
Industrial effluent as a substrate for glutaminase free L-asparaginase production from Pseudomonas plecoglossicida strain RS1; media optimization, enzyme purification and its characterization†
Ganeshan Shakambaria,
Babu M. Sumia,
Balasubramaniem Ashokkumarb,
Peramachi Palanivelua and
Perumal Varalakshmi*a
aDepartment of Molecular Microbiology, Madurai Kamaraj University, Madurai, India. E-mail: vara5277@gmail.com; Fax: +91-452-2459105; Tel: +91-452-2459115
bDepartment of Genetic Engineering, Madurai Kamaraj University, Madurai, India
First published on 30th April 2015
Abstract
Glutaminase free L-asparaginase is a vital enzyme because of its anticancer potential. A potent bacterium isolated from a marine environment which produces glutaminase free L-asparaginase using M-9 medium with L-asparagine, was identified as Pseudomonas plecoglossicida RS1 by 16S rRNA gene sequencing. Statistical modeling was employed to optimize the medium using sugar cane industry effluent as the sole substrate for L-asparaginase production. The enzyme activity of L-asparaginase was higher with M-9 medium containing 0.8% effluent (3.25 ± 0.12 IU mL−1) compared to M-9 medium containing 0.3% L-asparagine (0.73 ± 0.08 IU mL−1). The apparent Km and Vmax of the purified L-asparaginase was 2.25 ± 0.61 mM and 8.9 ± 0.81 IU mL−1 min−1 respectively and the optimal activity of L-asparaginase was at pH 8.5 and 55 °C. This study highlights the use of industrial effluent as an alternate to L-asparagine for the production of L-asparaginase and how to improve the cost effectiveness of this enzyme.
Introduction
L-Asparaginase (EC 3.5.1.1) has been used as therapeutic remedy for acute lymphoblastic leukemia (ALL) wherein lymphoblasts are auxotrophic for L-asparagine, and the enzyme diminishes the supply of exogenous asparagine, thus forcing the tumor cells into apoptosis.1,2 The antineoplastic activity of L-asparaginase has been explored from microbial sources such as Escherichia coli,3,4 Proteus vulgaris,5 Serratia marcescens,6,7 Bacillus sp.,8 Pseudomonas aeruginosa,9 Streptomyces sp.,10,11 Aspergillus terreus,12 and Erwinia carotovora.13The substrate specificity of L-asparaginase is characterized for both L-asparagine and L-glutamine where L-glutamine differs from L-asparagine by a single methyl group, consequently L-asparaginase treatment diminishes L-glutamine availability along with the concentration of L-asparagine, causing side effects like leucopenia, acute pancreatitis, hyperglycemia and neurological seizures in ALL patients.14–16 Hence glutaminase free L-asparaginase from microbial sources, with as little as possible cross reactivity, is a great matter of interest for researchers, but there has not been appreciable success. In addition, several L-asparaginases with potential catalytic properties were unsuccessful in preclinical trials, due to their therapeutic ineffectiveness.17 Therefore there is an imperative need to find glutaminase free L-asparaginase activity from a novel source which can be developed into targeted therapy and may become the best candidate for satisfactory treatment of ALL.
The fermentation process for the enhanced production of L-asparaginase was accomplished by supplementation with a variety of carbon and nitrogen sources to the media and this process may also be regulated by environmental and nutritional factors in Gram negative bacteria.18 In this perspective, it is essential to evaluate the microbial system that requires environmentally friendly nutritional requirements for growth and biocatalyst production. In recent years, many agro and industrial wastes are being used as alternate substrates for the production of proteins such as enzymes19 and lipids, etc.20 The selection of the wastes and their parameters including cost and availability are also very important for host enzyme production thus, in this study we have utilized sugar cane industry effluent as a substrate for L-asparaginase production since it is available in large quantities and can be obtained very easily. It is also noteworthy to mention that statistically designed experiments like Plackett–Burman Design (PBD) and Central Composite Design (CCD), which effectively embark upon resolving the problem involving upstream and downstream processing, thus reduce the inaccuracy of interactions between nutrient parameters, and also aid in achieving enhanced economical biocatalyst production.21,22 Hence, the present study was performed to attain improved production of L-asparaginase using industrial effluent as an alternate substrate to L-asparagine, by using bioprocess methodologies such as PBD and CCD. Purification and characterization of L-asparaginase was also performed. To the best of our knowledge, this is the first report highlighting sugar cane industry effluent being used as an alternate substrate to L-asparagine for the production of L-asparaginase.
Materials and methods
Chemicals
Zobell marine broth, nutrient broth, D-glucose, Na2HPO4, KH2PO4, CaCl2·2H2O, NaNO3, sarkosyl, sodium dodecyl sulfate and Tris base were purchased from Himedia, (India). L-Asparagine, L-glutamine, phenol red, DEAE sepharose, Readymix Taq Polymerase Kit and GenElute kit were obtained from Sigma Aldrich (USA). Nessler’s reagent was purchased from SD Fine Chemicals (India). Ammonium sulphate was purchased from MERCK (India).Bacterial culture and preliminary screening of L-asparaginase
Marine isolates from the Gulf of Mannar near Rameswaram, India were screened for the production of L-asparaginase, and the isolate positive for L-asparaginase was further identified based on its 16S rDNA sequence (SciGenome, India). Screening for L-asparaginase by a plate assay method was done using M-9 medium which consisted of D-glucose – 3 g, Na2HPO4 – 6 g, KH2PO4 – 3 g, MgSO4 – 0.5 g, NaCl – 0.5 g, L-asparagine – 3 g and 0.001% phenol red indicator.22Quantitative assay for L-asparaginase and L-glutaminase activity
A quantitative assay was performed for the estimation of L-asparaginase and L-glutaminase activity. 50 mL of M-9 medium containing the amino acids L-asparagine/L-glutamine with 2% (v/v) inoculum (Pseudomonas plecoglossicida RS1) were incubated at 37 °C for 48 h. The cell free supernatant was obtained by centrifugation at 8000 rpm for 10 min and was used as a source of crude enzyme to quantify the L-asparaginase/L-glutaminase activity by direct nesslerization, where the enzyme hydrolyzed product ammonia is detected and quantified based on a standard curve obtained using ammonium sulphate as a standard.3One unit of enzyme activity is defined as the amount of enzyme that produces 1 μmol of ammonia per minute at pH 8.6 and 37 °C.
The total protein content of the samples was determined according to the method by Folin–Lowry using bovine serum albumin as a standard. The specific activity of the enzyme is given by enzyme activity per milligram of protein, (IU per mg protein).
