
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aryl tosylates as non-ionic photoacid generators (PAGs): photochemistry and applications in cationic photopolymerizations†
Edoardo
Torti
a,
Gioia
Della Giustina
b,
Stefano
Protti
*a,
Daniele
Merli
c,
Giovanna
Brusatin
b and
Maurizio
Fagnoni
*a
aPhotogreen Lab, Department of Chemistry, University of Pavia, V. Le Taramelli 12, 27100 Pavia, Italy. E-mail: fagnoni@unipv.it; prottistefano@gmail.com; Fax: +39 0382 987323; Tel: +39 0382 987198
bDepartment of Industrial Engineering, University of Padova and INSTM, Via Marzolo 9, 35131 Padova, Italy
cDepartment of Chemistry, University of Pavia, V. Le Taramelli 12, 27100 Pavia, Italy
First published on 17th March 2015
Abstract
The irradiation of various substituted aryl tosylates was investigated in a solution and homolysis of the ArO–SO2C6H4CH3 bond was the path exclusively observed. The corresponding phenols and photo-Fries adducts were obtained and p-toluenesulfinic and p-toluenesulfonic acids were liberated. The nature and amount of the acid photoreleased were tuned by changing the reaction conditions and the nature and position of the aromatic substituents. In deaerated solutions p-toluenesulfinic acid was formed exclusively, whereas under oxygenated conditions the stronger p-toluenesulfonic acid was released, as shown by HPLC ion chromatography analyses. ArO–S bond photocleavage takes place from the singlet state, as confirmed by laser flash photolysis experiments, and competitive intersystem crossing can make the aryl tosylate unreactive when a nitro group is present. The application of these aryl tosylates as non-ionic photoacid generators (PAGs) in hybrid organic/inorganic sol–gel photoresists has been explored.
Introduction
The development of new compounds able to release acid upon light absorption (so-called photoacid generators, PAGs) is of crucial importance in the field of materials chemistry and in microelectronics, photonics, nanofabrications and sensors development.1 Aromatic onium salts (either iodonium or sulfonium) are widely used for this purpose as suitable “caged protons”.2 Despite the thermal stability of the latter compounds, their poor solubility in polymer matrices3 has led to an increasing demand for more soluble non-ionic PAGs for photo(nano)lithography applications. The compounds proposed for this role usually release a sulfonic acid upon photocleavage of a weak N–O bond in iminosulfonates,4,5 imidosulfonates5 (Scheme 1a) or related compounds.6 Such cleavage generates a RSO3˙ radical, which upon reaction with the medium, is able to form a strong sulfonic acid.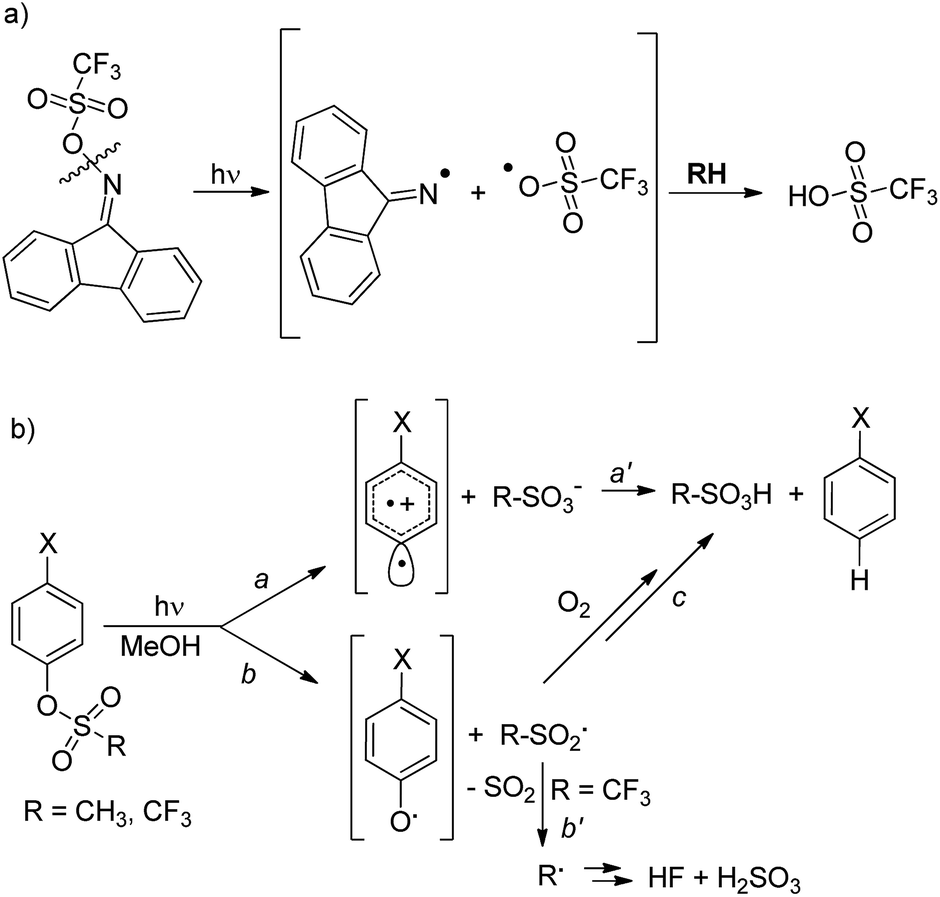 | ||
| Scheme 1 (a) Photorelease of sulfonic acid upon the photocleavage of a N–O bond according to ref. 5c. (b) Competitive pathways in the photorelease of acids from (substituted) phenyl mesylates and triflates. | ||
We recently reported, however, that sulfonic acids can be generated just as efficiently (>90%) by the irradiation of aryl mesylates and triflates, as shown in Scheme 1b.7 In this case, two mechanisms compete, namely, heterolysis of the Ar–OS bond (path a) and homolysis of the ArO–S bond (path b), depending on the substituent X present on the aromatic ring and on the sulfonic moiety.7,8 In the former case, a sulfonic acid is released directly (path a′), whereas in the latter case trapping of the generated RSO2˙ radical by oxygen leads to the formation of RSO3H (path c) provided that fragmentation of the sulfonyl radical intermediate is avoided. Fragmentation occurs with aryl triflates, with the liberation of weak acids, such as HF and H2SO3, in N2-equilibrated methanol (path b′, Scheme 1b), rather than sulfonic acids.7
Release of strong acids thus does not depend on initial homolytic or heterolytic cleavage, and other aryl sulfonates appeared to be worth evaluating as photoacid generators. Aryl tosylates constitute a worthwhile target, because they are crystalline compounds, readily prepared from inexpensive reagents and more stable toward hydrolysis than other commonly used sulfonates (e.g. triflates). In addition, aryl tosylates have exhibited higher thermal stability than the corresponding mesylates when heated in a poly(4-hydroxystyrene) matrix.9 In fact, nitrobenzyl tosylates10,11 and N-oxysuccinimidoarylsulfonates12 have previously been used for the photogeneration of p-toluenesulfonic acid (PTSA) and some iminotosylates are commercially available for such an application (e.g. the iminotosylate Irgacure PAG 121).
The photochemistry of aryl tosylates, however, has rarely been investigated13 and, to the best of our knowledge, the use of aryl tosylates as PAGs has only been tested with o-arenesulfonyloxyanilide derivatives, where photorelease of PTSA was found to be almost quantitative, albeit with very low efficiency (Φ−1ca. 0.05![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13b).
13b).
The aim of this work is exploration of the photochemistry of some substituted phenyl tosylates, with an investigation of their product distribution in solution, as well as a test of their use as photoinitiators for acid-induced polymerization in innovative epoxy-based hybrid organic–inorganic systems obtained via sol–gel processes.14 These systems exhibit several attractive properties such as mechanical robustness, high thermal and chemical stability and transparency, and have found increasing interest within the field of photo(nano)lithography.15 Their preparation proceeds by hydrolysis and condensation reactions of organically modified alkoxides with formulas RnSi(OR′)4−n with the addition of an acidic or basic catalyst. The presence of a non-hydrolyzable Si–C bond provides a stable linkage between the organic unit and the oxide matrix, resulting in an inorganic three-dimensional network with pendant moieties that can be derivatized (e.g. double bonds or, as in our case, acid-sensitive epoxy rings) and lead to crosslinking.
