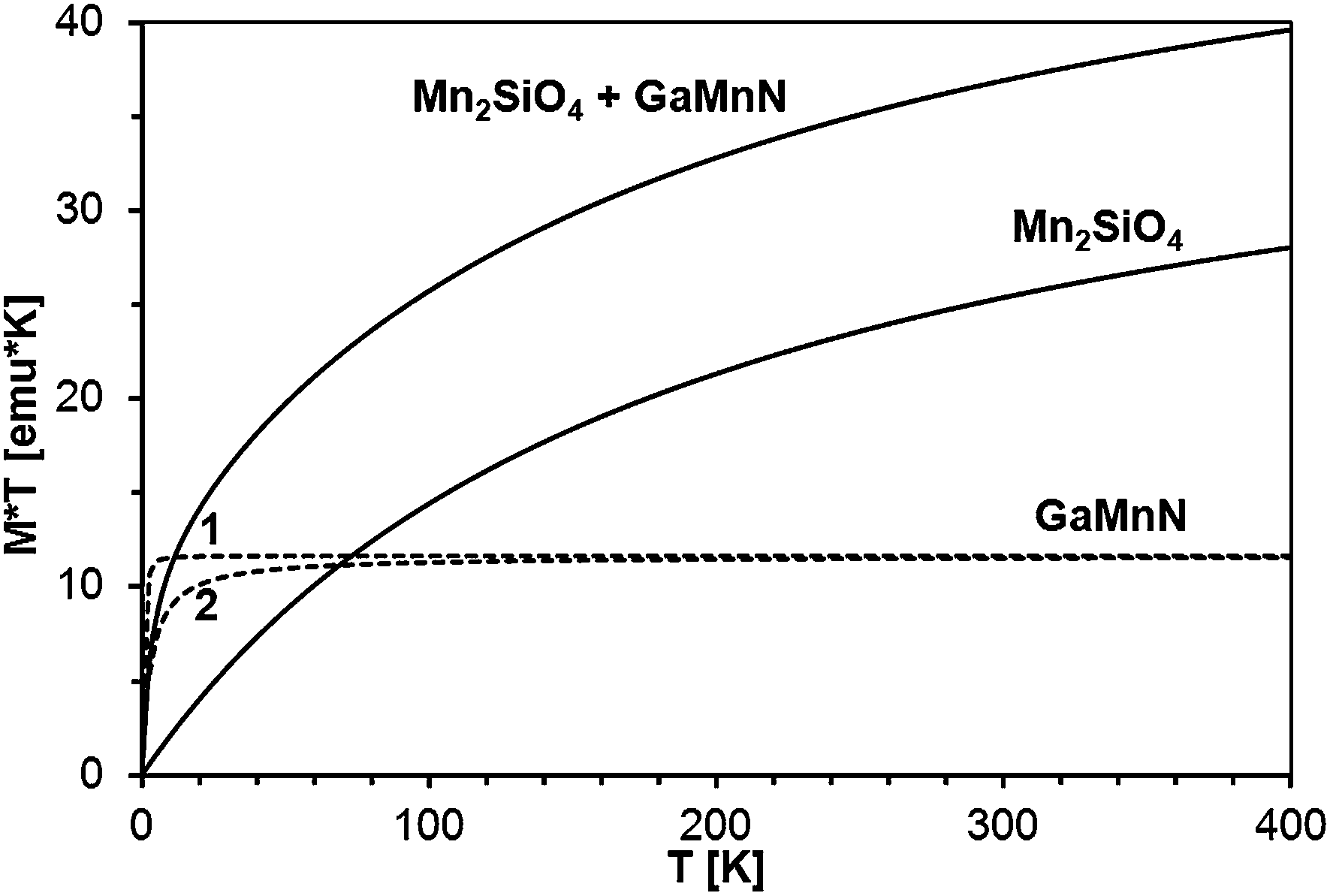 | ||
| Fig. 18 M × T vs. T for: noninteracting magnetic moments in GaMnN (dashed line 1, T0 = 0 K), antiferromagnetically interacting spins in GaMnN (dashed line 2, T0 = 3 K), AFM Mn2SiO4, sum of GaMnN and Mn2SiO4. | ||
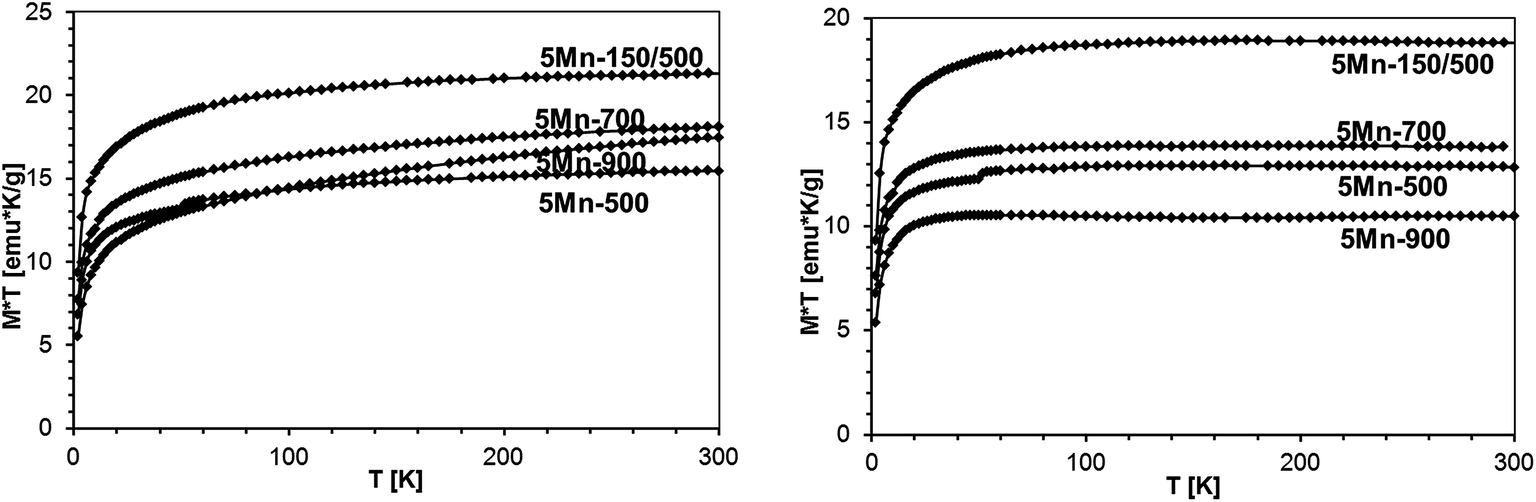 | ||
| Fig. 19 Left – M × T vs. T, B = 1 Tesla, for various samples with the initial 5 at.% Mn-content. Right – M × T vs. T after subtraction of the Mn2SiO4 contribution, as described in text. | ||
| 5Mn-150/500 | 5Mn-500 | 5Mn-700 | 5Mn-900 | |
|---|---|---|---|---|
| Mn-content in GaMnN [at.%] | 3.8 | 2.5 | 2.9 | 2.1 |
| wt (Mn2SiO4)/wt (sample) × 100% [wt%] | 0.5 | 1.0 | 1.7 | 1.4 |
We also note that in the group of the 5 at.% Mn-samples the highest amount of Mn2SiO4 was in 5Mn-700 (1.7 wt%) whereas the lowest was in 5Mn-150/500 (0.5 wt%). This is consistent with the view that the additional pyrolysis at 150 °C is efficient in removing more N(SiMe3) groups than otherwise rendering the associated Mn-centers to be more reactive towards incorporation in the condensing GaN lattice.
The M × T vs. T plots for powders prepared from the precursor with the 20 at.% Mn-content are shown in Fig. 20 where both the as-measured plots and the AFM-corrected plots are shown. The calculated Mn-contents in GaMnN (at.%) and Mn2SiO4 (wt%) are included in Table 6.