Analysis of the effect of pH, temperature and substrate for L-asparaginase production by Pseudomonas plecoglossicida RS1
The effects of different pH (6 to 8) medium and incubation temperature (20 °C, 37 °C and 55 °C) for L-asparaginase production were evaluated. Effluent from sugar cane industry was used for enhancing the production of L-asparaginase in M-9 medium that was free of L-asparagine and M-9 basal medium without any nitrogen source was used as a control. Protein concentrations were measured by Lowry’s method. Reducing sugar content was estimated by the DNSA method23 where, diluted effluent (3 mL) was added to 3 mL of dinitrosalicylic acid reagent and boiled for 10 min, cooled and then the absorbance was taken at 550 nm. The Biochemical Oxygen Demand (BOD) of the effluent was also estimated as per the stipulations of IS: 3025 (Part 44) – Reaffirmed 2003. Different concentrations of effluent (0.1% to 2%) were added to M-9 medium (50 mL) and each flask was provided with an equal amount (2%) of inoculum grown overnight (2 × 105 cfu mL−1). The flasks were incubated at static conditions at 37 °C for 48 h and the specific activity of the enzyme was quantified. The study was carried out in three independent triplicate analyses.Media optimization for the production of L-asparaginase by Pseudomonas plecoglossicida RS1 using Plackett–Burman Design
To enhance L-asparaginase production, Plackett–Burman design24 (PBD) was used to identify the important medium components and this was done using the software Design Expert version 9.0.7. In this design, the seven crucial components of the medium like glucose, Na2HPO4, KH2PO4, MgSO4·7H2O, NaCl, effluent and pH were selected as the parameters for the study and the responses were enzyme activity and the specific activity of L-asparaginase. Each variable was represented as high and low levels (ESI Table III†) as described by the Plackett–Burman design and a total of twelve experiments were carried out. All experiments were carried out in duplicates, and the mean values of L-asparaginase enzyme units and specific activity were taken as responses (Y). The process of optimization is defined in a first order polynomial model and is as follows:| Y = β0 + ∑βiXi (i = 1, 2, 3,…k) | (1) |
Based on the significant influence in the response, variables were further selected for CCD.
Central composite design
To optimize the levels and analyze the combined effect of the screened media components that were significant for the production of L-asparaginase, the CCD was used.8 According to this design, the total number of treatment combinations is 2k + 2k + n0 where ‘k’ is the number of independent variables and n0 is the number of replications of the experiments at the central point. Accordingly, there were a total of twenty experimental runs for the three components selected from the PBD, with six replicates at the central value. The others were performed in duplicates and the influence of each component was evaluated at five coded levels and responses were evaluated as enzyme activity and specific activity. For calculation, the variables Xi have been coded as xi.For predicting the optimum levels the quadratic equation was expressed as follows:
| Y = β0 + ∑βixi + ∑βiixi2 + ∑βijxixj | (2) |
Verification of the production media
The model was validated with the optimized medium M-9 medium (500 mL) consisting of the significant variables obtained from the CCD, inoculated with 2% (v/v) inoculum, incubated at 37 °C for 48 h in static conditions, and then the enzyme activity and specific activity were determined. The study was carried out in triplicates and the mean value was calculated.Purification and characterization of the enzyme
All the purification steps (ammonium sulphate precipitation, dialysis) were carried out at 4 °C, while the column purification of enzyme was done at 18 °C. The chromatographic purification steps were analysed for protein content at 280 nm (Biophotometer D30, Eppendorf, Germany).Ammonium sulfate precipitation
The purification of the crude enzyme obtained from 500 mL of optimized production media was initiated with ammonium sulfate precipitation. Powdered ammonium sulfate was added to achieve 20% saturation and increased successively to get the maximum precipitation of proteins (between 60–80% saturation). This mixture was left for 3 h, followed by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min and after this the precipitate was preserved. The precipitated protein was resuspended in 50 mM Tris–HCl buffer of pH 8.6 and further dialyzed (Dialysis membrane-110, Himedia) against the same buffer and incubated overnight in order to remove salt impurities, and analysed for the specific activity of L-asparaginase in the respective fractions. The dialyzed protein fraction was further purified using column purification.
000 rpm for 10 min and after this the precipitate was preserved. The precipitated protein was resuspended in 50 mM Tris–HCl buffer of pH 8.6 and further dialyzed (Dialysis membrane-110, Himedia) against the same buffer and incubated overnight in order to remove salt impurities, and analysed for the specific activity of L-asparaginase in the respective fractions. The dialyzed protein fraction was further purified using column purification.
DEAE sepharose ion exchange chromatography
For further purification, the dialyzed ammonium sulfate fraction was loaded on to a diethylaminoethyl (DEAE) sepharose column (1 cm × 50 cm) as 2% of the bed volume of 30 mL. The column was pre-equilibrated with two column volumes of 50 mM Tris–HCl (pH 9.6) before the loading of the sample, at a flow rate of 1 mL min−1. The adsorbed protein was eluted using a linear gradient of KCl (0–200 mM) in 50 mM Tris–HCl (pH 8.6). All the fractions were collected manually and assayed for protein content at 280 nm and L-asparaginase activity by the direct nesslerization method.3 The results were plotted as enzyme activity and protein absorbance (double Y plot) versus the eluted fraction, using statistical software Origin Professional version 6.0 Professional. The purified fractions of L-asparaginase were pooled and dialyzed with Tris–HCl (50 mM and pH 8.6) and were stored at −20 °C and this was used to further identify the molecular weight of L-asparaginase by native polyacrylamide gel electrophoresis and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).Electrophoretic analysis of purified protein
Native PAGE analysis of the purified L-asparaginase was performed on 7.5% polyacrylamide gel in Tris–glycine buffer at pH 8.8 and SDS PAGE was done using the Laemmli method (1970)25 with a 12% separating acrylamide gel (pH 8.8) and a 5% stacking gel (pH 6.8) containing 0.1% SDS. Electrophoresis was carried out using Tris–glycine buffer at pH 8.3 at 120 V for 3 h at room temperature (25 °C). The proteins in the gel were stained with Coomassie Brilliant Blue G-250 (SDFCL, India) for 3 h and destained until the dye was completely removed. The molecular weight and the subunit size of L-asparaginase were determined using the standard molecular weight markers of SDS-PAGE (Bio-Rad, USA) and native PAGE (Native Mark, Novex Life Technologies, USA).Effect of pH, temperature and effectors on activity and stability of enzyme