Results
We have examined a series of phenyl sulfonates substituted with both electron-donating and electron-withdrawing groups (1a–f in Chart 1). The silylated derivative 1g was also tested because we previously demonstrated that the presence of silicon-based substituents was able to influence the photoreactivity of aryl sulfonates.16,17 These were synthesized from the corresponding phenols.18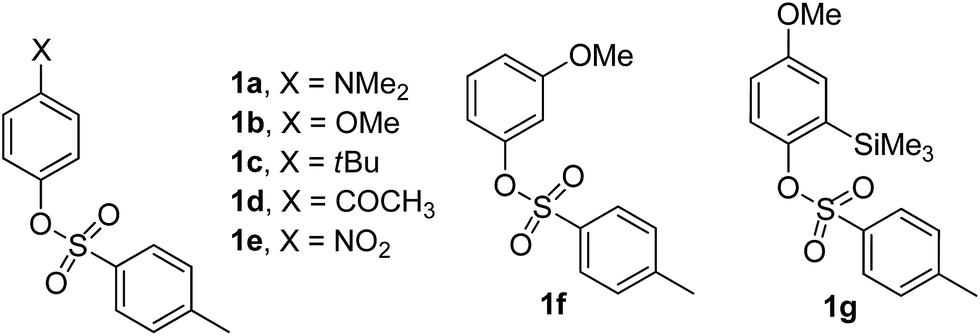 | ||
| Chart 1 | ||
The photophysical and photochemical properties of tosylates 1a–g are presented in Table S1 and Fig. S1–S7 (see ESI†). Two absorption maxima in the UV region around 260–290 nm (ε > 5000 L cm−1 mol−1) and 220–240 nm (ε > 104 L cm−1 mol−1) were observed for compounds 1a–c and 1e–g, whereas compound 1d exhibited a single significant absorption at 239 nm (ε = 17800 L cm−1 mol−1). All the tosylates investigated exhibited low (1a,b: ΦF < 0.01) or negligible (1c–g) fluorescence emission (Table S1†).
The photochemistry of 4-methoxyaryl tosylate 1b was investigated initially (Table 1) and its disappearance quantum yield (Φ−1) was measured. This turned out to be modest in all the solvents tested (Φ−1 = 0.07–0.14). Photolysis of 1b under UV irradiation (λ = 254 nm) caused ArO–S bond cleavage exclusively and 4-methoxyphenol 2b and o-tosylphenol 3b (a photo-Fries adduct) were formed in variable amounts depending on the solvent employed. Compound 3b was the predominant product formed in cyclohexane, whereas the amount of 2b increased with solvent proticity (methanol, 2-methoxyethanol) or in the presence of labile C–H bonds (e.g. in the case of THF, see Tables 1 and S2†). No products arising from Ar–OS bond cleavage, such as anisole or 1,4-dimethoxybenzene, were detected, in contrast to what was observed for other methoxyaryl sulfonates.7 Irradiation in neat acetone (a triplet sensitizer) likewise caused complete consumption of 1b, giving 2b as the main product. The same experiment carried out in oxygenated acetone led to no 1b consumption.
|
|
|||
|---|---|---|---|
| Solvent | Φ −1 | 2b (%) | 3b (%) |
| a A nitrogen-saturated solution of 1b (10−2 M) in the chosen solvent irradiated at 254 nm (4 × 15 W Hg lamps, 2 h) until >80% consumption of 1b was achieved. Product yields calculated on the basis of the amount of 1b consumed. b Disappearance quantum yield (Φ−1) measured for a 10−2 M 1b solution in the chosen solvent (λ = 254 nm, 1 × 15 W Hg lamp) using potassium ferrioxalate as actinometer. | |||
| Cyclohexane | — | 4 | 68 |
| CH2Cl2 | 0.08b | 12 | 33 |
| CH3COOCH2CH3 | — | 26 | 36 |
| THF | — | 39 | 24 |
| CH3COCH3 | — | 30 | 10 |
| CH3CN | 0.13b | 24 | 30 |
| CH3OCH2CH2OH | 0.07b | 34 | 29 |
| CH3OH | 0.14b | 41 | 28 |
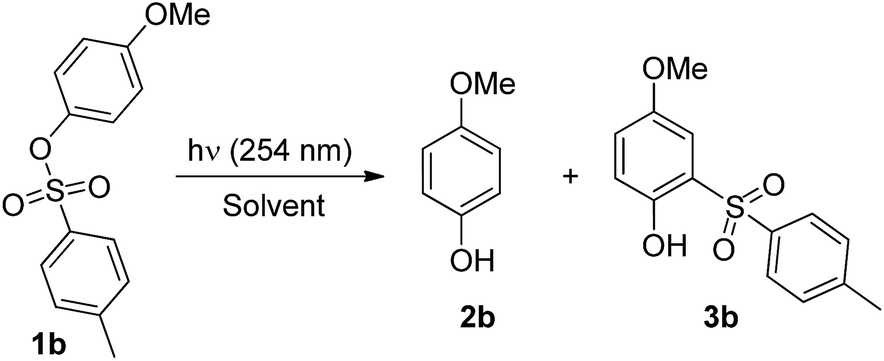
The photolyzed solution in methanol was subjected to potentiometric titration and ionic chromatography analyses, revealing that 4-tolylsulfinic acid was liberated in modest yield (ca. 40%, Fig. S10, ESI†).
