
Chlorine-functionalized reduced graphene oxide for methylene blue removal†
Chubei Wang*,
Jianwei Zhou and
Liangliang Chu
Institute of Energy and Fuel, Xinxiang University, Xinxiang, Henan 453003, China. E-mail: wangchubei@163.com; Fax: +86-373-3682028; Tel: +86-373-3682028
First published on 9th June 2015
Abstract
Chlorine-functionalized reduced graphene oxide was prepared by using graphene oxide and sulfuryl chloride as raw materials. It is firstly reported that the hydroxyl group in graphene oxide can be substituted by chlorine in sulfuryl chloride. The product has a wider pore size and average interlayer space, and its surface area can be easily utilized. The adsorption data indicate that the material has high adsorption capacity on methylene blue. The adsorption process follows a second-order rate and Bangham diffusion kinetic model. The adsorption of methylene blue preferably fitting the Langmuir adsorption isotherm suggests monolayer coverage of the adsorbed molecules. The adsorption capacity of the product for methylene blue is 221.4 mg g−1 at room temperature, which is significantly higher than those of reduced graphene oxide (80.0 mg g−1) and active carbon (46.7 mg g−1).
1 Introduction
Graphene composites have potential applications in the wastewater treatment system. The synergistic effects of graphene nanosheets and modifications improve their properties.1,2 There are many non-covalent modifications.3,4 Pillared graphene was prepared by Külaots and Ye,5,6 Külaots found that pillaring was a more effective method than crumpling for increasing the surface area. However, non-covalent interactions (van der Waals, electrostatic or π–π stacking) are weak, leading to unstable systems.Graphene oxide (GO), derived from the oxidation of graphite, possesses abundant oxygenated functional groups, which offer reactive sites for covalent modification. The epoxide groups of GO undergo a nucleophilic substitution reaction with N-containing compounds to form composites.7–11 The carboxyl groups can react with amine, secondary amine and other group.12–18 The hydrogen of phenolic hydroxyl in GO can be substituted.19–22 Hydroxyl group also can be initiated to grow polymer brushes form GO and reduced graphene oxide (RGO).23–25 GO sheets also can be modified with siloxane coupling agent.26–31 GO was functionalized with amino acids by the reaction between carboxylic groups, epoxides and the amine groups in amino acids.32,33 The GO was treated with chloroacetic acid to convert the hydroxyl and epoxide groups to carboxyl groups.34,35 Toluene-2,4-diisocyanate was applied to create anchor site on GO, which can lead to the derivatization of carboxyl and hydroxyl functional groups.36 In all, the chemical properties of these groups in GO are like those in organic compounds.
In this work, GO was treated by sulfuryl chloride (SO2Cl2) and then it was reduced by thermal reduction, the product was the chlorine-functionalized reduced graphene oxide (ClRGO). The OH groups in GO behave like those of phenol in substitution reactions. As well known, phenolic hydroxyl group can not react with sulfuryl chloride. To the best of our knowledge, there has been little report about that OH group in GO is substituted by chlorine in sulfuryl chloride.
2 Experimental
2.1 Materials
Pristine graphite was purchased from Qingdao BCSM Co., Ltd., (Qingdao, China), and sulfuryl chloride (SO2Cl2) was supplied by Shanghai Chemical Reagent Company (Shanghai, China). All other chemicals were of analytical grade and were purchased from Beijing Chemical Reagents Company (Beijing, China). All chemicals were used without further purification.2.2 Synthesis of ClRGO and RGO
A mixture of GO (0.50 g) and acetonitrile (30 mL) was sonicated for 60 min. Water bath sonication was performed using a JL-60 DTH sonicator (100 W). Sulfuryl chloride (2.00 g, about 1.2 mL) was added to the GO solution at 75 °C under stirring for 2 h. The mixture was heated and refluxed for 5 h. The mixture was cooled down to ambient temperature, and then the mixture was poured in water (100 mL). The product was collected by filtration and washed several times with deionized water. The ClRGO was dried at 50 °C for 12 h to yield a black powder (0.31 g).GO (0.20 g) was dispersed in water (500 mL) with the aid of ultrasonication, and then hydrazine hydrate (80 wt%, 20 mL) was added at 100 °C for 24 h.37–39 The product was collected by filtration and washed several times with deionized water. The RGO was dried at 50 °C for 10 h to yield a loose black powder (0.12 g).
