
2,4-Bis(4-aryl-1,2,3-triazol-1-yl)pyrrolo[2,3-d]pyrimidines: synthesis and tuning of optical properties by polar substituents†
Jonas Buceviciusa,
Lina Skardziuteb,
Jelena Dodonovaa,
Karolis Kazlauskasb,
Gintautas Bagdziunasc,
Saulius Jursenasb and
Sigitas Tumkevicius*a
aVilnius University, Department of Organic Chemistry, Naugarduko 24, LT-03225 Vilnius, Lithuania. E-mail: sigitas.tumkevicius@chf.vu.lt
bInstitute of Applied Research, Vilnius University, Sauletekio 9-III, LT-10222 Vilnius, Lithuania
cKaunas University of Technology, Department of Polymer Chemistry and Technology, Radvilenu 19, LT-50254 Kaunas, Lithuania
First published on 22nd April 2015
Abstract
2,4-Bis(4-aryl-1,2,3-triazol-1-yl)pyrrolo[2,3-d]pyrimidines as D–π–A–π–D chromophores were successfully prepared by CuAAC reaction of 2,4-diazido-7-methylpyrrolo[2,3-d]pyrimidine with ethynylarenes in dichloromethane in the presence CuI/DIPEA/AcOH as a catalyst system. The incorporation of small polar substituents enabled tuning of the energy of frontier orbitals and thus the FMOs energy gap by up to 0.9 eV, while the incorporation of bulky steric substituents resulted in narrowing of the energy gap by up to 0.4 eV. Owing to electron-accepting properties of pyrrolo[2,3-d]pyrimidine core extending to triazole moieties the compounds with electron-donating groups showed expressed intramolecular charge transfer character (ICT) of the excited states which was proved by solvatochromic dynamics and supported by DFT calculations. The optimization of ICT reduced radiative and non-radiative deactivation pathways resulted in enhancement of fluorescence quantum yield up to 73%.
Introduction
Over recent years, organic molecules with a π-conjugated backbone have received much attention in both academic and industry due to their applications in a wide range of electronic and optoelectronic devices.1 Introduction of heteroaryl moieties into the backbone of extended π-systems considerably influences molecular orbitals, stereochemical structure and linking topology of the substituents. In this regard, pyrimidine ring containing heterocycles owing to their aromaticity, significant π-deficiency, and ability of nitrogen atoms to take part in the chelation processes, are desired structural units to be incorporated in more complex organic structures targeted for numerous applications.2 Pyrimidine moiety has already been incorporated into linear, star- and banana-shaped oligomers that show good light emitting properties and two-photon absorption.3 Moreover, pyrimidine ring is frequently used in push–pull structures as strong electron-withdrawing unit and together with electron-donating moieties results in intramolecular charge transfer character, which plays a key role in most of the important applications.3a,d,e,g–h Particularly, the applications of intramolecular charge transfer complexes has recently attracted attention due to invention of delayed fluorescence emitters, possessing nitrogen heterocycles.4 Another important use of donor–acceptor structures is fluorescence bio-material labelling and metal ions sensing.5 Generally, these push–pull chromophores have an A–π–D structure, where A is an electron-withdrawing moiety and D is an electron-donating group connected by a π-conjugated linker. Since the discovery of Cu(I)-catalyzed Huisgen azide–alkyne cycloaddition (CuAAC) by Sharpless6 and Meldal7 many examples that incorporate the 1,2,3-triazole moiety have been reported. CuAAC provides 1,4-disubstituted 1,2,3-triazoles with such efficiency and scope that transformation is usually referred as “click” chemistry and is one of the most atom-economical transformations.8 Various 1,2,3-triazoles have found application in fields of bioconjugation,9 material science,10 chemical sensors11 and drug discovery.12 More recently, the 1,2,3-triazole unit as a linker in the conjugated backbone of fluorescent and two-photon absorption compounds has been investigated.3e,f,13 Structure–photophysical properties relationships of some push–triazole–pull chromophores revealed the 1,2,3-triazole is better π-conjugated linker in terms of both, quantum yields and Stokes shifts than the triple bond.3e,f Recently, we have shown that pyrrolo[2,3-d]pyrimidines bearing π-conjugated aromatic assemblies as side branches exhibit strong UV-blue fluorescence and are promising candidates as fluorescent functional materials.14 Continuing our work aimed to the search of novel fluorescent materials we became interested in the synthesis of pyrrolo[2,3-d]pyrimidine derivatives with a 1,2,3-triazole linker between the aromatic side chains and heterocyclic scaffold. In the current literature triazolylpyrrolo[2,3-d]pyrimidines have received very little attention. To the best of our knowledge, there are only few reports on the synthesis of pyrrolo[2,3-d]pyrimidines containing 1,2,3-triazole moiety. Using azide–alkyne ligation reaction 1,2,3-triazole moiety was constructed at the C5 or N7 position of the pyrrolopyrimidine ring12b,15 and none of these reactions were carried out using azidopyrrolo[2,3-d]pyrimidines as reactive partners.The aim of this work is to describe the synthesis of new D–π–A–π–D chromophores that contain pyrrolo[2,3-d]pyrimidine system as electron-attracting part and substituted aromatic moieties as the electron-donating parts. The central pyrrolo[2,3-d]pyrimidine core and the external aromatic parts are linked by the π-conjugated electron accepting 1,2,3-triazole rings. The impact of the polar and steric substituents on the photophysical properties of the synthesized D–π–A–π–D chromophores were assessed by thorough analysis of their optical and electrochemical properties in various surroundings and the results of DFT modelling. Substituent induced altering of the excited state recombination pathways is discussed with special emphasis on the enhancement of fluorescence efficiency.
Results and discussion
Synthesis
The starting material – 2,4-diazido-7-methylpyrrolo[2,3-d]pyrimidine (2) was obtained in 78% yield by nucleophilic substitution of 2,4-dichloro-7-methylpyrrolo[2,3-d]pyrimidine (1) with NaN3 in DMF at ambient temperature (Scheme 1). It should be mentioned that diazide 2 is rather unstable in daylight and at elevated temperatures, but can be stored for a long period of time in the dark below 5 °C. Azides of nitrogen heterocycles bearing azido group in an adjacent position to an endocyclic nitrogen atom can undergo spontaneous cyclization to form a tetrazole ring. The azide-tetrazole equilibrium is often observed in π-deficient heterocycles such as azidopyrimidines16 and azidopurines.9g,17 Earlier reports on the synthesis of 4-azidopyrrolo[2,3-d]pyrimidines18 indicate that these compounds in a solid state exist as a tetrazole tautomer. However, in the IR spectrum of compound 2 two absorbtion bands of both azido groups at 2152 cm−1 and 2115 cm−1 in KBr, and 2147 cm−1 and 2132 cm−1 in CHCl3 are observed. 1H NMR spectrum of compound 2 recorded in CDCl3 indicated that compound 2 exists mainly in one tautomeric form in the solution. A ratio of diazide and another possible tetrazole tautomer was found to be 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively. These data suggest that in a solid state and in the chloroform solution diazido form of compound 2 predominates.
:1, respectively. These data suggest that in a solid state and in the chloroform solution diazido form of compound 2 predominates.
