
Simple and expeditious pinacol coupling of non usual α,β-unsaturated carbonyl compounds in water†‡
Muriel Billamboza,
Nicolas Sottoa,
Carole Chevrin-Villettea and
Christophe Len*ab
aSorbonne Universités, Université de Technologie de Compiègne, Ecole Supérieure de Chimie Organique et Minérale, EA 4297 Transformations Intégrées de la Matière Renouvelable, Centre de Recherches de Royallieu, CS 60319, F-60205 Compiègne cedex, France. E-mail: christophe.len@utc.fr; Fax: +33 (0)3 44 97 15 91; Tel: +33 (0)3 44 23 88 28
bDepartement of Chemistry, University of Hull, Hull 6 HU6 7RX, UK
First published on 15th May 2015
Abstract
Using zinc (0) in a 5% v AcOH aqueous solution allowed the efficient pinacol coupling of aliphatic or aromatic unusual, α,β-unsaturated carbonyl compounds such as citral A in good to excellent yields (56–99%). It can also be successfully applied to acetophenone.
Pinacol coupling is one of the most used methods for the formation of diols via carbon–carbon bonds. Although discovered in 1859,1 this well documented reaction2 is still studied and finds many applications as the vicinal diols obtained are useful intermediates for the construction of biologically important natural or synthetic products.3 These diols can also be transformed to numerous high value products, such as epoxides, ketones, aminoalcohols…
To perform this reaction, a variety of methods have been developed using mainly aromatic carbonyl compounds as starting materials. Among them, the most reliable systems use either simple low-valent metals such as Mg,4 Mn,5 Zn,6 Al,7 Zn–Cu couple,8 or transition metal complexes such as Ti-, V- or Zr-derivatives.9 Pinacol-type couplings were also developed by using toxic or expensive promoters (In,10 Sm,11 Ga,12 and other metals13), in particular for the development of reaction with aliphatic derivatives that are commonly inert under classical conditions.2a,7b,9a,14
Nowadays, due to the depletion of fossil resources, chemists are strongly encouraged to develop safer protocols, less hazardous synthesis, catalysis for the production of high value products and the use of benign solvents or renewable feedstocks. Starting form biosourced α,β-unsaturated carbonyl compounds such as citral, myrtenal, farnesal…, pinacol couplings are rarely studied. The corresponding diols were obtained in low yields and no report deals with their coupling in water as green solvent. For example, the reductive homo-coupling of geranial was described by electrochemical reduction in EtOH-pH5 buffer in around 35% yield.15 Moreover, starting from crotonaldehyde in presence of Al/InCl3 in THF, the diol was prepared in 45% yield.16 Otherwise, pinacol coupling of trans hex-2-enal was performed by TiI4 in EtCN with 16% yield.17
The aim of this work was to develop a green, simple and efficient methodology to form vicinal diols from natural green note aldehydes and more widely α,β-unsaturated derivatives 1 (Fig. 1). Crotonaldehyde 1a affording the corresponding diol 2a was selected as model substrate for our study.
 | ||
| Fig. 1 α,β-Unsaturated derivatives 1, crotonaldehyde 1a and the diol 2a. | ||
Initially, the effect of solvent changes on the pinacol coupling of 1a at room temperature in presence of 2 equivalents of zinc was examined. The results are summarized in Table 1.
| Entry | Solvent | Additive (eq.) | Time (h) | 2a (%) |
|---|---|---|---|---|
| 1 | THF/H2O | None | 15 | Traces |
| 2 | THF/H2O | HCl (2eq.) | 15 | 1 |
| 3 | THF/H2O | H2SO4 (2eq.) | 15 | 4 |
| 4 | THF/H2O | AcOH (2eq.) | 15 | 36 |
| 5 | THF/H2O | NH4Cl sat. | 15 | 76 |
| 6 | H2O | None | 15 | 11 |
| 7 | H2O | NH4Cl sat. | 15 | 27 |
| 8 | H2O | AcOH (2eq.) | 0.3 | 99 |
| 9 | EtOH | AcOH (2eq.) | 0.3 | 99 |
| 10 | EtOH/H2O | AcOH (2eq.) | 15 | 39 |
The previously described medium THF/water18 was initially tested. Without acidic additive or in the presence of strong acids, the pinacol coupling is very slow (Table 1, entries 1–3). For this medium, weak acids such as NH4Cl or AcOH proved to be best promoters (Table 1, entries 4 and 5). Protic solvents (water, ethanol or a mixture of water and ethanol; Table 1, entries 6–10) are ideal to conduct pinacol coupling reactions, especially in presence of AcOH as additive. Total conversions and excellent yields in only 20 minutes were obtained (Table 1, entries 8 and 9). In the topic of green chemistry, organic syntheses in water present many advantages in terms of safety and environmental aspects compared to organic solvents. Moreover, in the case of radical reactions, it was proved that solvent plays a crucial role and that water allows control of radical reactions.19 As a consequence, water was selected as solvent for further optimization. Then, metallic source was screened (Table 2).