The optimum enzyme activity was estimated at various pHs ranging from 4 to 9.5. The pH stability studies were performed by incubating the enzyme substrate system with either acetate buffer (pH 4 to 6), potassium phosphate (pH 6.0–8.0) or Tris–HCl (pH 8.0–9.5). In order to determine the pH at which the enzyme can be stored with optimum activity, enzyme in the absence of substrate was stored with buffers of different pHs for 24 h at 4 °C and then the enzyme activity was determined by direct nesslerization reaction and the results of enzyme activity versus pH plotted on a graph indicated the pH stability for the purified L-asparaginase. Similarly, the optimum temperature for the enzyme was evaluated by incubating the enzyme substrate system at different temperatures ranging from 0 °C to 80 °C and the enzyme activity was estimated. Furthermore, the enzyme activity in the presence of various effectors (Na+, K+, Mg2+, Ca2+, Mn2+, Zn2+, Hg2+, Fe3+, ethylene diamine tetra acetic acid (EDTA), L-cystine, L-histidine, mercaptoethanol and sodium dodecyl sulphate) was calculated after a 30 min exposure of the enzyme to each effector mentioned.14,26,27 The relative activity was given as the percentage ratio of the activity of enzyme incubated with the effectors and enzyme without effectors, under standard conditions used as a control.Kinetic parameters of L-asparaginase
Purified enzyme was suspended in 50 mM Tris–HCl buffer pH 8.6 with different concentrations of the substrate L-asparagine ranging from 0.01 mM to 10 mM, and the Michaelis constant (Km) and maximal velocity (Vmax) were calculated by non linear regression analysis. The reaction velocity (V) reported as (IU mL−1 min−1) is the mean of at least three measurements that were normalized to a blank, which contained no enzyme. The obtained enzyme activity (IU mL−1 min−1) was plotted against the substrate concentration (mM) using the software Graph Pad Prism Version 6 to obtain the Michaelis–Menten plot.Results and discussion
Screening and production of L-asparaginase by marine bacteria
Fifteen bacterial colonies were isolated from a marine water sample collected from Gulf of Mannar, Tamil Nadu, India and were screened for their potential for L-asparaginase activity with M-9 medium containing L-asparagine (0.3% w/v) using the phenol red plate method.22 Among the 15 isolates, bacterial strain RS1 was found to be positive for L-asparaginase by developing a pink colouration and was further identified as Pseudomonas plecoglossicida by 16S rRNA sequence analysis which showed 98% sequence similarity and 99% query coverage to Pseudomonas plecoglossicida strain FPC951 with BLAST analysis (http://www.ncbi.nlm.nih.gov/) (GenBank accession number: KJ508408) (ESI Fig. A†). Furthermore, the L-asparaginase produced by Pseudomonas plecoglossicida RS1 showed an enzyme activity of 0.87 ± 0.2 IU mL−1 min−1 with a protein content of 1.21 ± 0.193 mg mL−1 and a specific activity of 0.64 ± 0.13 in 50 mL of M-9 medium with 0.3% L-asparagine as a nitrogen source and incubated at 37 °C for 48 h. Similarly, glutaminase activity was estimated using L-glutamine as substrate instead of L-asparagine by direct nesslerization, no ammonia was detected and thus it can be concluded that this enzyme was free of L-glutaminase activity. Furthermore, the pH and temperature of the medium were optimized for the production of L-asparaginase by this strain and it was noticed that the maximum specific activity was obtained at pH 6.5 and at 37 °C, while growth at 50 °C did not occur (ESI Table I†).Production of L-asparaginase using industrial effluent as a substrate
Wastes from agriculture and industry have been explored for bio energy and bio fuel production by microorganisms, yielding methane, ethanol and hydrogen peroxide as by-products.28 A variety of cheap agro and industrial wastes like corn cobs, coconut oil cake, leguminous crops and palm oil effluent are being used as substrates for the production of enzymes like L-asparaginase29,30 and lipases.31 Sugar cane industry effluent has many applications including the possibility of acting as a biocatalyst for microbial fuel cells and for aconitic and lactic acid recovery. Hence, in this study an attempt was made to exploit sugar cane industry effluent as a cheaper alternate substrate to L-asparagine for the enhanced production of L-asparaginase. Based on the parameters like the presence of L-asparagine (Thin layer Chromatography) and the other parameters (pH (4.85), reducing sugar content (0.43 ± 0.03 mg mL−1), protein content (40.66 ± 3.8 mg mL−1) and BOD (76.6 mg L−1)) the effluent was chosen as an alternate substrate for the production of L-asparaginase (ESI Table II†). Further HPLC analysis and FT-IR analysis of the effluent was done and the presence of organic nitrogen was confirmed in form of amides and amines (ESI Fig. B and C†). In order to ensure that the microbial load of the effluent did not interfere with the enzyme activity of Pseudomonas plecoglossicida RS1, the effluent was sterilized by autoclaving at 15 psi for 15 minutes, thrice, on three successive days (to kill the germinating spore cells). Complete sterilization was confirmed by determining the total viable count (TVC) after each autoclaving cycle until the microbial load was brought to zero and thus it was ensured that the enzyme production was solely contributed by the isolate Pseudomonas plecoglossicida RS1. The medium containing 1% effluent was capable of producing enzyme with a higher specific activity (1.9 fold) than the medium containing 1% L-asparagine, whereas no activity was detected in the M-9 medium without a nitrogen source, which was the control (Table 1).| Media M-9 | Enzyme activity (IU mL−1 min−1) | Protein content of crude enzyme (mg mL−1) | Specific activity (IU mg−1) | |
|---|---|---|---|---|
| L-Asparagine (% w/v) | 0 | 0.00 | 0.008 ± 0.005 | 0 |
| 0.1 | 0.61 ± 0.03 | 0.55 ± 0.06 | 1.02 ± 0.41 | |
| 0.3 | 0.87 ± 0.20 | 1.21 ± 0.19 | 0.64 ± 0.13 | |
| 1.0 | 1.54 ± 0.90 | 2.3 ± 0.30 | 0.62 ± 0.3 | |
| 1.5 | 0.76 ± 0.20 | 2.32 ± 0.28 | 0.32 ± 0.06 | |
| 2 | 0.58 ± 0.12 | 2.33 ± 0.23 | 0.24 ± 0.03 | |
| Effluent (% v/v) | 0.1 | 0.31 ± 0.006 | 0.66 ± 0.06 | 0.47 ± 0.05 |
| 0.5 | 0.88 ± 0.35 | 0.77 ± 0.12 | 1.11 ± 0.27 | |
| 1 | 1.43 ± 0.64 | 1.15 ± 0.06 | 1.21 ± 0.46 | |
| 1.5 | 0.53 ± 0.06 | 1.49 ± 0.14 | 0.37 ± 0.08 | |
| 2 | 0.71 ± 0.45 | 1.68 ± 0.31 | 0.49 ± 0.38 |
The high protein content of the effluent, substantiated by the presence of organic nitrogen shown by FT-IR and HPLC analysis, mimicks the natural amino acid asparagine, (ESI Fig. B and C†) and this means that the effluent might have served as a rich nitrogen source for the enhanced production of L-asparaginase by Pseudomonas plecoglossicida RS1. It has also been reported previously that nitrogen supplementation had significantly influenced the production of L-asparaginase.31 It has been documented that sugar cane industry effluent rich in organic nitrogen content influences the production of commercially important products from microorganisms.19,20 However, this is the first study highlighting that sugar cane industry effluent as an alternate substrate to L-asparagine would enhance L-asparaginase production and that it may pave a way to reduce the production cost for commercialization.
Media optimization by Plackett–Burman design
To enhance the potential of Pseudomonas plecoglossicida RS1 for L-asparaginase production, the nutrient factors such as L-asparagine, glucose, KH2PO4, and MgSO4·7H2O, pH and effluent were chosen for this optimization study using the statistical models PBD and CCD. Critical factors in the fermentation process and their significant interaction with each other for practical application and validation in commercial production of this enzyme were also analyzed. Both PBD and CCD have been used previously for increasing the yield of L-asparaginse in Bacillus licheniformis and Pectobacterium carotovorum MTCC 1428.8,32PBD was used with seven factors, which were, glucose, Na2HPO4, KH2PO4, MgSO4·7H2O, NaCl, effluent and pH, involving 12 trials and two levels of concentration, for the response in terms of the enzyme activity (IU mL−1) and specific activity (IU mg−1). Variation in these factors revealed the significance of media optimization in achieving higher L-asparaginase productivity and the influence of all the factors on enzyme activity which varied from 1.17 IU mL−1 to 3.63 IU mL−1 and the specific activity which varied from 0.48 IU mg−1 to 1.68 IU mg−1 (Table 2).
