
Lipase immobilized in microemulsion based organogels (MBGs) as an efficient catalyst for continuous-flow esterification of protected fructose†
Felipe K. Sutiliab,
Daniel de O. Nogueiraa,
Selma G. F. Leiteb,
Leandro S. M. Mirandaa and
Rodrigo O. M. A. de Souza*a
aBiocatalysis and Organic Synthesis Group, Chemistry Institute, Federal University of Rio de Janeiro, CEP 22941 909, Rio de Janeiro, Brazil. E-mail: rodrigosouza@iq.ufrj.br
bEscola de Química, Federal University of Rio de Janeiro, CEP 22941 909, Rio de Janeiro, Brazil
First published on 15th April 2015
Abstract
Sugar-based organogels are important esters for the cosmetic, food and pharmaceutical industries due to their intrinsic properties. Chemical routes to obtain these molecules suffer from low yields and undesirable side products. In this way, biocatalysis can be an interesting and efficient alternative, which meets the green chemistry principles. Herein we report our results on the development of a continuous flow process for the production of sugar-based surfactants mediated by immobilized enzymes in microemulsion based organogels, leading to the desired product in high productivities (66.8 to 88.1 g of ester per h per g of lipase).
1. Introduction
The importance of sugar-based esters as biodegradable and odorless surfactants is already known in the literature and its application in innumerable areas has grown during the recent years, including the pharmaceutical, cosmetics and food industry.1–4Biocatalysis is an important alternative to the traditional chemical synthesis since it can meet the green chemistry criteria, important in the development of sustainable processes. The synthesis of sugar-based esters is an interesting subject in which biocatalysis can help in bringing some greenness to the inefficient chemical synthesis.1,3,5–14 Traditionally, high temperatures in the presence of alkaline catalysts and toxic solvents are used, resulting in high-energy consumption, formation of undesirable by-products and low regioselectivity towards the desired product.
Lipases are triacylglycerol acyl hydrolytic enzymes that have found use as hydrolysis, esterification and transesterification reaction catalysis. Upon immobilization, supported enzymes can provide an easily separable and reusable system (together with enhanced product recovery), which boasts of enhanced resistance to deactivation as compared to free enzymes. Immobilization has several implications when generating increasingly stable biocatalysts compatible with continuous processing technologies. Various strategies to immobilize enzymes on a number of supports have been reported. These range from the more extended and widely employed physical methods (e.g. adsorption, entrapping and/or electrostatic immobilization) to chemical protocols (e.g. covalent immobilization).15,16
In our continuous work on the development of more efficient process for sugar-based esters synthesis,9,17 here in we report our results on the immobilization of Cal B in microemulsion based organogels (MBGs) and its application on the development of biocatalyzed continuous-flow process for production of sugar-based surfactants. MBGs are formed by the gelling agent, such as cellulose derivatives, which fully retains the surfactant, water and enzyme components and can be handled as an immobilized biocatalyst that facilitates the diffusion of non-polar substrates and products, being in this way an eligible catalyst for continuous-flow heterogeneous process.
2. Materials and methods
2.1 Materials
Lipase B from Candida antarctica (CaL) and bis-(2-ethylhexyl) sodium sulfosuccinate (AOT) were supplied by Fluka, Basel, Switzerland. Toluene was purchase from Tedia and no further purification was done. Hydroxy-propyl-methyl cellulose (HPMC) (3500–5600 cP) and palmitic acid (≥98%) were purchased from Sigma, Germany and Vetec, Brazil respectively.2.2 Synthesis of 2,3:4,5-O-diisopropylidene-β-d-frutopyranose (1)
In a 2000 mL reactor was added 30 g (44.8 mmol) of 1 and 400 mL of acetone being vigorously stirred (mechanical stirrer) at 5 °C for 15 min. Then, 16 mL of concentrate sulfuric acid (H2SO4) was slowly added to the reaction mixture. The solution was kept under stirring for 150 min. Subsequently, the reaction mixture was cooled (0–10 °C) in ice bath and neutralized with 50% NaOH (w/v). The pH was adjusted with saturated sodium carbonate. The final mixture was filtered through paper to remove the sodium carbonate and unreacted sugar and subsequently, the solvent was evaporated under reduced pressure. The solid formed was diluted with 400 mL of dichloromethane. A 0.5 M H2SO4 solution was added and stirred vigorously for 120 min. The organic phase was separated and washed consecutively with sodium bicarbonate (NaHCO3) and water and dried with anhydrous sodium sulfate (Na2SO4). The solvent was evaporated under reduced pressure until obtaining a white solid which was recrystallized in hexane with 30% final yield after filtration through activated charcoal.92.3 Batch reaction procedure
A stock solution containing palmitic acid and 1 in n-heptane or in toluene in proportion 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (5–100 mM) was prepared. In 4 mL glass reactors were put 0.75 g of MBG (containing 30–180 μg of lipase) and 2 mL of palmitic acid and 1 (1:1) in a desired solvent with the respective concentration according design of experiments. Molecular sieves were used at concentration according DOE (10–20% p/v). The flasks were incubated at 40–55 °C at 50–250 rpm.