2.3 Characterization
Scanning electron microscopy (SEM) and energy-dispersive X-ray (EDX) spectroscopy were performed using Hitachi S-4800 field emission and FEI-Quanta 200 scanning electron microscopes, respectively. High-resolution TEM (HRTEM) was performed on a JEOL JEM-2011 electron microscope operated at 200 kV, equipped with an Oxford Link ISIS energy dispersive X-ray spectroscopy (EDX) system, and a Gatan 794 camera. Fourier-transform infrared (FTIR) spectra were obtained on an FTS-40 (Bio-Rad, CA, USA). Raman spectra were recorded on a Renishaw InVia multi-channel confocal microspectrometer with 532 nm excitation laser. X-ray diffraction (XRD) measurements were obtained on an X'pert PRO diffractometer using Co Kα radiation. X-ray photoelectron spectroscopy (XPS) with monochromatized Al Kα X-ray (hν = 1486.6 eV) radiation (Thermo Fisher Scientific Co., ESCALAB 250, USA) was used to investigate the surface properties of the ClRGO. The shift in binding energies was corrected using the C 1s signal at 284.6 eV as the internal standard. Nitrogen sorption measurements were performed with ASAP 2020 V3.01H (Micromeritics, USA). Specific surface areas, pore volumes, and pore size distributions were calculated using the Brunauer–Emmett–Teller (BET) and density functional theory (DFT) models from the adsorption branches. Prior to measurement, the samples were outgassed under vacuum at 250 °C for ca. 10 h until the pressure was less than 5 μm Hg.2.4 Adsorption
Batch adsorption experiments were performed using 500 mL glass bottles with a mixture of adsorbent (approximately 2 mg) and methylene blue (MB) solution (400 mL, 6 mg L−1 to 14 mg L−1). The glass bottles were sealed with Teflon. The temperature of the solution was controlled to the desired value with a variation of ±0.02 °C by adjusting the flow rate of thermostatically controlled water through an external glass-cooling spiral. The mixture was sampled in a series of intervals, and the concentration of the organic compound remaining in the mixture was measured after centrifugation. The concentration was measured by a UV/vis spectrometer. Values were calculated using the standard spectrophotometric method at the maximum absorbance of each compound.3 Results and discussion
3.1 Structural features of the ClRGO
The morphologies and microstructures of ClRGO are characterized by SEM and TEM in Fig. 1. The presence of wrinkles and folds on the sheet might be a characteristic feature of few-layered sheets in Fig. 1a and b. The atomic-scale roughness might result from the chlorine-containing functional groups and residual oxygenated functional groups attached over ClRGO sheets. Thus, ClRGO is expected to be porous. Some stacking pores are shown in Fig. 1c. The corresponding EDX spectrum revealed the presence of C, O and Cl, which imply that some OH groups in GO was substituted by chlorine. The atomic percentages of elements in ClRGO are in Table S1.† Compared with phenolic compounds, GO has extended conjugation. Charge is easy to disperse in a conjugated system, and then the intermediate is more stable. GO can be reduced by thermal reduction at about 100 °C, this fact also confirms that the OH group in GO can leave. The OH group in GO might leave, and might be substituted by chlorine. | ||