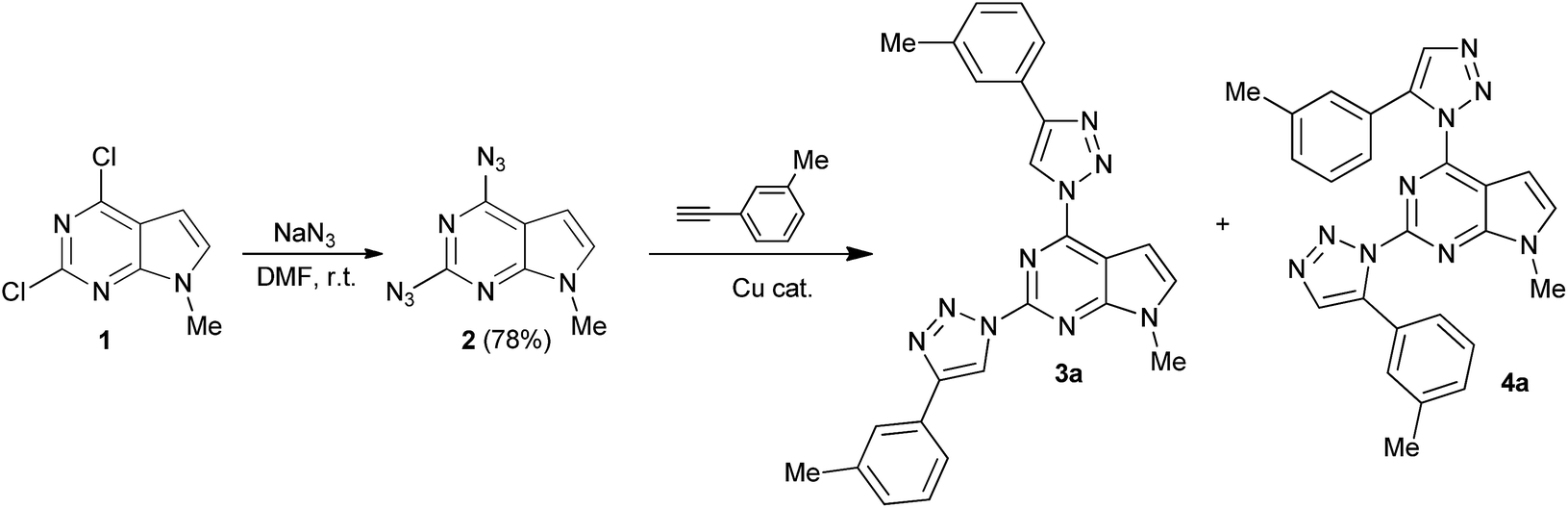 | ||
| Scheme 1 Synthesis of 2,4-diazido-7-methylpyrrolo[2,3-d]pyrimidine (2) and model CuAAC reaction of 2 with 3-ethynyltoluene. | ||
Further, on the example of reaction of diazide 2 with 3-ethynyltoluene formation of 2,4-bis(triazolyl)pyrrolo[2,3-d]pyrimidine was investigated (Table 1). It was found that no ligation of diazide 2 with 3-ethynyltoluene occurred using well established CuSO4/Na ascorbate catalyst system at ambient temperature in t-BuOH/H2O (Table 1, entry 1). The target compound 3a was obtained in 18% yield only when the reaction temperature was raised up to 70 °C (Table 1, entry 2). Low yield, probably, arises from low stability of diazide 2 at elevated temperatures. Another popular catalyst system CuI/NEt3 was examined in different solvents. Cycloaddition reaction did not occur in DMF, DMSO and THF at room temperature (Table 1, entries 3–5). However, in dichloromethane CuAAC reaction took place to give a mixture of two regioisomers 3a and possibly, 4a in overall 30% yield with a ratio 71:29 as determined from 1H NMR spectra (Table 1, entry 6). Addition of acetic acid, which is known to accelerate protonation of C–Cu bond,19 raised the overall yield up to 48% (Table 1, entry 7). Switching base from TEA to DIPEA afforded a mixture of 3a and 4a in overall 78% yield (Table 1, entry 8). In all cases stated above regioisomeric ratio in the cycloaddition reaction of 2 with 3-ethynyltoluene was almost the same, so regioselectivity of this reaction seems to be not affected by the reaction conditions. To our knowledge, there are only few reports on the formation of 1,5-isomers in CuAAC reaction.17,20 1,5-Disubstituted 1,2,3-triazoles as by-products can be formed by a competitive non-catalyzed thermal Huisgen cycloaddition, therefore, experiments were conducted in the absence of copper source and additives. However, no traces of the reaction products were observed even after 7 days (Table 2, entries 9 and 10). This indicates that both isomers are formed by CuAAC pathway. Perhaps, presence of copper catalyst lowers activation energy barriers of the formation of both, 1,4- and 1,5-disubstituted isomers.21
| Entry | Copper source/additivea | Solvent | Temp. (°C), time (h) | 3a:4ab |
Overall yield, % (3a + 4a) |
|---|---|---|---|---|---|
| a Reaction conditions: azide 2 (0.46 mmol), alkyne (1.4 mmol), copper source (20 mol%), trialkylamine (0.51 mmol), AcOH (0.51 mmol), solvent (5 mL).b 1H NMR ratio of the crude product.c Yield of 1,4-isomer 3a. | |||||
| 1 | CuSO4/Na ascorbate | t-BuOH/H2O 2:1 |
rt, 72 h | — | No reaction |
| 2 | CuSO4/Na ascorbate | t-BuOH/H2O 2:1 |
70, 48 h | — | 18c |
| 3 | CuI/NEt3 | DMSO | rt, 48 h | — | Decomposition products of 2 |
| 4 | CuI/NEt3 | DMF | rt, 48 h | — | Decomposition products of 2 |
| 5 | CuI/NEt3 | THF | rt, 96 h | — | Trace |
| 6 | CuI/NEt3 | CH2Cl2 | rt, 96 h | 71:29 |
30 |
| 7 | CuI/NEt3/AcOH | CH2Cl2 | rt, 72 h | 73:27 |
48 |
| 8 | CuI/DIPEA/AcOH | CH2Cl2 | rt, 72 h | 70:30 |
78 |
| 9 | — | CH2Cl2 | rt, 168 h | — | No reaction |
| 10 | DIPEA/AcOH | CH2Cl2 | rt, 168 h | — | No reaction |
| Calculated shifts, δ, ppm | Experimental shifts, δ, ppm | |||||
|---|---|---|---|---|---|---|
| 3d | 4d | 5d | 6d | 3d | 4d | |
| H′ | 9.28 | 7.73 | 7.66 | 9.15 | 8.93 | 7.80 |
| H′′ | 8.98 | 7.53 | 8.90 | 7.69 | 8.72 | 7.52 |
Theoretically, the cycloaddition reaction of diazide 2 with alkynes can lead to the formation of four isomeric triazoles. The regiochemistries of the formed 2,4-bis(triazolyl)pyrrolo[2,3-d]pyrimidines were assigned on the basis of the characteristic 1H NMR chemical shifts for positions C4–H and C5–H of the triazole moieties. Here, structural assignment of the formed regioisomers is illustrated on the example of compound 3d, because none of the 5-H/4-H peaks of 1,2,3-triazole in 1H NMR spectra of its isomeric mixture were overlapped. For clarity, in Fig. 1 structures of all possible isomers of 4-(dimethylamino)phenyl derivative 3d–6d are presented. It is known that 5-H signal in the 1H NMR spectra of 1,4-disubstituted-1,2,3-triazoles is usually observed in lower fields than 4-H peak of 1,5-disubstituted-1,2,3-triazoles.13d,17,22
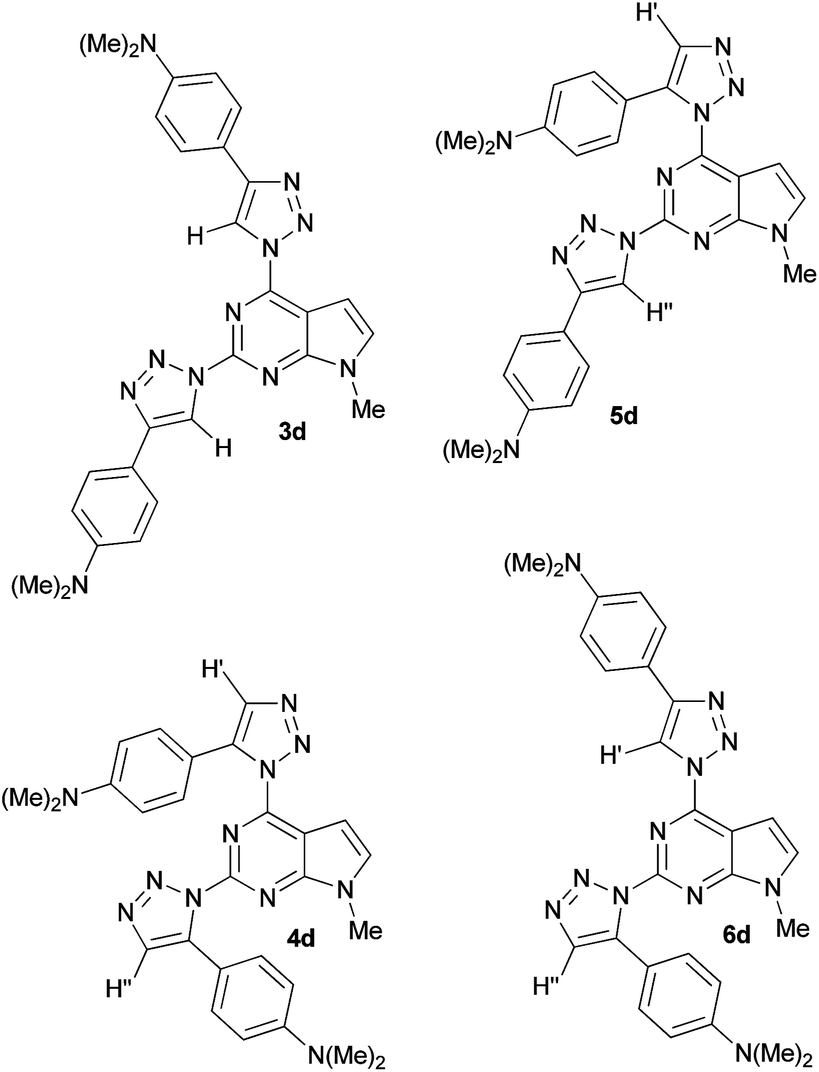 | ||
| Fig. 1 Structures of possible isomers 3d–6d of ligation reaction of 2 with 4-(dimethylamino)phenylethyne. | ||
Analogous relationship was observed in the 1H NMR spectra of compounds 3 and 4. For example, in the 1H NMR spectrum of a mixture of 3d and 4d both 5-H peaks of isomer 3d are observed at 8.93 ppm and 8.72 ppm, while the 4-H peaks of 4d are located at 7.80 ppm and 7.52 ppm, respectively (Table 2). It is logical to assume that in case of any “mixed” isomer (5d or 6d) only one of the peaks would be shifted to higher fields. In addition, geometries of all regioisomers 3d–6d were optimized by DFT/B3LYP/6-311G** level of theory and shielding values of the corresponding 4-H and 5-H of 1,2,3-triazole moieties were computed by GIAO method. Comparison of the calculated chemical shifts of 4-H and 5-H of the triazole rings for compounds 3d–6d with the experimental values (Table 2) also indicates that in the reaction of diazide 2 with 4-(dimethylamino)phenylethyne the corresponding 1,4-disubstituted (3d) and 1,5-disubstituted 1,2,3-triazoles (4d) were formed.