:1 (5–100 mM) was prepared. In 4 mL glass reactors were put 0.75 g of MBG (containing 30–180 μg of lipase) and 2 mL of palmitic acid and 1 (1:1) in a desired solvent with the respective concentration according design of experiments. Molecular sieves were used at concentration according DOE (10–20% p/v). The flasks were incubated at 40–55 °C at 50–250 rpm.
2.4 Continuous flow experiments
Reaction mixture (palmitic acid and 1 in the appropriate solvent) was initially stirred for 5 min at the appropriate reaction temperature. Esterification reactions were performed in a packed-bed reactor (Asia Flow Reactor) equipped with an Omnifit column (4.71 mL) containing 3.4 g of the desired MBG (0.6 mg of lipase). For each flow tested (0.1 mL min−1 to 0.6 mL min−1), first only the pure solvent was pumped through the system until the instrument had achieved the desired reaction parameters and stable processing was ensured, after which the reaction mixture was pumped through the system.2.5 Conversion analysis
Process conversions were measured by determining the residual fatty acid by applying a modified Lowry and Tinsley method.18 In eppendorf tube was placed 0.3 mL of reaction medium, 1 mL heptane, 0.3 mL of 5% copper acetate–pyridine solution (pH 6) and stirred vigorously for 30 s. The supernatant was measured by spectrophotometer UV/715 nm visible wavelength. Each reaction was measured in triplicate and conversion calculations were based on the analytical curve with the acid used. This methodology for determination of residual acid is aligned with the analytical chromatographic methods, being widely used.2.6 Factorial designs for optimization of the system
First, we carried out a fractional factorial design 25−1 to determine the optimal conditions for esterification reaction in batch conditions catalyzed by Cal B immobilized in MBG for designed solvent, with the independent variables temperature, amount of enzyme, substrate concentration, amount of molecular sieves and stirring, varying in two levels and three replications of the central point. The fractional factorial design (FFD) is presented in ESI† where shows the variables with the respective levels used.2.7 Preparation of microemulsion-based organogels (MBGs)
The microemulsion organogels were prepared as described by Xenakis and coworkers.21,22 The appropriate amount of lipase CaL B (5–30 mg mL−1) was dissolved in 0.2 M Tris/HCl buffer pH 7.5 solution. The system was carefully homogenized until the formation of a clear solution indicating the formation of the microemulsion. Microemulsion-based organogels (MBGs) were prepared by introducing appropriate amounts of AOT microemulsion containing lipase to a second solution of HPMC in water.83. Results and discussion
Experimental design is already known over literature as an important tool to help optimization of reaction conditions once it is possible to determine the influence of different parameters in order to maximize yields. With this in mind, we decided to investigate the main variables that impact yield in the esterification reaction between 1 and palmitic acid (2) catalyzed by Cal B immobilized in microemulsion based organogel (MBG), under batch conditions. This immobilized enzyme was already reported by our group as an efficient support for monoacylglycerol production under batch conditions.8A 25−1 fractional factorial design (FFD) was performed and several independent variables were tested in two levels: temperature, amount of supported enzyme, substrate concentration, amount of molecular sieves and stirring. Aiming to improvement the yield esterification reaction molecular sieves were added to the system due the raise in the water content that favors hydrolysis over synthesis.19
Since reaction time usually corresponds to a variable, which exerts greatest influence on the reaction course, masking real effects upon other variables, we started by evaluating its influence on ester production independently. The esterification reaction between palmitic acid (2) and 1 catalyzed by Cal B immobilized in MBG (180 μg of lipase) was monitored for 2.5 hours under the following conditions: atmospheric pressure, 40 °C, 10% (w/v) of molecular sieves, 40 mM of palmitic acid and 1 (1:1) and 150 rpm of stirring rate, using heptane or toluene as solvent and conversions monitored by Lowry and Tinsley method as shown in Fig. 1.