| Fig. 1 Images of ClRGO. Image (SEM) (a) and images (TEM) of ClRGO (b and c). | ||
FTIR spectra further show substitution and reduction of GO to ClRGO in Fig. 2a. ClRGO shows the skeletal vibration of unoxidized graphitic domains at 1580 cm−1, alkoxy C–O at 1092 cm−1 and C–Cl at 653 cm−1. The bond of carbon and chlorine forms in ClRGO. RGO shows the stretching vibrations of –OH groups at 3380 cm−1, skeletal vibration of unoxidized graphitic domains at 1580 cm−1, and alkoxy C–O at 1120 cm−1. The Raman spectra of ClRGO present a broadened G band at 1587 cm−1 in Fig. 2b, because of the formation of a conjugated system. The G band at about 1580 cm−1 is associated with the vibration of sp2 carbon atoms. The D band at about 1350 cm−1 related to the vibration of sp3 carbon atoms of defects and disorder. The intensity of the D band at 1346 cm−1 of ClRGO increases substantially, indicating the decrease in size of the in-plane sp2 domains, possibly due to the extensive substitution. There are more sp3 carbon atoms in the ClRGO than these in RGO. When the hydroxyl group is substituted by chlorine and GO is reduced, the intensity of the D band increases further. The GO has more sp3 carbon atoms than ClRGO, some sp3 carbon atoms in ClRGO become sp2 carbon atoms. This can also be due to defects introduced into the ClRGO during preparation.40,41 The ID/IG ratio of the ClRGO is approximately 1.34, which is higher than that of RGO (1.15). This result indicates that more graphene domains are formed on the ClRGO sheet when the oxygenated functional groups on sheet are removed.
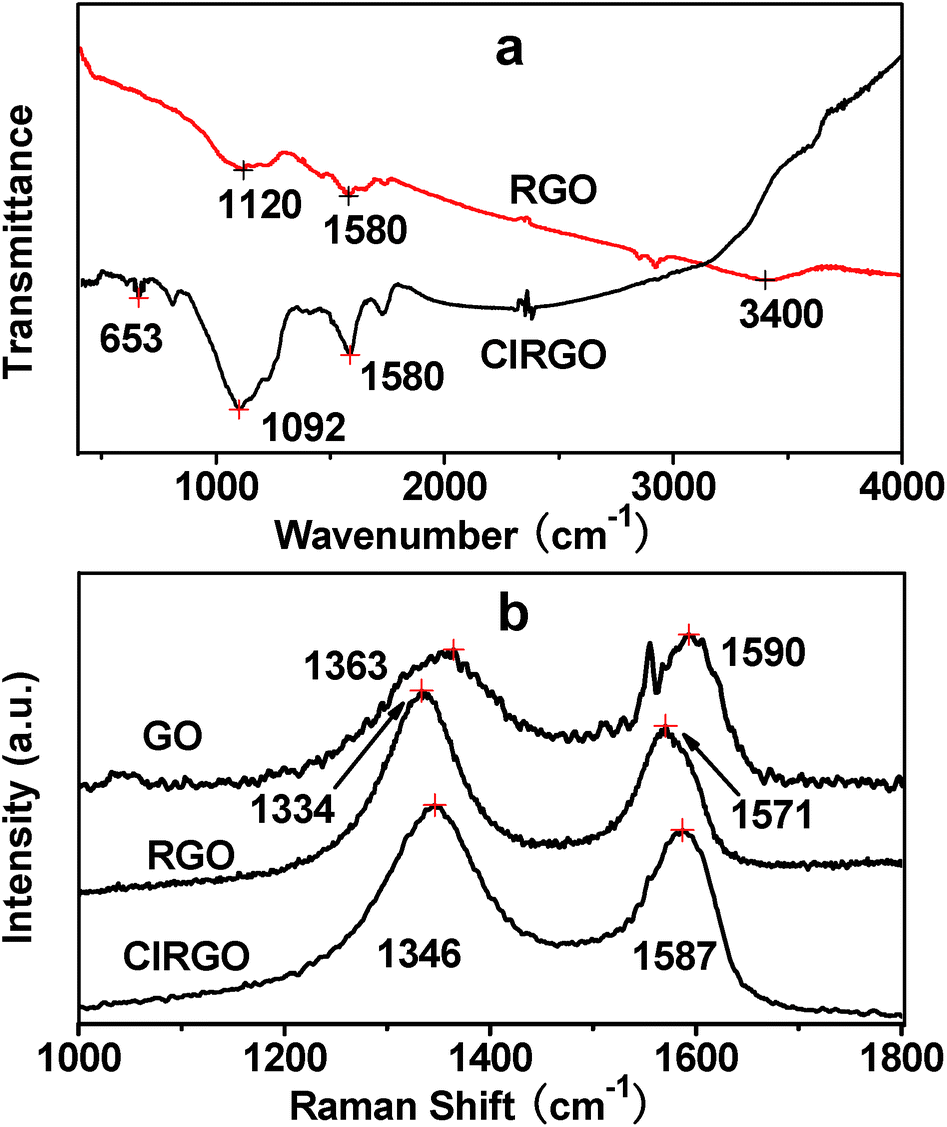 | ||
| Fig. 2 FTIR spectra of RGO and ClRGO (a), Raman spectra of GO, RGO and ClRGO (b). | ||
The XRD patterns of RGO and ClRGO are shown in Fig. 3a. ClRGO exhibits the intensity and broadness of the peak at 2θ = 24.8°, the average interlayer space of ClRGO is about 0.41 nm. The characteristic peak of RGO is at 27.6° (0.36 nm). Some OH group in graphene oxide is substituted by chlorine. The amount of OH group and hydrogen bond between sheets decrease, which weaken attractive force between sheets. Moreover, chlorine might also act as spacer and/or pillaring agent. The height of spacer or pillaring agent is about 0.33 nm (see ESI, Fig. S1†), which is about equal to the interlayer spacer of graphite. It might prevent graphene sheet close to each other, and then the interlayer space of ClRGO is widened.
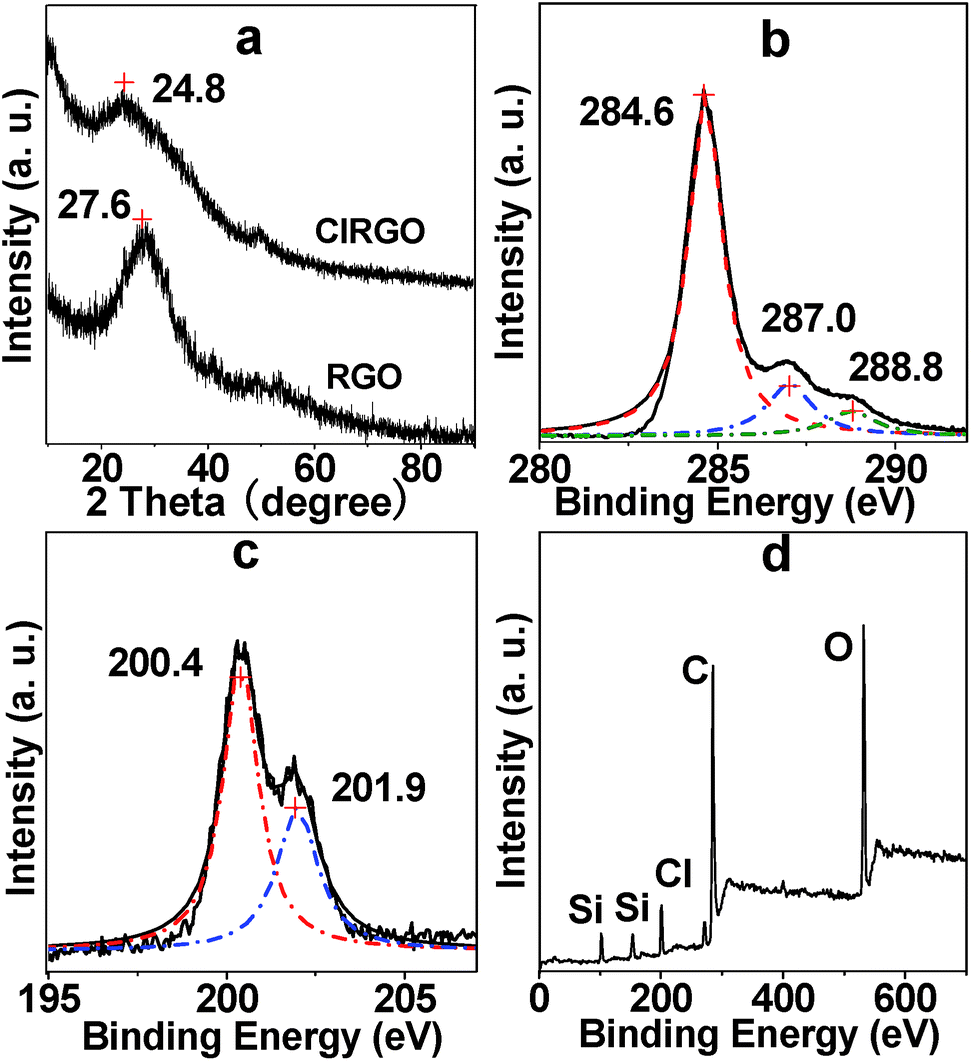 | ||
| Fig. 3 XRD patterns of ClRGO and RGO (a), core-level XPS spectra of C 1s (b) and Cl 2p (c) of ClRGO, XPS survey spectra (d) of ClRGO. | ||
The composites of the ClRGO were further analyzed by XPS. The core-level XPS signals of C 1s are shown in Fig. 3b. The peak centered at approximately 284.6 eV originates from the graphitic sp2 carbon atoms, whereas that located at 287.0 eV is caused by C–Cl and C–O. The peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in carboxylic acid is found at 288.8 eV. The peaks at the 200.4 and 201.9 eV are assigned to Cl 2p3/2 and Cl 2p1/2 in Fig. 3c. The XPS survey spectra of the ClRGO show the presence of C, O and Cl elements in Fig. 3d. The core-level XPS signals of C 1s are similar to those of RGO, which also indicate that ClRGO is fully reduced.