Having established that the optimal CuAAC reaction conditions were CuI/DIPEA/AcOH/CH2Cl2 we next turned our attention to the scope of the cycloaddition of diazide 2 with a range of arylethynes. In most cases, alkynes underwent ligation with diazide 2 to give mixtures of bis-triazoles 3 and 4 with overall yields varying from 62% to 89% (Table 3). Cycloaddition of 2 proceeds faster with arylethynes bearing electron-withdrawing substituents (Table 3, entries 5–7). Regioselectivity favors the formation of 1,4-isomer. The lowest regioselectivity (63:37) and the lowest yield of 1,4-isomer was obtained when 4-(dimethylamino)phenylethyne bearing strong electron donating group was employed in the reaction (Table 3, entry 4).
|
||||
|---|---|---|---|---|
| Entry | Reaction time, h | Regioisomeric ratioa (3:4) |
Overall yieldb (3 + 4), % | Yieldc, % |
| a Determined from the 1H NMR spectra of obtained isomeric mixture.b Yield of crude mixture of isomers.c Isolated yield.d Formation of compounds 4 was not observed. | ||||
| 1 | 72 | 70:30 |
78 | 46 (3a) |
| 2 | 48 | —d | — | 48 (3b) |
| 3 | 72 | 83:17 |
79 | 58 (3c) |
| 4 | 72 | 63:37 |
74 | 35 (3d) |
| 5 | 12 | —d | — | 61 (3e) |
| 6 | 12 | 95:5 |
89 | 79 (3f) |
| 7 | 8 | —d | — | 75 (3g) |
| 8 | 120 | 79:21 |
68 | 38 (3h) |
| 9 | 144 | 85:51 |
62 | 53 (3i) |
| 9 (4i) | ||||
| 10 | 96 | 90:10 |
78 | 43 (3j) |
| 11 | 96 | —d | — | 56 (3k) |
| 12 | 120 | —d | — | 60 (3l) |

However, in the reaction of 2 with phenyl-, p-fluorophenyl, p-cianophenyl, 9,9-dihexyl-2-fluorenyl- and 7,7-dimethylbenzo[c]fluoren-5-ylethynes formation of 1,5-disubstituted triazoles was not detected by NMR technique (Table 3, entries 2, 5, 7, 11, 12). Due to similar Rf values of isomeric triazoles 3 and 4 mixtures could not be separated into both individual isomers by column chromatography. Only one pure 1,5-disubstituted triazole 4i was isolated in 9% yield (Table 3, entry 9). Nevertheless, all major 1,4-disubstituted triazoles 3a–l were isolated and purified by fractional crystallization of the crude mixture of isomers from 2-PrOH or toluene.
Molecular orbital calculations and cyclic voltammetry
To gain insight into the effect of various substituents on the geometrical and electronic properties of the chromophores density functional theory (DFT) calculations were carried out on the synthesized compounds 3a–l by using Gaussian 09 program package.23 The geometries of compounds were optimized using B3LYP exchange–correlation hybrid functional together with the 6-311G** basis set. To get correctly optimized structures potential energy surface scan of dihedral angle at the position 4 of the pyrrolo[2,3-d]pyrimidine was performed with a step of 5 degrees. The scan revealed two local minima at dihedral angle at 40° and 320° degrees and global minimum positioned at coplanar geometry with anti-orientation of the 1,2,3-triazole ring (Fig. 2). 1,2,3-Triazole ring at the position 2 of the heterocycle can accept syn- or anti-conformation (in respect of the pyrrolo[2,3-d]pyrimidine ring). Potential energy comparison of both structures indicated that anti–anti conformation of triazole rings is energetically more favored than anti–syn conformation by 0.64 kcal mol−1 (Fig. 2).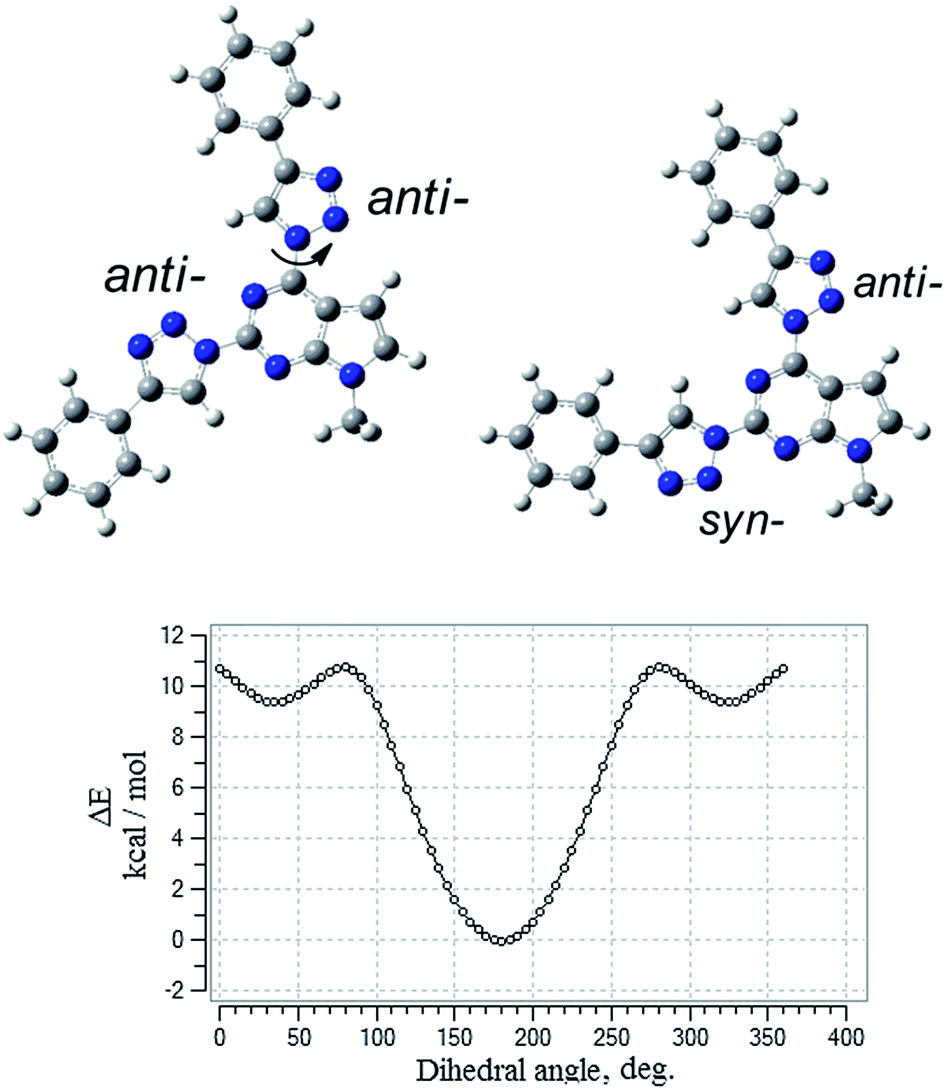 | ||
| Fig. 2 Anti–anti, anti–syn conformations and potential energy surface scan of compound 3b. | ||
Examination of geometry of the optimized structures of the studied systems revealed that compounds possessing phenyl substituent at 1,2,3-triazole linker are almost coplanar (3a–g, 3k), while compounds with more bulky aryl fragments exhibit twisted geometry. For molecules 3h and 3l, naphthyl and benzofluorenyl fragments are out of plane of the rest of the molecule (dihedral angle 34.4–36.3°) and for 3i, j carbazolyl moieties are twisted by 57.4–58.6°.
Optimised structures of compounds 3b and 3i are given in Fig. 3 (see ESI† for the rest compounds).
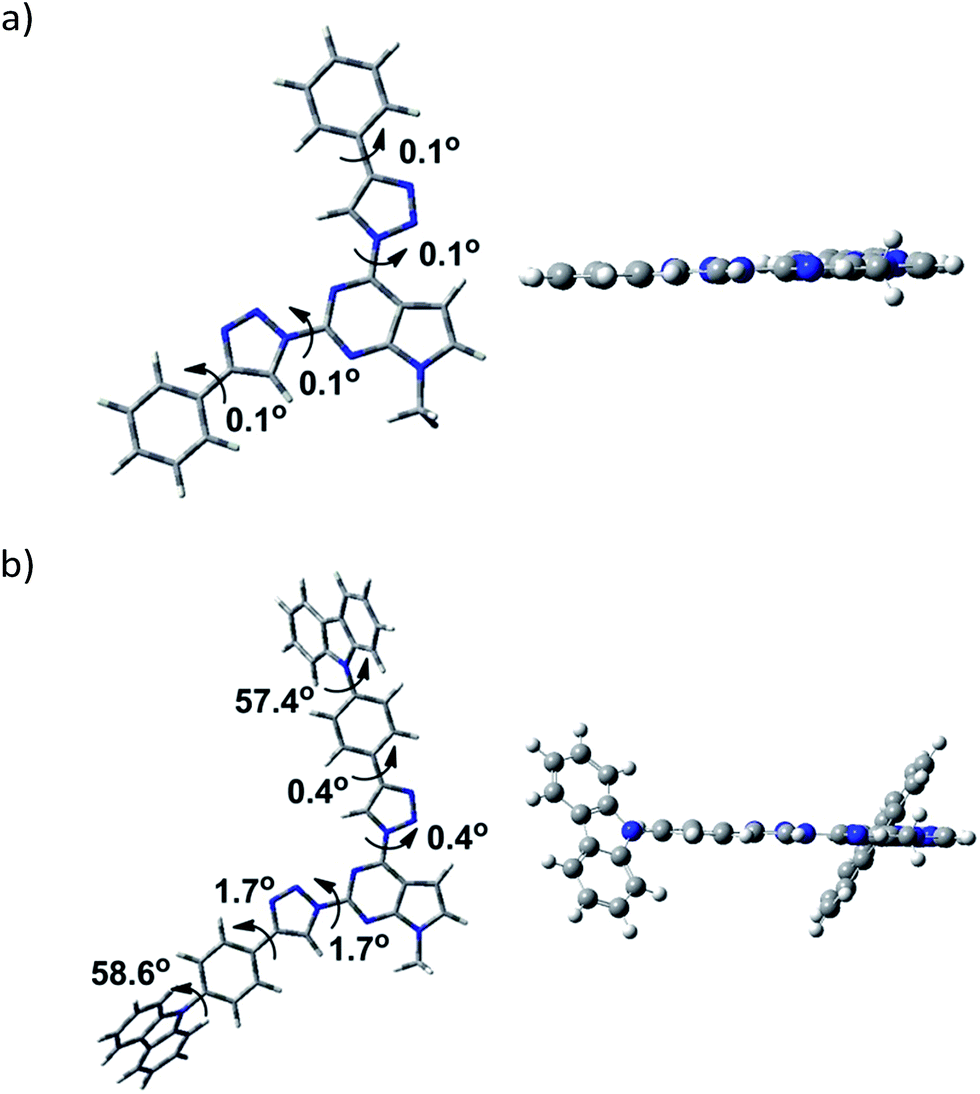 | ||
| Fig. 3 Optimized structure geometries (a) compound 3b; (b) compound 3i. | ||
The DFT computed localization of frontier molecular orbitals (FMOs) of compounds 3a–l is presented in Fig. 4. A comparison of electronic structures revealed that the LUMO of all the homologues is localized on pyrrolo[2,3-d]pyrimidine core involving conjugated co-planar 1,2,3-triazolyl moieties. The main difference of the electronic structure is revealed in the HOMO state. For molecules possessing small slightly electron-donating substituents (3a–b) or electron-withdrawing substituents (3e–g) HOMO is localized over the entire molecule, enclosing the pyrrolo[2,3-d]pyrimidine core. Introduction of substituents with increasing electron-donating properties (3c–d) results in enhanced intramolecular charge transfer. On the other hand, the introduction of bulky naphthyl (3h), carbazolyl (3i–j) and benzo[c]fluorenyl (3l) fragments, resulting in twisted geometry of the molecules, yields non-symmetric localization of the HOMO on the sole substituent attached at the position 4 of the pyrrolopyrimidine moiety and thus, manifests strong intramolecular charge transfer character. Compound 3k possessing bulky fluorene moieties and nevertheless, showing planar geometry exhibits intermediate properties of HOMO localization, comprising both substituents at the 2nd and 4th positions of the pyrimidine ring and partially extending to the pyrrolo[2,3-d]pyrimidine core.