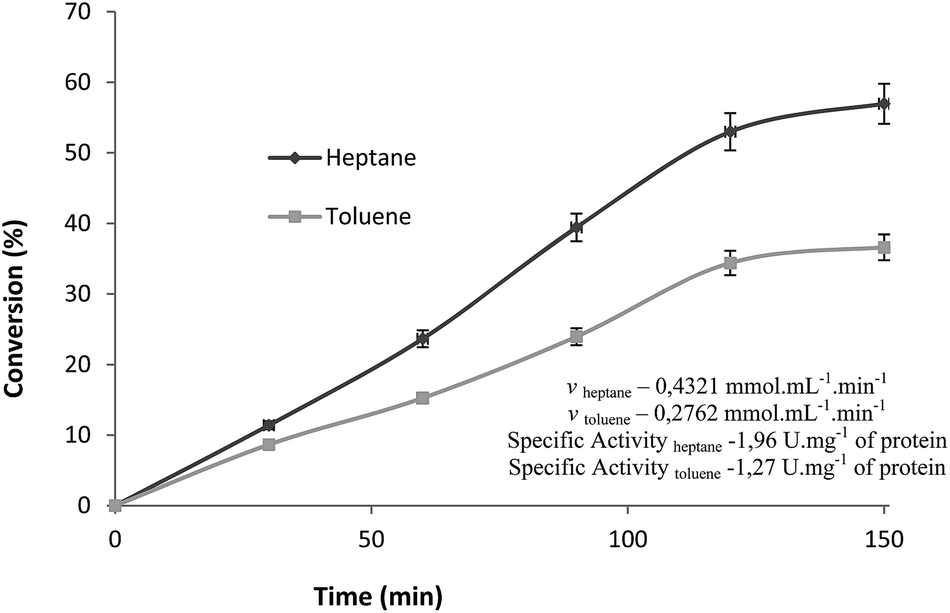 | ||
| Fig. 1 Conversion of esterification (%) in different reaction times. | ||
Either in heptane as in toluene, the best conversion of linear phase was obtained after 2 hours of reaction with (53% and 34%, respectively). Therefore, to study the influence of the above mentioned variables in the designs of experiments, the reaction time adopted for esterification reaction was 2 hours.
In a FFD 25−1, the main effects can be calculated and used to indicate which variables must be included in the following design as well as to define the new levels of the variables. Moderate results on the esterification reaction between 1 and the palmitic acid (2) were observed in the fractional factorial design study (see ESI for further details†).
Table 1 shows the estimated effect for independents variables used in the FFD 25−1 for both solvents. As can be observed in Table 1 for the esterification performed in heptane, the amount of enzyme, substrate concentration and stirring were variables, which presented p < 0.05 and thus selected to be optimized in a complete design (CCRD). Temperature and molecular sieve had no significance and therefore kept constant at 50 °C and 15%, respectively, in a subsequent design (CCRD).
| Variables | Effect | p value | ||
|---|---|---|---|---|
| Heptane | Toluene | Heptane | Toluene | |
| a Statistically significant at 95% of confidence level. | ||||
| Mean | 56.2 | 40.5 | <0.0001a | <0.0001a |
| Curvature | −21.6 | −11.4 | 0.005a | 0.01a |
| Temperature (T) | 0.8 | 9.7 | 0.3 | 0.002a |
| Amount of enzyme | −6.7 | −1.4 | 0.008a | 0.1 |
| Substrate concentration | −7.8 | −31.9 | 0.006a | 0.0002a |
| Molecular sieve | 2.1 | −2.3 | 0.08 | 0.04a |
| Stirring | 8.1 | 0.8 | 0.006a | 0.2 |
For the esterification reactions performed in toluene, temperature, substrate concentration and molecular sieve were significant variables (p < 0.05) and therefore used in the CCRD. The amount of enzyme and stirring had no significance in toluene and therefore kept constant in 105 μg and 150 rpm, respectively. The curvature was also significant for both solvents used (p = 0.0054 and 0.0131 for heptane and toluene, respectively) and additional axial points are needed to construct a quadratic model.