O in carboxylic acid is found at 288.8 eV. The peaks at the 200.4 and 201.9 eV are assigned to Cl 2p3/2 and Cl 2p1/2 in Fig. 3c. The XPS survey spectra of the ClRGO show the presence of C, O and Cl elements in Fig. 3d. The core-level XPS signals of C 1s are similar to those of RGO, which also indicate that ClRGO is fully reduced.
The porosity of the ClRGO was also confirmed by nitrogen sorption. Nitrogen adsorption–desorption isotherm exhibits a characteristic IUPAC type IV curve in Fig. 4a. The surface areas of ClRGO, RGO, and commercial activated carbon (AC) are 341.7, 319.9, and 420.4 m2 g−1, respectively, as obtained by fitting the isotherm to the BET model. Some pores and slits with small width in RGO type of materials are inaccessible to probe molecules used in surface area analysis (e.g. N2, CO2).5 RGO has little spacers and/or pillaring agents, some surface area can not be analyze, while ClRGO has spacers and/or pillaring agents, more surface area can be analyzed. The porosities of ClRGO, RGO, and AC can be further confirmed by porosity distribution analysis based on the original DFT in Fig. 4b. The incremental pore volume of ClRGO is higher than those of RGO and AC within the ranges of 15 nm to 50 nm and 50 nm to 120 nm, respectively. The ClRGO might have relatively more macropores and mesopores. Moreover, the mesopores of the ClRGO have wider width than those of the RGO. RGO sheets are smooth and can close to each other. RGO can substantially form micropores but only a few mesopores (Fig. 4b). ClRGO sheets with chlorine might be rough as shown in Fig. 1. Roughness, wrinkles, folds, and twists act as spacer and/or pillaring agent. Compared with RGO, ClRGO has more spacers and/or pillaring agents on ClRGO sheets, more mesopores with wide pore size and more macropores are formed between ClRGO sheets. The interlayer space of ClRGO is wider, which also improve the pore size. The stacked pore size of ClRGO is obviously improved, which might enhance adsorption capacity.
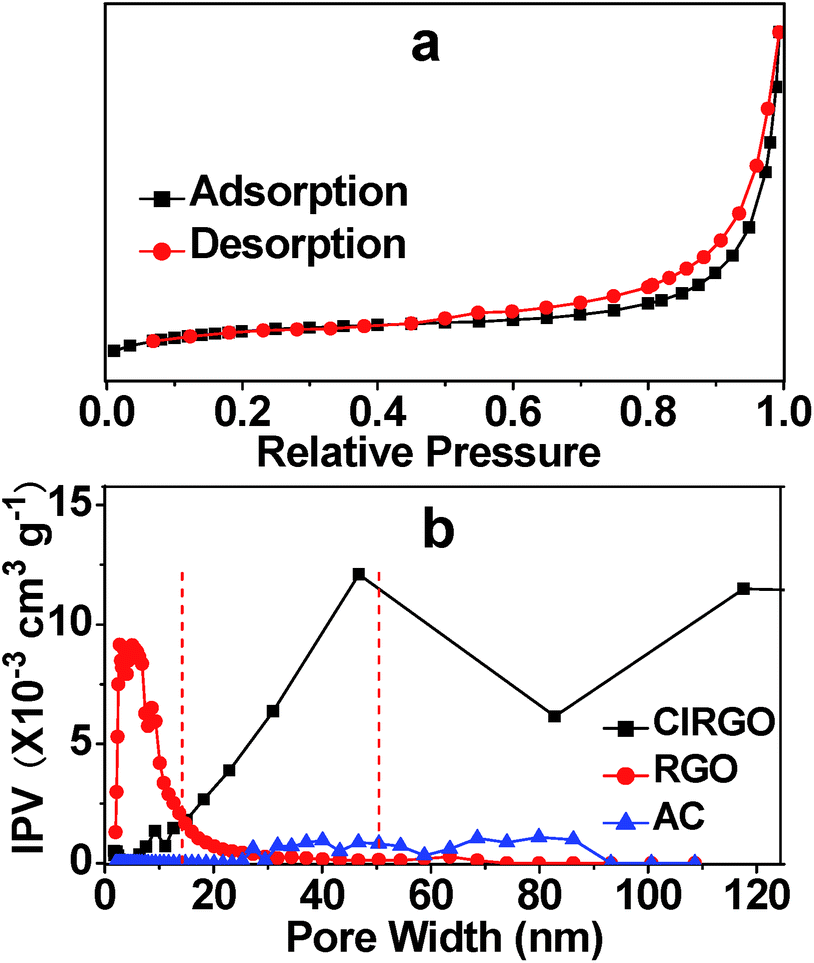 | ||
| Fig. 4 Typical nitrogen adsorption and desorption of ClRGO (a) and pore size distribution curves of ClRGO, RGO, and AC (b). IPV = incremental pore volume. | ||
3.2 MB adsorption and adsorption mechanism