| Entry | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a For typical conditions: 1a (2.3 mmol) was reacted with metal (2 eq.) in 5% v AcOH in water during 20 minutes at rt. | ||||||
| Metal | Zn | Mg | Al | Mn | Fe | Cu |
| Yield of 2a (%) | 99 | 6 | 0 | 7 | 0 | 0 |
Among the 6 tested metals, only zinc, a cheap readily available metal, permitted to afford the corresponding diol 2a in good yield (Table 2).
It was selected for further studies. As an acidic medium is required to maintain good kinetics, a screening of mineral and organic acids was then realized (Table 3). In our hands, yields were lower than 63% except for acetic acid (Table 3, entry 1).
| Entry | Acid (2 eq.) | 2a (%) |
|---|---|---|
| a All reactions are carried out in presence of 2eq. of acid in water (5 mL total volume for 2.3 mmol of 1a) during 20 minutes in presence of 2 eq. of zinc. | ||
| 1 | AcOH | 99 |
| 2 | Citric acid | 40 |
| 3 | CH3SO3H | 52 |
| 4 | Tartaric acid | 63 |
| 5 | H2SO4 | 30 |
| 6 | HCl | 29 |
| 7 | NH4Cl | 33 |
Combining all results, expeditious, simple and eco-compatible conditions were found for the efficient pinacol coupling of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 equivalents of zinc in a 5% v AcOH/H2O medium during 20 minutes at room temperature. This new methodology was then applied to various α,β-unsaturated aldehydes and ketones as presented in Table 4.
:2 equivalents of zinc in a 5% v AcOH/H2O medium during 20 minutes at room temperature. This new methodology was then applied to various α,β-unsaturated aldehydes and ketones as presented in Table 4.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | No. | Substrate 1 | Results (%) | Sela (%) | dl/mesob | |||||
| R1 | R2 | R3 | R4 | Conv. | 2 | 3 | ||||
| a Selectivity is defined as 2/(2 + 3).b The dl/meso ratio was determined by 1H NMR of the crude product.c For entry 11, the major product resulted from a cascade of reductive coupling and cyclization of chalcone, as previously described20 (product 4). | ||||||||||
| 1 | a | CH3 | H | H | H | 100 | 99 | 0 | 100 | 42/58 |
| 2 | b | CH3–(CH2)2 | H | H | H | 100 | 79 | 21 | 79 | 37/63 |
| 3 | c | CH3–(CH2)6 | H | H | H | 94 | 78 | 10 | 89 | 42/58 |
| 4 | d | CH3–(CH2)4–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– CH– |
H | H | H | 63 | 61 | 0 | 100 | 0/100 |
| 5 | e | Ph | H | H | H | 91 | 63 | 28 | 70 | 43/57 |