The amount of molecular sieves added to the reaction mixture presented different significance for the solvents used. As can be observed in Table 1, molecular sieves were significant in the reactions performed in toluene while for the reaction in heptane was not significant (p = 0.0803 and 0.0467 for heptane and toluene, respectively). This probably happen, because the residual water present in solvents is different. Hydrolysis reactions can interfere in a competitive manner, hampering the conversion of esterification.20
Following the first factorial design mentioned previously, a central composite rotatable design (CCRD) was employed to find the optimal conditions that maximize yield of the esterification reaction in both solvents (heptane and toluene). The experimental matrix with their values coded (see ESI for further details†) and the respective conversion for esterification performed in heptane are presented in the Table 2. In the CCRD, both selected variables were varied at five levels resulting in 17 experiments, including eight factorial points and three central points allowing us to calculate the experimental error. To fit a second order model, four axial points with the same distance from the central point were added at the matrix for this design.
| Variables | Conversionb (%) | |||
|---|---|---|---|---|
| Entry | Amount of enzyme | Substrate concentration | Stirring | |
| a Conditions maintained constant: temperature (50 °C) and amount of molecular sieves (15%).b Measured by Lowry–Tinsley method. | ||||
| 1 | −1 | −1 | −1 | 61.9 |
| 2 | −1 | −1 | +1 | 63.6 |
| 3 | −1 | +1 | −1 | 35.5 |
| 4 | −1 | +1 | +1 | 49.4 |
| 5 | +1 | −1 | −1 | 71.8 |
| 6 | +1 | −1 | +1 | 70.4 |
| 7 | +1 | +1 | −1 | 29.1 |
| 8 | +1 | +1 | +1 | 26.2 |
| 9 | −1.68 | 0 | 0 | 66.7 |
| 10 | 1.68 | 0 | 0 | 67.7 |
| 11 | 0 | −1.68 | 0 | 88.3 |
| 12 | 0 | 1.68 | 0 | 19.0 |
| 13 | 0 | 0 | −1.68 | 62.5 |
| 14 | 0 | 0 | 1.68 | 71.0 |
| 15 | 0 | 0 | 0 | 60.6 |
| 16 | 0 | 0 | 0 | 61.6 |
| 17 | 0 | 0 | 0 | 61.6 |
The best conversion could be obtained by the optimization of reaction conditions affording the desired product in 88.3% of conversion (entry 11, Table 2). When comparing entries 11 and 12, the negative effect of substrate concentration can be observed. Increasing the substrate concentration from 5 to 30 mM, the from 88.3 to 19%. This statement can be confirmed when observing the estimated effects (see ESI for further details†), where the variable substrate concentration was most statistically significant among all independent variables since it has a value of p < 0.05.
The data presented on Table 2, (entry 14) shows that using CaL-B immobilized in MBG a productivity up to 66.82 g of ester per h per g of lipase can be obtained, a higher productivity when compared with our previous work with commercial lipase immobilized from Rhizomucor miehei for the same reaction (8.1 of ester per h per g of lipase), a productivity almost nine times bigger than the immobilized commercial lipase.9 This high catalytic activity of the lipase can be attributed to good performance of the enzyme at the interface of lipid/water, besides the double protective effect against the deleterious effects of solvent propitiated when the enzyme encapsulated in reverse micelles.21,22
Similar to the procedure adopted to the reactions performed in heptane, variables with higher influence in the esterification process in toluene were used in a central composite design rotatable (CCRD) used to obtain the optimum conditions for the fructose ester synthesis. Table 3 shows the experimental factorial design 23 for the reactions in toluene with the corresponding conversions. Again, the first 8 experiments are sufficient for to determine the mathematical model and statistical parameters related to 23 designs, from entries 9 to 14 are shown the axial points for the construction of the quadratic model and entries from 15 to 17 are the central points of the triplicate to obtain the experimental error.