With these results in hand, we proceeded to investigate the photorelease of sulfinic and sulfonic acids from tosylates 1a–g in methanol under both nitrogen- and oxygen-saturated conditions (Tables 2 and 3, Fig. S8–S19†). Under deaerated conditions, the Φ−1 measured was in the 0.1–0.15 range except for 1d, in which the MeCO-group provides higher photoreactivity (Φ−1 = 0.29), and 1e which is virtually photostable under these conditions (Table 2). It is notable that the presence of a trimethylsilyl group makes the tosylate more photoreactive (compare the Φ−1 value of 1b with 1g, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| 1 | λ max a (nm), ε/(L mol−1 cm−1) | Φ −1 b | Photoproductc, (%) | H+d (%) | Acidse (% yield) |
| a Maximum absorption wavelength and molar absorption coefficient. b Disappearance quantum yield measured for a 10−2 M 1a–g solution in MeOH (λ = 254 nm, 1 × 15 W Hg lamp) using potassium ferrioxalate as actinometer. c Isolated yields. 2 h irradiation time (complete consumption of 1a–g). d Determined by potentiometric titration. e Determined by HPLC ion chromatography analyses. f No acid released. g A similar Φ−1 value was measured under oxygen-saturated conditions. h Formic acid (<1%) detected. i No photoproducts detected by GC analysis. | |||||
| 1a | 231, 8241 | 0.11 | 3a, 100 | f | f |
| 264, 10 287 | |||||
| 317, 1171 | |||||
| 1b | 227, 21 190 | 0.14g | 2b, 41 | 40 | CH3C6H4SO2H, 38 |
| 274, 2423 | 3b, 28 | CH3C6H4SO3H, < 1h | |||
| 1c | 227, 15 060 | 0.14 | 2c, 52 | 50 | CH3C6H4SO2H, 48 |
| 263, 1230 | 3c, 18 | ||||
| 1d | 232, 28 320 | 0.29g | 2d, 73 | 67 | CH3C6H4SO2H, 45h |
| CH3C6H4SO3H, 29 | |||||
| 1e | 227, 7980 | <0.01 | i | f | f |
| 264, 6350 | |||||
| 1f | 224, 20 940 | 0.11 | 2f, 52 | 49 | CH3C6H4SO2H, 47 |
| 273, 3057 | |||||
| 1g | 228, 17 100 | 0.21 | 2g, 21 | 32 | CH3C6H4SO2H, 22h |
| 285, 2010 | 3g, 18 | ||||
| ArOTs | H+b (% yield) | Anionsc (% yield) |
|---|---|---|
| a A 10−2 M solution of tosylates 1a–g in oxygen-saturated methanol irradiated for 2 h at 254 nm (4 × 15 W Hg lamps). b Determined by potentiometric titration. c Determined by means of HPLC ion chromatography analyses. d CH3C6H4SO2H (<4%) was likewise formed. e No consumption of 1e was observed. | ||
| 1a | 46 | CH3C6H4SO3H (53) |
| 1b | 82 | CH3C6H4SO3H (82)d |
| 1c | 96 | CH3C6H4SO3H (90) |
| 1d | 100 | CH3C6H4SO3H (100) |
| 1e | e | e |
| 1f | 99 | CH3C6H4SO3H (95) |
| 1g | 84 | CH3C6H4SO3H (77) |
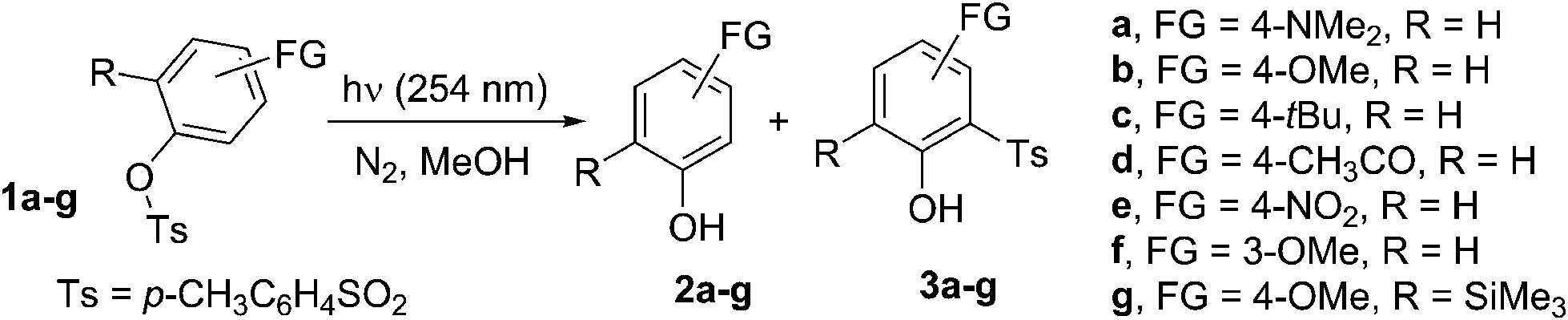
Cleavage of the sulfonyl group upon UV light absorption led either to phenols 2 or to photo-Fries adducts 3, in analogy with what had previously been observed for 1b. Formation of 3 was exclusive when a strong electron-donating group (FG = NMe2) was present, negligible with electron-withdrawing groups (e.g. MeCO, Table 2) or of the same magnitude as 2 with less strongly donating groups (FG = OMe, tBu). Photocleavage of the ArO–S bond occurred independently of the nature and position of the substituents present on the aromatic ring (except for the case of unreactive 1e). Formation of photo-Fries products, on the contrary, depended on the position, as shown by the different results for isomeric 1b and 1f (no Fries adduct in the latter case). In some cases, the reaction was very clean as demonstrated by the UV-monitored conversion of 1d to p-acetylphenol 2d (see Fig. S20 and S21†). With respect to the release of acid, p-toluenesulfinic acid was formed in variable amounts in nitrogen-saturated methanol, with the single exception of tosylate 1d, for which a mixture of sulfinic acid and PTSA was observed. As expected, the higher the amount of phenol 3 formed, the smaller was the amount of acid released (Table 2). Notably, the disappearance quantum yield (Φ−1) values for 1b and 1d were not affected by the presence of oxygen (Table 2).
On the other hand, under oxygenated conditions, a large amount of PTSA was released depending on the tosylate used (up to quantitative yield for 1d) and the corresponding sulfinic acid was found only as a minor product in the case of 1b. The lowest yield of released PTSA was found in the photolysis of 1a (Table 3).
Some mechanistic studies were carried out with tosylates 1b and 1d as well as the photostable 1e. Laser flash photolysis at 266 nm of an argon-saturated solution of 1b (ca. 10−4 M) in methanol revealed the rapid formation (within a few ns) of two intense absorption bands with λmax = 330 nm and 410 nm, respectively, with a weaker, broad absorption located at longer wavelengths (580–700 nm, Fig. 1a). The time evolution of these absorption bands indicated the presence of different transient species, with the predominant contribution from a long-lived intermediate (τ = 24 μs, k = 1.5 × 1010 M−1 s−1) in both bands (see also Fig. S10†). Kinetic analysis revealed the presence of a second short-lived (τ = 4.1 μs) species absorbing at 330 nm.
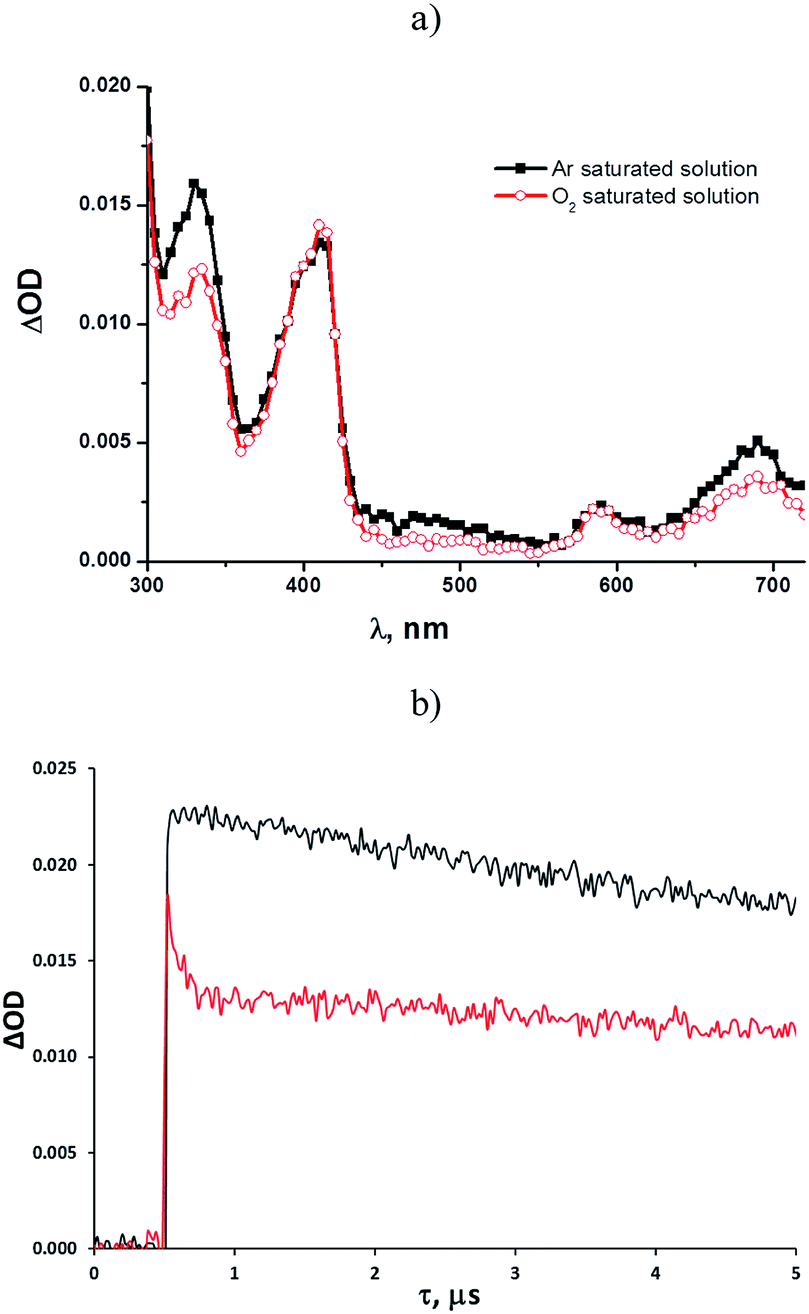 | ||
| Fig. 1 Transient spectra of a solution of 1b (10−4 M) in (a) argon-saturated methanol and O2-saturated methanol recorded 0.1 μs after a 20 ns laser pulse (λ = 266 nm); (b) profile of the absorbance change observed at 330 nm for a solution of 1b in argon-saturated (black) and O2-saturated (red) methanol. | ||
Shifting to an oxygen-saturated solution resulted in a slight modification of the transient absorption profile, with quenching of that portion of the 330 nm absorption attributable to the short-lived species (see Fig. 1a and b), while the kinetics of the remaining absorption bands remained almost unchanged (Fig. 1a). A residual absorption around 300–350 nm, along with the broad band at 580–700 nm, was observed 150 μs after the laser pulse (see Fig. S22†).