| Std | A – glucose (g per 100 mL) | B – Na2HPO4 (g per 100 mL) | C – KH2PO4 (g per 100 mL) | D – MgSO4 (g per 100 mL) | E – NaCl (g per 100 mL) | F – effluent (mL per 100 mL) | G – pH | Enzyme activity | Enzyme activity | Specific activity | Specific activity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Actual values (IU mL−1) | Predicted values (IU mL−1) | Actual values (IU mg−1) | Predicted values (IU mg−1) | ||||||||
| 1 | 0.4 | 0.7 | 0.2 | 0.06 | 0.06 | 1.2 | 6.5 | 3.34 ± 0.47 | 3.30 ± 0.53 | 0.96 ± 0.03 | 1.05 ± 0.19 |
| 2 | 0.2 | 0.7 | 0.4 | 0.04 | 0.06 | 1.2 | 7.1 | 1.43 ± 0.42 | 1.46 ± 0.53 | 0.48 ± 0.01 | 0.49 ± 0.19 |
| 3 | 0.4 | 0.5 | 0.4 | 0.06 | 0.04 | 1.2 | 7.1 | 1.17 ± 0.02 | 1.46 ± 0.53 | 0.54 ± 0.01 | 0.49 ± 0.19 |
| 4 | 0.2 | 0.7 | 0.2 | 0.06 | 0.06 | 0.8 | 7.1 | 2.21 ± 0.18 | 2.57 ± 0.53 | 0.98 ± 0.06 | 1.14 ± 0.19 |
| 5 | 0.2 | 0.5 | 0.4 | 0.04 | 0.06 | 1.2 | 6.5 | 1.43 ± 0.18 | 2.17 ± 0.53 | 0.58 ± 0.06 | 0.76 ± 0.19 |
| 6 | 0.2 | 0.5 | 0.2 | 0.06 | 0.04 | 1.2 | 7.1 | 2.29 ± 0.93 | 2.57 ± 0.53 | 1.0 ± 0.07 | 0.78 ± 0.19 |
| 7 | 0.4 | 0.5 | 0.2 | 0.04 | 0.06 | 0.8 | 7.1 | 3.14 ± 0.97 | 2.57 ± 0.53 | 1.0 ± 0.05 | 1.14 ± 0.19 |
| 8 | 0.4 | 0.7 | 0.2 | 0.04 | 0.04 | 0.8 | 6.5 | 2.99 ± 0.48 | 3.29 ± 0.53 | 1.32 ± 0.02 | 1.41 ± 0.19 |
| 9 | 0.4 | 0.7 | 0.4 | 0.04 | 0.04 | 0.8 | 7.1 | 1.87 ± 0.50 | 1.46 ± 0.53 | 0.90 ± 0.01 | 0.85 ± 0.19 |
| 10 | 0.2 | 0.7 | 0.4 | 0.06 | 0.04 | 0.8 | 6.5 | 1.90 ± 0.11 | 2.17 ± 0.53 | 0.97 ± 0.05 | 1.12 ± 0.19 |
| 11 | 0.4 | 0.5 | 0.4 | 0.06 | 0.06 | 0.8 | 6.5 | 3.12 ± 0.51 | 2.17 ± 0.53 | 1.36 ± 0.08 | 1.12 ± 0.19 |
| 12 | 0.2 | 0.5 | 0.2 | 0.04 | 0.04 | 0.8 | 6.5 | 3.63 ± 0.18 | 3.29 ± 0.53 | 1.68 ± 0.07 | 1.41 ± 0.19 |
The order of significance among the variables and the most effective factors showing high level of significance were indicated in the Pareto chart (ESI Fig. D†). The positive and the negative influence of the factors on L-asparaginase specific activity are also depicted further, the reference line at 2.3 (t-value limit) shows the significance level of the factors and the effect of the variables extending beyond this line were found to be highly significant by the t-values. L-Asparaginase activity was influenced greatly by KH2PO4 and pH, while specific activity was greatly influenced by the factors: KH2PO4, effluent and pH. Thus these factors were selected for further optimization by the CCD whereas the other factors: glucose, Na2HPO4, MgSO4·7H2O and NaCl were used at their normal central values for the further studies.
ANOVA of the design (Table 3) also substantiated the findings that the three factors: KH2PO4, effluent concentration and pH were significant with the p-value < 0.05 (95% confidence) and the final equation was given with coefficient values for each of the three factors. The negative sign indicates a negative effect of the factor on the specific activity, while a positive coefficient indicates a positive influence in both enzyme and specific activity.
| Analysis of variance (ANOVA) table (specific activity) | Analysis of variance (ANOVA) table (enzyme activity) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Source | Sum of squares | Df | Mean square | F value | p-value Prob > F | Confidence level (%) | Sum of squares | Df | Mean square | F value | p-value Prob > F | Confidence level (%) |
| a Factor not significant for this response. | ||||||||||||
| Model | 1.06 | 3 | 0.35 | 9.38 | 0.005 | 99.5 | 5.28 | 2 | 2.64 | 9.39 | 0.006 | 99.4 |
| C – KH2PO4 | 0.24 | 1 | 0.24 | 6.85 | 0.031 | 96.9 | 3.74 | 1 | 3.74 | 13.30 | 0.005 | 99.5 |
| F – effluent | 0.37 | 1 | 0.21 | 10.74 | 0.011 | 98.9 | —a | —a | —a | —a | —a | —a |
| G – pH | 0.21 | 1 | 0.21 | 5.75 | 0.043 | 95.7 | 1.54 | 1 | 1.54 | 5.47 | 0.044 | 95.6 |
The Model F-value of 9.38 and 9.39 (Table 3) declares the model to be significant, and only a 0.54% chance that a “Model F-Value” so large could be due to noise. Values of “Prob > F” less than 0.0500 indicate the model terms are significant. In this case, C, F, G are significant model terms and values greater than 0.1000 indicate the model terms are not significant. As per the suggestions from earlier reports, confidence levels, (which is the P value expressed in percent) higher than 70% are acceptable, likewise in this experiment, variables with confidence levels greater than 90% were considered as significant.33,34
Final equations in terms of actual factors:
| Enzyme activity (IU mL−1) = 12.17 − 5.58 × C − 1.19 × G | (3) |
| Specific activity (IU mg−1) = 5.32 − 1.45 × C − 0.91 × F − 0.44 × G | (4) |
Therefore, these three factors were used for the next optimization experiment using CCD, while the factors identified as insignificant were used at their normal central values.
