DOI:
10.1039/C5RA06991B
(Paper)
RSC Adv., 2015,
5, 42131-42134
A concise protecting-group-free synthesis of cephalosporolides E and F†
Received
17th April 2015
, Accepted 27th April 2015
First published on 27th April 2015
Abstract
A concise protecting-group-free synthesis of cephalosporolides E and F has been described. The key steps involve the one-pot conversion of L-mannonic-γ-lactone to γ-vinyl-β-hydroxy-γ-lactone, cross-metathesis and Wacker-type oxidative spiroketalization. The internal olefin served as a latent keto functionality with excellent delivery of a regioselective keto group for spiroketalization. The synthetic strategy is protecting-group-free and marks the shortest route so far to cephalosporolides E and F.
Introduction
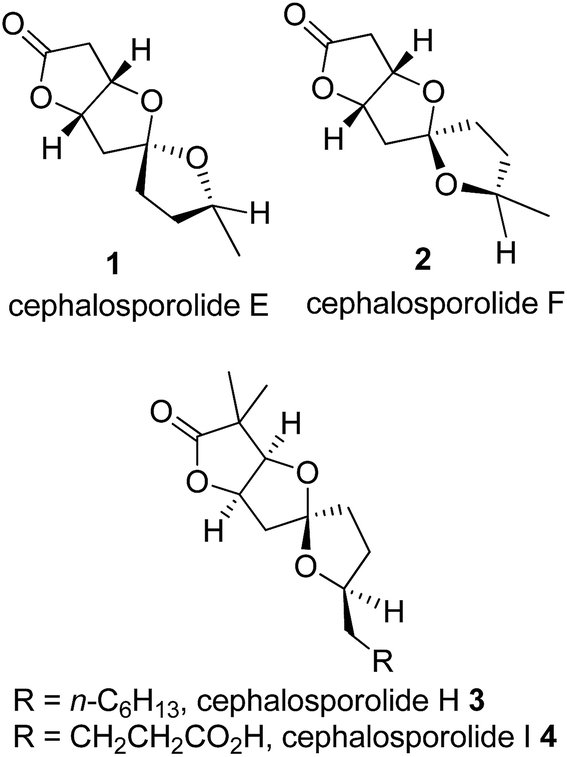
The 5,5-spiroketal natural products cephalosporolides E 1 and F 2 (Fig. 1) were isolated in 1985 by Hanson and co-workers1a from the fungus Cephalosporium aphidicola and later by Rukachaisirikul and co-workers from the entomopathogenic fungus Cordyceps militaris BCC 2816.1b These are epimers at the spirocenter with other features being common. They belong to a bigger family of 5,5-spiroketal natural products like penisporolides A and B,2 cephalosporolides H and I (3 and 4, Fig. 1),3 symbiospirol A4 and ascospiroketal B5 to name a few. These molecules with a rigid spiroketal structure are known to exhibit significant biological activities. This has prompted many to design new strategies toward their total synthesis. The total synthesis of unnatural enantiomers of cephalosporolides E and F was first reported by Ramana and co-workers6 followed by our first synthesis of the natural antipodes.7 Since then a few more syntheses of 1 and 2 or analogs are reported,8 the significant being the chelation-controlled epimerization to the spiroketals involving Zn-salts giving a diasteroselectivity as high as 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 8b for the spiroketal center.
:1 8b for the spiroketal center.
 |
| | Fig. 1 The cephalosporolides E, F, H, and I. | |
With our continued interest in the synthesis of several γ-lactone based natural products,9 our own synthesis7,10 of cephalosporolides E, F and H and inclination to protecting-group-free synthesis9f,h we embarked on a concise strategy to achieve the synthesis of cephalosporolides E and F from L-mannonic-γ-lactone without involving protecting groups.