With acetyl derivative 1d (ca. 10−4 M in methanol, see Fig. 2a–c), two absorption bands (λmax = 330 nm and 410 nm), along with a broad signal (580–700 nm, Fig. 2a), were again detected under argon-saturated conditions. Kinetic analysis revealed the presence of three different transient species, for which the longest-lived (τ = 33 μs) represented the main contributor to both maxima, whereas two short-lived bands were observed at 340 (τ = 2.9 μs) and 420 nm (τ = 0.7 μs), respectively.
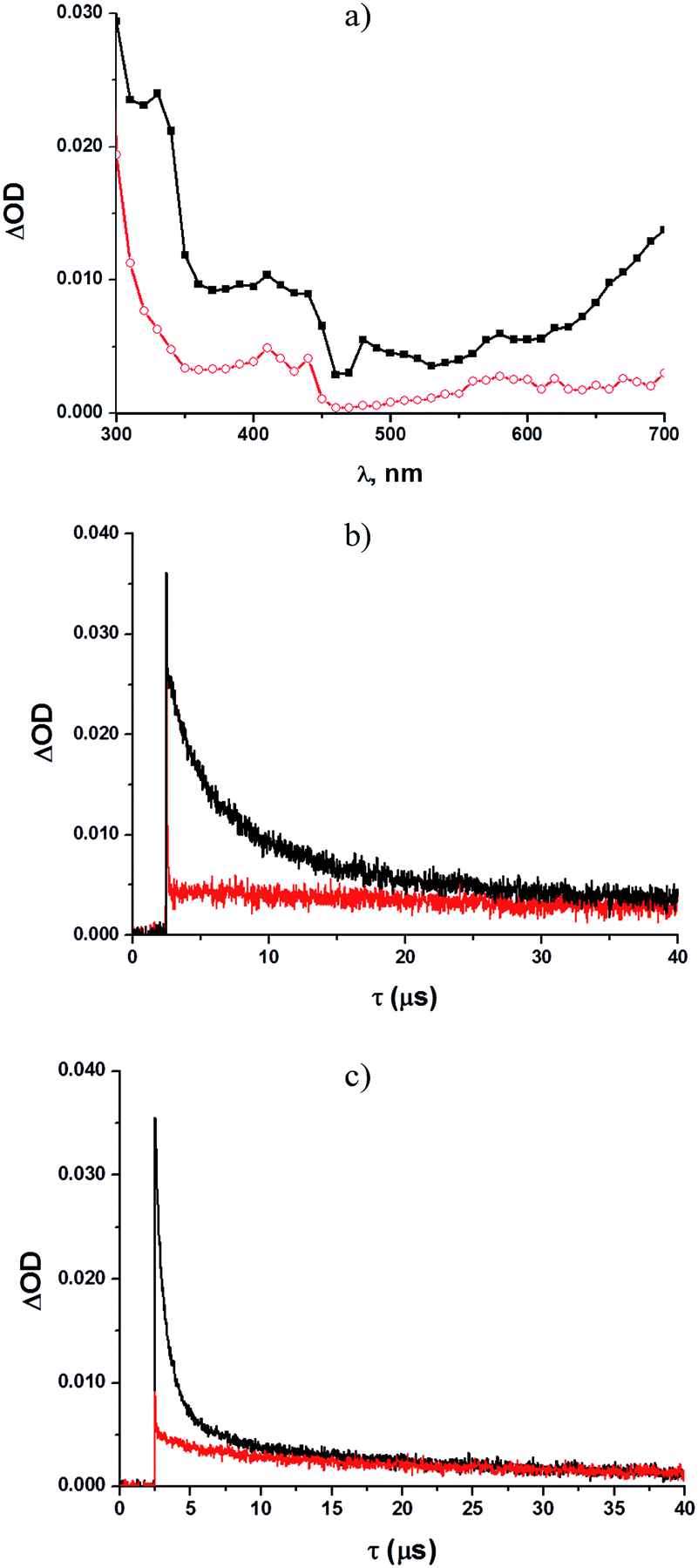 | ||
| Fig. 2 Transient spectra of a solution of 1d (10−4 M) in (a) argon-saturated methanol (black) and O2-saturated methanol (red) recorded 0.1 μs after a 20 ns laser pulse (λ = 266 nm); Profile of the absorbance change observed at (b) 340 nm and (c) 420 nm for a solution of 1d in argon-saturated (black) and O2-saturated (red) methanol. | ||
The latter was the main contributor to the broad absorption band at 580–700 nm. Shifting to an oxygen-saturated solution resulted in the quenching of the two short-lived intermediates (see Fig. 2b and c), whereas no significant effects were observed for the longest-lived species. In contrast, no significant transient species were observed with the nitro derivative 1e.
The results obtained in solution encouraged us to determine the efficiency of the photoactive aryl tosylates 1a–d and 1f–g in photoinduced cationic polymerization processes. With this aim, their inclusion in a hybrid organic–inorganic photoresist (G8Ge2), obtained via acid-catalyzed condensation of 3-glycidyloxypropyltrimethoxysilane (GPTMS) and germanium tetraethoxide (TEOG) in 2-methoxyethanol, was planned.19 Preliminary experiments to determine the effect of the photoresist components were first carried out on 1d, for which the generation of PTSA was most efficient in methanol. Irradiation of 1d was carried out in oxygen-saturated 2-methoxyethanol and PTSA was photoreleased in 71% yield, analogous to what was found in neat oxygen-saturated methanol. The presence of GPTMS (0.73 M), germanium tetraethoxide (TEOG, 0.18 M) and H2O (2.55 M) produced comparable results (PTSA yield = 57%).20
At this point, tests were carried out on the assembled G8Ge2 containing acid-sensitive epoxy functionalities obtained via sol–gel techniques from GPTMS and TEOG (molar ratio: 80:20) in 2-methoxyethanol.21 The synthesized aryl tosylates were then incorporated in the G8Ge2 system and the resulting sol was spin-coated on silicon (100) substrates. The films obtained were then exposed to UV light (Hg–Xe UV spot light source) at increasing doses and the structural modifications induced by aryl tosylates were investigated by means of both UV and FT-IR analysis.