| Variables | Conversionb (%) | |||
|---|---|---|---|---|
| Entry | Temperature | Substrate concentration | Molecular sievesc | |
| a Conditions maintained constant: amount of enzyme (105 μg) and stirring (150 rpm).b Measured by Lowry–Tinsley method.c w/v. | ||||
| 1 | −1 | −1 | −1 | 54.8 |
| 2 | −1 | −1 | +1 | 60.0 |
| 3 | −1 | +1 | −1 | 34.8 |
| 4 | −1 | +1 | +1 | 47.4 |
| 5 | +1 | −1 | −1 | 53.0 |
| 6 | +1 | −1 | +1 | 61.7 |
| 7 | +1 | +1 | −1 | 34.8 |
| 8 | +1 | +1 | +1 | 44.8 |
| 9 | −1.68 | 0 | 0 | 41.2 |
| 10 | 1.68 | 0 | 0 | 32.1 |
| 11 | 0 | −1.68 | 0 | 71.2 |
| 12 | 0 | 1.68 | 0 | 33.9 |
| 13 | 0 | 0 | −1.68 | 27.7 |
| 14 | 0 | 0 | 1.68 | 41.9 |
| 15 | 0 | 0 | 0 | 42.5 |
| 16 | 0 | 0 | 0 | 41.2 |
| 17 | 0 | 0 | 0 | 41.2 |
The data presented in Table 3 shows a positive effect of the molecular sieves content on reaction system. When we observe the experiments in entries 13 and 14, both reactions were carried out at the same conditions, only changing the amount of molecular sieve from 10 to 20%, increasing the conversion from 27.7% to 41.9.
Again the negative influence of substrate concentration can be observed in entries 11 and 12. The increased of substrate concentration (from 5 to 30 mM) generates a decreased in the conversion from 71.2% to 33.9%.
The experimental data have been adjusted to the propose model and adequacy was performed by the analysis of variance and parameter R2. Eqn (1) and (2) represent the mathematical model for the conversion of 2,3:4,5-O-diisopropylidene-β-D-fructopyranose palmitate catalyzed by MBG carried out in heptane and toluene, respectively.
| Y = 60.91 − 0.82 × E − 17.87 × S − 5.01 × S2 + 1.88 × St − 5.79 × E × S − 2.49 × E × St + 1.35 × S × St | (1) |
| Y = 40.36 − 1.31 × T − 9.55 × S + 5.73 × S2 − 4.41 × MS | (2) |
The computed F values from the analysis of variance (ANOVA) for the two models, was highly significant, larger than theirs tabulated F (see ESI for further details†), showing the validity of the experimental model. The goodness of the model can be checked by the determination (R2). The determination coefficient (R2 = 0.90 and 0.89 for reactions in heptanes and toluene, respectively) implies that the sample variation of 90 and 89% for ester production is attributed to the independent variables and can be accurately explained by the model.