| 6 | f | CH3 | H | CH3 | H | 100 | 90 | 8 | 92 | 0/100 |
| 7 | g | CH3–CH2 | H | CH3 | H | 100 | 65 | 0 | 100 | 0/100 |
| 8 | h |  |
CH3 | H | H | 100 | 56 | 0 | 100 | 39/61 |
| 9 | i |  |
93 | 67 | 10 | 87 | 0/100 | |||
| 10 | j |  |
80 | 71 | 6 | 92 | 0/100 | |||
| 11 | k | Ph | H | H | Ph | 100 | — | — | ND | NDc |
| 12 | l | 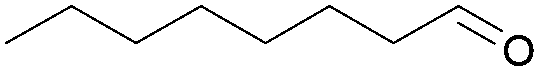 |
0 | — | — | — | — | |||

As depicted, α,β-unsaturated aldehydes reacted smoothly under these conditions to conduct to the desired pinacols in good to excellent yields. Various types of compounds have been used as substrates such as linear saturated enals 1a–c, unsaturated enals 1d–e or disubstituted enals 1f–j. Comparison with an α,β-unsaturated ketone 1k and an aliphatic aldehyde 1l has been also made. Linear saturated enals 1a–c reacted smoothly under the optimized conditions (Table 4, entries 1–3). Crotonaldehyde (1a) was totally converted with excellent yield and selectivity (Table 4, entry 1). Long chain linear derivatives: trans hex-2-enal (1b) and trans dec-2-enal (1c) led to the desired pinacol products in 79% and 78% yield, respectively (Table 4, entries 2 and 3). It should be noticed that non negligible amounts of the reduction side products, i.e. the allylic alcohols 3b and 3c, have been isolated in 21% and 10% yield, respectively. For this linear saturated enals, no diastereoselectivity has been observed during the reaction as pinacols 2a–c are obtained as a mixture of dl and meso isomers with a slightly excess of the meso isomer (Table 4, entries 1–3). When using unsaturated enals 1d–e, lower yields were obtained (Table 4, entries 4 and 5, 61 and 63% yield, respectively). This lower reactivity can be explained by a higher stability of the ketyl radical. Cinnamaldehyde (1e), as an aromatic α,β-unsaturated aldehyde, gave 2e in only 63% yield with a non negligible amount of the reduction product 3e (Table 4, entry 5). This can be the result of an enhancement of the radical stability by the aromatic moiety. Disubstituted enals 1f–j also reacted fast under the optimized conditions, but with slightly lower yields than their less hindered analogues (Table 4, entries 6–10). For these derivatives, the increase of the steric hindrance around the carbonyl group has a dramatic effect on the dl/meso ratio. The 2f and 2g meso isomers, with a methyl group on the α position, are