Central composite design: response surface methodology
Based on the results obtained from the PBD, three factors: KH2PO4, effluent and pH were identified as critical components and CCD was performed using Design Expert 9.0 (Table 4) in order to analyse the combined effect of nutrients and to obtain a quadratic equation in terms of these factors while other factors were added as in the un-optimized medium. Twenty runs were performed with six replicates at the central value of the three factors: KH2PO4, effluent and pH (0.3 g per 100 mL, 1 mL per 100 mL, 6.8), respectively in the design, while for all other runs duplicates were performed, and the mean value of response for each run in terms of enzyme activity and specific activity obtained after 48 h incubation at 37 °C, was reported. The CCD result with a higher specific activity (3.31 ± 0.47 IU mL−1) of L-asparaginase in the 1st run consisted of KH2PO4 (0.2 g mL−1), effluent (0.8 mL) and pH (6.5) (Table 4). The data obtained from the CCD was fitted with a second order polynomial equation in order to elaborate the interaction of nutrients for L-asparaginase production by the following equations.| Enzyme activity (IU mL−1) = −137.27 − (18.43 × A) − (56.52 × B) + (51.68 × C) + (9.57 × A × B) + (1.66 × A × C) + (10.45 × B × C) − (3.89 × A2) − 10.14 × B2 − 4.67 × C2 | (5) |
| Specific activity (IU mg−1) = −51.92 − (13.31 × A) − (52.35 × B) + (25.12 × C) + (2.66 × A × B) + (2.10 × A × C) + (6.97 × B × C) − (6.978 × A2) + (0.62 × B2) − (2.46 × C2) | (6) |
| Std | Factor 1 | Factor 2 | Factor 3 | Observed response | Predicted response | Observed response | Predicted response |
|---|---|---|---|---|---|---|---|
| A: KH2PO4 g per 100 mL | B: effluent mL | C: pH | Enzyme activity IU mL−1 | Enzyme activity IU mL−1 | Specific activity IU mg−1 | Specific activity IU mg−1 | |
| 1 | 0.2 | 0.8 | 6.5 | 3.31 ± 0.47 | 3.65 ± 0.51 | 2.54 ± 0.09 | 2.56 ± 0.19 |
| 2 | 0.4 | 0.8 | 6.5 | 2.84 ± 0.22 | 3.17 ± 0.51 | 1.98 ± 0.15 | 2.23 ± 0.19 |
| 3 | 0.2 | 1.2 | 6.5 | 0.62 ± 0.05 | 0.85 ± 0.51 | 0.34 ± 0.03 | 0.45 ± 0.19 |
| 4 | 0.4 | 1.2 | 6.5 | 0.83 ± 0.26 | 1.14 ± 0.51 | 0.36 ± 0.09 | 0.33 ± 0.19 |
| 5 | 0.2 | 0.8 | 7.1 | 1.69 ± 0.35 | 1.73 ± 0.51 | 1.06 ± 0.11 | 1.19 ± 0.19 |
| 6 | 0.4 | 0.8 | 7.1 | 1.34 ± 0.17 | 1.46 ± 0.51 | 1.12 ± 0.11 | 1.11 ± 0.19 |
| 7 | 0.2 | 1.2 | 7.1 | 1.43 ± 0.06 | 1.44 ± 0.51 | 0.90 ± 0.13 | 0.75 ± 0.19 |
| 8 | 0.4 | 1.2 | 7.1 | 1.92 ± 0.33 | 1.93 ± 0.51 | 0.81 ± 0.14 | 0.89 ± 0.19 |
| 9 | 0.13 | 1 | 6.8 | 2.87 ± 0.37 | 2.67 ± 0.51 | 1.36 ± 0.09 | 1.34 ± 0.19 |
| 10 | 0.47 | 1 | 6.8 | 2.98 ± 0.32 | 2.69 ± 0.51 | 1.30 ± 0.04 | 1.17 ± 0.19 |
| 11 | 0.3 | 0.66 | 6.8 | 2.94 ± 0.35 | 2.62 ± 0.51 | 2.68 ± 0.06 | 2.5 ± 0.19 |
| 12 | 0.3 | 1.33 | 6.8 | 0.84 ± 0.49 | 0.67 ± 0.51 | 0.51 ± 0.21 | 0.55 ± 0.19 |
| 13 | 0.3 | 1 | 6.2 | 2.63 ± 1.20 | 2.08 ± 0.51 | 1.34 ± 0.12 | 1.18 ± 0.19 |
| 14 | 0.3 | 1 | 7.3 | 1.06 ± 0.12 | 1.13 ± 0.51 | 0.46 ± 0.07 | 0.48 ± 0.19 |
| 15 | 0.3 | 1 | 6.8 | 2.91 ± 2.91 | 2.79 ± 0.51 | 1.52 ± 0.08 | 1.45 ± 0.19 |
| 16 | 0.3 | 1 | 6.8 | 2.95 ± 0.35 | 2.79 ± 0.51 | 1.24 ± 0.04 | 1.45 ± 0.19 |
| 17 | 0.3 | 1 | 6.8 | 3.72 ± 0.21 | 2.79 ± 0.51 | 1.54 ± 0.15 | 1.45 ± 0.19 |
| 18 | 0.3 | 1 | 6.8 | 2.51 ± 0.28 | 2.79 ± 0.51 | 1.48 ± 0.12 | 1.45 ± 0.19 |
| 19 | 0.3 | 1 | 6.8 | 2.45 ± 0.07 | 2.79 ± 0.51 | 1.44 ± 0.15 | 1.45 ± 0.19 |
| 20 | 0.3 | 1 | 6.8 | 2.10 ± 0.31 | 2.79 ± 0.51 | 1.50 ± 0.06 | 1.45 ± 0.19 |
The observed values of enzyme activity and specific activity of L-asparaginase were close to those predicted by the design model. Thus these factors were effective in achieving high production of the enzyme. Similar studies of interaction between 3 g L−1 of KH2PO4 and 0.5 g L−1 of MgSO4·7H2O in M-9 medium had shown maximum L-asparaginase activity by Bacillus licheniformis RAM-8.8 Also, optimization for large scale production of L-asparaginase with a carbon and nitrogen source along with a little amount of mineral nutrients to achieve maximum production has also been reported.11
The significance of the model was evaluated based on Fisher’s F-test value as given by ANOVA (Table 5). Here, the Model F-value of 32.63 showed the model is significant, with the small chance (0.01%) that this large “Model F-Value” is due to noise. In this case B, C, BC and C2 were significant model terms for the response specific activity and for enzyme activity, C and BC were significant as their “Prob > F” was less than 0.0500. The goodness of fit of the model, given by the R squared value 0.967, (closer to 1 for specific activity) and 0.84 (for enzyme activity) indicated the reliability of the model and hence the design is effectively conveyed in a quadratic model. Moreover, the R squared value is comparable to the values previously reported.11
| Source | Analysis of variance (ANOVA) table (specific activity) | Analysis of variance (ANOVA) table (enzyme activity) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sum of squares | Df | Mean square | F value | p-value Prob > F | Sum of squares | Df | Mean square | F value | p-value Prob > F | |||
| Model | 7.46 | 9 | 0.83 | 32.63 | <0.0001 | Significant | 13.62 | 9 | 1.51 | 5.92 | 0.005 | Significant |
| A – KH2PO4 | 0.03 | 1 | 0.03 | 1.34 | 0.2741 | 0.001 | 1 | 0.001 | 0.002 | 0.96 | ||
| B – effluent | 4.62 | 1 | 4.62 | 181.70 | <0.0001 | 4.59 | 1 | 4.58 | 17.95 | 0.001 | ||
| C – pH | 0.58 | 1 | 0.58 | 22.82 | 0.0007 | 1.08 | 1 | 1.08 | 4.24 | 0.06 | ||
| AB | 0.023 | 1 | 0.02 | 0.89 | 0.3670 | 0.29 | 1 | 0.29 | 1.14 | 0.31 | ||
| AC | 0.03 | 1 | 0.03 | 1.26 | 0.2879 | 0.02 | 1 | 0.019 | 0.07 | 0.78 | ||
| BC | 1.40 | 1 | 1.40 | 55.08 | <0.0001 | 3.14 | 1 | 3.14 | 12.30 | 0.005 | ||
| A2 | 0.07 | 1 | 0.07 | 2.76 | 0.1275 | 0.02 | 1 | 0.021 | 0.08 | 0.77 | ||
| B2 | — | 1 | — | 0.35 | 0.5654 | 2.37 | 1 | 2.3716 | 9.28 | 0.01 | ||
| C2 | 0.17 | 1 | 0.70 | 27.73 | 0.0004 | 2.55 | 1 | 2.55 | 9.98 | 0.01 | ||
| Lack of fit | 0.05 | 5 | 0.01 | 3.33 | 0.11 | Not significant | 0.98 | 5 | 0.19 | 0.62 | 0.69 | Not significant |
The fitted responses for the analyzed regression model were plotted as a three dimensional graph with the pair wise combination of the three factors for L-asparaginase production (Fig. 1). The contour plot coupled with three dimensional surface plots explains the interaction of the parameters in terms of the selected responses. In Fig. 1a, the effect of KH2PO4 and effluent on enzyme activity and specific activity is seen. It can be inferred that KH2PO4 has very little effect on both enzyme activity and specific activity when the concentration is varied, however among the different concentrations of effluent used, from 1.2 (% v/v) to 0.66 (% v/v) the maximum activity was found at 0.8 (% v/v) of effluent. Similarly, from Fig. 1b and c, it can be seen that higher enzyme activity and specific activity is observed at acidic pH (6.5) with the interaction of KH2PO4 at 0.2 g (% w/v) and effluent 0.8 (% v/v).