Fig. 1 shows the fitted response surface graphs construed for esterification reaction performed in heptane (a) and toluene (b). The negative effect of substrate concentration variable can be seen in both graphs. In the fitted response surface for toluene (b) when increased the amount of molecular sieve increased the conversion, showing the positive effect this variable (Fig. 2).
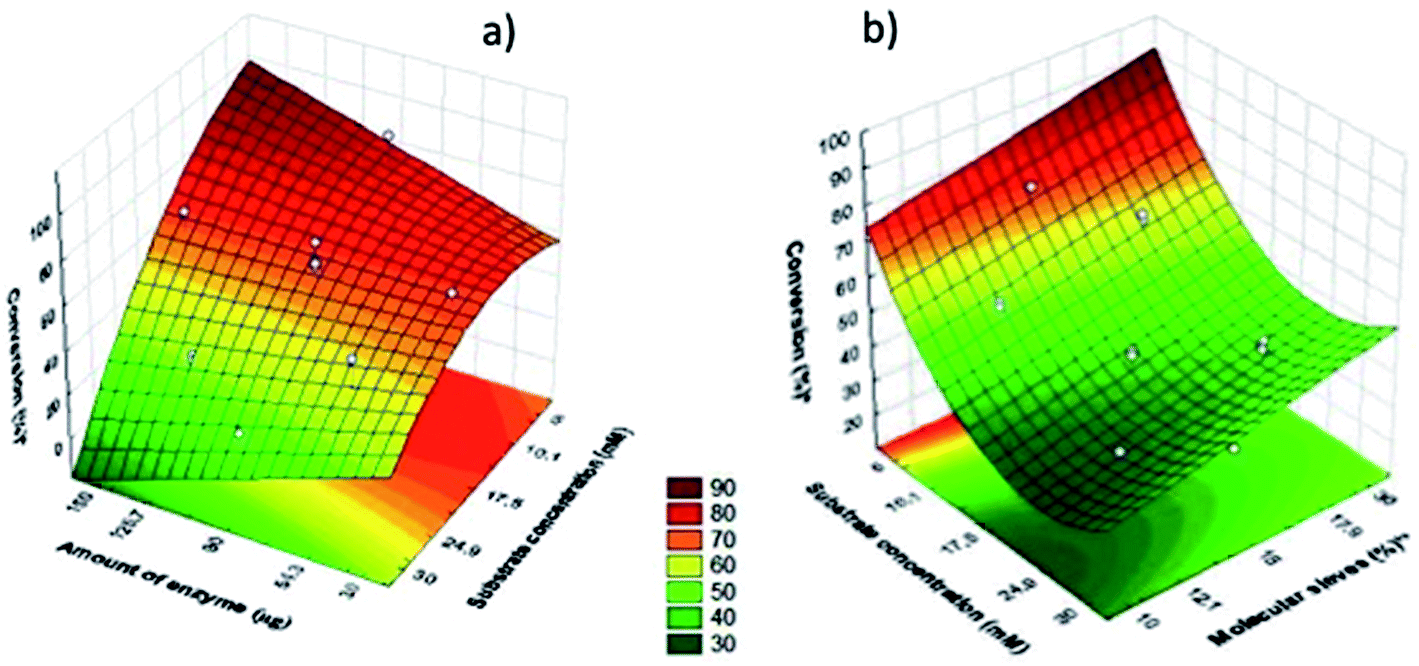 | ||
| Fig. 2 Surface response for the esterification reaction catalyzed by Cal B immobilized in MBG performed in heptane (a) and toluene (b). | ||
In order to improve the productivity of MBG, we developed a continuous flow methodology to prepare ester of 1 taking optimized batch conditions as a starting point. The packed bed was filled with MBG containing the Cal B lipase and the reaction performed in heptane with different flow rates according to the desired residence time (Table 4).
|
|
|---|---|
| Flow rate (residence time) | Conversionb (%) |
| a Conditions tested were 5 mM stock solution of a mixture (1:1) of 1 and palmitic acid at 50 °C (600 μg of enzyme, reactor volume = 4.71 mL).b Measured by Lowry–Tinsley method. |
|
| 0.1 mL min−1 (47.1 min) | 90.2 ± 4.3 |
| 0.3 mL min−1 (15.7 min) | 25 ± 2.6 |
| 0.6 mL min−1 (7.8 min) | 16.1 ± 2.4 |
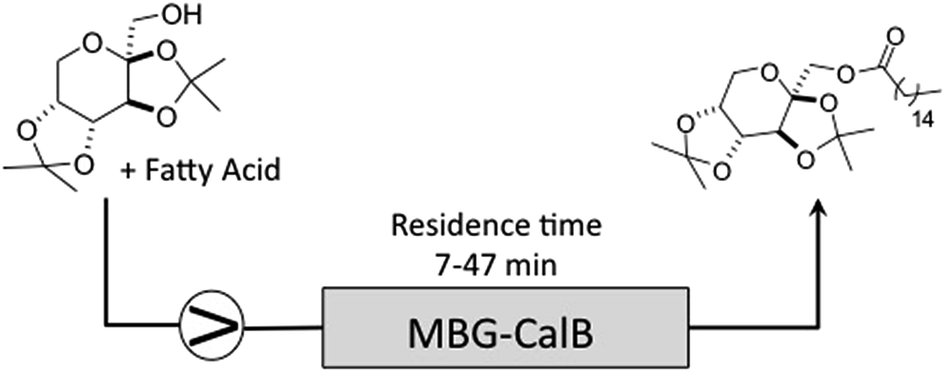
Tests in toluene under optimal condition obtained in batch were also performed, but showed no conversion. When toluene was submitted in flow, a degradation of the MBG was observed. It is not clear how this deleterious effect of toluene on gel is working, however a similar phenomenon was noticed by Rees et al. and Soni et al.23,24
4. Conclusion
In conclusion, we have developed an efficient protocol for synthesis of fructose ester catalyzed by lipase Cal B immobilized in microemulsion-based organogels in batch using design of experiments. The mathematic model proposed for batch reactions, suggested a satisfactorily representation of the process and good correlation among the experimental results and the theoretical values predicted by the model equation were achieved. Heptane was the best solvent for the esterification reaction in batch and continuous flow condition. The best reaction conditions were translated to the continuous flow approach and the results presented indicate that the reaction productivity increased of 66.8 to 88.1 g of ester per h per g of lipase, for the batch and continuous flow conditions, respectively.References
- N. d. A. Sampaio Neta, J. C. Sousa dos Santos, S. d. O. Sancho, S. Rodrigues, L. R. Barros Goncalves, L. R. Rodrigues and J. A. Teixeira, Food Hydrocolloids, 2012, 27, 324 CrossRef CAS PubMed.