 | ||
| Fig. 4 Calculated spatial distributions of HOMO and LUMO of compounds 3a–l. | ||
Comparison of calculated energies of FMO's for compounds 3a–g revealed the impact of polar substituents (Fig. 5). The energy of the HOMO level steadily decreases from −5.08 eV to −6.69 eV going from the most electron-donating substituents to the most electron-withdrawing substituents (compound 3d and compound 3e, respectively). The energy of the LUMO state also steadily decreases with substituent polarity from −1.95 eV to −2.71 eV. The resulting energy gap increases from 3.13 eV to 4.04 eV. Incorporation of the bulky substituents (Fig. 5) results in the increase of the HOMO energy with increasing size of aryl substituents (from −5.85 eV to −5.5 eV for compounds 3h and 3l). The energy of the LUMO state slightly decreases with increasing size of the conjugated system from −2.24 eV to −2.29 eV, thus the corresponding band gap decreases from 3.61 eV to 3.21 eV.
 | ||
| Fig. 5 Diagram of calculated FMOs energy values of compounds 3a–l. | ||
The cyclic voltammetry (CV) measurements were carried out with a glassy carbon electrode in DCM or DMF solutions containing 0.10 M tetrabutylammonium hexafluorophosphate as electrolyte and Ag/Ag+ as the reference electrode at the 50 mV s−1 scan rate. Each measurement was calibrated with ferrocene as internal standard. The LUMO energies were calculated from the first signal onset of reduction using formula LUMO = −(1.19 × E(red)onsvsFc + 4.78).24 All compounds exhibited one irreversible reduction wave. HOMO values were calculated from the optical band gaps and LUMO values using formula EHOMO = ELUMO − Eoptg. Typical voltammogram of compound 3e is shown in Fig. 6 (for compounds 3a–d and 3g–l see ESI†).
 | ||
| Fig. 6 Typical voltammogram of compound 3e (Fc/Fc+ as ref.). | ||
The obtained data of CV measurements are summarized in Table 4. Variation of the experimentally obtained values induced by electronic effects of substituents is significantly smaller due to an opposite effect resulting from the polarity of the surrounding media. Note, that the cyclic voltammetry results were obtained in dichloromethane or in highly polar DMF surrounding, in contrast to the theoretical calculations simulating vacuum conditions. Thus, the shift of the LUMO level energy induced by substituent polarity is, probably, diminished by the solvatic shift of energy levels (Table 4).
| Compound | ELUMOa, eV | EHOMOb, eV | Eoptgc, eV |
|---|---|---|---|
| a Calculated by the formula: ELUMO = −(1.19 × E(red)onsvsFc + 4.78).b Calculated by the formula: EHOMO = ELUMO − Eoptg.c Obtained from the intersection of UV/vis and fluorescence spectra.d CV measurements were performed in CH2Cl2.e Could not be measured because of very low solubility.f CV measurements were performed in DMF. | |||
| 3ad | −2.16 | −5.54 | 3.38 |
| 3bd | −2.18 | −5.58 | 3.40 |
| 3cd | −2.24 | −5.50 | 3.26 |
| 3dd | −2.17 | −5.04 | 2.87 |
| 3ed | −2.23 | −5.60 | 3.37 |
| 3fd | —e | —e | 3.35 |
| 3gd | −2.30 | −5.72 | 3.42 |
| 3hf | −2.30 | −5.67 | 3.37 |
| 3if | −2.34 | −5.59 | 3.25 |
| 3jf | −2.35 | −5.7 | 3.35 |
| 3kd | −2.64 | −5.96 | 3.32 |
| 3ld | −2.26 | −5.40 | 3.14 |
Optical properties
Optical properties of the synthesized compounds 3a–l were assessed by performing absorption and fluorescence spectroscopy, fluorescence lifetime and fluorescence quantum yield measurements in CHCl3, THF and DMF solutions. Absorption and fluorescence data of 2,4-bis(4-aryl-1,2,3-triazol-1-yl)pyrrolo[2,3-d]pyrimidines (3a–l) are collected in Table 5. The studied compounds exhibit strong UV absorption in dilute solutions with their absorption maxima positioned in a region from 245 to 365 nm. Most of the synthesized compounds show two specific absorption bands located at around 260 nm and 310 nm, characteristic to pyrrolo[2,3-d]pyrimidine units (not observed in DMF because of UV cut-off value 268 nm).25| 3a | 3b | 3c | 3d | 3e | 3f | 3g | 3h | 3i | 3j | 3k | 3l | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a ΦF values obtained by the integrated sphere method with 4% error are provided.b Radiative and non-radiative decay time constants are calculated as τ/ΦF and τ/(1 − ΦF), respectively. | |||||||||||||
| CHCl3 | λabs, nm | 255 | 264 | 268 | 308 | 249 | 253 | 277 | 245 | 247 | 253 | 245 | 246 |
| 309 | 311 | 318 | 365 | 312 | 310 | 311 | 298 | 293 | 293 | 293 | 334 | ||
| 313 | 314 | 319 | 348 | ||||||||||
| 329 | 326 | ||||||||||||
| 341 | 340 | ||||||||||||
| λem, nm | 402 | 402 | 405 | 530 | 403 | 404 | 408 | 404 | 453 | 454 | 422 | 459 | |
| ΦFa, % | 52 | 49 | 27 | 52 | 49 | 44 | 52 | 27 | 55 | 6 | 34 | 28 | |
| Stokes shift, nm | 93 | 91 | 87 | 165 | 91 | 93 | 97 | 106 | 112 | 114 | 103 | 111 | |
| τ, ns | 5.2 | 5.4 | 2.9 | 12.3 | 5.8 | 6.7 | 6.4 | 3.5 | 9.0 | 17.8 (86%) | 2.6 | 3.3 | |
| 3.4 (10%) | |||||||||||||
| 0.1 (4%) | |||||||||||||
| τRb, ns | 10.9 | 10.7 | 8.7 | 23.6 | 11.4 | 12.0 | 13.4 | 4.7 | 16.4 | — | 7.6 | 11.9 | |
| τNRb, ns | 10.0 | 11.1 | 4.2 | 25.6 | 11.9 | 15.3 | 12.4 | 12.8 | 20.0 | — | 3.9 | 4.6 | |
| THF | λabs, nm | 256 | 262 | 266 | 310 | 250 | 255 | 270 | 247 | 249 | 251 | 246 | 246 |
| 309 | 309 | 321 | 366 | 312 | 312 | 290 | 297 | 295 | 295 | 293 | 334 | ||
| 312 | 312 | 320 | 348 | ||||||||||
| 324 | 328 | ||||||||||||
| 342 | 341 | ||||||||||||
| λem, nm | 416 | 417 | 418 | 569 | 418 | 422 | 422 | 418 | 459 | 422 | 417 | 455 | |
| ΦFa, % | 55 | 73 | 31 | 38 | 61 | 43 | 67 | 32 | 50 | 17 | 32 | 25 | |
| Stokes shift, nm | 107 | 108 | 97 | 203 | 106 | 110 | 132 | 121 | 117 | 81 | 97 | 107 | |
| τ, ns | 7.3 | 7.5 | 3.8 | 9.8 | 7.8 | 7.8 | 7.5 | 5.5 | 8.7 | 15.2 (71%) | 3.0 | 3.7 | |
| 1.3 (17%) | |||||||||||||
| 0.3 (12%) | |||||||||||||
| τRb, ns | 13.2 | 10.3 | 12.2 | 25.9 | 12.8 | 18.2 | 11.1 | 17.1 | 17.6 | — | 9.3 | 14.8 | |
| τNRb, ns | 16.1 | 27.8 | 5.5 | 15.9 | 20.0 | 13.7 | 22.8 | 8.0 | 17.6 | — | 4.4 | 4.9 | |
| DMF | λabs, nm | 311 | 311 | 320 | 309 | 311 | 314 | 279 | 299 | 298 | 297 | 295 | 335 |
| 370 | 312 | 315 | 316 | 324 | 348 | ||||||||
| 330 | 326 | ||||||||||||
| 345 | 346 | ||||||||||||
| λem, nm | 424 | 423 | 436 | 650 | 431 | 436 | 435 | 432 | 530 | 435 | 436 | 492 | |
| ΦFa, % | 71 | 62 | 21 | 3 | 56 | 50 | 56 | 31 | 27 | 5 | 41 | 19 | |
| Stokes shift, nm | 113 | 112 | 116 | 279 | 120 | 122 | 123 | 133 | 185 | 89 | 112 | 144 | |
| τ, ns | 7.3 | 9.8 | 6.3 | 2.2 | 10.4 | 10.6 | 9.8 | 7.7 | 12.5 (74%) | 1.8 (44%) | 6.3 | 4.8 | |
| 20.4 (26%) | 0.5 (56%) | ||||||||||||
| τRb, ns | 10.2 | 15.8 | 30.2 | 75.0 | 18.5 | 21.2 | 17.6 | 24.9 | — | — | 15.4 | 25.5 | |
| τNRb, ns | 25.1 | 25.8 | 8.0 | 2.3 | 23.6 | 21.2 | 22.3 | 11.2 | — | — | 10.7 | 6.0 | |
Compounds 3c–d, possessing electron donating substituents show decreasing oscillator strength and a red-shift of the lowest energy absorption band, which is attributed to the ICT behaviour, in accordance with quantum chemical calculations. Strong electron-donating dimethylamino group (compound 3d) caused a red shift of the absorption bands to 365 nm. The absorption spectra of compounds 3i–j, bearing bulky carbazole fragments show absorption bands at 290 nm and 340 nm, which are assigned to the carbazolyl moieties.26 Solvent polarity has no significant influence on the absorption maxima of the studied compounds indicating small dipole moment in the ground state.