 | ||
| Fig. 1 Three dimensional response surface curves for L-asparaginase production in terms of enzyme units per mL (IU mL−1) and specific activity (IU mg−1), showing interactions between the media components of M-9 medium. (a) KH2PO4 and effluent concentration when pH was kept constant, (b) KH2PO4 and pH when effluent concentration was kept constant, and (c) effluent concentration and pH when KH2PO4 concentration was constant. | ||
Verification of the model
The verification of the model was performed to validate the above model for the reproducibility of the results. The significant increase in the enzyme activity (as the response) obtained from the media components of standard order 1 of the CCD design consisted of KH2PO4 (0.2 g% w/v), effluent (0.8 mL% v/v) and pH (6.5). Hence the run with the above components at optimum values would be considered as the optimized media and all other runs of the design that had not shown significant increase in the response were neglected for further verification. The enzyme activity (3.25 ± 0.11 IU mL−1) and specific activity (2.77 ± 0.06 IU mg−1) obtained after optimization were consistent and very close to the predicted model (3.65 ± 0.51 IU mL−1 and 2.56 ± 0.19 IU mg−1) with a 4.5 fold increase in enzyme activity and 4.3 fold increase in specific activity compared to the M-9 medium with 0.3% L-asparagine (enzyme activity 0.73 ± 0.08 IU mL−1 and specific activity 0.64 ± 0.03 IU mg−1).Purification and characterization of enzyme
The purification process of L-asparaginase sometimes may lead to loss in its activity and makes it worthless in commercial applications. Hence, L-asparaginase activity was continuously analysed in all the purification steps while all purification steps were carried out at 4 °C and the column chromatographic runs were analyzed for protein content at 280 nm, while the enzyme activity was estimated by direct nesslerization. Ammonium sulphate precipitation (80%) had retained the enzyme activity and after the dialysis against Tris–HCl buffer at pH 8.6 the activity enzyme was also found to be stable. The enzyme was further purified to homogeneity by DEAE Sepharose chromatography, which was verified by both native PAGE and SDS PAGE analysis (Fig. 2). The fold purification of L-asparaginase was 4.36 with 19% recovery and the summary of fold purification achieved is given in Table 6.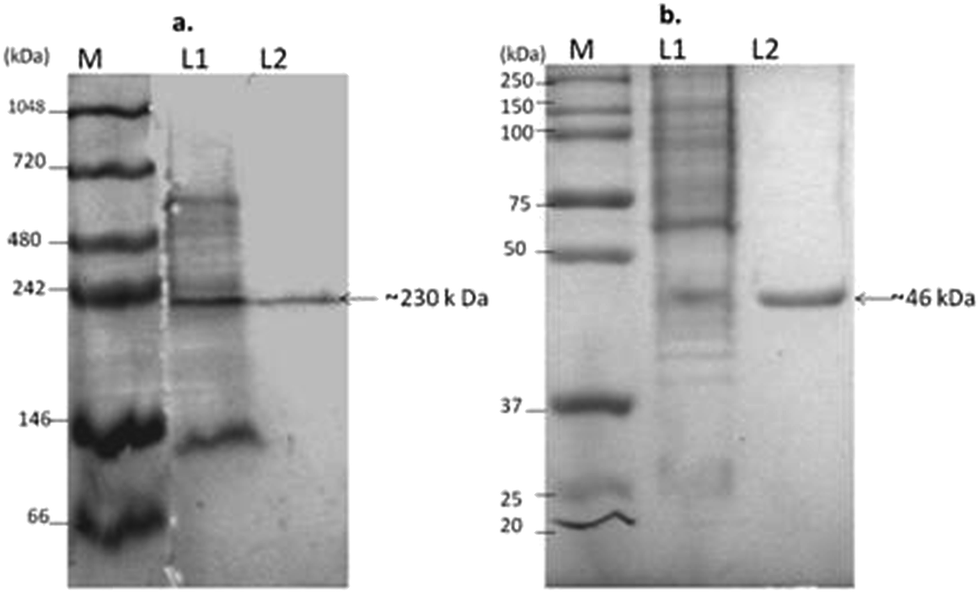 | ||
| Fig. 2 Electrophoretic analysis of L-asparaginase produced by Pseudomonas plecoglossicida RS1. (a) Native PAGE on 7.5% resolving gel, and (b) SDS PAGE on 12% resolving gel. M – marker, L1 – crude enzyme, L2 – purified enzyme. | ||
| Enzyme fraction | Vol (mL) | Total units (IU) | Total protein (mg) | Specific activity (IU mg−1) | Fold purification | % yield |
|---|---|---|---|---|---|---|
| Crude enzyme | 100 | 211.30 | 97.90 | 2.16 ± 0.03 | — | 100 |
| Ammonium sulphate ppt. (80%) | 18 | 55.01 | 16.00 | 3.44 ± 0.30 | 1.59 | 26 |
| DEAE sepharose | 6 | 41.13 | 4.32 | 9.43 ± 0.40 | 4.36 | 19 |
The purity and approximate size of L-asparaginase from DEAE Sepharose was assessed by native PAGE, revealing that the single distinct band of the protein at ∼230 kDa was compared with multiple bands in the crude fraction. Also SDS PAGE analysis showed the size of the subunit of the protein as a single band with a molecular weight of ∼46 kDa, which showed evidence that L-asparaginase is a homo-pentamer. The L-asparaginase of the Gram negative bacteria Serratia sp. exists as pentamer or hexamer with an average molecular weight of 170 to 180 kDa as reported by Stern et al., (1976).35 Whereas the L-asparaginase was found to be a homotetramer with identical sub units (38000 Da) in E. coli.36 The molecular size and subunits of L-asparaginase in Pseudomonas vary from a single subunit of 34 kDa to 33 kDa under non-denaturing and denaturing conditions respectively.14 L-Asparaginase also exists as a monomer with a size of 160 kDa in Pseudomonas as reported by El-Bessoumy et al., (2004).9 Thus, L-asparaginase shows a wide structural variation in the subunits of the above mentioned bacteria.