obtained selectively (Table 4, entries 6 and 7). It is noteworthy than citral A (1h), a long chain hindered natural aliphatic α,β-unsaturated aldehyde reacted well under these conditions to conduct to a mixture of meso and dl isomers (Table 4, entry 8). To our knowledge, it is the first time that citral A (1g) proved to be a good substrate for pinacol coupling in water. Moreover, natural cyclic α,β-unsaturated aldehydes such as myrtenal (1i) and perillaldehyde (1j) were also successfully converted into pinacol coupling products 2i and 2j with a total selectivity for the meso isomers (Table 4, entries 9 and 10). The reactivity towards pinacol coupling of these cyclic hindered compounds was never described before. For this family of hindered enals 1f–j, it is notable that a substituent on the α position led to total selectivity towards the meso isomer. Less reactive α,β-unsaturated ketone, chalcone 1k, was then submitted to these optimized reaction conditions without success (Table 4, entry 11).
In fact, the previously described cyclodimerization product 4, resulting from a reductive coupling/cyclization cascade (Fig. 2), is mainly isolated in 65% yield.20 As expected, aliphatic aldehyde such as octanal (1l) did not coupled under these simple conditions (Table 4, entry 12).
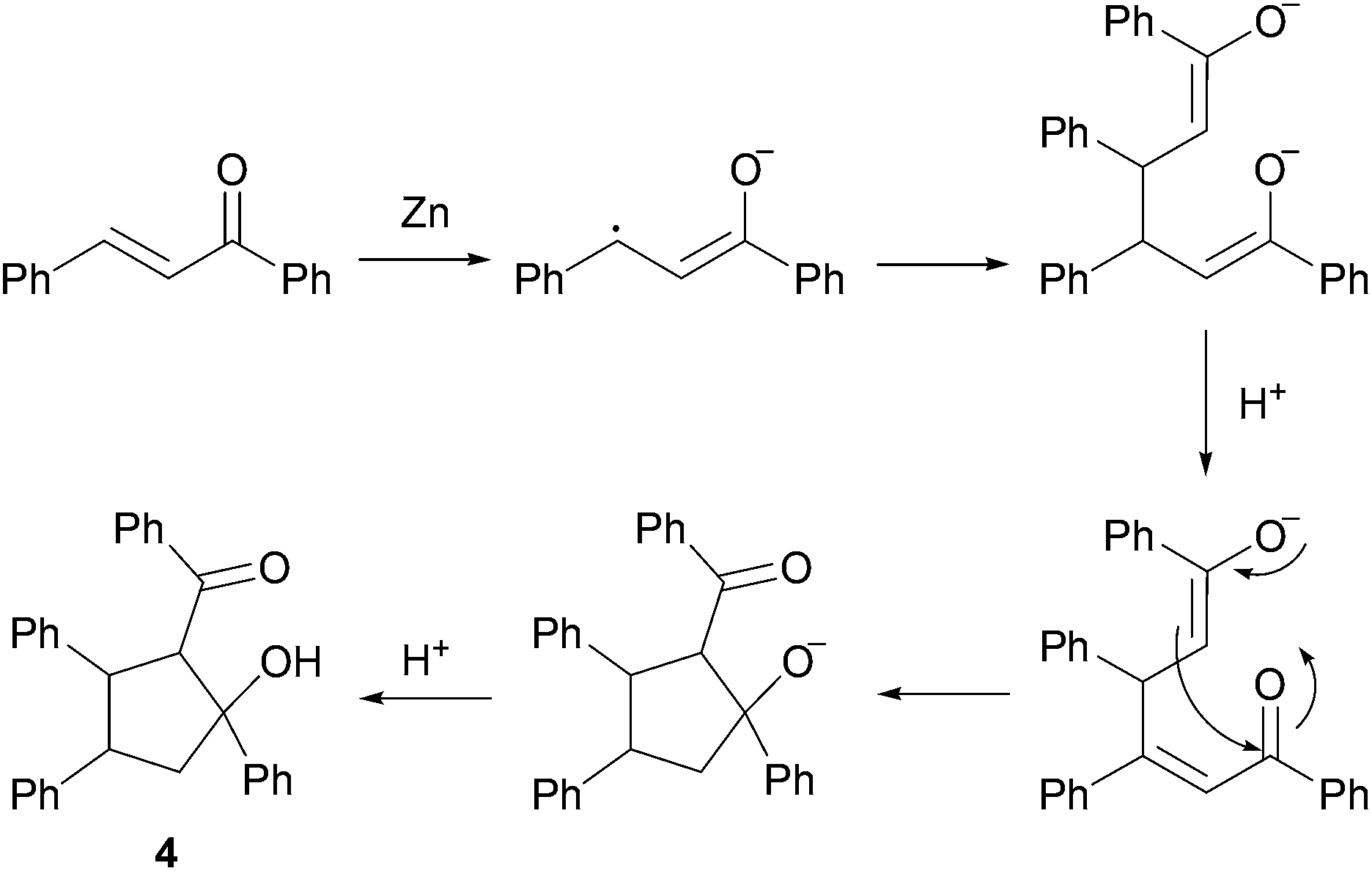 | ||
| Fig. 2 Product 4 formation pathway. | ||
This simple and efficient methodology was then extended to classical aromatic carbonyl compounds (Table 5). Aromatic aldehydes reacted fast under these conditions, but, as observed for cinnamaldehyde (1e) (Table 4, entry 5), the amount of reduction products 3m–3s is high (Table 5, entries 1–7) and sometimes mainly obtained (Table 5, entries 3 and 6). One possible explanation is that kinetics of reaction is too high to let the possibility to ketyl radicals to meet and coupled effectively. For acetophenone derivatives, which are less reactive in single electron transfer, this medium is very interesting and promoted efficiently the ketyl radicals formation and its coupling, allowing the formation of pinacol 2t and 2u from acetophenone (1t) and 4-chloroacetophenone (1u) with 75% yield (Table 5, entries 8 and 9). Much hindered ketone, exemplified by benzophenone (1v), did not coupled under these conditions and only reduction product 3v is obtained in low yield after 20 minutes (Table 5, entry 10).