All compounds possessing substituted phenyl fragments show broad structureless fluorescence spectra in the region from 402 nm to 649 nm depending on the electronic nature of the substituents and the polarity of the surrounding media. The normalized absorption and fluorescence spectra of compounds 3b–d in CHCl3, THF, DMF are presented in Fig. 7 (see ESI† for compounds 3a, 3e–l). Weak λem dependence on solvent polarity in the range from 402 nm to 436 nm was observed for compounds bearing phenyl, m-tolyl and electron-withdrawing substituents in a para-position of aromatic rings of the 2,4-bis(triazolyl)pyrrolo[2,3-d]pyrimidines (3a, b, 3e–g).
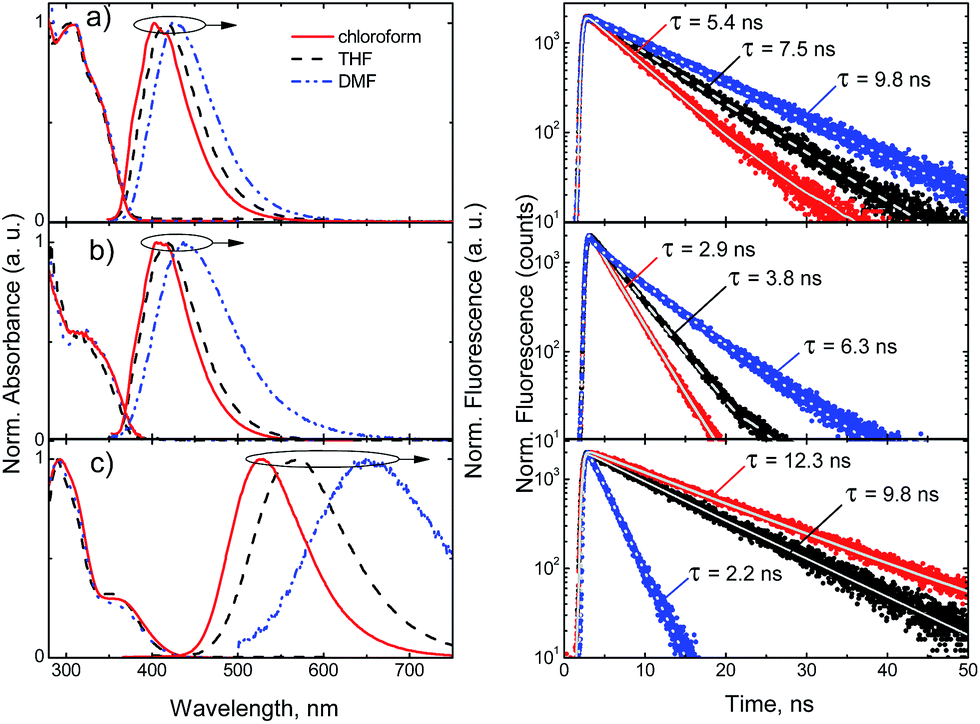 | ||
| Fig. 7 Normalized absorption and fluorescence spectra (left) and fluorescence decay profiles (right) of compounds 3b (a), 3c (b) and 3d (c) in chloroform, THF and DMF solutions (c = 10−5 M). | ||
However, incorporation of electron-donating methoxy (compound 3c) group results in broadening of fluorescence spectra. Moreover, dimethylamino substituent (compound 3d) results in prominent red-shift from 402 nm up to 530 nm. Thus, broadening and enhanced Stokes shift from 91 nm (3b in CHCl3) to 165 nm (3d in CHCl3) of the fluorescence spectra, this is due to the strong charge transfer character of the excited state. In accordance with the CT nature of transitions the enhancement of surrounding media polarity results in even higher red-shift of the fluorescence spectra up to 650 nm and increased Stokes shift – 279 nm.
Compounds 3h, 3k and 3l bearing naphthyl, fluorenyl and benzofluorenyl fragments show steady red-shifting of the fluorescence bands from 402 nm to 459 nm in the solvent of low polarity, due to extension of π-conjugated system. All these compounds show minor solvatochromic changes with increasing polarity of the surrounding media. Compound 3i, bearing bulky electron donating carbazole moiety, shows red-shifted broad fluorescence band at 453 nm in chloroform and at 530 nm in polar DMF. Interestingly, its isomer – compound 3j, bearing carbazole fragment at the meta-position of the benzene ring, reveals the dual-fluorescence properties in polar DMF surrounding showing both locally excited band at 435 nm, typical for non-polar media, and an additional CT band at around 550 nm (Fig. 8).
 | ||
| Fig. 8 Normalized absorption and fluorescence spectra (left) and fluorescence decay profiles (right) of compounds 3i (a) and 3j (b) in chloroform, THF and DMF solutions (c = 10−5 M). | ||
Most of the compounds demonstrated efficient blue fluorescence with quantum yields up to 71%. Compounds bearing neutral and electron-withdrawing substituents (3a–b, 3e–g) demonstrated fluorescence quantum yields varying from 44% to 52% in solvent of low polarity (CHCl3), while the higher polarity of the surrounding media results in slightly higher efficiencies from 50% to 71%. Much more prominent variation depending on solvent polarity is observed for the excitation relaxation rates. The single exponential profiles of the decay transients enabled the estimation of radiative and non-radiative decay lifetimes. For compounds 3a–b, 3e–g, both the radiative and non-radiative decay lifetimes show almost two-fold increase in polar DMF solvent, as compared to less polar chloroform, which is evident by significant slowdown of the fluorescence decay transients (see Fig. 7). The prominent variation of both radiative and non-radiative decay lifetimes indicates on charge transfer induced intramolecular twisting reactions, determined by solvation shell.27 This effect is even more evident for compounds 3c and 3d bearing electron donating substituents, with pronounced intramolecular charge transfer. Here, the radiative decay lifetimes increase more than 3 times in the solutions of polar DMF, while in the case of compounds with non-polar and electron withdrawing moieties, with less pronounced CT nature, the increase of the radiative decay lifetime was about 30%. The non-radiative decay rates are 2 or 3 times higher in the polar surrounding for all the compounds, with the exception of dimethylamino group bearing compound 3d, where the non-radiative decay rate undergoes a 10-fold increase in polar surrounding. Thus, the resulting fluorescence quantum yield of differently substituted derivatives, comprising electron deficient pyrrolo[2,3-d]pyrimidine core, depends on competition between both, decreased radiative and non-radiative decay rates.
Compounds 3h–k bearing non-polar bulky aryl substituents revealed similar trends of solvent polarity dependencies of the radiative and non-radiative decay rates. Two-fold increase of the radiative decay lifetime accompanied by the significant increase of the non-radiative decay lifetime from 1.5 to 2.5 times results in slightly lower fluorescence quantum yields ranging from 19% to 41%. Incorporation of electron donating carbazole units at the para-position of the phenyl fragments (compound 3i) results in the fluorescence quantum yield of 55%, which decreases to 27% in polar surroundings. Interestingly, the attachment of the carbazole unit at the meta position of the phenyl fragment (compound 3j) results in a dramatic decrease of the fluorescence efficiency to 6%, which might be attributed to the broken symmetry of donor and acceptor moieties, resulting in dual-fluorescence nature of the excited states, prominent not only in the fluorescence spectra but also in the non-exponential decay transients in both non-polar and polar surrounding media.