Results and discussion
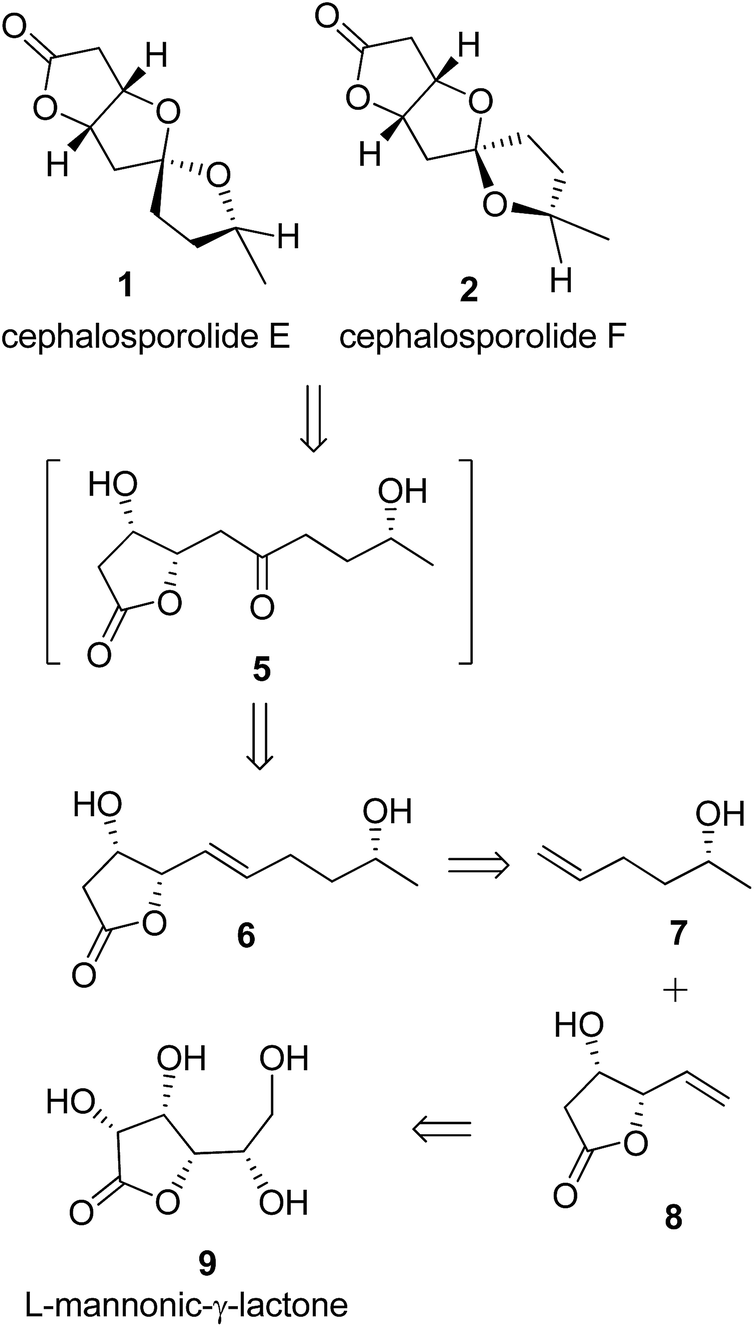
As shown in our retrosynthetic design (Scheme 1), we visualized the γ-vinyl-β-hydroxy-γ-lactone 8 (obtained by a one-pot procedure from L-mannonic-γ-lactone, 9)11a to represent the lactone portion of 1 and 2. If the spirocenter can be generated through ketalization by suitably placed hydroxyl groups, the compound 6 seemed the obvious choice as the required keto group (of 5) can be generated in situ through Wacker-type oxidation. Our recent hetero-atom directed regioselective Wacker-type oxidation of related olefin supported the anticipated regioselectivity for keto group.12 The olefin compound 6 can easily be obtained by cross-metathesis of 7 with 8. The synthetic design would prove to be the shortest route known so far for cephalosporolides E and F synthesis and is notably protecting-group-free.