- J. Tsukamoto, S. Haebel, G. P. Valenca, M. G. Peter and T. T. Franco, J. Chem. Technol. Biotechnol., 2008, 83, 1486 CrossRef CAS PubMed.
- A. V. Paula, D. Urioste, J. C. Santos and H. F. de Castro, J. Chem. Technol. Biotechnol., 2007, 82, 281 CrossRef CAS PubMed.
- K. Hill and O. Rhode, Fett/Lipid, 1999, 25 CrossRef.
- S.-H. Pyo and D. G. Hayes, J. Am. Oil Chem. Soc., 2009, 86, 521 CrossRef CAS PubMed.
- S.-H. Pyo and D. G. Hayes, J. Am. Oil Chem. Soc., 2008, 85, 1033 CrossRef CAS PubMed.
- A. Ducret, A. Giroux, M. Trani and R. Lortie, J. Am. Oil Chem. Soc., 1996, 73, 109 CrossRef CAS.
- J. I. Itabaiana, K. M. Gonçalves, M. Zoumpanioti, I. C. R. Leal, L. S. M. Miranda, A. Xenakis and R. O. M. A. d. Souza, Org. Process Res. Dev., 2014, 18, 1372 CrossRef.
- F. K. Sutili, H. S. Ruela, S. G. F. Leite, L. S. M. Miranda, I. C. R. Leal and R. O. M. A. d. Souza, J. Mol. Catal. B: Enzym., 2013, 85–86, 37 CrossRef CAS PubMed.
- K. Zhu, J. Lu, J. Hu, H. Wang, N. Xu, P. Han and P. Wei, Asian J. Chem., 2011, 23, 5367 CAS.
- P. P. Dandekar and V. B. Patravale, Indian J. Chem. Technol., 2009, 16, 317 CAS.
- M. Habulin, S. Sabeder and Z. Knez, J. Supercrit. Fluids, 2008, 45, 338 CrossRef CAS PubMed.
- I. S. Yoo, S. J. Park and H. H. Yoon, J. Ind. Eng. Chem., 2007, 13, 1 CAS.
- J. F. Kennedy, H. Kumar, P. S. Panesar, S. S. Marwaha, R. Goyal, A. Parmar and S. Kaur, J. Chem. Technol. Biotechnol., 2006, 81, 866 CrossRef CAS PubMed.
- U. T. Bornscheuer, Angew. Chem., Int. Ed., 2003, 42, 3336 CrossRef CAS PubMed.
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844 RSC.
- H. S. Ruela, F. K. Sutili, I. C. R. Leal, N. M. F. Carvalho, L. S. M. Miranda and R. O. M. A. d. Souza, Eur. J. Lipid Sci. Technol., 2013, 115, 464 CrossRef CAS PubMed.
- R. R. Lowry and I. J. Tinsley, J. Am. Oil Chem. Soc., 1976, 470 CrossRef CAS.
- N. Paludo, J. S. Alves, C. Altmann, M. A. Z. Ayub, R. Fernandez-Lafuente and R. C. Rodrigues, Ultrason. Sonochem., 2015, 22, 89 CrossRef CAS PubMed.
- S. Y. In, S. J. Park and H. H. Yoon, J. Ind. Eng. Chem., 2007, 13, 1 Search PubMed.
- H. Stamatis, A. Xenakis and F. N. Kolisis, Biotechnol. Adv., 1999, 17, 293 CrossRef CAS.
- M. Zoumpanioti, H. Stamatis and A. Xenakis, Biotechnol. Adv., 2010, 28, 395 CrossRef CAS PubMed.
- G. D. Rees, M. G. Nascimento, T. R.-J. Jenta and B. H. Robinson, Biochim. Biophys. Acta, 1991, 1073, 493 CrossRef CAS.
- K. Soni and D. Madamwar, Process Biochem., 2001, 36, 607 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04686f |
| This journal is © The Royal Society of Chemistry 2015 |