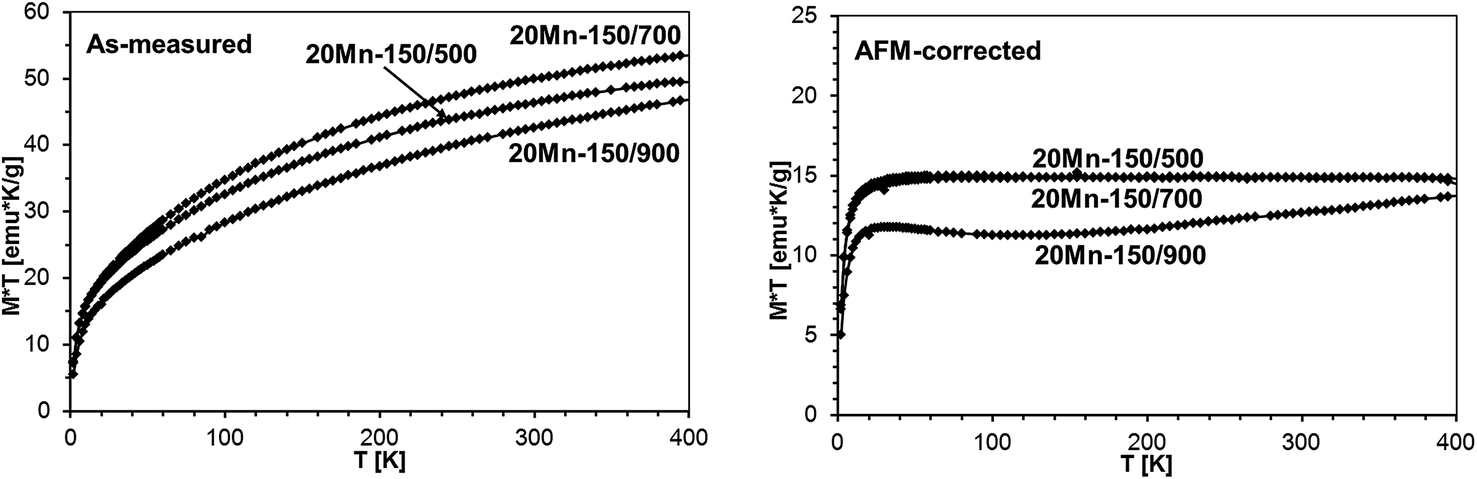 | ||
| Fig. 20 M × T vs. temperature, B = 1 Tesla, for powders 20Mn-150/500, 20Mn-150/700, and 20Mn-150/900. The bunch of curved lines on the left side are as-measured plots, the prevailingly flat lines on the right side are AFM-corrected plots. | ||
| 20Mn-150/500 | 20Mn-150/700 | 20Mn-150/900 | |
|---|---|---|---|
| Mn-content in GaMnN [at.%] | 3.0 | 3.1 | 2.4 |
| wt (Mn2SiO4)/wt (sample) × 100% [wt%] | 6.3 | 7.0 | 5.6 |
As may be seen from comparison of Tables 5 and 6, the concentration of the paramagnetic GaMnN does not seem to depend much on the initial Mn-content and with one exception of 3.8 at.% for 5Mn-150/500 is confined to values in the range 2–3 at.%. On the other hand, the higher initial Mn-content of 20 at.% is associated with significant increases of the antiferromagnetic Mn2SiO4 by-product from 1.7 wt% up to 7 wt%. The pyrolysis at 700 °C appears to yield the powders with optimal properties – high Mn-contents and chemically stable. Increasing pyrolysis temperature to 900 °C results in the relatively most stable powders but with somewhat reduced Mn-contents in GaMnN in the range 2.1–2.4 at.%. An interesting observation is associated with the potential impact on product/magnetic properties of the first-stage low temperature pyrolysis at 150 °C. The properties of the pair of the related powders, i.e., 5 Mn-500 and 5Mn-150/500, point out to a great significance of the factor and this has prompted us to an extended study addressing it to be published elsewhere.