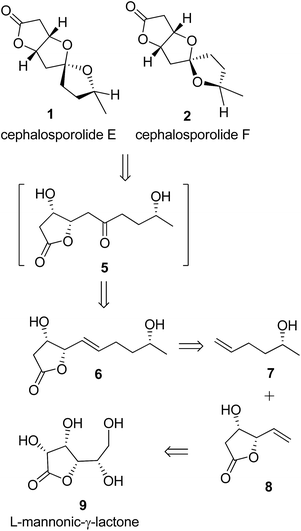 |
| | Scheme 1 Retrosynthesis of cephalosporolides E 1 and F 2. | |
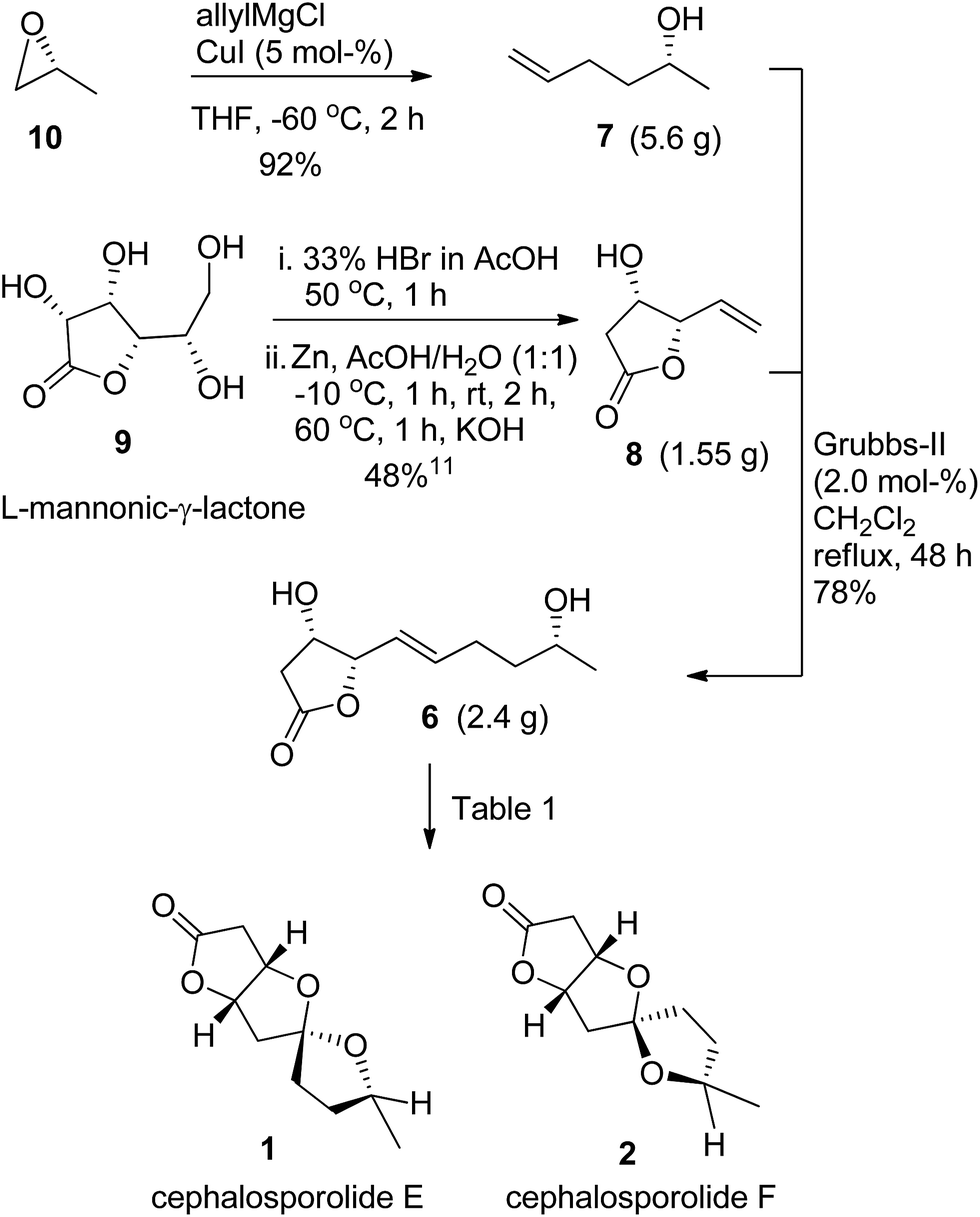
The forward synthesis is shown in Scheme 2. The allyl magnesium chloride/CuI opening of known epoxide 1013 delivered the olefin partner 7 in excellent yield of 92%. The γ-vinyl-β-hydroxy-γ-lactone 8 was prepared from L-mannonic-γ-lactone following our reported literature procedure for its enantiomer.11 The cross-metathesis of 8 with 7 using the Grubbs-II catalyst (2.0 mol%) provided the key olefin compound 6 in 78% yield with E-selectivity.14 The compound 6 was prepared on 2.4 g scale. The olefin 6 was subjected to Wacker-type oxidation with concomitant spiroketalization as shown in Table 1. We screened four Pd-catalysts (10 mol%, entries 1–4) using CuCl (1.5 equiv.) in DMF:H2O (4:1) as solvent at 60 °C and found PdCl2 to be superior (entry 1). Pd/C did not work for this reaction and compound 6 was recovered over 75% with partial decomposition. The screening of common solvents used in Wacker oxidation (entries 5–8) showed DMF:H2O to be better (entry 1). We also considered changing PdCl2 loading (entries 9–11) and found 20 mol% to give improved yield of 1 and 2 in 60% and 28% respectively (entry 9). The increase in catalyst loading to 30 mol% did not affect the yield much (entry 10), while the lowering to 5 mol% gave diminished yields (entry 11). The lowering of CuCl concentration to 1.0 equiv. gave inferior results (entry 12). Thus the use of PdCl2 (20–30 mol%) and CuCl (1.5 equiv.) gave the best results. A reaction of 6 (4.0 mmol scale) using the reaction conditions as in entry 9 delivered 1 and 2 in 56% and 25% yields respectively, indicating a good scalability of the reaction (entry 13).