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | N° | Substrate 1 | Results (%) | Sela (%) | dl/mesob | |||||
| R1 | R2 | R3 | R4 | Conv. | 2 | 3 | ||||
| a Selectivity is defined as 2/(2 + 3).b The dl/meso ratio was determined by 1H NMR of the crude product. | ||||||||||
| 1 | m | H | H | H | H | 96 | 51 | 45 | 53 | 38/62 |
| 2 | n | CH3 | H | H | H | 80 | 54 | 26 | 67 | 48/52 |
| 3 | o | Cl | H | H | H | 80 | 13 | 67 | 16 | 26/74 |
| 4 | p | Br | H | H | H | 64 | 40 | 24 | 62 | 27/73 |
| 5 | q | NO2 | H | H | H | 46 | 0 | 0 | Reduction of NO2 | |
| 6 | r | H | Cl | Cl | H | 90 | 38 | 52 | 42 | 16/84 |
| 7 | s | 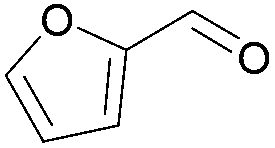 |
100 | 51 | 39 | 57 | 32/68 | |||
| 8 | t | H | H | H | CH3 | 80 | 75 | 0 | 100 | 56/44 |
| 9 | u | Cl | H | H | CH3 | 80 | 75 | 4 | 95 | 57/43 |
| 10 | v | Ph | H | H | Ph | 10 | 0 | 10 | 0 | — |
Conclusions
A simple, green and expeditious methodology for pinacol coupling of α,β-unsaturated aldehydes and acetophenone derivatives has been established in 5% v AcOH in water. Zinc as cheap readily available reductant and acetic acid as promoter are used to obtain high value added pinacol products in only 20 minutes at room temperature in good to excellent yields. Natural compounds such as citral A, myrtenal or perillaldehyde proved to react smoothly under these conditions. Works are still in progress to enhance the selectivity of the coupling reaction for aromatic compounds and α,β-unsaturated ketones.Acknowledgements
This work was supported, as part of the Investments for the Future, by the French Government under the reference ANR-001-01.Notes and references
- (a) R. Fitting, Justus Liebigs Ann. Chem., 1859, 110, 23 CrossRef PubMed; (b) G. Zhao, C. Yang, L. Guo, H. Sun, R. Lin and W. Xia, J. Org. Chem., 2012, 77, 6302 CrossRef CAS PubMed; (c) J.-T. Li, C. Du and X.-L. Zhai, Synth. Commun., 2012, 42, 820 CrossRef CAS PubMed.
- For recent reviews, see: (a) M. Meciarova, S. Toma and P. Babiak, Chem. Pap., 2001, 55, 302 CAS; (b) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed; (c) S. P. Morcillo, D. Miguel, A. G. Campana, L. Alvares de Cienfuegos, J. Justicia and J. M. Cuerva, Org. Chem. Front., 2014, 1, 15 RSC; (d) T. Hirao, Pure Appl. Chem., 2005, 77, 1539 CrossRef CAS; (e) B. S. Terra and F. Macedo Jr, ARKIVOC, 2012, 134 CAS; (f) P. G. Steel, J. Chem. Soc., Perkin Trans. 1, 2001, 2727 RSC; (g) A. Chatterjee and N. N. Joshi, Tetrahedron, 2006, 62, 12137 CrossRef CAS PubMed; (h) J.-T. Li, S.-X. Wang, G.-F. Chen and T.-S. Li, Curr. Org. Synth., 2005, 2, 415 CrossRef CAS.
- (a) T. Wirth, Angew. Chem., Int. Ed. Engl., 1996, 35, 61 CrossRef CAS PubMed; (b) M. Avalos, R. Babiano, P. Cintas, J. L. Jimenze and J. C. Palacios, Recent Res. Dev. Org. Chem., 1997, 1, 159 CAS.
- (a) W.-C. Zhang and C.-J. Li, J. Chem. Soc., Perkin Trans. 1, 1998, 3131 RSC; (b) M. Mecarova and S. Toma, Green Chem., 1999, 1, 257 RSC; (c) W.-C. Zhang and C.-J. Li, J. Org. Chem., 1999, 64, 3230 CrossRef CAS PubMed; (d) H. Maekawa, Y. Yamamoto, H. Shimada, K. Yoneruma and I. Nishiguchi, Tetrahedron Lett., 2004, 45, 3869 CrossRef CAS PubMed; (e) A. Fürstner, R. Csuk, C. Rohrer and H. Weidmann, J. Chem. Soc., Perkin Trans. 1, 1988, 1729 RSC.
- (a) C.-J. Li, Y. Meng and X.-H. Yi, J. Org. Chem., 1997, 62, 8632 CrossRef CAS; (b) R. D. Rieke and S.-H. Kim, J. Org. Chem., 1998, 63, 5235 CrossRef CAS.
- (a) B. K. Hazarika and D. K. Dutta, Synth. Commun., 2011, 41, 1088 CrossRef CAS PubMed; (b) J.-H. So, M.-K. Park and P. J. Boudjouk, J. Org. Chem., 1988, 53, 5871 CrossRef CAS.