4. Conclusions
The ammonolysis of the new molecular precursor system made of gallium tris(dimethyl)amide Ga(NMe2)3 and manganese bis(trimethylsilyl)amide Mn[N(SiMe3)2]2, initial Mn-contents from 0.1 to 50 at.%, yields bimetallic precursors that upon nitriding pyrolysis in the range of 500–900 °C result in GaN/Mn nanopowders. They are structurally based on the various GaN lattices as the only crystalline phases and imply Mn-doping towards GaMnN. However, transamination reactions are incomplete under a range of applied compositions and conditions and, consequently, some MnN(SiMe3)2 groups are retained, later decomposed to amorphous Mn–N–Si–C species that, eventually, may oxidize in air mainly to the manganese orthosilicate Mn2SiO4 by-product. The use of the precursors with higher initial Mn-contents causes increased precursor losses due to sublimation during pyrolysis as well as the formation of larger proportions of the residual by-products whereas having no significant impact on the extent of Mn-doping. Some observations linked to low temperature pyrolysis stages offer potentials for further optimization of the synthesis via a more efficient ammonolysis towards purer powders.The results of magnetic measurements confirm the general utility of the applied anaerobic synthesis route for the incorporation of Mn-ions into paramagnetic lattices of GaMnN that are free of FM precipitations typical for other synthesis methods. The effective Mn-concentrations in the GaN lattice are in the range of 2–4 at.%. Attempts to get higher Mn-concentrations by using the bimetallic precursor with increased Mn-contents from 5 to 20 and 50 at.% failed in reaching the goal while yielded higher proportions of the AFM by-product Mn2SiO4, an artefact of an incomplete ammonolysis of the precursor. Further work on better tuning the synthesis is in progress including a study on structural and magnetic aspects of high-pressure and high-temperature sintering of such GaN/Mn nanopowders.
Acknowledgements
This study was supported by the Polish National Science Centre – NCN, Grant no. 2011/01/B/ST5/06592. On-call help with the Raman study from Prof. M. Sitarz and Mr P. Jelen, Faculty of Materials Engineering and Ceramics, AGH-UST, is gratefully acknowledged.References
- (a) T. Dietl and H. Ohno, Rev. Mod. Phys., 2014, 86, 187–251 CrossRef CAS; (b) T. Jungwirth, J. Wunderlich, V. Novak, K. Olejnik, B. L. Gallagher, R. P. Campion, K. W. Edmonds, A. W. Rushforth, A. J. Ferguson and P. Nemec, Rev. Mod. Phys., 2014, 86, 855–896 CrossRef CAS; (c) A. Hirohata and K. Takanashi, J. Phys. D: Appl. Phys., 2014, 47, 193001 CrossRef; (d) K. Varaprasad, K. Ramam, G. S. M. Reddy and R. Sadiku, RSC Adv., 2014, 4, 60363–60370 RSC; (e) J. M. Li, X. L. Zeng, D. P. Wei, Y. B. Hu and Z. A. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 4490–4497 CrossRef CAS PubMed; (f) S. S. Khludkov, I. A. Prudaev and O. P. Tolbanov, Russ. Phys. J., 2013, 55, 903–909 CrossRef CAS; (g) J. M. Li, X. L. Zeng, G. Q. Wua and Z. A. Xu, CrystEngComm, 2012, 14, 525–532 RSC.
- (a) J. Borysiuk, P. Caban, W. Strupiński, S. Gierlotka, S. Stelmakh and J. F. Janik, Cryst. Res. Technol., 2007, 42, 1291–1296 CrossRef CAS; (b) J. F. Janik, M. Drygaś, C. Czosnek, B. Pałosz, S. Gierlotka, S. Stelmakh, E. Grzanka, G. Kalisz, A. Swiderska-Środa, M. Leszczyński, G. Nowak and R. Czernecki, Polish Patent PL378458, February 29, 2012.
- For example, see: (a) M. Kaminska, S. Podsiadlo, P. Dominik, K. Wozniak, L. Dobrzycki, R. Jakiela, A. Barcz, M. Psoda, J. Mizera, R. Bacewicz, M. Zajac and A. Twardowski, Chem. Mater., 2007, 19, 3139–3143 CrossRef; (b) S. Podsiadlo, T. Szyszko, W. Gebicki, J. Gosk, R. Bacewicz, L. Dobrzycki, K. Wozniak, M. Zajac and A. Twardowski, Chem. Mater., 2003, 15, 4533–4535 CrossRef CAS; (c) A. Wolos, M. Palczewska, M. Zając, J. Gosk, M. Kaminska, A. Twardowski, M. Bockowski, I. Grzegory and S. Porowski, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 115210–115217 CrossRef.
- M. Zając, J. Gosk, E. Grzanka, S. Stelmakh, M. Palczewska, A. Wysmołek, K. Korona, M. Kamińska and A. Twardowski, J. Alloys Compd., 2008, 456, 324–338 CrossRef.