In the present investigation, pseudo-first-order, pseudo-second-order, intraparticle diffusion and Bangham diffusion kinetic models were tested to obtain the rate constants, equilibrium adsorption capacity, and adsorption mechanism.42,43The adsorption curves of MB with ClRGO, RGO and AC are shown in Fig. 5a. The removal of organic compound attains equilibrium after approximately 70 h. For the ClRGO, the adsorption capacity for MB is 221.4 mg g−1, which is considerably larger than that of RGO (80.0 mg g−1) and AC (46.7 mg g−1). Although RGO and ClRGO have similar surface areas, ClRGO has a large adsorption capacity. This phenomenon may be due to the fact that pore blockage may occur for adsorbents with small pore diameters because of the aggregation of bulky molecules in the pore orifice.44,45 Therefore, the full surface area of RGO cannot be utilized, reducing the effectiveness of adsorption. ClRGO has more mesopores and macropores, and wider pore size than RGO. The results show that a large surface area is not vital for high adsorption capacities as is commonly believed; instead, large pore sizes are principally important for effective dye uptake, which is consistent with previous reports.19,21
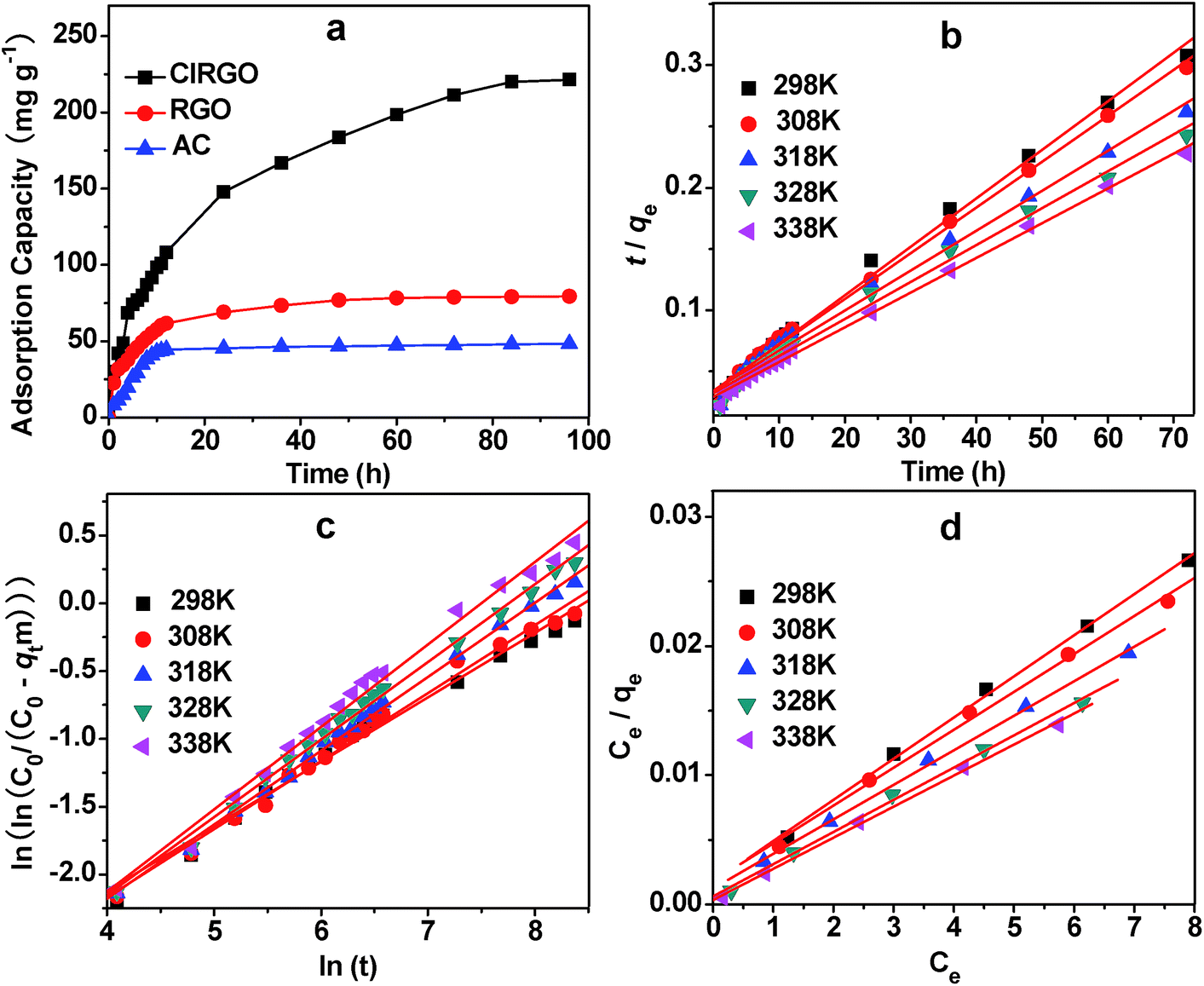 | ||
| Fig. 5 Adsorption of MB by ClRGO, RGO, and AC (a). Conditions: concentration = 8 mg L−1 and room temperature. Adsorption kinetics of MB by ClRGO for pseudo-second order (b). Conditions: concentration = 8 mg L−1. Bangham's diffusion plots for the removal of MB at different temperature (c). Concentration = 8 mg L−1. Langmuir plot (Ce/qe vs. Ce) for MB adsorption by ClRGO at different temperatures (d). Concentration: concentration = 6 mg L−1 to 14 mg L−1. | ||
The fitting results obtained from different models are summarized in Table 1. The correlation coefficients for the pseudo-first-order models are 0.985, 0.974, 0.989, 0.966, and 0.986. Significant differences exist between the calculated qcal values and experimental qexp values. For the MB with high correlation coefficients of R2 = 0.995, 0.996, 0.993, 0.993, 0.997 (fitting curve is shown in Fig. 5b), the pseudo-second-order model provides good correlation for the adsorption of organic compounds on ClRGO. For the pseudo-second-order model, the calculated qcal values agree well with the experimental qexp values.