Fig. 3a illustrates the UV-vis spectra of the hybrid samples before and after addition of 1d to the sample. An absorption band at 240 nm, due to the presence of aryl tosylate 1d (1%), was apparent and was markedly reduced within the first minute of UV irradiation. Among the vibrational modes that characterize the epoxy moiety in G8Ge2, the signals assigned to C–H stretching at 3060 and 3000 cm−1 were chosen to determine the degree of polymerization (see Fig. 3b).19
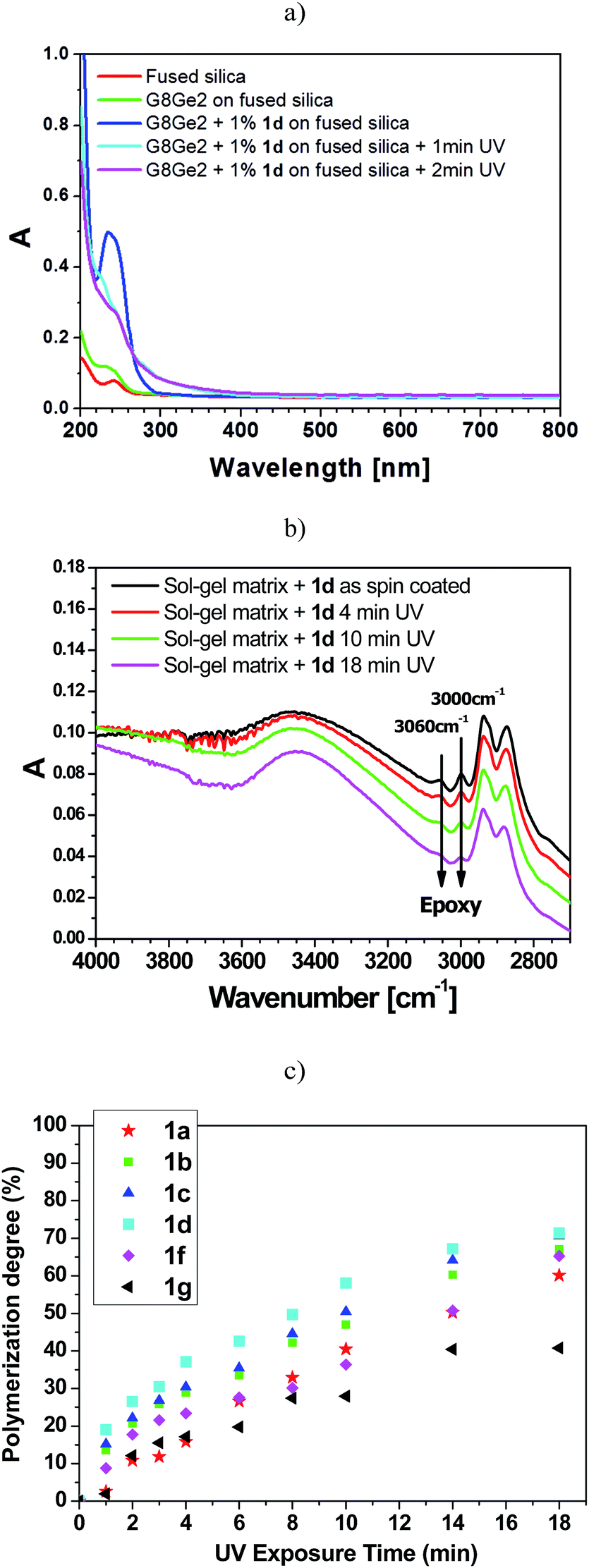 | ||
| Fig. 3 (a) UV-vis spectra of the photoresist G8Ge2 in the 200–800 nm region in the absence and presence of 1% tosylate 1d before and after UV irradiation; (b) FTIR spectra of G8Ge2 films in the 4000–2600 cm−1 region with 1% molar concentration of 1d before and after UV irradiation. Samples were spin-coated on fused silica slides and silicon, respectively; and (c) degree of photopolymerization of epoxy groups versus UV curing time, achieved by 1% molar addition of aryl tosylates 1a–d, 1f–g to G8Ge2 sol–gel matrix. | ||
As shown in Fig. 3b for the G8Ge2/1d system, UV irradiation caused a decrease in absorbance in the bands at 3060 and 3000 cm−1, indicating gradual ring-opening of the epoxy moiety with subsequent formation of an interpenetrating organic network and densification of the exposed area. The degree of photopolymerization induced by phenyl tosylates 1a–d and 1f–g was then measured by monitoring the evolution of epoxy ring-opening as a function of the UV dose. The degree of polymerization (at least within the first three minutes of UV exposure, Fig. 3c) roughly followed the amount of PTSA photoreleased under oxygenated conditions (with 1d the best and 1a the worst of the series, see Table 3).
It is noteworthy that a degree of polymerization above 60% was achieved in all cases (except 1g) after 18 min UV exposure. Photodegradation of the PAG (see Fig. 3a) was essentially complete in 1–2 min, while epoxy polymerization required several minutes (no polymerization took place when UV curing was carried out in the absence of 1a–g).
Discussion
The photochemistry of phenyl 4-methylbenzenesulfonate was investigated in 1966 by Havinga et al., who observed homolytic cleavage of the ArO–S bond.22 More recently, our research group noticed competition between ArO–S bond homolytic cleavage and Ar–OS heterolysis taking place in aryl triflates, mesylates,7 nonaflates23 and imidazylates.24 For the substrates examined in this paper, the homolytic pathway is exclusive, with subsequent formation of a phenoxy (I)/paratoluenesulfonyl (II) radical pair (see Scheme 2 below, path a).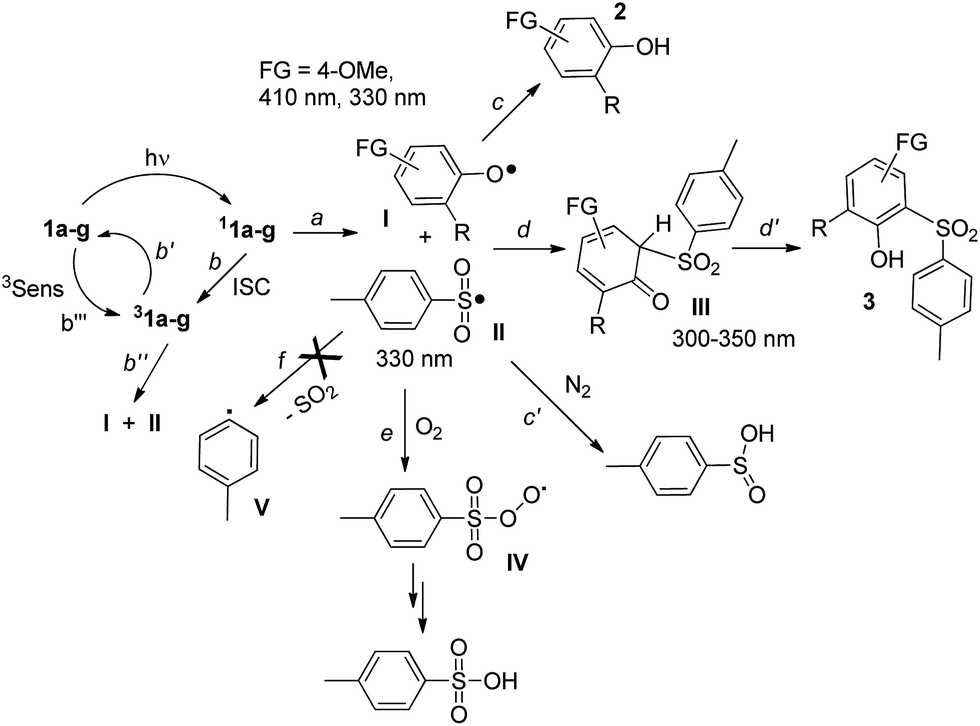 | ||
| Scheme 2 Photoreactivity of aryl tosylates 1a–g. | ||
Laser flash photolysis analyses of 1b in methanol (Fig. 1a) showed two significant absorption bands at 330 and 410 nm. The oxygen-sensitive signal dominating at 330 nm was recognized as due to the 4-methylbenzenesulfonyl radical II, as previously suggested,12,25 whereas the long-lived intermediate with a maximum at 410 nm (and the portion of the 330 nm absorption not quenched by oxygen) can be confidently attributed to the 4-methoxyphenoxy radical Ib.26 In the case of 1d, however, LFP revealed the presence of an additional transient signal, assigned to triplet 31d (absorption at 420 and 580–700 nm), accompanying the two radicals absorbing at 330 and 410 nm.