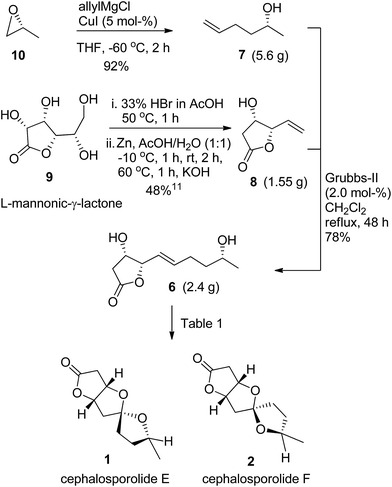 |
| | Scheme 2 Synthesis of cephalosporolides E and F. | |
Table 1 Wacker-type oxidative-spiroketalization of 6 to 1 and 2a
| Entry |
Pd-catalyst (x mol%) |
CuCl (y equiv.) |
Solvent |
Isolated yield of 1 and 2 |
| Reaction conditions: substrate 6 (0.5 mmol), Pd-catalyst (x mol%), CuCl (y equiv.), DMF:H2O (4:1, 5.0 mL), at 60 °C under O2, 2 h. Reaction on 4.0 mmol of 6. |
| 1 |
PdCl2 (10) |
1.5 |
DMF:H2O |
1 (42%), 2 (18%) |
| 2 |
Pd(OAc)2 (10) |
1.5 |
DMF:H2O |
1 (18%), 2 (8%) |
| 3 |
Pd/C (10) |
1.5 |
DMF:H2O |
— |
| 4 |
Pd(OCOCF3)2 (10) |
1.5 |
DMF:H2O |
1 (22%), 2 (10%) |
| 5 |
PdCl2 (10) |
1.5 |
DMA:H2O |
1 (32%), 2 (12%) |
| 6 |
PdCl2 (10) |
1.5 |
THF:H2O |
1 (12%), 2 (8%) |
| 7 |
PdCl2 (10) |
1.5 |
CH2Cl2:H2O |
1 (28%), 2 (16%) |
| 8 |
PdCl2 (10) |
1.5 |
tBuOH:H2O |
1 (30%), 2 (18%) |
| 9 |
PdCl2 (20) |
1.5 |
DMF:H2O |
1 (60%), 2 (28%) |
| 10 |
PdCl2 (30) |
1.5 |
DMF:H2O |
1 (61%), 2 (28%) |
| 11 |
PdCl2 (5) |
1.5 |
DMF:H2O |
1 (32%), 2 (15%) |
| 12 |
PdCl2 (20) |
1.0 |
DMF:H2O |
1 (48%), 2 (23%) |
| 13b |
PdCl2 (20) |
1.5 |
DMF:H2O |
1 (56%), 2 (25%) |
Conclusions
In conclusion an efficient and concise strategy for the synthesis of cephalosporolides E and F has been developed. The Wacker-type oxidative-spiroketalization efficiently delivered both 1 and 2 in good yields. The internal olefin served as latent keto functionality offering excellent heteroatom directed regioselectivity in Wacker-type oxidation. The synthesis is completed in 22.8% and 10.5% over all yields for 1 and 2 respectively from 9 and marks the shortest route to these molecules. It is also notably a protecting-group-free synthesis. The strategy has potential for synthesis of other 5,5-spiroketal natural products.