Effect of pH, temperature and metal ions on activity and stability of purified L-asparaginase
Characterization of the purified enzyme was performed using different effectors like pH, temperature, and metal ions. The maximum enzyme activity (6.45 ± 0.87 IU mL−1) was observed at pH 8.5 whereas the enzyme activity decreased in acidic conditions. The stability of the enzyme (residual activity) when stored at 4 °C for 24 h with buffers of different pH (4–9.5), was maximum (100%) at 8.5 and decreased significantly in the acidic range, whereas it retained up to 80% activity at pH 9.5 (Fig. 3a). It was also already reported that the L-asparaginase activity was stable at pH 7.5 to 9 in Pectobacterium carotovorum MTCC1428,26 Pseudomonas aeruginosa 500719 and Pseudomonas aeruginosa 10145.34 Likewise the optimum temperature for enzyme activity (10.74 ± 0.08 IU mL−1) was found at 55 °C for Pseudomonas plecoglossicida RS1 while at a temperature beyond 15 °C or 55 °C the enzyme activity decreased to 50% (Fig. 3b). This is a noteworthy feature to compare with the previous reports of stability of L-asparaginases beyond 45 °C or 60 °C, where 75% of enzyme activity was lost.26 The impact of various effectors on the enzyme were also demonstrated using different effectors like: Na+, K+, Mg2+, Ca2+, Mn2+, Zn2+, Hg2+, Fe3+, EDTA, L-cystine, L-histidine, mercaptoethanol and SDS. Of the various effectors Na+ and mercaptoethanol slightly enhanced enzyme activity where as Hg2+, Ca2+ and Zn2+ had shown a highly detrimental effect on activity compared to the control and other effectors (Fig. 3c) and this effect could be due to the presence of sulfhydryl group(s) in the enzyme substrate complex. These results were corroborated with Erwinia carotovora L-asparaginase activity in the presence of thiol compounds as reported by Warangkar and Khobragade (2010).26 Furthermore, EDTA did not result in a large inhibition, indicating that the enzyme may not be a metalloprotein as suggested by Kumar et al., (2011).27
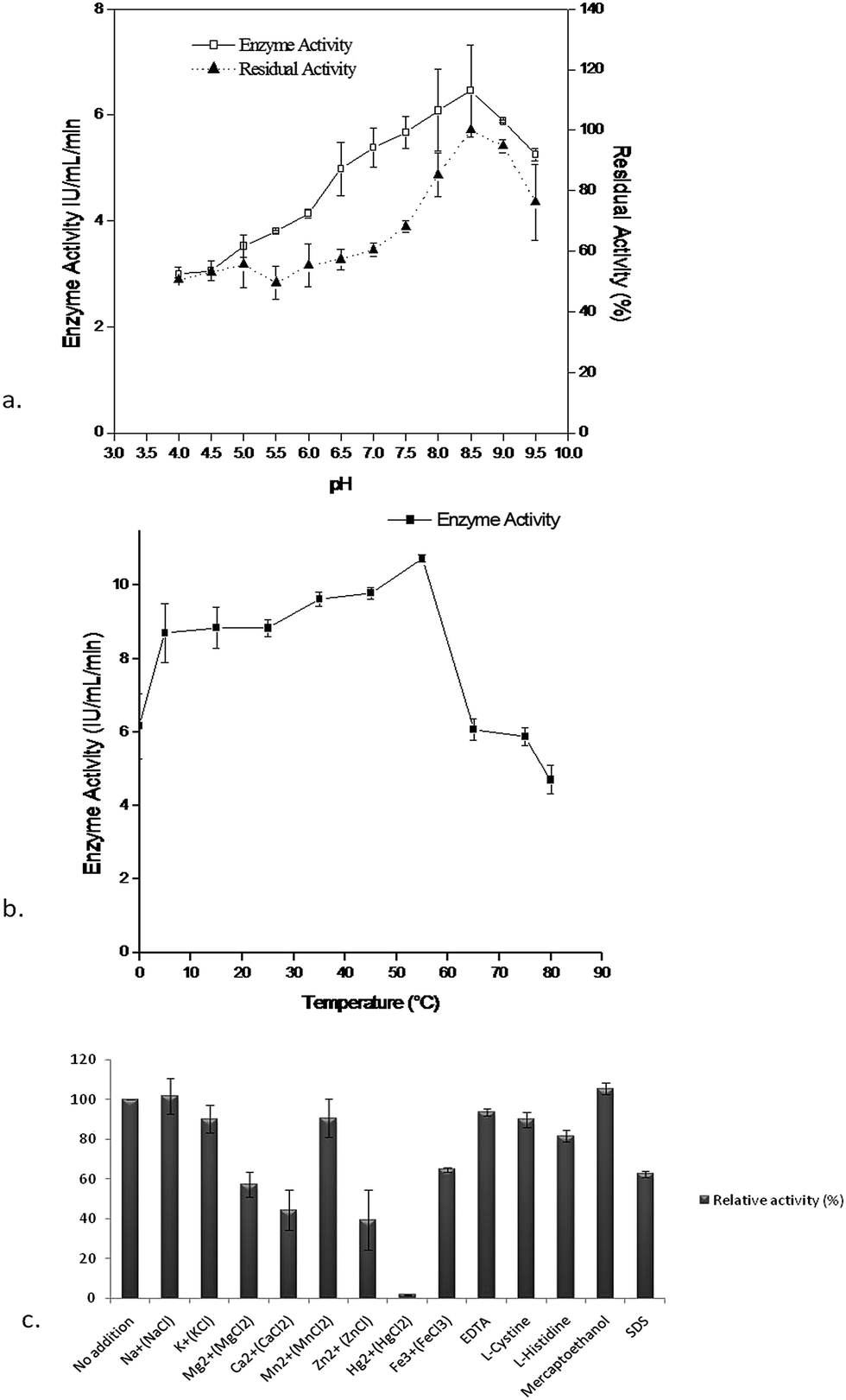 | ||
| Fig. 3 (a) Effect of pH of assay buffer at 37 °C on enzyme activity and stability of purified L-asparaginase when stored at 4 °C for 24 h at different pH 3.5 to 9.5. (b) Effect of temperature on L-asparaginase activity at different temperature from 0 to 80 °C after 30 min incubation. (c) Influence of various effectors on the activity of purified L-asparaginase. | ||
Kinetic parameters of L-asparaginase
In order to find the affinity between the enzyme and the substrate, determination of Km and Vmax values are important. Obviously, L-asparaginases from different microorganisms may also differ in their substrate affinities and that would execute different physiological functions in enzyme activity. The Km of the purified L-asparaginase produced by Pseudomonas plecoglossicida RS1 was 2.25 ± 0.61 mM and its Vmax was 8.95 ± 0.8 IU mL−1 min−1 (Fig. 4). Km values for L-asparaginase produced by C. glutamicum and E. coli (2.5 mM and 3.5 mM) were reported by Willis and Woolfolk, (1974)37 while the lower values of Km (0.074 mM) by Vibrio succinogenes and 0.657 mM by Pectobacterium carotovorum MTCC 1428 were also reported previously by Kumar et al., (2011).27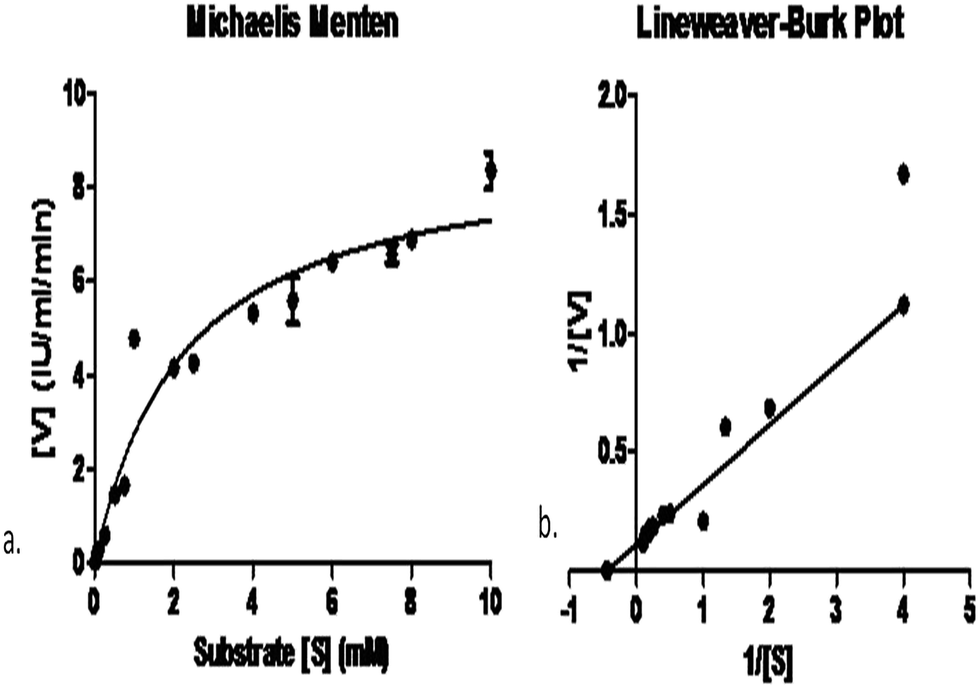 | ||
| Fig. 4 (a) Michaelis–Menten plot of reaction velocities (V) given by enzyme activity per minute per mL of the enzyme vs. substrate conc. (S) given by mM. Parameters Vmax 8.95 IU mL−1 min−1 and Km 2.25 mM were calculated by non-linear regression analysis. (b) Corresponding Lineweaver–Burk plot. | ||
Conclusion
This study demonstrated glutaminase free L-asparaginase production by Pseudomonas plecoglossicida RS1 in M-9 medium provided with sugar cane industry effluent as an alternate substrate to L-asparagine. The media optimization studies using PBD and CCD also enhanced production and the enzyme was further purified to homogeneity with a molecular size of ∼230 kDa on native PAGE and a subunit size of ∼46 kDa on SDS PAGE. The enzyme characterization studies exhibited effective activity and stability to a wide range of pH, temperature and effectors. Conclusively, glutaminase free L-asparaginase can be produced using effluent as an alternate substrate for cost effectiveness and its therapeutic applications. The therapeutic application of the enzyme produced can further be evaluated by testing on T-cell cancer cell lines and by animal model studies.Author contributions
PV and BA designed the study, analysed the data and wrote the manuscript. GS and SMB performed the experiments, analysed the data and wrote the manuscript. PP was involved in the purification and characterization experiments. All the authors have contributed to writing the manuscript and have approved the final manuscript.Acknowledgements
Authors thank the DST-SERB-SB/EMEQ-128/2013 for the financial support to PV; CSIR-UGC (no. 17-06/2012(i) EU-V) fellowship to GS and DST-PURSE program for instrumentation facility.Notes and references
- J. D. Broome, J. Exp. Med., 1968, 127, 1055 CrossRef CAS.