Photo-Fries reactions are known to arise mainly from excited singlets, although triplets have occasionally been suggested.27 Our data show that cleavage of the ArO–S bond in the excited singlet is the pathway exclusively observed upon direct irradiation. A triplet is not involved, as confirmed by the lack of oxygen quenching of photocleavage for 1b and 1d (Table 2) and as further supported by LFP experiments. As for compound 1d, none of the photoreactivity normally observed in triplet aromatic ketones (e.g. hydrogen abstraction) was found, even in an excellent hydrogen-donating solvent such as methanol. Triplets play a role only when cleavage from singlets does not take place, as in the case of 1e where efficient intersystem crossing leads to a very short-lived triplet (lifetime in the τ = 350–900 ps range in EtOH28), thus preventing any photo-Fries reaction, in analogy with other nitroaryl esters (Scheme 2, paths b and b′).29 On the other hand, acetone-sensitized experiments on 1b revealed that in some cases, when triplets were populated by sensitization, a photo-Fries reaction also took place (paths b′′′, b′′), but was completely inhibited in oxygen-saturated solution.
Finally, in contrast to what was reported for other aryl sulfonates such as mesylates and triflates, heterolysis of the Ar–OS bond7,8,13a,16 plays no role in the photochemical behavior of these substrates, even in the presence of acetone as a triplet sensitizer.
When generated in N2-saturated methanol, the resulting phenoxy and sulfonyl radical intermediates can follow two competing pathways. The first is escape from the solvent cage followed by hydrogen abstraction from the medium to give phenol 2 and paratoluenesulfinic acid, respectively (paths c and c′). On the other hand, direct recombination of the two radicals results in formation of the Fries rearrangement product 3, via the cyclohexadienone intermediate III (paths d and d′). In the case of 1b, the residual, persistent absorption at 300–350 nm and 600–700 nm (see Fig. S22 in ESI† and Fig. 1a) found in LFP experiments could be assigned to III, in analogy with what has already been reported about the photo-Fries rearrangement of naphthyl30a and phenyl acetates.30b,c
With the single exception of the nitroaryl tosylate 1e, all the sulfonates examined displayed comparable photoreactivity in solution (Φ−1 = 0.11–0.29, the highest value measured for tosylate 1d), but different product distributions. We reasoned that the stability of the phenoxy radical I and accordingly the O–H bond dissociation energy (BDE) of 2 could play a role in determining the fate of the reaction. As is apparent from Table 4, the lower the O–H BDE (e.g. 321.3 kJ mol−1 in the case of amino tosylate 1a), the higher is the amount of the Fries adduct 3, whereas formal hydrolysis to 2 is the sole reaction promoted for tosylates with electron-withdrawing substituents and stronger O–H bonds (e.g. BDE = 380.3 kJ mol−1 for 1d).
Thus, competition between paths c, c′ and d, d′ (Scheme 2) depends on the nature and stability of the radical I. Recombination to III (paths d and d′) is favored for electron-rich tosylates such as the amino derivative 1a, making the photoinduced release of PTSA inaccessible. On the other hand, although the presence of electron-withdrawing groups is desirable to maximize the yield of acid generated, moving to a stronger EWG (e.g. in the nitro derivative 1e) completely inhibits the overall photoreactivity. A peculiar case is represented by 1g. As previously observed in related sulfonate esters (mesylates, triflates),16 the presence of a silicon-based substituent ortho to the sulfonate moiety significantly increased the overall photoreactivity.16 The same behaviour was found here, although the cleavage shifted from heterolysis of an Ar–OS bond (in the cases of mesylate and triflate)16 to ArO–S homolysis (in the case of tosylate). The presence of oxygen promoted efficient trapping of the ArSO2˙ radical II (Scheme 2, path e), prior to recombination with the phenoxy radical, and PTSA was obtained via the corresponding arylperoxysulfonyl radical IV.25 Both experimental and spectroscopic data exclude SO2 loss from II to afford the phenyl radical V (path f), in contrast to results previously reported for aryl sulfonyl radicals in acetonitrile.25
Most of the aryl tosylates tested were found to be suitable PAGs for the release of PTSA to be used for cationic ring-opening of epoxy groups in sol–gel systems. When included in photoresists, compounds 1a–g were the only species responsible for the absorption of light (see Fig. 3a). Acid release preceded polymerization (see the different rates observed for 1d conversion and induced polymerization, see Fig. 3a and c), which is consistent with the key role of the substrates examined as PAGs. In this study, the efficiency of organic crosslinking of G8Ge2 acid-labile moieties depended on the tosylate used, the most favourable case being the acetyl derivative 1d. Furthermore, in contrast to what was observed for other aryl sulfonates,7 where competing pathways affected the yield and quality of the generated acids, photodecomposition of aryl tosylates proceeds via an unambiguous mechanism, and strong PTSA was the only acid obtained during irradiation in oxygenated solutions.
Conclusion
Summing up, the photochemistry of substituted phenyl tosylates proceeds exclusively via ArO–S bond homolytic cleavage to generate a phenoxy/sulfonyl radical pair. Depending on the reaction conditions, either paratoluenesulfinic or paratoluenesulfonic acid can be released, the latter being obtained exclusively under oxygenated conditions. It can be noted that acids are photoreleased even in the solid state. The results highlight the potential of aryl tosylates as a promising class of non-ionic PAGs, able to promote cationic polymerization of epoxy groups in hybrid sol–gel photoresists. Furthermore, structural changes can result in a decrease in the solubility of exposed areas in organic or suitable solvents. This peculiarity can be exploited for photo(nano)lithography on sol–gel materials, which plays an important role in the realization of many devices.32Experimental
General information
NMR spectra were recorded on a 300 MHz spectrometer. Attributions were made on the basis of 1H and 13C NMR, as well as DEPT-135 experiments; chemical shifts are reported in ppm downfield from TMS. The photochemical reactions were performed using nitrogen- or oxygen-saturated solutions in quartz tubes in a multi-lamp reactor fitted with 4 × 15 W Hg lamps (emission centered at 254 nm) for irradiation. The reaction course was followed by GC analyses and the products formed were identified and quantified by comparison with authentic samples. Work-up of the photolytes involved concentration in vacuo and chromatographic separation using silica gel. Solvents of HPLC purity were employed in the photochemical reactions. Quantum yields were measured at 254 nm (1 Hg lamp, 15 W).The acidity released was determined from 5 mL of the photolysed solutions by dilution with water (25 mL) and titration with aqueous 0.1 M NaOH by means of a potentiometer equipped with a pH glass combined electrode module. para-Toluenesulfonic (PTSA), para-toluenesulfinic and formic acid were determined via HPLC ion chromatography and quantified by means of a calibration curve obtained with commercially available samples.
Nano-to-microsecond transient absorption experiments were performed using a nanosecond laser flash photolysis apparatus equipped with a 20 Hz Nd:YAG laser (20 ns, 1 mJ at 266 nm) and a 150 W Xe flash lamp as the probe light. Samples were placed in a quartz cell (10 × 10 mm section) at a concentration adjusted to obtain an OD value of 1.0 at 266 nm.
Phenols 2b–f, 3-glycidyloxypropyltrimethoxysilane (GPTMS) and germanium tetraethoxide (TEOG) were commercially available and used as received. 4-N,N-Dimethylaminophenol 2a13a and 4-methoxy-2-trimethylsilylphenol 2g16 were prepared by known procedures.