| Models and equations | Temperature | Parameters | R2 | ||
|---|---|---|---|---|---|
| K1 (×10−2 min−1) | qcal (mg g−1) | qexp (mg g−1) | |||
| a Concentration = 8 mg L−1. | |||||
| Pseudo-first-order ln(qe − qt) = ln(qe) − k1t | 298 K | 3.29 | 178.1 | 248.9 | 0.985 |
| 308 K | 2.96 | 195.0 | 268.7 | 0.974 | |
| 318 K | 2.94 | 234.1 | 305.9 | 0.989 | |
| 328 K | 2.73 | 267.6 | 332.7 | 0.966 | |
| 338 K | 2.82 | 256.3 | 355.6 | 0.986 | |
| Models and equations | Temperature | Parameters | R2 | ||
|---|---|---|---|---|---|
| K2 (×10−4 g mg−1 min−1) | qcal (mg g−1) | qexp (mg g−1) | |||
| Pseudo-second-order t/qt = 1/k2qe2 + t/qe | 298 K | 4.91 | 250.0 | 248.9 | 0.995 |
| 308 K | 4.04 | 270.3 | 268.7 | 0.996 | |
| 318 K | 3.18 | 303.0 | 305.9 | 0.993 | |
| 328 K | 2.80 | 333.3 | 332.7 | 0.993 | |
| 338 K | 2.71 | 357.1 | 355.6 | 0.997 | |
| Models and equations | Temperature | Parameters | R2 | |
|---|---|---|---|---|
| kdif (mg g−1 min−1/2) | C (mg g−1) | |||
| Intraparticle diffusion qt = kdift1/2 + C | 298 K | 24.59 | 42.3 | 0.952 |
| 308 K | 26.78 | 36.7 | 0.956 | |
| 318 K | 31.02 | 31.0 | 0.974 | |
| 328 K | 33.54 | 33.8 | 0.972 | |
| 338 K | 36.14 | 37.6 | 0.949 | |
| Models and equations | Temperature | Parameters | R2 | |
|---|---|---|---|---|
| k0 | α | |||
Bangham diffusion ln(ln(C0/(C0 − mqt))) = ln(k0m/V) + α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(t) ln(t) |
298 K | 0.0894 | 0.476 | 0.985 |
| 308 K | 0.0767 | 0.502 | 0.991 | |
| 318 K | 0.0637 | 0.546 | 0.997 | |
| 328 K | 0.0588 | 0.573 | 0.996 | |
| 338 K | 0.0521 | 0.609 | 0.991 | |
The intraparticle diffusion models have low correlation (0.952, 0.956, 0.974, 0.972, and 0.949), indicating that these models are not suitable for describing the MB adsorption on ClRGO. kdif increases but C decreases with temperature. These results indicate that the rate of MB removal increases with increasing temperature. None of the plots passes through the origin, indicating that the intraparticle diffusion is part of the adsorption but is not the only rate-controlling step.
Since the uptake of MB on ClRGO slowed down during the later stages of the adsorption, Bangham diffusion model was tested for its applicability for MB adsorption on ClRGO. The parameters α and k0 were calculated from the slope and intercept for five different temperatures and are displayed along with the R2 values (Fig. 5c, and Table 1). The plots reveal that the lines at 298 K are not straight for ClRGO indicating that pore diffusion is not solely rate-determining in Fig. 5c. The diffusion speed of MB in solution might also affect adsorption. At high temperatures, such as 308, 318, 328, and 338 K, the R2 values (0.991, 0.997, 0.996, and 0.991) are high. The diffusion speed of MB in solution is high, pore diffusion mainly affect adsorption. Bangham diffusion kinetic model was found applicable to this adsorption system in terms of relatively high regression values.