- E. A. Boyse, L. J. Old, H. A. Campbell and L. T. Mashburn, J. Exp. Med., 1966, 125, 17 CrossRef.
- L. T. Mashburn and J. C. Wriston, Arch. Biochem. Biophys., 1964, 105, 450 CrossRef CAS.
- Y. Ghasemi, A. Ebrahiminezhad, S. R. Amini, G. Zarrini, M. B. Ghoshoon, M. J. Raee, M. H. Morowvat, F. Kafilzadeh and A. Kazemi, Am. J. Biochem. Biotechnol., 2008, 4, 422 CrossRef CAS.
- T. Tosa, R. Sano, K. Yamamoto and M. Nakamura, Am. Soc. Microbiol., 1971, 387 CAS.
- C. K. Venil, K. Nanthakumar, K. Karthikeyan and P. Lakshmanaperumalsamy, Iran. J. Biotechnol., 2009, 7, 1 Search PubMed.
- S. Ghosh, S. Murthy, S. Govindasamy and M. Chandrasekaran, Sustainable Chem. Processes, 2013, 1, 9 CrossRef.
- R. V. Mahajan, S. Saran, K. Kameswaran, V. Kumar and R. K. Saxena, Bioresour. Technol., 2012, 125, 11 CrossRef CAS PubMed.
- A. A. El-Bessoumy, M. Sarhan and J. Mansour, J. Biochem. Mol. Biol., 2004, 37, 387 CrossRef CAS.
- S. Amena, N. Vishalakshi, M. Prabhakar, A. Dayanand and K. Lingappa, Braz. J. Microbiol., 2010, 41, 173 CrossRef CAS PubMed.
- R. Usha, K. K. Mala, C. K. Venil and M. Palaniswamy, Pol. J. Microbiol., 2011, 60, 213 CAS.
- B. Gurunathan and R. Sahadevan, J. Microbiol. Biotechnol., 2012, 22, 923 CrossRef CAS.
- J. B. Howard and F. H. Carpenter, J. Biol. Chem., 1972, 247, 1020 CAS.
- S. Manna, A. Sinha, R. Sadhukhan and S. L. Chakrabarty, Curr. Microbiol., 1995, 3, 291–298 CrossRef.
- G. Ollenschläger, E. Roth, W. Linkesch, S. Jansen, A. Simmel and B. Mödder, Eur. J. Clin. Invest., 1988, 18, 512 CrossRef PubMed.
- L. N. Ramya, M. Doble, V. P. B. Rekha and K. K. Pulicherla, Appl. Biochem. Biotechnol., 2012, 167, 2144 CrossRef CAS PubMed.
- U. K. Narta, S. S. Kanwar and W. Azmi, Crit. Rev. Oncol. Hematol., 2007, 61, 208 CrossRef PubMed.
- S. Kumar, K. Pakshirajan and V. Venkata Dasu, Process Biochem., 2010, 45, 223 CrossRef CAS PubMed.
- S. R. Rao, R. S. Prakasham, K. Krishna Prasad, S. Rajesham, P. N. Sharma and V. Rao, Process Biochem., 2004, 39, 951 CrossRef.
- R. Karpagam, K. J. Raj, B. Ashokkumar and P. Varalakshmi, Bioresour. Technol., 2015, 188, 177 CrossRef CAS PubMed.
- R. S. Prakasham, S. Rao, S. R. Rao and P. N. Sarma, Biotechnol. Prog., 2005, 21, 1380 CrossRef CAS PubMed.
- R. Gulati, R. K. Saxena and R. Gupta, Lett. Appl. Microbiol., 1997, 23, 6 Search PubMed.
- J. B. Sumner, J. Biol. Chem., 1921, 47, 5 Search PubMed.
- R. L. Plackett and J. P. Burman, Biometrika, 1946, 33, 305 CrossRef PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680 CrossRef CAS PubMed.
- S. C. Warangkar and C. N. Khobragade, Enzyme Res., 2010, 165878 Search PubMed.
- S. Kumar, V. Venkata Dasu and K. Pakshirajan, Appl. Biochem. Biotechnol., 2011, 163, 327 CrossRef CAS PubMed.
- D. Pant, A. Singh, G. Van Bogaert, S. I. Olsen, P. S. Nigam, L. Dielsa and K. Vanbroekhovena, RSC Adv., 2012, 2, 1248 RSC.
- E. A. Makky, Y. C. Loh and M. R. Karim, Biocatal. Agric. Biotechnol., 2014, 3, 265 Search PubMed.
- A. Mishra, Appl. Biochem. Biotechnol., 2006, 135, 33 CrossRef CAS.
- A. Salihu, M. Z. Alam, M. I. AbdulKarim and H. M. Salleh, Afr. J. Biotechnol., 2011, 10, 18704 CAS.
- S. Kumar, K. Pakshirajan and V. Venkata Dasu, Appl. Microbiol. Biotechnol., 2009, 4, 477 CrossRef PubMed.
- R. A. Stowe and R. P. Mayer, Ind. Eng. Chem., 1966, 58, 36 CrossRef CAS.
- Y. R. Abdel-Fattah and Z. A. Olama, Process Biochem., 2002, 38, 115 CrossRef CAS.
- M. L. Stern, A. W. Phillips and A. J. Gottlieb, J. Bacteriol., 1976, 125, 719 CAS.
- H. J. Muller and J. Boos, Crit. Rev. Oncol. Hematol., 1998, 28, 97 CrossRef CAS.
- R. C. Willis and C. A. Woolfolk, J. Bacteriol., 1974, 118, 231 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: phylogenetic tree, characterization of effluent, Paretos chart. See DOI: 10.1039/c5ra05507e |
| This journal is © The Royal Society of Chemistry 2015 |