Synthesis of aryl tosylates 1a–g
Compounds 1a–g were synthesized by adapting a known procedure.18 To a solution of the chosen phenol (20 mmol) in dichloromethane (80 mL) and triethylamine (15 mL) at room temperature, p-toluenesulfonyl chloride (24 mmol) was added portionwise and the resulting mixture was stirred overnight. Water (25 mL) was then added and the resulting mixture was stirred for an additional 3 h. The mixture was then extracted with ethyl acetate (4 × 50 mL) and the organic layers were reunited and washed with water (3 × 50 mL), 10% aqueous HCl (3 × 150 mL, except in the case of 1a), water (2 × 150 mL), saturated aqueous NaHCO3 (2 × 150 mL) and brine (2 × 100 mL), and dried over Na2SO4. The solvent was evaporated under vacuum. The resulting residue was purified by column chromatography or recrystallization.:1). Colorless solid, mp = 60–62 °C. 1g: 1H NMR (CDCl3, δ) 7.85–7.35 (AA′BB′ system, 4H), 6.95 (m, 2H), 6.75 (dd, 1H, J = 3 Hz and 9 Hz), 3.80 (s, 3H), 2.45 (s, 3H), 0.30 (s, 9H). 13C NMR (CDCl3, δ) 157.0, 148.4, 145.0, 134.2, 134.0, 129.7 (CH), 128.2 (CH), 121.0 (CH), 120.5 (CH), 114.5 (CH), 55.4 (CH3), 21.6 (CH3), −0.7 (CH3). IR (neat, ν/cm−1): 2956, 1467, 1370, 1194, 1179, 1157, 1093, 1037. Anal. calcd for C17H22O4SSi: C, 58.25; H, 6.33. Found: C, 58.2; H, 6.2.
Preparative irradiations
:1) to afford 330 mg 4-(N,N-dimethylamino)-2-(p-toluenesulfonyl)phenol (3a, colorless solid, 75% yield, mp = 133–135 °C). 3a: 1H NMR (CDCl3, δ): 8.55 (s, 1H), 7.80–7.30 (AA′BB′ system, 4H), 6.90–6.80 (m, 3H), 2.80 (s, 6H), 2.40 (s, 3H). 13C NMR (CDCl3, δ) 146.0, 145.0, 144.4, 138.9, 129.9 (CH), 126.7 (CH), 123.5, 121.8 (CH), 119.6 (CH), 110.9 (CH), 41.0 (CH3), 21.5 (CH3). IR (neat, ν/cm−1): 3377, 2924, 1503, 1139, 1090, 963. Anal. calcd for C15H17NO3S: C, 61.83; H, 5.88; N, 4.81. Found: C, 61.9; H, 6.1; N, 4.9.
:1) to give a mixture of 4-methoxy-2-(p-toluenesulfonyl)phenol (3b, 44 mg, 32% yield based on consumed 1b) and unreacted 1b (3 mg). 3b: 1H NMR (CDCl3, from the mixture, δ) 7.85–7.30 (AA′BB′, 4H), 7.15 (d, 1H, J = 3 Hz), 7.05 (dd, 1H, J = 3 and 9 Hz), 6.95 (d, 1H, J = 9 Hz), 6.80 (s, 1H), 3.70 (s, 3H), 2.40 (s, 3H). 13C NMR (CDCl3, from the mixture) δ: 153.0, 149.7, 144.8, 138.4, 130.0 (CH), 126.8 (CH), 123.7 (CH), 123.4, 120.1 (CH), 111.2 (CH), 55.7 (CH3), 21.5 (CH3).
:2) to afford 143 mg of a mixture of 4-tert-butylphenol (2c, 79 mg, 23%) and 4-tert-butyl-2-(p-toluenesulfonyl)phenol (3c, 64 mg, 19% yield). 3c: 1H NMR (from the mixture, CDCl3) δ: 7.80–7.35 (AA′BB′, 4H), 7.65 (d, 1H, J = 3 Hz), 7.48 (dd, 1H, J = 3 and 9 Hz), 7.35 (s, 1H), 6.90 (d, 1H, J = 9 Hz), 3.80 (s, 3H), 2.45 (s, 3H). 13C NMR (from the mixture, CDCl3) δ: 153.4, 143.9, 138.4, 133.5 (CH), 130.0 (CH), 129.7, 126.7 (CH), 125.0 (CH), 123.4, 118.6 (CH), 34.2, 31.1 (CH3). IR of the mixture (neat, ν/cm−1): 3392, 2963, 1516, 1227, 1141, 831.
:1), to afford a mixture of 22 mg 4-methoxy-2-(p-toluenesulfonyl)-5-trimethylsilylphenol (3g, 14% yield based on consumed 1g) along with 6 mg unreacted 1g. 3g: 1H NMR (from the mixture, CDCl3, δ) 9.05 (s, 1H), 7.85–7.30 (AA′BB′, 4H), 7.15 (d, 1H, J = 3 Hz), 7.10 (d, 1H, J = 3 Hz), 3.80 (s, 3H), 2.40 (s, 3H), 0.30 (s, 9H). 13C NMR (from the mixture, CDCl3, δ) 153.9, 152.6, 144.6, 136.4, 132.3, 129.9 (CH), 126.7 (CH), 126.4 (CH), 122.1, 111.6 (CH), 55.8 (CH3), 21.5 (CH3), −1.3 (CH3).
Use of aryl tosylates 1a–g as PAGs in polymerization processes
The detailed protocol for the synthesis of the G8Ge2 system has already been described.21 As indicated above, GPTMS and TEOG are the main precursors of the sol and react in a molar ratio of 80:20 under acidic conditions to generate the G8Ge2 system with a final concentration of 150 (SiO2 + GeO2) g L−1. Aryl tosylates 1a–d, 1f–g (1% mol with respect to the concentration of the starting material GPTMS) were incorporated in the sol. Films were deposited by spin-coating (spinning rate: 2000 rpm, spinning time: 30 s) on silicon (100) substrates and fused silica slides to give a thickness of about 1 μm.
The samples obtained were pre-baked for 30 min at 80 °C to remove residual solvent and irradiated with UV light for an increasing curing time using a Hg–Xe UV spot light source, enhanced in the UV region (254–365 nm range), with a power density of around 150 mW cm−2 on the sample surface. The progress of the photoinduced epoxy polymerization was followed by means of Fourier transform infrared spectroscopy (FT-IR) analyses. The analyses were performed in transmission mode within the 400–4000 cm−1 range with 4 cm−1 resolution for a total of 32 scans. To evaluate and compare the efficiency of the different PAGs in the solid state, the epoxy polymerization degree for each PAG/sol–gel matrix system was assessed by collecting progressive FT-IR spectra with increasing UV exposure time (1–18 min); the area under the bands at 3000–3060 cm−1, related to C–H stretching in the epoxy ring, was calculated by a Gaussian peak fitting procedure (Microcal Origin software).
Acknowledgements
S.P. acknowledges MIUR, Rome (FIRB-Futuro in Ricerca 2008 project RBFR08J78Q) for financial support and Prof. A. Albini (University of Pavia) and Prof. P. Hoggard (Santa Clara University) for fruitful discussions. This work has been supported by the Fondazione Cariplo (grant no. 2012-0186).Notes and references
- R. Ayothi, Y. Yi, H. B. Cao, W. Yueh, S. Putna and C. K. Ober, Chem. Mater., 2007, 19, 1434–1444 CrossRef CAS; S.-Y. Moon and J.-M. Kim, J. Photochem. Photobiol., C, 2007, 8, 157–173 Search PubMed; M. Shirai and M. Tsunooka, Bull. Chem. Soc. Jpn., 1998, 71, 2483–2507 CrossRef; M. Shirai and M. Tsunooka, Prog. Polym. Sci., 1996, 21, 1–45 CrossRef; J. M. Frechét, Pure Appl. Chem., 1992, 64, 1239–1248 CrossRef.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- K. L. Covert and D. J. Russell, J. Appl. Polym. Sci., 1993, 49, 657–671 CrossRef CAS PubMed.
- J. Lalevée, X. Allonas, J.-P. Fouassier, M. Shirai and M. Tsunooka, Chem. Lett., 2003, 32, 178–179 CrossRef.