Conclusions
In summary, novel D–π–A–π–D type chromophores – 2,4-bis(4-aryl-1,2,3-triazol-1-yl)pyrrolo[2,3-d]pyrimidines were prepared by CuAAC reaction of 2,4-diazido-7-methylpyrrolo[2,3-d]pyrimidine with diverse ethynylarenes at room temperature in dichloromethane using CuI/DIPEA/AcOH catalyst system. It was demonstrated that in the CuAAC reaction of 2,4-diazidopyrrolo[2,3-d]pyrimidine along with 1,4-disubstituted 1,2,3-triazoles some amounts of 1,5-disubstituted isomers were formed. The synthesized 2,4-bis(4-aryl-1,2,3-triazol-1-yl)pyrrolopyrimidines exhibit efficient fluorescence in the range from 402 nm to 650 nm. The introduction of various substituents enables tuning of HOMO and LUMO energies: the attachment of polar substituents in the para position of phenyl group with increasing electron-donating character results in lower HOMO and LUMO energies (from −6.69 eV to −5.08 eV for HOMO and from −2.71 eV to −1.95 eV for LUMO), while the addition of more bulky aryl substituents caused the reduction of HOMO energy (from −5.85 eV to −5.50 eV). The DFT calculations revealed that LUMO of all the derivatives are located on the pyrrolo[2,3-d]pyrimidine moiety and includes triazole fragments. This leads to the extension of the LUMO localization and enhanced electron-accepting properties of heterocyclic part of molecules. The bulky aryl substituents show mostly electron donating character and influence the energy levels of HOMO state. Variation of the size, polarity and geometry of the substituents alters the charge transfer character of the transitions, influencing the properties of the fluorescence spectra and fluorescence quantum yield. Intramolecular charge transfer has a considerable influence on the dynamics of radiative and non-radiative decay in polar surroundings. Both processes slow down and the competition between the radiative and non-radiative decay pathways results in smaller fluorescence quantum yields for compounds possessing electron-donating substituents or substituents with twisted geometry. The highest quantum yield of 73% was observed for a pyrrolo[2,3-d]pyrimidine derivative 3b with non-polar methyl group in medium polarity solvent – THF. Compound 3d bearing strong electron-donating dimethylamino group exhibited the largest Stokes shift of 279 nm in DMF solution.The synthesized novel D–triazole–A–triazole–D chromophores show potential for applications as blue emitters in OLED technologies, while the sensitivity of their emission properties to the polarity of surrounding media implies potential applications for biomaterial labeling and metal ions sensing.
Experimental section
General remarks
Melting points were determined in open capillaries with a digital melting point IA9100 series apparatus (ThermoFischer Scientific). All reactions and purity of the synthesized compounds were monitored by TLC using Silica gel 60 F254 aluminum plates (Merck). Visualization was accomplished by UV light. Column chromatography was performed using Silica gel 60 (0.040–0.063 mm) (Merck). NMR spectra were recorded on a Bruker Ascend 400 spectrometer (400 MHz and 100 MHz for 1H and 13C, respectively). 1H NMR and 13C NMR were referenced to residual solvent peaks. Infrared spectra (IR) were recorded on an IR spectrophotometer Spectrum BX II (Perkin Elmer). High Resolution Mass Spectrometry (HRMS) analyses were carried out on a quadrupole, time-of-flight mass spectrometer (microTOF-Q II, Bruker Daltonik GmbH, Bremen, Germany) or on Dual-ESI Q-TOF 6520 (Agilent Technologies) mass spectrometers. All quantum chemical calculations were carried out using Gaussian 09 program package. Geometries were optimized at DFT/B3LYP/6-311G** level of theory. Structures for calculations of shielding values were further optimized by applying self-consistent reaction field (SCRF) under the polarizable continuum model (IEFPCM) incorporating DMSO as solvent, absolute shielding values were calculated by GIAO method. TMS was used as a reference in calculating 1H and 13C chemical shifts from absolute shielding values. Electrochemical investigation was carried out using BioLogic SAS and a micro-AUTOLAB Type III potentiostat-galvanostat. The solutions of the synthesized compounds with the concentration of 10−5 M were used for cyclic voltamperometry (CV) measurements. The experiments were calibrated with the standard ferrocene/ferrocenium redox system. The absorption spectra were recorded on a Perkin-Elmer UV-vis-NIR spectrophotometer Lambda 950. Fluorescence of the sample solutions was excited by 320 nm wavelength light-emitting diode and measured using back-thinned CCD spectrometer (Hamamatsu PMA-11). The fluorescence quantum yield (ΦF) of the solutions was estimated by comparing wavelength-integrated fluorescence intensity of the solution with that of the reference. Quinine sulfate in 0.1 M H2SO4 was used as a reference. Optical densities of the reference and the sample solutions were ensured to be below 0.05 to avoid reabsorption effects. Estimated quantum yield was verified by using an alternative method of an integrating sphere (Sphere Optics), which was coupled to the CCD spectrometer by an optical fiber. The obtained ΦF values measured by both methods coincide within 4% error. The fluorescence quantum yield values obtained by the integrated sphere method are provided in Table 5. Fluorescence transients of the sample solutions were measured using time-correlated single photon counting system (PicoQuant PicoHarp 300).General procedure for the synthesis of compounds 3a–l
A mixture of compound 2 (100 mg, 0.46 mmol), corresponding alkyne (1.4 mmol), CuI (17 mg, 0.09 mmol), DIPEA (89 μL, 0.51 mmol) and acetic acid (29 μL, 0.51 mmol) in CH2Cl2 (5 mL) was stirred at room temperature. End of the reaction was monitored by TLC. Then the reaction mixture was poured into water (20 mL) and extracted with CH2Cl2 (3 × 10 mL). The extracts were combined, dried over Na2SO4 and filtered. After removal of the solvent the residue was purified by column chromatography on silica gel (eluent – hexane/EtOAc) to give a crude isomeric mixture. Compounds 3a–l were purified by recrystallization from 2-PrOH or toluene.104 and 8320). 1H NMR (400 MHz, CDCl3): δ 2.49 (6H, s, 2 × CH3), 4.07 (3H, s, NCH3), 7.22–7.30 (2H, m, ArH), 7.40–7.48 (3H, m, ArH, 5-H), 7.49 (1H, d, J = 3.6 Hz, 6-H), 7.84 (2H, t, J = 8.8 Hz, ArH), 7.90 (2H, s, ArH), 8.91 (1H, s, triazole-H), 9.14 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 21.4, 31.9, 103.2, 106.9, 117.5, 118.3, 123.11, 123.16, 126.6, 126.7, 128.7, 128.9, 129.34, 129.36, 129.6, 129.8, 132.3, 138.6, 138.7, 147.1, 147.6, 147.7, 147.9, 154.1. HRMS (ESI): calculated for C25H21N9Na [M + Na]+ = 470.1812; found: 470.1824.827 and 6182). 1H NMR (400 MHz, CDCl3): δ 3.03 (6H, s, N(CH3)2), 3.05 (6H, s, N(CH3)2), 4.02 (3H, s, NCH3), 6.81 (2H, d, J = 8.8 Hz, ArH), 6.84 (2H, d, J = 8.8 Hz, ArH), 7.35 (1H, d, J = 3.2 Hz, 5-H), 7.43 (1H, d, J = 3.2 Hz, 6-H), 7.87 (2H, d, J = 8.8 Hz, ArH), 7.90 (2H, d, J = 8.8 Hz, ArH), 8.72 (1H, s, triazole-H), 8.94 (1H, s, triazole-H). 13C NMR (100 MHz, d6-DMSO, 80 °C): δ 31.8, 40.41, 40.43, 103.4, 106.9, 112.3, 112.4, 115.7, 116.7, 117.5, 118.1, 127.0, 127.1, 132.0, 147.4, 147.9, 148.1, 148.3, 150.6, 150.7, 154.1. HRMS (ESI): calculated for C27H28N11 [M + H]+ = 506.2524; found: 506.2536.602 and 7777). 1H NMR (400 MHZ, CDCl3): δ 4.02 (3H, s, NCH3), 7.20 (4H, m, ArH), 7.38 (1H, d, J = 3.6 Hz, 5-H), 7.41 (1H, d, J = 3.6 Hz, 6-H), 7.98 (4H, m, ArH), 8.82 (1H, s, triazole-H), 9.04 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 31.9, 103.9, 107.0, 115.9 (d, J = 21 Hz), 116.1 (d, J = 21 Hz), 117.4, 118.1, 125.7 (d, J = 4 Hz), 126.2 (d, J = 4 Hz), 127.8 (d, J = 8 Hz), 127.9 (d, J = 8 Hz), 132.5, 146.8, 147.10, 147.16, 147.6, 154.1, 162.9 (d, J = 247 Hz), 163.1 (d, J = 247 Hz). HRMS (ESI): calculated for C23H15F2N9Na [M + Na]+ = 478.1311; found: 478.1321.761 and 8730). 