- (a) S.-Z. Yuan, Z.-Y. Wang and Z. Li, Chin. J. Chem., 2006, 24, 141 CrossRef CAS PubMed; (b) Y. Mitoma, I. Hashimoto, C. Simion, M. Tashiro and N. Egashira, Synth. Commun., 2008, 38, 3243 CrossRef CAS PubMed; (c) S. Bhar and C. Panja, Green Chem., 1999, 1, 253 RSC; (d) A. A. P. Schreibmann, Tetrahedron Lett., 1970, 11, 4271 CrossRef and references herein.
- P. Delair and J.-L. Luche, J. Chem. Soc., Chem. Commun., 1989, 398 RSC.
- (a) J. L. Oller-Lopez, A. G. Campana, J. M. Cuerva and J. E. Oltra, Synthesis, 2005, 2619 CAS; (b) A. Clerici and O. Porta, J. Org. Chem., 1982, 47, 2852 CrossRef CAS; (c) F. R. Askham and K. M. Carroll, J. Org. Chem., 1993, 58, 7328 CrossRef CAS; (d) T. Hirao, M. Asahara, Y. Muguruma and A. Ogawa, J. Org. Chem., 1998, 63, 2812 CrossRef CAS; (e) T. Hirao, B. Hatano, Y. Imamoto and A. Ogawa, J. Org. Chem., 1999, 64, 7665 CrossRef CAS.
- H. J. Lim, G. Keum, S. B. Kang, B. Y. Chung and Y. Kim, Tetrahedron Lett., 1998, 39, 4367 CrossRef CAS.
- S. Matsukawa and Y. Hinakubo, Org. Lett., 2003, 5, 1221 CrossRef CAS PubMed.
- Z. Y. Wang, S. Z. Yuan, Z. G. Zha and Z. D. Zhang, Chin. J. Chem., 2003, 21, 1231 CrossRef CAS PubMed.
- (a) R. L. Halterman, J. P. Porterfield and S. Mekala, Tetrahedron Lett., 2009, 50, 7172 CrossRef CAS PubMed; (b) T. Hirao, Pure Appl. Chem., 2005, 77, 1539 CrossRef CAS; (c) P. G. Steel, J. Chem. Soc., Perkin Trans. 1, 2001, 2727 RSC; (d) S. Arai, Y. Sudo and A. Nashida, Chem. Pharm. Bull., 2004, 52, 287 CrossRef CAS.
- (a) B. P. Mundy, R. Srinivasa, Y. Kim and T. Dolph, J. Org. Chem., 1982, 47, 1657 CrossRef CAS; (b) J. C. Johnston, J. D. Faulkner, L. Mandell and R. A. Day Jr, J. Org. Chem., 1976, 41, 2611 CrossRef CAS.
- J. C. Johnston, J. D. Faulkner, L. Mandell and R. A. Day Jr, J. Org. Chem., 1976, 41, 2611 CrossRef CAS.
- S. Ohtaka, K. Mori and S. Uemura, Heteroat. Chem., 2001, 12, 309 CrossRef CAS PubMed.
- M. Shimizu, H. Goto and R. Hayakawa, Org. Lett., 2002, 4, 4097 CrossRef CAS PubMed.
- M. Billamboz, J.-C. Legeay, F. Hapiot, E. Monflier and C. Len, Synthesis, 2012, 137 CAS.
- (a) H. Yorimitsu, T. Nakamura, H. Shinokubo, K. Oshima, K. Omoto and H. Fujimoto, J. Am. Chem. Soc., 2000, 122, 11041 CrossRef CAS; (b) M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415 RSC.
- (a) G. Zhao, C. Yang, L. Guo, H. Sun, R. Lin and W. Xia, J. Org. Chem., 2012, 77, 6302 CrossRef CAS PubMed; (b) J.-T. Li, C. Du and X.-L. Zhai, Synth. Commun., 2012, 42, 820 CrossRef CAS PubMed.
Footnotes |
| † This work was performed, in partnership with the SAS PIVERT, within the frame of the French Institute for the Energy Transition Institut pourla Transition Energétique (ITE) P.I.V.E.R.T. (http://www.institut-pivert.com) selected as an Investment for the Future (“Investissements d’Avenir”). |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, products description and 1H and 13C NMR spectra. See DOI: 10.1039/c5ra03870g |
| This journal is © The Royal Society of Chemistry 2015 |