- (a) J. P. Malval, S. Suzuki, F. M. Savary, X. Allonas, J. P. Fouassier, S. Takahara and T. Yamaoka, J. Phys. Chem. A, 2008, 112, 3879–3885 CrossRef CAS PubMed; (b) M. Shirai, T. Yatsuo and M. Tsunooka, J. Photopolym. Sci. Technol., 1996, 9, 273–276 CrossRef CAS; (c) M. Shirai and H. Okamura, Prog. Org. Coat., 2009, 64, 175–181 CrossRef CAS PubMed; (d) L. Steidl, S. J. Jhaveri, R. Ayothi, J. Sha, J. D. McMullen, S. Y. C. Ng, W. R. Zipfel, R. Zentel and C. K. Ober, J. Mater. Chem., 2009, 19, 505–513 RSC.
- M. Ikbal, R. Banerjee, S. Atta, A. Jana, D. Dhara, A. Anoop and N. D. P. Singh, Chem.–Eur. J., 2012, 18, 11968–11975 CrossRef CAS PubMed and references therein.
- M. Terpolilli, D. Merli, S. Protti, V. Dichiarante, M. Fagnoni and A. Albini, Photochem. Photobiol. Sci., 2011, 10, 123–127 CAS.
- D. Ravelli and M. Fagnoni, in CRC Handbook of Organic Photochemistry and Photobiology, ed. A. Griesbeck, M. Oelgemoeller and F. Ghetti, CRC Press, 3rd edn, 2012, pp. 393–417 and references therein Search PubMed.
- G. G. Barclay, D. R. Medeiros and R. F. Sinta, Chem. Mater., 1995, 7, 1315–1324 CrossRef CAS.
- F. M. Houlihan, T. X. Neenan, E. Reichmanis, J. M. Kometani, L. F. Thompson, T. Chin and O. Nalamasu, J. Photopolym. Sci. Technol., 1990, 3, 259–273 CrossRef CAS.
- K. E. Uhrich, E. Reichmanis and F. A. Baiocchi, Chem. Mater., 1994, 6, 295–301 CrossRef CAS; F. M. Houlihan, A. Shugard, R. Gooden and E. Reichmanis, Macromolecules, 1988, 21, 2001–2006 CrossRef.
- F. Ortica, C. Coenjarts, J. C. Scaiano, H. Liu, G. Pohlers and J. F. Cameron, Chem. Mater., 2001, 13, 2297–2304 CrossRef CAS.
- (a) M. De Carolis, S. Protti, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2005, 44, 1232–1236 CrossRef CAS PubMed; (b) M. Ikbal, A. Jana, N. D. P. Singh, R. Banerjee and D. Dhara, Tetrahedron, 2011, 67, 3733–3742 CrossRef CAS PubMed.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS; C. Sanchez, B. Lebeau, F. Ribot and M. In, J. Sol-Gel Sci. Technol., 2000, 19, 31–38 CrossRef.
- G. Brusatin and G. Della Giustina, J. Sol-Gel Sci. Technol., 2011, 60, 299–314 CrossRef CAS PubMed.
- E. Abitelli, S. Protti, M. Fagnoni and A. Albini, J. Org. Chem., 2012, 77, 3501–3507 CrossRef CAS PubMed.
- S. Crespi, D. Ravelli, S. Protti, A. Albini and M. Fagnoni, Chem.–Eur. J., 2014, 20, 17572–17578 CrossRef CAS PubMed.
- Z.-Y. Tang and Q.-S. Hu, J. Am. Chem. Soc., 2004, 126, 3058–3059 CrossRef CAS PubMed.
- B. Alonso, D. Massiot, F. Babonneau, G. Brusatin, G. Della Giustina, P. Innocenzi and T. Kidchob, Chem. Mater., 2005, 17, 3172–3180 CrossRef CAS.
- A significant amount of formate ion, up to 2 mg L−1 was observed, probably due to the degradation of G8Ge2.
- G. Della Giustina, G. Brusatin, M. Guglielmi and F. Romanato, Mater. Sci. Eng., C, 2007, 27, 1382–1385 CrossRef CAS PubMed.
- J. L. Stratenus and E. Havinga, Recl. Trav. Chim. Pays-Bas, 1966, 85, 434–436 CrossRef CAS PubMed.
- C. Raviola, V. Canevari, S. Protti, A. Albini and M. Fagnoni, Green Chem., 2013, 15, 2704–2708 RSC.
- H. Qrareya, S. Protti and M. Fagnoni, J. Org. Chem., 2014, 79, 11527–11533 CrossRef CAS PubMed.
- C. Coenjarts, F. Ortica, J. Cameron, G. Pohlers, A. Zampini, D. Desilets, H. Liu and J. C. Scaiano, Chem. Mater., 2001, 13, 2305–2312 CrossRef CAS.
- L. Johnstonn, N. Mathivan, F. Negri and W. Siebrand, Can. J. Chem., 1993, 71, 1655–1662 CrossRef CAS; D. Shukla, N. P. Schepp, N. Mathivan and L. J. Johnston, Can. J. Chem., 1997, 75, 1820–1829 CrossRef.
- See for example: Y. Kageyama, R. Ohshima, K. Sakurama, Y. Fujiwara, Y. Tanimoto, Y. Yamada and S. Aoki, Chem. Pharm. Bull., 2009, 57, 1257–1266 CrossRef CAS; I. F. Molokov, Y. P. Tsentalovich, A. V. Yurkovskaya and R. Z. Sagdeev, J. Photochem. Photobiol., A, 1997, 110, 159–165 CrossRef; A. K. Zarkadis, V. Georgakilas, G. P. Perdikomatis, A. Trifonov, G. G. Gurzadyan, S. Skoulika and M. G. Siskosa, Photochem. Photobiol. Sci., 2005, 4, 469–480 Search PubMed.
- M. Takezaki, N. Hirota and M. Terazima, J. Phys. Chem. A, 1997, 101, 3443–3448 CrossRef CAS.
- See for instance G. M. Coppinger and E. R. Bell, J. Phys. Chem., 1966, 70, 3479–3489 CrossRef CAS; M. M. Miranda and F. Galindo, in Photochemistry of Organic Molecules in Isotropic and Anisotropic Media, ed. V. Ramamurthy and K. S. Schanze, Marcel Dekker Inc., New York-Basel, 2003 Search PubMed.
- (a) M. Gohdo, T. Takamasu and M. Wakasa, Phys. Chem. Chem. Phys., 2011, 13, 755–761 RSC; (b) M. C. Jiménez, M. A. Miranda, J. C. Scaiano and R. Tormosa, Chem. Commun., 1997, 1487–1488 RSC; (c) C. E. Kalmus and D. M. Hercules, J. Am. Chem. Soc., 1974, 96, 449–456 CrossRef CAS.
- C. X. Xue, R. S. Zhang, H. X. Liu, X. J. Yao, M. C. Liu, Z. D. Hu and B. T. Fan, J. Chem. Inf. Comput. Sci., 2004, 44, 669–677 CrossRef CAS PubMed.
- L. Brigo, E. Zanchetta, G. Della Giustina and G. Brusatin, Proc. SPIE, 2014, 9161 Search PubMed.
- R. H. Munday, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 2754–2755 CrossRef CAS PubMed.
- J.-i. Kuroda, K. Inamoto, K. Hiroya and T. Doi, Eur. J. Org. Chem., 2009, 2251–2261 CrossRef CAS PubMed.
- J. H. Choi, B. C. Lee, H. W. Lee and I. Lee, J. Org. Chem., 2002, 67, 1277–1281 CrossRef CAS PubMed.
- H.-D. Choi, P.-J. Seo and B.-W. Son, Arch. Pharmacal Res., 2002, 25, 786–789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of 1H and 13C NMR spectra of compounds 1a–g, 3a–c, 3g, potentiometric titration of photolysed solutions of 1a–g in methanol. See DOI: 10.1039/c5ra03522h |
| This journal is © The Royal Society of Chemistry 2015 |