1H NMR (400 MHz, CDCl3): 4.09 (3H, s, NCH3), 7.47 (1H, d, J = 3.6 Hz, 5-H), 7.50 (1H, d, J = 3.6 Hz, 6-H), 7.78–7.81 (4H, m, ArH), 8.17 (2H, d, J = 8.0 Hz, ArH), 8.19 (2H, d, J = 8.0 Hz, ArH), 9.01 (1H, s, triazole-H), 9.26 (1H, s, triazole-H). 13C NMR could not be obtained because of very low solubility in common deuterated solvents. HRMS (ESI): calculated for C25H15F6N9Na [M + Na]+ = 578.1247; found: 578.1248.300 and 10716). 1H NMR (400 MHz, d6-DMSO, 80 °C): δ 4.01 (3H, s, NCH3), 7.23 (1H, d, J = 3.2 Hz, 5-H), 7.86 (1H, d, J = 3.2 Hz, 6-H), 7.95 (4H, d, J = 6.8 Hz, ArH), 8.23 (4H, m, ArH), 9.62 (1H, s, triazole-H), 9.73 (1H, s, triazole-H). 13C NMR (100 MHz, d6-DMSO, 80 °C): δ 32.0, 102.0, 107.3, 111.4, 111.8, 118.90, 118.96, 121.3, 122.3, 126.8, 127.0, 133.33, 133.36, 134.5, 135.0, 135.1, 145.8, 145.9, 146.7, 147.4, 154.6. HRMS (ESI): calculated for C25H15N11Na [M + Na]+ = 492.1404; found: 492.1411.158 and 12867). 1H NMR (400 MHz, CDCl3): δ 4.07 (3H, s, NCH3), 7.44 (1H, d, J = 3.6 Hz, 5-H), 7.54 (1H, d, J = 3.6 Hz, 6-H), 7.55–7.61 (6H, m, ArH), 7.86–7.97 (6H, m, ArH), 8.49–8.54 (2H, m, ArH), 8.96 (1H, s, triazole-H), 9.19 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 32.0, 103.4, 107.1, 120.7, 121.6, 125.33, 125.37, 125.38, 125.4, 126.1, 126.2, 126.82, 12.97, 126.98, 127.4, 127.5, 127.6, 128.50, 128.56, 129.3, 129.5, 131.09, 131.2, 132.6, 133.92, 133.94, 147.22, 147.27, 147.3, 147.8, 154.33. HRMS (ESI): calculated for C31H22N9 [M + H]+ = 520.1993; found: 520.2005.093, 25031, 17628, 17233 and 16471). 1H NMR (400 MHz, d6-DMSO): δ 4.04 (3H, s, NCH3), 7.29–7.34 (5H, m, 5-H, carbazole-H), 7.45–7.52 (8H, m, ArH, carbazole-H), 7.86 (4H, t, J = 8 Hz, carbazole-H), 7.97 (1H, d, J = 3.6 Hz, 6-H), 8.28 (4H, d, J = 7.6 Hz, ArH), 8.38–8.43 (4H, m, carbazole-H), 9.87 (1H, s, triazole-H), 9.99 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 32.0, 103.4, 107.3, 109.7, 109.8, 117.9, 118.7, 120.15, 120.19, 120.41, 120.42, 123.53, 123.56, 123.61, 126.06, 126.08, 127.54, 127.59, 127.6, 128.6, 129.1, 132.7, 138.0, 138.2, 140.6, 140.7, 147.1, 147.3, 147.4, 147.7, 154.3. HRMS (ESI): calculated for C47H31N11Na [M + Na]+ = 772.2656; found: 772.2659.488, 26491, 12013, 11255 and 10549). 1H NMR (400 MHz, CDCl3): δ 3.98 (3H, s, NCH3), 7.25–7.28 (5H, m, 5-H, carbazole-H), 7.36–7.46 (10H, m, carbazole-H, ArH), 7.54 (1H, d, J = 8.0 Hz, carbazole-H), 7.58 (1H, d, J = 8.0 Hz, carbazole-H), 7.68 (1H, d, J = 8.0 Hz, carbazole-H), 7.72 (1H, J = 8.0 Hz, carbazole-H), 8.08–8.14 (6H, m, ArH, 6-H, carbazole-H), 8.23 (1H, s, ArH), 8.88 (1H, s, triazole-H), 9.12 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 31.9, 103.3, 107.1, 109.72, 109.74, 118.2, 119.0, 120.02, 120.07, 120.24, 120.25, 123.3, 123.4, 124.57, 124.58, 124.9, 125.0, 125.9, 126.0, 127.1, 127.3, 130.5, 130.6, 131.5, 132.0, 132.5, 138.3, 138.5, 140.7, 140.8, 146.84, 146.89, 147.0, 147.3, 154.1. HRMS (ESI): calculated for C47H31N11Na [M + Na]+ = 772.2656; found 772.2653.769, 23487 and 26245). 1H NMR (400 MHz, CDCl3): δ 0.67–0.79 (20H, m, 4CH2CH3), 1.06–1.16 [24H, m, 4(CH2)3–], 2.02–2.16 (8H, m, 4CH2), 4.10 (3H, s, NCH3), 7.34–7.41 (6H, m, ArH), 7.44 (1H, d, J = 3.6 Hz, 5-H), 7.52 (1H, d, J = 3.6 Hz, 6-H), 7.77 (2H, d, J = 7.2 Hz, ArH), 7.85 (2H, t, J = 8.0 Hz, ArH), 7.99–8.08 (4H, m, ArH), 9.00 (1H, s, triazole-H), 9.24 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 14.0, 22.6, 23.8, 29.7, 31.5, 40.4, 40.5, 55.3, 103.4, 107.2, 117.5, 118.4, 119.8, 119.9, 120.1, 120.2, 120.42, 120.48, 122.9, 124.94, 124.95, 126.85, 126.86, 127.3, 127.4, 128.1, 128.6, 132.4, 140.5, 140.6, 141.7, 142.0, 147.4, 147.9, 148.2, 148.4, 148.6, 151.01, 151.07, 151.62, 152.67, 154.28. HRMS (ESI): calculated for C61H74N9 [M + H] = 932.6062; found 932.6056.131, 20724 and 25790). 1H NMR (400 MHz, CDCl3): δ 1.64 (6H, s, 2CH3), 1.65 (6H, s, 2CH3), 4.07 (3H, s, NCH3), 7.40–7.75 (12H, m, ArH, 5-H, 6-H), 8.01 (1H, s, ArH), 8.06 (1H, s, ArH), 8.36–8.42 (2H, m, ArH), 8.54 (1H, d, J = 8.4 Hz, ArH), 8.61 (1H, d, J = 8.4 Hz, ArH), 8.84–8.90 (2H, m, ArH), 8.99 (1H, s, triazole-H), 9.25 (1H, s, triazole-H). 13C NMR (100 MHz, CDCl3): δ 26.7, 31.9, 46.9, 103.4, 107.1, 120.9, 121.9, 122.5, 122.6, 122.8, 122.9, 123.3, 123.4, 124.2, 124.3, 125.83, 126.0, 126.5, 126.6, 126.74, 126.79, 126.81, 126.85, 126.87, 127.23, 127.26, 127.3, 130.0, 130.1, 131.2, 131.4, 132.6, 134.3, 134.7, 139.7, 139.8, 147.3, 147.5, 147.7, 151.6, 151.7, 154.25, 154.27, 154.9, 155.0. HRMS (ESI): calculated for C49H38N9 [M + H]+ = 752.3245; found 752.3252.Acknowledgements
The research was supported by the Research Council of Lithuania (grant no. MIP-027/2013).Notes and references
- (a) M. T. Bernius, M. Inbasekaran, J. O'Brien and W. S. Wu, Adv. Mater., 2000, 12, 1737 CrossRef CAS; (b) P. K. H. Ho, J. S. Kim, J. H. Burroughes, H. Becker, S. F. Y. Li, T. M. Brown, F. Cacialli and R. H. Friend, Nature, 2000, 404, 481 CrossRef CAS PubMed; (c) M. Gross, D. C. Muller, H. G. Nothofer, U. Scherf, D. Neher, C. Brauchle and K. Meerholz, Nature, 2000, 405, 661 CrossRef CAS PubMed; (d) A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS; (e) G. Hughes and M. R. Bryce, J. Mater. Chem., 2005, 15, 94 RSC; (f) W. Y. Wong, G. J. Zhou, X. M. Yu, H. S. Kwok and Z. Lin, Adv. Funct. Mater., 2007, 17, 315 CrossRef CAS PubMed; (g) Y. Tao, Q. Wang, C. Yang, Q. Wang, Z. Zhang, T. Zou, J. Qin and D. A. Ma, Angew. Chem., Int. Ed., 2008, 47, 8104–8107 CrossRef CAS PubMed.
- (a) S. Achelle and C. Baudequin, in Targets in Heterocyclic Systems, ed. O. A. Attanasi and D. Spinelli, Royal Society of Chemistry, London, 2013, vol. 17, p. 1 Search PubMed; (b) S. Achelle and N. Ple, Curr. Org. Synth., 2012, 9, 163 CrossRef CAS; (c) S. J. Su, C. Cai and J. Kido, Chem. Mater., 2011, 23, 274 CrossRef CAS; (d) C. Hadad, S. Achelle, I. Lopez-Solera, J. C. Garcia-Martinez and J. Rodriguez-Lopez, Dyes Pigm., 2013, 97, 230 CrossRef CAS PubMed; (e) S. S. Mati, S. Chall, S. Konar, S. Rakchit and S. C. Bhattacharya, Sens. Actuators, B, 2014, 201, 204 CrossRef CAS PubMed; (f) J. Weng, Q. Mei, Q. Ling, Q. Fan and W. Huang, Tetrahedron, 2012, 68, 3129 CrossRef CAS PubMed.
- For recent review on pyrimidine rings in conjugated materials, see: S. Achelle and N. Ple, Curr. Org. Synth., 2012, 9, 163; For examples, see: (a) S. Achelle, A. Barsella, C. Baudequin, B. Caro and F. Robin-Le Guen, J. Org. Chem., 2012, 77, 4087 CrossRef CAS PubMed; (b) J. Weng, Q. Mei, Q. Ling, Q. Fan and W. Huang, Tetrahedron, 2012, 68, 3129 CrossRef CAS PubMed; (c) I. Malik, Z. Ahmed, S. Reimann, I. Ali, A. Villinger and P. Langer, Eur. J. Org. Chem., 2011, 2088 CrossRef CAS PubMed; (d) C. Hadad, S. Achelle, J. C. Garcia-Martinez and J. Rodriguez-Lopez, J. Org. Chem., 2011, 76, 3837 CrossRef CAS PubMed; (e) A.-S. Cornec, C. Baudequin, C. Fiol-Petit, N. Ple, G. Dupas and Y. Ramondenc, Eur. J. Org. Chem., 2013, 1908 CrossRef CAS PubMed; (f) C. Denneval, O. Moldovan, C. Baudequin, S. Achelle, P. Baldeck, N. Ple, M. Darabantu and Y. Ramondenc, Eur. J. Org. Chem., 2013, 5591 CrossRef CAS PubMed; (g) D. Chen, C. Zhong, X. Dong, Z. Liu and J. Qin, J. Mater. Chem., 2012, 10, 4343 RSC; (h) L. Skardziute, J. Dodonova, A. Voitechovicius, J. Jovaisaite, R. Komskis, A. Voitechoviciute, J. Bucevicius, K. Kazlauskas, S. Jursenas and S. Tumkevicius, Dyes Pigm., 2015, 118, 118 CrossRef CAS PubMed.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed; (b) C. Adachi, Jpn. J. Appl. Phys., 2014, 53, 60101 CrossRef; (c) Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931 CrossRef CAS PubMed.