Experimental section
General information
Flasks were oven- or flame-dried and cooled in a desiccator. Dry reactions were carried out under an atmosphere of N2. Solvents and reagents were purified by standard methods. Thin-layer chromatography was performed on EM250 Kieselgel 60 F254 silica gel plates. The spots were visualized by staining with KMnO4 or by UV lamp. 1H NMR and 13C NMR were recorded with Bruker AVANCE III 400 or 500 and 100 or 125 MHz, respectively, and chemical shifts are based on TMS peak at δ = 0.00 ppm for proton NMR and CDCl3 peak at δ = 77.00 ppm (t) in carbon NMR. IR samples were prepared by evaporation from CHCl3 on CsBr plates. High-resolution mass spectra were obtained using positive electrospray ionization by TOF method. Optical rotations were measured with a Jasco P-2000 digital polarimeter.
(R)-(−)-5-Hexen-2-ol (7)
Allyl magnesium chloride (61 mL, 121.55 mmol, 2.0 M solution in THF) was added dropwise to a suspension of CuI (579 mg, 3.04 mmol, 5.0 mol%) in dry ether (75 mL) at −60 °C. It was stirred at the same temperature for 30 min. The solution of epoxide 10 (3.53 g, 60.78 mmol) in ether (5 mL) was added. It was stirred for 2 h and then quenched with saturated aq. NH4Cl solution (10 mL). The reaction mixture was extracted with ether (2 × 60 mL). The combined organic extracts were washed with brine (15 mL), dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography using petroleum ether/Et2O (2:1) to give 7 (5.6 g, 92%) as colorless oil. [α]25D −10.2 (c 2.0, CHCl3). IR (neat): νmax = 3440, 3021, 1585, 1410, 1221, 1115, 669 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.89–5.77 (m, 1H), 5.06–4.92 (m, 2H), 3.81 (quin, J = 6.2 Hz, 1H), 2.15–2.09 (m, 2H), 1.99 (br. s, 1H), 1.57–1.48 (m, 2H), 1.17 (d, J = 6.2 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 138.5, 114.7, 67.6, 38.2, 30.1, 23.4 ppm.
(4S,5S)-4-Hydroxy-5-vinyldihydrofuran-2(3H)-one (8)
The title compound was prepared from L-mannonic-γ-lactone 9 following similar one-pot procedure reported by us for its enantiomer.9h,11 A reaction on 9 (4.5 g, 25.26 mmol) delivered 8 (1.55 g, 48%) as colorless oil. [α]25D −46.1 (c 1.0, CHCl3). Lit.11a −44 (c 1.56, CHCl3). Other spectral data is same as its enantiomer.9h
(4S,5S)-4-Hydroxy-5-[(R,E)-5-hydroxyhex-1-enyl]dihydrofuran-2(3H)-one (6)
To a degassed solution of 8 (1.97 g, 15.37 mmol) and 7 (2.31 g, 23.06 mmol, 1.5 equiv.) in dry CH2Cl2 (200 mL) was added Grubbs-II catalyst (261 mg, 0.307 mmol, 2.0 mol%) at room temperature and the mixture refluxed for 48 h. The mixture was cooled and filtered through a small pad of celite and the filtrate concentrated. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (3:2) to give 6 (2.4 g, 78%) as colorless oil. [α]25D = −24.3 (c = 1.75, CHCl3). IR (CHCl3): νmax = 3435, 3018, 2969, 2931, 1769, 1404, 1376, 1327, 1163, 1076, 1014, 968, 907, 847, 667 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.97–5.90 (m, 1H), 5.66–5.61 (m, 1H), 4.82 (dd, J = 3.6 Hz, 1H), 4.44 (t, J = 4.9 Hz, 1H), 3.78 (q, J = 6.2 Hz, 1H), 2.77 (dd, J = 17.7, 5.4 Hz, 1H), 2.57 (dd, J = 17.7, 1.0 Hz, 1H), 2.23 (q, J = 6.4 Hz, 2H), 1.60–1.51 (m, 2H), 1.18 (d, J = 6.2 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 176.1, 137.7, 122.3, 85.0, 69.7, 67.5, 38.8, 37.5, 28.9, 23.6 ppm. HRMS (ESI-TOF) calcd for [C10H16O4 + Na]+ 223.0938, found 223.0944.