The Langmuir, Freundlich, and Dubinin–Radushkevich (D–R) isotherm models are applied to simulate and understand the adsorption mechanism of MB at different temperatures,42 the fitting results from different models are summarized in Table 2.
| Models and equations | Constants | 298 K | 308 K | 318 K | 328 K | 338 K | |
|---|---|---|---|---|---|---|---|
| Langmuir: Ce/qe = 1/qmb + Ce/qm, RL = 1/(1 + bC0) | qm (mg g−1) | 312.5 | 344.8 | 370.4 | 400.1 | 416.7 | |
| b (L mg−1) | 1.61 | 1.88 | 2.25 | 4.17 | 6.00 | ||
| RL | C0 = 6 mg L−1 | 0.094 | 0.081 | 0.069 | 0.038 | 0.027 | |
| C0 = 8 mg L−1 | 0.072 | 0.062 | 0.053 | 0.029 | 0.020 | ||
| C0 = 10 mg L−1 | 0.058 | 0.051 | 0.043 | 0.023 | 0.016 | ||
| C0 = 12 mg L−1 | 0.049 | 0.042 | 0.036 | 0.020 | 0.014 | ||
| C0 = 14 mg L−1 | 0.042 | 0.037 | 0.031 | 0.017 | 0.012 | ||
| R2 | 0.998 | 0.996 | 0.998 | 0.997 | 0.998 | ||
| Freundlich: ln(qe) = ln(KF) + (1/n)ln(Ce) | KF (mg g−1 (L mg−1)1/n) | 229.5 | 250.9 | 285.4 | 321.4 | 350.5 | |
| n−1 | 0.121 | 0.109 | 0.105 | 0.102 | 0.092 | ||
| R2 | 0.981 | 0.921 | 0.984 | 0.987 | 0.977 | ||
| Dubinin–Radushkevich: ln(qe) = ln(qs) − Bε2, ε = RTln(1 + 1/Ce), E = (2B)−1/2 |
qs (mol g−1) | 0.90 | 0.95 | 1.07 | 1.16 | 1.22 | |
| B (×10−2 mol2 kJ−2) | 9.2 | 8.5 | 6.97 | 1.66 | 0.98 | ||
| E (kJ mol−1) | 2.3 | 2.4 | 2.7 | 5.5 | 7.1 | ||
| R2 | 0.794 | 0.789 | 0.923 | 0.830 | 0.919 | ||
The Langmuir model parameters and the statistical fits of the adsorption data are shown in Fig. 5d. The high regression coefficient confirms that the Langmuir isotherm best represents the equilibrium adsorption of MB to ClRGO at different temperatures. The excellent fit of the Langmuir isotherm to the experimental data at all temperatures confirms that the adsorption is monolayer. The adsorption of each molecule has equal activation energy, and the sorbate–sorbate interaction is negligible. The constant RL lies within the favorable limit (between 0 and 1), indicating a favorable process. In addition, qm increases with temperature. The Langmuir constant (b) also increases with temperature. Overall, the information obtained specifies an endothermic nature of the existing process. The monolayer adsorption capacity of graphene for MB as obtained from the Langmuir isotherm at 338 K is 357.1 mg g−1, and the experimental qexp value is 355.6 mg g−1.
KF and n are the Freundlich parameters related to adsorption capacity and adsorption intensity, respectively. The value of 1/n is lower than 1, indicating a normal Langmuir isotherm.
The D–R model is related to the porous structure of the sorbent and apparent energy of adsorption, the model is also used to distinguish between physical and chemical adsorption of MB.42 The magnitude of E is lower than 8.0 kJ mol−1 at all tested temperatures, indicating that the adsorption mechanism is physical adsorption.
4 Conclusions
Hydroxyl group in graphene oxide can be substituted by chlorine in sulfuryl chloride. ClRGO will be an important intermediate for preparing Grignard reagents and other compounds. ClRGO has wider pore size and average interlayer space than RGO, and it enhances capability for MB. Adsorption data indicate that the material has high adsorption capacity on methylene blue. The adsorption process follows second-order rate and Bangham diffusion kinetic model. The adsorption of MB preferably fitting the Langmuir adsorption isotherm suggests monolayer coverage of adsorbed molecules.Acknowledgements
This work was supported by the Education Department of Henan Province (no. 12B210022), the Science and Technology Bureau of Xinxiang (no. 13SF39), and the Innovation Fund of Xinxiang University (no. 12ZB09 and 12ZB10).References
- C. Chao, J. D. Liu, J. T. Wang, Y. W. Zhang, B. Zhang, Y. T. Zhang, X. Xiang and R. F. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 10559 CAS.
- C. Chao, B. Zhang, R. Zhai, X. Xiang, J. D. Liu and R. F. Chen, ACS Sustainable Chem. Eng., 2014, 2, 396 CrossRef CAS.
- C. X. Gui, Q. Q. Wang, S. M. Hao, J. Qu, P. P. Huang, C. Y. Cao, W. G. Song and Z. Z. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 14653 CAS.
- C. Zhang, R. Z. Zhang, Y. Q. Ma, W. B. Guan, X. L. Wu, X. Liu, H. Li, Y. L. Du and C. P. Pan, ACS Sustainable Chem. Eng., 2015, 3, 396 CrossRef CAS.
- F. Guo, M. Creighton, Y. T. Chen, R. Hurt and I. Külaots, Carbon, 2014, 66, 476 CrossRef CAS PubMed.
- Z. M. Wu, L. Zhang, Q. Q. Guan, P. Ning and D. Q. Ye, Chem. Eng. J., 2014, 258, 77 CrossRef CAS PubMed.
- G. G. Liu, S. Gui, H. Zhou, F. T. Zeng, Y. H. Zhou and H. Q. Ye, Dalton Trans., 2014, 43, 6977 RSC.
- Y. S. Fu, J. W. Zhu, C. Hu, X. D. Wu and X. Wang, Nanoscale, 2014, 6, 12555 RSC.
- M. M. Sk and C. Y. Yue, RSC Adv., 2014, 4, 19908 RSC.
- G. Erdenedelger, T. Lee, T. D. Dao, J. S. Kim, B. S. Kim and H. M. Jeong, J. Mater. Chem. A, 2014, 2, 12526 CAS.