- (a) A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC; (b) V. D. Suryawanshi, A. H. Gore, P. R. Dongare, P. V. Anbhule, S. R. Patil and G. B. Kolekar, Spectrochim. Acta, Part A, 2013, 114, 681 CrossRef CAS PubMed; (c) Q. Meng, D. H. Kim, X. Bai, L. Bi, N. J. Turro and J. Ju, J. Org. Chem., 2006, 71, 3248 CrossRef CAS PubMed; (d) D. A. Berry, K.-Y. Jung, D. S. Wise, A. D. Sercel, W. H. Pearson, H. Mackie, J. B. Randolph and R. J. Somers, Tetrahedron Lett., 2004, 45, 2457 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67(9), 3057 CrossRef PubMed.
- (a) V. D. Bock, H. Hiemstra and J. H. Maarseveen, Eur. J. Org. Chem., 2006, 51 CrossRef CAS PubMed; (b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108(8), 2952 CrossRef CAS PubMed.
- (a) J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. J. Carell, Org. Lett., 2006, 8, 3639 CrossRef CAS PubMed; (b) R. Franke, C. Dol and J. Eichler, Tetrahedron Lett., 2005, 46, 4479 CrossRef CAS PubMed; (c) H. Jang, A. Farman, J. M. Holub and K. J. Kirshenbaum, Org. Lett., 2005, 7, 1951 CrossRef CAS PubMed; (d) J. M. Holub, H. Jang and K. Kirshenbaum, Org. Biomol. Chem., 2006, 4, 1497 RSC; (e) F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207 CrossRef CAS PubMed; (f) A. Kovalovs, I. Novosjolova, E. Bizdena, I. Bizane, L. Skardziute, K. Kazlauskas, S. Jursenas and M. Turks, Tetrahedron Lett., 2013, 54, 850 CrossRef CAS PubMed; (g) I. Novosjolova, E. Bizdena and M. Turks, Tetrahedron Lett., 2013, 54, 6557 CrossRef CAS PubMed; (h) C. Dyrager, K. Börjesson, P. Dinér, A. Elf, B. L. Albinsson, M. Wilhelmsson and M. Grøtli, Eur. J. Org. Chem., 2009, 1515 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- For recent review see Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848; For examples see (a) M. Collot, A. Lasoroski, A. I. Zamaleeva, A. Feltz, A. Vuilleumier and J. M. Mallet, Tetrahedron Lett., 2013, 69, 10482 CrossRef CAS PubMed; (b) H. Singh, J. Sindhu and J. M. Khurana, Sens. Actuators, B, 2014, 192, 536 CrossRef CAS PubMed; (c) L. Cao, R. Jiang, Y. Zhu, X. Wang, Y. Li and Y. Li, Eur. J. Org. Chem., 2014, 2687 CrossRef CAS PubMed.
- (a) H. Zhang, L. Zhou, S. J. Coats, T. R. McBrayer, P. M. Tharnish, L. Bondada, M. Detorio, S. Amichai, M. D. Johns, T. Whitaker and R. F. Schinazi, Bioorg. Med. Chem. Lett., 2011, 21, 6788 CrossRef CAS PubMed; (b) P. Chittepu, V. R. Sirivolu and F. Seela, Bioorg. Med. Chem., 2008, 16, 8427 CrossRef CAS PubMed.
- (a) S. S. Bag and R. J. Kundu, J. Org. Chem., 2011, 76, 3348 CrossRef CAS PubMed; (b) D. Schweinfurth, K. I. Hardcastle and U. H. F. Bunz, Chem. Commun., 2008, 2203 RSC; (c) J. Shi, L. Liu, J. He, X. Meng and Q. Guo, Chem. Lett., 2007, 36, 1142 CrossRef CAS; (d) P. D. Jarowski, Y.-L. Wu, B. Schweizer and F. Diederich, Org. Lett., 2008, 10, 3347 CrossRef CAS PubMed; (e) D. J. V. C. van Stennis, O. R. P. David, G. P. F. van Srijdonck, J. H. Van Maarseveen and J. N. H. Reek, Chem. Commun., 2005, 4332 Search PubMed; (f) M. Juricek, P. H. J. Kouwer and A. E. Rowan, Chem. Commun., 2011, 47, 8740 RSC; (g) P. D. Zoon, I. H. M. Van Stokkum, M. Parent, O. Mongin, M. Blanchard-Desce and A. M. Brouwer, Phys. Chem. Chem. Phys., 2010, 12, 2706 RSC; (h) D. Urankar, A. Pevec, I. Turel and J. Kosmrlj, Cryst. Growth Des., 2010, 10, 4920 CrossRef CAS; (i) M. Parent, O. Mongin, K. Kamada, C. Katan and M. Blanchard-Desce, Chem. Commun., 2005, 2029 RSC.
- (a) S. Tumkevicius, J. Dodonova, K. Kazlauskas, V. Masevicius, L. Skardziute and S. Jursenas, Tetrahedron Lett., 2010, 51, 3902 CrossRef CAS PubMed; (b) J. Dodonova, L. Skardziute, K. Kazlauskas, S. Jursenas and S. Tumkevicius, Tetrahedron, 2012, 68, 329 CrossRef CAS PubMed; (c) L. Skardziute, K. Kazlauskas, J. Dodonova, J. Bucevicius, S. Tumkevicius and S. Jursenas, Tetrahedron, 2013, 69, 9566 CrossRef CAS PubMed.
- (a) M. E. Di Francesco, S. Avolio, M. Pompei, S. Pesci, E. Monteagudo, V. Pucci, C. Giuliano, F. Fiore, R. Michael and V. Summa, Bioorg. Med. Chem., 2012, 20, 4801 CrossRef CAS PubMed; (b) C. P. Lawson, A. Dierckx, F.-A. Miannay, E. Wellner, L. M. Wilhelmsson and M. Grotli, Org. Biomol. Chem., 2014, 12, 5158 RSC; (c) S. A. Ingale and F. Seela, Tetrahedron, 2014, 70, 380 CrossRef CAS PubMed.
- (a) A. S. Cornec, C. Baudequin, C. Fiol-Petit, N. Plé, G. Dupas and Y. Ramondenc, Eur. J. Org. Chem., 2013, 1908 CrossRef CAS PubMed; (b) L. I. Nilsson, A. Ertan, D. Weigelt and J. M. J. Nolsoë, J. Heterocycl. Chem., 2010, 47, 887 CrossRef CAS PubMed; (c) C. Denneval, O. Moldovan, C. Baudequin, S. Achelle, P. Baldeck, N. Plé, M. Darabantu and Y. Ramondenc, Eur. J. Org. Chem., 2013, 5591 CrossRef CAS PubMed.
- M. K. Lakshman, M. K. Singh, D. Parrish, R. Balachandran and B. W. Day, J. Org. Chem., 2010, 75, 2461 CrossRef CAS PubMed.
- (a) E. G. Paronikyan and A. S. Noravyan, Chem. Heterocycl. Compd., 1995, 31(5), 621 CrossRef; (b) C. G. Dave and R. D. Shah, J. Heterocycl. Chem., 1998, 35, 1295 CrossRef CAS PubMed; (c) C. G. Dave and R. D. Shah, Molecules, 2002, 7, 554 CrossRef CAS PubMed; (d) N. D. Desai, Synth. Commun., 2006, 36, 2169 CrossRef CAS.
- C. Shao, X. Wang, Q. Zhang, S. Luo, J. Zhao and Y. Hu, J. Org. Chem., 2011, 76, 6832 CrossRef CAS PubMed.
- P. M. Chandrika, T. Yakaiah, G. Gayatri, K. P. Kumar, B. Narsaiah, U. S. N. Murthy and A. R. Ram Rao, Eur. J. Med. Chem., 2010, 45, 78 CrossRef PubMed.
- D. Cantillo, M. Avalos, R. Babiano, P. Cintas, J. L. Jimenez and J. C. Palacios, Org. Biomol. Chem., 2011, 9, 2952 CAS.
- (a) G. Alonso, M. T. Garcia-Lopez, R. Madroner and M. J. Rico, Heterocycl. Chem., 1970, 7, 1269 CrossRef CAS PubMed; (b) A. Bolje, D. Urankar and J. Kosmrlj, Eur. J. Org. Chem., 2014, 8167 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. I. Djurovich, E. I. Mayo, S. R. Forrest and M. E. Thompson, Org. Electron., 2009, 10, 515 CrossRef CAS PubMed.
- F. Sella and H. Steker, Liebigs Ann. Chem., 1984, 1719 CrossRef PubMed.
- J. Simokaitiene, S. Grigalevicius, J. V. Grazulevicius, R. Rutkaite, K. Kazlauskas, S. Jursenas, V. Jankauskas and J. Sidaravicius, J. Optoelectron. Adv. Mater., 2006, 8(2), 876 CAS.
- Z. R. Grabowski and K. Rotkiewicz, Chem. Rev., 2003, 103, 3899 CrossRef PubMed.
- H. Xie, L. Zeng, S. Zeng, X. Lu, G. Zhang, X. Zhao, N. Cheng, Z. Tu, Z. Li, H. Xu, L. Yang, X. Zhang, M. Huang, J. Zhao and W. Hu, Eur. J. Med. Chem., 2012, 52, 205 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05482f |
| This journal is © The Royal Society of Chemistry 2015 |