- J. F. Janik, M. Drygaś, C. Czosnek, M. Kamińska, M. Palczewska and R. T. Paine, J. Phys. Chem. Solids, 2004, 65, 639–645 CrossRef CAS.
- J. B. Gosk, M. Drygaś, J. F. Janik, M. Palczewska, R. T. Paine and A. Twardowski, J. Phys. D: Appl. Phys., 2006, 39, 3717–3725 CrossRef CAS.
- H. Li, B. Song, H. Q. Bao, G. Wang, W. J. Wang and X. L. Chen, J. Magn. Magn. Mater., 2009, 321, 222–225 CrossRef CAS.
- Y. Y. Wei, J. M. Guo, D. L. Hou and S. Qiao, Solid State Commun., 2008, 148, 234–236 CrossRef CAS.
- U. Kim, H. K. Seong, M.-H. Kim and H. J. Choi, Mater. Lett., 2009, 63, 97–99 CrossRef CAS.
- M. H. Ham and J. M. Myoung, Appl. Phys. Lett., 2006, 89, 173117 CrossRef.
- J. F. Janik, R. L. Wells, J. L. Coffer, J. V. St. John, W. T. Pennington and G. L. Schimek, Chem. Mater., 1998, 10, 1613–1622 CrossRef CAS.
- (a) D. C. Bradley, M. B. Hursthouse, K. M. A. Malik and R. Moseler, Transition Met. Chem., 1978, 3, 253–254 CrossRef CAS; (b) H. Chen, M. M. Olmstead, D. C. Pestana and P. P. Power, Inorg. Chem., 1991, 30, 1783–1787 CrossRef CAS.
- M. Yarema, R. Caputo and M. V. Kovalenko, Nanoscale, 2013, 5, 8398–8410 RSC , and references therein.
- J. F. Janik and R. L. Wells, Chem. Mater., 1996, 8, 2708–2711 CrossRef CAS.
- (a) J. F. Janik, E. N. Duesler and R. T. Paine, Inorg. Chem., 1987, 26, 4341–4345 CrossRef CAS; (b) R. T. Paine, J. F. Janik and C. K. Narula, Mater. Res. Soc. Symp. Proc., 1988, 121, 461 CrossRef CAS; (c) J. F. Janik, E. N. Duesler and R. T. Paine, Inorg. Chem., 1988, 27, 4335–4338 CrossRef CAS.
- (a) K. J. L. Paciorek, J. H. Nakahara and S. R. Masuda, Inorg. Chem., 1990, 29, 4252–4255 CrossRef CAS; (b) F. C. Sauls, W. J. Hurley, L. V. Interrante, P. S. Marchetti and G. E. Maciel, Chem. Mater., 1995, 7, 1361–1368 CrossRef CAS.
- J. W. Hwang, J. P. Campbell, J. Kozubowski, S. A. Hanson, J. F. Evans and W. L. Gladfelter, Chem. Mater., 1995, 7, 517–525 CrossRef CAS.
- (a) J. B. Gosk, Cryst. Res. Technol., 2001, 36, 197–231 CrossRef CAS; (b) J. B. Gosk, Cryst. Res. Technol., 2000, 35, 101–116 CrossRef CAS.
- (a) A. Pokhrel, Z. P. Degregorio, J. M. Higgins, S. N. Girard and S. Ji, Chem. Mater., 2013, 25, 632–638 CrossRef CAS; (b) V. N. Men'shov and V. V. Tugushev, J. Exp. Theor. Phys., 2011, 113, 121–137 CrossRef.
- (a) R. Pompe, Thermochim. Acta, 1976, 16, 295–300 CrossRef CAS; (b) R. Marchand, C. R. Seances Acad. Sci., Ser. C, 1971, 272, 1654–1656 CAS; (c) M. Kretschmer, Arch. Eisenhuttenwes., 1976, 47, 15–20 CAS.
- (a) L. Stolt, O. Thomas and F. M. d'Heurle, J. Appl. Phys., 1990, 68, 5133–5139 CrossRef CAS; (b) M. H. Ham, J. W. Lee, K. J. Moon, J. H. Choi and J. M. Myoung, J. Phys. Chem. C, 2009, 113, 8143–8146 CrossRef CAS.
- F. Takanoa, H. Akinaga, H. Ofuchi, S. Kuroda and K. Takita, J. Appl. Phys., 2006, 99, 08J506 Search PubMed.