- F. Zhang, B. Wang, S. F. He and R. L. Man, J. Chem. Eng. Data, 2014, 59, 1719 CrossRef CAS.
- M. Jana, P. Khanra, N. C. Murmu, P. Samanta, J. H. Lee and T. Kuila, Phys. Chem. Chem. Phys., 2014, 16, 7618 RSC.
- R. Rajesh, S. S. Kumar and R. Venkatesan, New J. Chem., 2014, 38, 1551 RSC.
- R. K. Yadav, J. O. Baeg, A. Kumar, K. Kong, G. H. Oh and N. J. Park, J. Mater. Chem. A, 2014, 2, 5068 CAS.
- W. Ai, X. H. Cao, Z. P. Sun, J. Jiang, Z. Z. Du, L. H. Xie, Y. L. Wang, X. J. Wang, H. Zhang, W. Huang and T. Yu, J. Mater. Chem. A, 2014, 2, 12924 CAS.
- H. Y. Qin, T. Gong, Y. J. Cho, C. G. Lee and T. Kim, Polym. Chem., 2014, 5, 4466 RSC.
- J. Yang, J. X. Wu, Q. F. Lü and T. T. Lin, ACS Sustainable Chem. Eng., 2014, 2, 1203 CrossRef CAS.
- T. Wu, X. Y. Xu, L. Zhang, H. B. Chen, J. P. Gao and Y. Liu, RSC Adv., 2014, 4, 7673 RSC.
- C. B. Wang, J. Ni, J. W. Zhou, J. L. Wen and X. B. Lü, RSC Adv., 2013, 3, 23139 RSC.
- C. B. Wang, J. W. Zhou, J. Ni, Y. L. Cheng and H. Li, Chem. Eng. J., 2014, 253, 130 CrossRef CAS PubMed.
- C. B. Wang, J. Ni and J. W. Zhou, Mater. Tech., 2014, 29, 252 CrossRef CAS PubMed.
- B. T. McGrail, B. J. Rodier and E. Pentzer, Chem. Mater., 2014, 26, 5806 CrossRef CAS.
- J. Chen, P. Xiao, J. C. Gu, Y. J. Huang, J. W. Zhang, W. Q. Wang and T. Chen, RSC Adv., 2014, 4, 44480 RSC.
- X. B. Zhao and P. Liu, Langmuir, 2014, 30, 13699 CrossRef CAS PubMed.
- Z. Z. Liu, S. J. Zhu, Y. J. Li, Y. S. Li, P. Shi, Z. Huang and X. Y. Huang, Polym. Chem., 2015, 6, 311 RSC.
- C. J. Madadrang, H. Y. Kim, G. H. Gao, N. Wang, J. Zhu, H. Feng, M. Gorring, M. L. Kasner and S. F. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 1186 CAS.
- M. Z. Iqbal, M. S. Katsiotis, S. M. Alhassan, M. W. Liberatore and A. A. Abdala, RSC Adv., 2014, 4, 6830 RSC.
- H. L. Su, Z. F. Li, Q. S. Huo, J. Q. Guan and Q. B. Kan, RSC Adv., 2014, 4, 9990 RSC.
- M. A. Nasseri, A. Allahresani and H. Raissi, RSC Adv., 2014, 4, 26087 RSC.
- S. Verma, M. Aila, S. Kaul and S. L. Jain, RSC Adv., 2014, 4, 30598 RSC.
- H. M. Liu, Y. Guo, X. S. Wang, Y. K. Li, X. J. Liang and X. Liu, Anal. Methods, 2015, 7, 135 RSC.
- H. J. Zhu, Y. J. Zhang, L. L. Zhang, T. Yu, K. Zhang, H. Jiang, L. J. Wu and S. H. Wang, J. Mater. Chem. C, 2014, 2, 7126 RSC.
- W. J. Liu, Y. K. Wang and Z. H. Li, Chem. Commun., 2014, 50, 10311 RSC.
- P. Kumar, B. Sain and S. L. Jain, J. Mater. Chem. A, 2014, 2, 11246 CAS.
- P. Kumar, G. Singh, D. Tripathi and S. L. Jain, RSC Adv., 2014, 4, 50331 RSC.
- A. Gupta, G. Chen, P. Joshi, S. Tadigadapa and P. C. Eklund, Nano Lett., 2006, 6, 2667 CrossRef CAS PubMed.
- C. Gomez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499 CrossRef CAS PubMed.
- H. A. Becerril, J. Man, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270 CrossRef CAS PubMed.
- F. B. Meng, H. Ishida and X. B. Liu, RSC Adv., 2014, 4, 9471 RSC.
- Z. H. Ni, H. M. Wang, Y. Ma, J. Kasim, Y. H. Wu and Z. X. Shen, ACS Nano, 2008, 2, 1033 CrossRef CAS PubMed.
- S. Vasudevan and J. Lakshmi, RSC Adv., 2012, 2, 5234 RSC.
- C. Aharoni, S. Sideman and E. Hoffer, J. Chem. Technol. Biotechnol., 1979, 29, 404 CrossRef CAS PubMed.
- M. Valix, W. H. Cheng and G. McKay, Langmuir, 2006, 22, 4574 CrossRef CAS PubMed.
- H. L. Parker, A. J. Hunt, V. L. Budarin, P. S. Shuttleworth, K. L. Miller and J. H. Clark, RSC Adv., 2012, 2, 8992 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5ra05618g |
| This journal is © The Royal Society of Chemistry 2015 |