Cephalosporolides E and F (1 and 2)
Reaction conditions as in Table 1, entry 9: to a suspension of PdCl2 (17.7 mg, 0.1 mmol, 20 mol%) and CuCl (74.3 mg, 0.75 mmol, 1.5 equiv.) in DMF/H2O (5 mL, 4:1) under oxygen atmosphere at room temperature was added 6 (100.1 mg, 0.5 mmol). The reaction mixture was stirred at 60 °C for 2 h. The mixture was cooled and filtered through a short pad of silica gel and the filtrate concentrated. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (4:1) to afford 2 (27.7 mg, 28%). Further elution with petroleum ether/EtOAc (3:1) gave 1 (59.5 mg, 60%). The reaction on 6 (801 mg, 4.0 mmol, Table 1, entry 13) delivered 1 (444 mg, 56%) and 2 (198.2 mg, 25%).
Data for 1. White solid, mp 97–99 °C. [α]25D = 49.2 (c = 0.25, CHCl3). IR (CHCl3): νmax = 3020, 2975, 1779, 1460, 1348, 1303, 1193, 1157, 1099, 1057, 919, 826, 667 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.12 (t, J = 5.8 Hz, 1H), 4.84 (td, J = 6.6, 1.5 Hz, 1H), 4.12–4.16 (m, 1H), 2.71 (dd, J = 18.6, 7.4 Hz, 1H), 2.61 (dd, J = 18.6, 1.5 Hz, 1H), 2.41 (d, J = 14.2 Hz, 1H), 2.01–2.12 (m, 4H), 1.38–1.45 (m, 1H), 1.18 (d, J = 6.1 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 175.9, 115.0, 83.4, 77.2, 75.0, 41.6, 37.6, 34.1, 31.2, 20.9 ppm. HRMS (ESI-TOF) calcd for [C10H14O4 + H]+ 199.0970, found: 199.0975.
Data for 2. White solid, mp 61–63 °C. [α]25D = −69.1 (c = 0.15, CHCl3). IR (CHCl3): νmax = 3016, 2973, 2934, 1779, 1455, 1404, 1338, 1272, 1195, 1167, 1097, 1059, 927, 864, 667 cm−1. 1H NMR (400 MHz, CDCl3/TMS) δ = 5.11–5.06 (m, 1H), 4.79 (t, J = 4.4 Hz, 1H), 4.21–4.17 (m, 1H), 2.73 (dd, J = 18.4, 5.3 Hz, 1H), 2.66 (d, J = 18.0 Hz, 1H), 2.51 (dd, J = 14.9, 6.7 Hz, 1H), 2.31 (dd, J = 14.9, 2.1 Hz, 1H), 2.16–2.11 (m, 1H), 2.06–2.01 (m, 1H), 1.96 (dd, J = 12.4, 7.7 Hz, 1H), 1.75–1.67 (m, 1H), 1.27 (d, J = 6.1 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ = 175.7, 115.4, 83.8, 76.8, 76.5, 42.1, 36.9, 35.9, 32.4, 22.7. HRMS (ESI-TOF) calcd for [C10H14O4 + H]+ 199.0970, found 199.0967.
Acknowledgements
We thank the Board of Research in Nuclear Sciences (BRNS), Government of India (Basic Sciences, Grant no. 2013/37C/59/BRNS/2443) for financial support. D.A.C. and P.K. thanks the Council of Scientific and Industrial Research (CSIR), New Delhi for research fellowships.
Notes and references
-
(a) M. J. Ackland, J. R. Hanson, P. B. Hitchcock and A. H. Ratcliffe, J. Chem. Soc., Perkin Trans. 1, 1985, 843–847 RSC;
(b) V. Rukachaisirikul, S. Pramjit, C. Pakawatchai, M. Isaka and S. Supothina, J. Nat. Prod., 2004, 67, 1953–1955 CrossRef CAS PubMed.
- X. Li, I. Sattler and W. Lin, J. Antibiot., 2007, 60, 191–195 CrossRef CAS PubMed.
- X. Li, Y. Yao, Y. Zheng, I. Sattler and W. Lin, Arch. Pharmacal Res., 2007, 30, 812–815 CrossRef CAS.
- Y. Tsunematsu, O. Ohno, K. Konishi, K. Yamada, M. Suganuma and D. Uemura, Org. Lett., 2009, 11, 2153–2156 CrossRef CAS PubMed.