- (a) Y. Hasegawa, M. Iimura and S. Yajima, J. Mater. Sci., 1980, 15, 720–728 CrossRef CAS; (b) H. B. Li, L. T. Zhang, L. F. Cheng, Y. G. Wang, Z. J. Yu, M. H. Huang, H. B. Tu and H. P. Xia, J. Mater. Sci., 2008, 43, 2806–2811 CrossRef CAS.
- (a) D. Su, Y. Li, F. Hou and X. Yan, J. Am. Ceram. Soc., 2014, 97, 1311–1316 CrossRef CAS; (b) S. Li and Y. Zhang, J. Non-Cryst. Solids, 2012, 358, 54–57 CrossRef CAS.
- N. V. Chukanov, in Infrared spectra of mineral species: Extended library, Springer Geochemistry/Mineralogy, 2014, vol. 1, p. 376 Search PubMed.
- (a) W. Limmer, W. Ritter, R. Sauer, B. Mensching, C. Liu and B. Rauschenbach, Appl. Phys. Lett., 1998, 72, 2589–2591 CrossRef CAS; (b) S. N. Waheeda, N. Zainal, Z. Hassan, S. V. Novikov, A. V. Akimov and A. J. Kent, Appl. Surf. Sci., 2014, 317, 1010–1014 CrossRef CAS; (c) X. G. Gao, B. Y. Man, M. Liu, C. Yang, C. S. Chen, C. Zhang and Z. C. Sun, J. Magn. Magn. Mater., 2015, 378, 447–450 CrossRef CAS; (d) M. Zajac, R. Doradzinski, J. Gosk, J. Szczytko, M. Lefeld-Sosnowska, M. Kaminska, A. Twardowski, M. Palczewska, E. Grzanka and W. Gebicki, Appl. Phys. Lett., 2001, 78, 1276–1278 CrossRef CAS; (e) Y. Y. Yu, R. Zhang, X. Q. Xiu, Z. L. Xie, H. Q. Yu, Y. Shi, B. Shen, S. L. Gu and Y. D. Zheng, J. Cryst. Growth, 2004, 269, 270–275 CrossRef CAS; (f) L. L. Guo, Y. H. Zhang and W. Z. Shen, Appl. Phys. Lett., 2006, 89, 161920 CrossRef.
- J. L. Coffer, T. W. Zerda, R. Appel, R. L. Wells and J. F. Janik, Chem. Mater., 1999, 11, 20–22 CrossRef CAS.
- M. Zając, J. Gosk, M. Kamińska, A. Twardowski, T. Szyszko and S. Podsiadło, Appl. Phys. Lett., 2001, 79, 2432–2434 CrossRef.
- M. Zajac, J. Gosk, E. Grzanka, M. Kaminska, A. Twardowski, B. Strojek, T. Szyszko and S. Podsiadło, J. Appl. Phys., 2003, 93, 4715–4717 CrossRef CAS.
- J. A. Gaj, R. Planel and G. Fishman, Solid State Commun., 1993, 88, 927–930 CrossRef.
- (a) R. L. Aggarwal, S. N. Jasperson, J. Stankiewicz, Y. Shapira, S. Foner, B. Khazai and A. Wold, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 6907–6913 CrossRef CAS; (b) A. Twardowski, M. von Ortenberg, M. Demianiuk and R. Pauthenet, Solid State Commun., 1984, 51, 849–852 CrossRef CAS; (c) A. Twardowski, P. Świderski, M. von Ortenberg and R. Pauthenet, Solid State Commun., 1984, 50, 509–513 CrossRef CAS.
- (a) R. P. Santoro, R. E. Newnham and S. Nomura, J. Phys. Chem. Solids, 1966, 27, 655–666 CrossRef CAS; (b) W. Lottermoser and H. Fuess, Phys. Status Solidi A, 1988, 109, 589–595 CrossRef CAS; (c) I. S. Hagemann, P. G. Khalifah, A. P. Ramirez and R. J. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R771–R774 CrossRef CAS.
Footnote |
| † This paper is dedicated to Prof. Robert T. Paine, UNM, Albuquerque, New Mexico, USA, on the occasion of his retirement. |
| This journal is © The Royal Society of Chemistry 2015 |