- S. F. Seibert, A. Krick, E. Eguereva, S. Kehraus and G. M. Konig, Org. Lett., 2007, 9, 239–242 CrossRef CAS PubMed.
- C. V. Ramana, S. B. Suryawanshi and R. G. Gonnade, J. Org. Chem., 2009, 74, 2842–2845 CrossRef CAS PubMed.
- R. A. Fernandes and A. B. Ingle, Synlett, 2010, 158–160 CrossRef CAS PubMed.
-
(a) M. A. Brimble, O. C. Finch, A. M. Heapy, J. D. Fraser, D. P. Furkert and P. D. O'Connor, Tetrahedron, 2011, 67, 995–1001 CrossRef CAS PubMed;
(b) S. M. Tlais and G. B. Dudley, Beilstein J. Org. Chem., 2012, 8, 1287–1292 CrossRef CAS PubMed;
(c) S. Chang and R. Britton, Org. Lett., 2012, 14, 5844–5847 CrossRef CAS PubMed;
(d) L. Song, Y. Liu and R. Tong, Org. Lett., 2013, 15, 5850–5853 CrossRef CAS PubMed;
(e) O. C. Finch, D. P. Furkert and M. A. Brimble, Tetrahedron, 2014, 70, 590–596 CrossRef CAS PubMed;
(f) C. N. Kona and C. V. Ramana, Tetrahedron, 2014, 70, 3653–3656 CrossRef CAS PubMed;
(g) O. Cortezano-Arellano, L. Quintero and F. Sartillo-Piscil, J. Org. Chem., 2015, 80, 2601–2608 CrossRef CAS PubMed.
-
(a) R. A. Fernandes and A. K. Chowdhury, J. Org. Chem., 2009, 74, 8826–8829 CrossRef CAS PubMed;
(b) R. A. Fernandes, A. B. Ingle and V. P. Chavan, Tetrahedron: Asymmetry, 2009, 20, 2835–2844 CrossRef CAS PubMed;
(c) R. A. Fernandes and A. B. Ingle, Tetrahedron Lett., 2011, 52, 458–460 CrossRef CAS PubMed;
(d) R. A. Fernandes and A. K. Chowdhury, Eur. J. Org. Chem., 2011, 1106–1112 CrossRef CAS PubMed;
(e) R. A. Fernandes, M. B. Halle, A. K. Chowdhury and A. B. Ingle, Tetrahedron: Asymmetry, 2012, 23, 60–66 CrossRef CAS PubMed;
(f) R. A. Fernandes and P. Kattanguru, J. Org. Chem., 2012, 77, 9357–9360 CrossRef CAS PubMed;
(g) R. A. Fernandes, V. P. Chavan, S. V. Mulay and A. Manchoju, J. Org. Chem., 2012, 77, 10455–10460 CrossRef CAS PubMed;
(h) R. A. Fernandes and P. Kattanguru, Asian J. Org. Chem., 2013, 2, 74–84 CrossRef CAS PubMed;
(i) R. A. Fernandes, P. H. Patil and A. K. Chowdhury, Eur. J. Org. Chem., 2013, 237–243 Search PubMed.
- R. A. Fernandes and M. B. Halle, Asian J. Org. Chem., 2013, 2, 593–599 CrossRef CAS PubMed.
- The lactone 8 is prepared from L-mannonic-γ-lactone, see:
(a) J. Song and R. I. Hollingsworth, Tetrahedron: Asymmetry, 2001, 12, 387 CrossRef CAS;
(b) It was prepared in 48% yield by us following our modified procedure used for the synthesis of its antipode from D-glucono-δ-lactone.9h.
-
(a) V. Bethi, P. Kattanguru and R. A. Fernandes, Eur. J. Org. Chem., 2014, 3249–3255 CrossRef CAS PubMed;
(b) A day before the submission of this work a similar strategy was employed in the synthesis of cephalosporolide H and I, see: J. Li, C. Zhao, J. Liu and Y. Du, Tetrahedron, 2015 DOI:10.1016/j.tet.2015.04.025.
- The epoxide was prepared on gram scale following literature procedure, see: S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef CAS PubMed.
- The Z-olefin was seen in trace amount in the 1H NMR spectra.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all the compounds. See DOI: 10.1039/c5ra06991b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.