
Correlating experimental electrochemistry and theoretical calculations in 2′-hydroxy chalcones: the role of the intramolecular hydrogen bond†
Maximiliano Martínez-Cifuentes*a,
Ricardo Salazarb,
Carlos A. Escobarc,
Boris E. Weiss-Lópezd,
Leonardo S. Santosa and
Ramiro Araya-Maturana*ef
aLaboratorio de Síntesis Asimétrica, Instituto de Química de los Recursos Naturales, Universidad de Talca, Casilla 747, Talca, Chile. E-mail: mmartinez@ug.uchile.cl; Tel: +56-9-81640195
bLaboratorio de Electroquímica MedioAmbiental, LEQMA, Departamento de Química de los Materiales, Facultad de Química y Biología, Universidad de Santiago de Chile, USACh, Casilla 40, Correo 33, Santiago, Chile
cDepartamento de Ciencias Químicas, Universidad Andres Bello, Av. República 275, Santiago, Chile
dDepartamento de Química, Facultad de Ciencias, Universidad de Chile, Casilla 653, Santiago, Chile
eDepartamento de Química Orgánica y Fisicoquímica, Facultad de Ciencias Químicas Y Farmacéuticas, Universidad de Chile, Casilla 233, Santiago 1, Chile. E-mail: raraya@ciq.uchile.cl; Tel: +56-2-2978-2874
fInstituto de Química de los Recursos Naturales, Universidad de Talca, Casilla 747, Talca, Chile
First published on 3rd June 2015
Abstract
In this work we present a study on the molecular structure and electrochemical behavior of a series of methoxylated 2′-hydroxychalcones, whose antitumor activity has been previously described. Cyclic voltammetry was used to quantitatively characterize the formation and stability of the anion radicals. The molecular structure of the neutral compounds and their anion radicals, particularly the intramolecular hydrogen bonds (IHB), were investigated through density functional theory (DFT) calculations. Geometrical and frontier orbital changes in the anion, relative to the neutral species, were examined and the adiabatic and vertical electron affinities (AEA, VEA) as well as vertical detachment energy (VDE) were calculated. Natural bond orbital (NBO) analysis was used to obtain insights into the electronic characteristics of the IHB and the results were correlated with 1H-NMR chemical shifts. A direct relation among the substitution pattern on rings A and B, the strength of the IHBs and the reduction potentials was found. NBO energies (ΔE(2)ij) show that the main contributions to the stabilization of the IHBs arises from LP → σ* interactions. The strength of IHBs, given by ΔE(2)ij, exhibit a notable quantitative correlation with the experimental reduction potential, which, at least to the best of our knowledge, has not been described before for any type of molecule. The results show the importance of the methoxy substitution pattern on the IHB and redox properties of these compounds. Our findings have potential implications in the design of antitumor chalcones.
1. Introduction
Studies concerning the chemistry of molecular radical ions has been an issue of great interest in physical organic chemistry for many years.1–3 It is of fundamental importance to understand their formation, properties and behavior, because radical species participate in several important biochemical process, such as redox enzymatic reactions4,5 and DNA strand break,4,6–8 among others. At the same time, chemical reactions mediated by radical species have received great attention for decades and remain a very active field of research.9–11In this work the center of attention is a series of chalcone derivatives. These molecules are known to have beneficial effects, such as antioxidant and tumor cell growth inhibitory activities, as well as apoptosis inductors in several cancer cell lines. These activities explain the great attention received by chalcones as chemo-preventive agents.12 Particularly interesting is their ability to modify the mitochondrial membrane potential. On the above, it was found that chalcones with fewer hydroxyl groups on the aromatic rings were more effective as mitochondrial uncouplers.13 More recently, the role of the methoxylation in the inhibition potency and cytotoxicity of chalcones towards breast cancer cells has been critically studied.14 Furthermore, it has been demonstrated that 2′-hydroxychalcones induce apoptosis and inhibit invasion of human breast cancer cells.15
It has been suggested that the radical anion of 2′-hydroxychalcones play a central role in the catalyzed and uncatalyzed reactions with prenylflavonoid dienes,16,17 to give biologically active prenylflavonoid derivatives.18,19 The above has prompted the development of biomimetic total synthesis, mostly via catalysis, of these natural products.17,20,21
Few studies that directly relate electrochemical properties with theoretical calculations have been carried out in chalcone type structures. We found a computational study about the electron affinities of substituted chalcones and their comparison with experimental reduction potentials that showed a highly linear correlation, however none of these molecules presented IHB.22 On the other hand, oxidation potential of hydroxychalcones were studied by cyclic voltammetry and some thermodynamic and kinetic parameters compared with theoretical calculations.23 Among their results, the authors claim that the IHB, present in some of the studied structures, does not modify the redox properties, but it does affect the molecular conformation, maintaining planar both, the neutral and the radical species.23
In this work, we study the influence of molecular structure, particularly the role of IHB, on the reduction potential, formation and stability of the radical anion of 2′-hydroxychalcones of pharmacological interest. In a previous study, cyclic voltammetry was employed to quantitatively characterize the formation and stability of the anion radical from three 2′-hydroxychalcones.24
In this work, the molecular structure and electrochemical behaviour of six methoxylated 2′-hydroxychalcones (Fig. 1), which have been previously reported as antitumoral agents, were examined in detail.14,25 First, cyclic voltammetry is used to quantitatively characterize the formation and stability of the anion radical of the series Ch4, Ch5 and Ch6 and the results compared with three previously studied molecules (Ch1–Ch3).24 Then, the molecular structures of all six compounds, particularly their intramolecular hydrogen bond (IHB) O2′–H2′⋯O7′ in the neutral species and in their radical anions, were investigated through DFT calculations. The strength of IHB O2′–H2′⋯O7′ was experimentally assessed through 1H-NMR chemical shift of H2. A comparison of geometrical parameters and frontier orbitals between the neutral and the anion radical is presented, in particular, adiabatic and vertical electron affinities (AEA, VEA) as well as vertical detachment energies (VDE) were calculated. Finally, natural bond orbital (NBO) analysis was used to get insight about the electronic characteristics of the IHB present in these molecules.
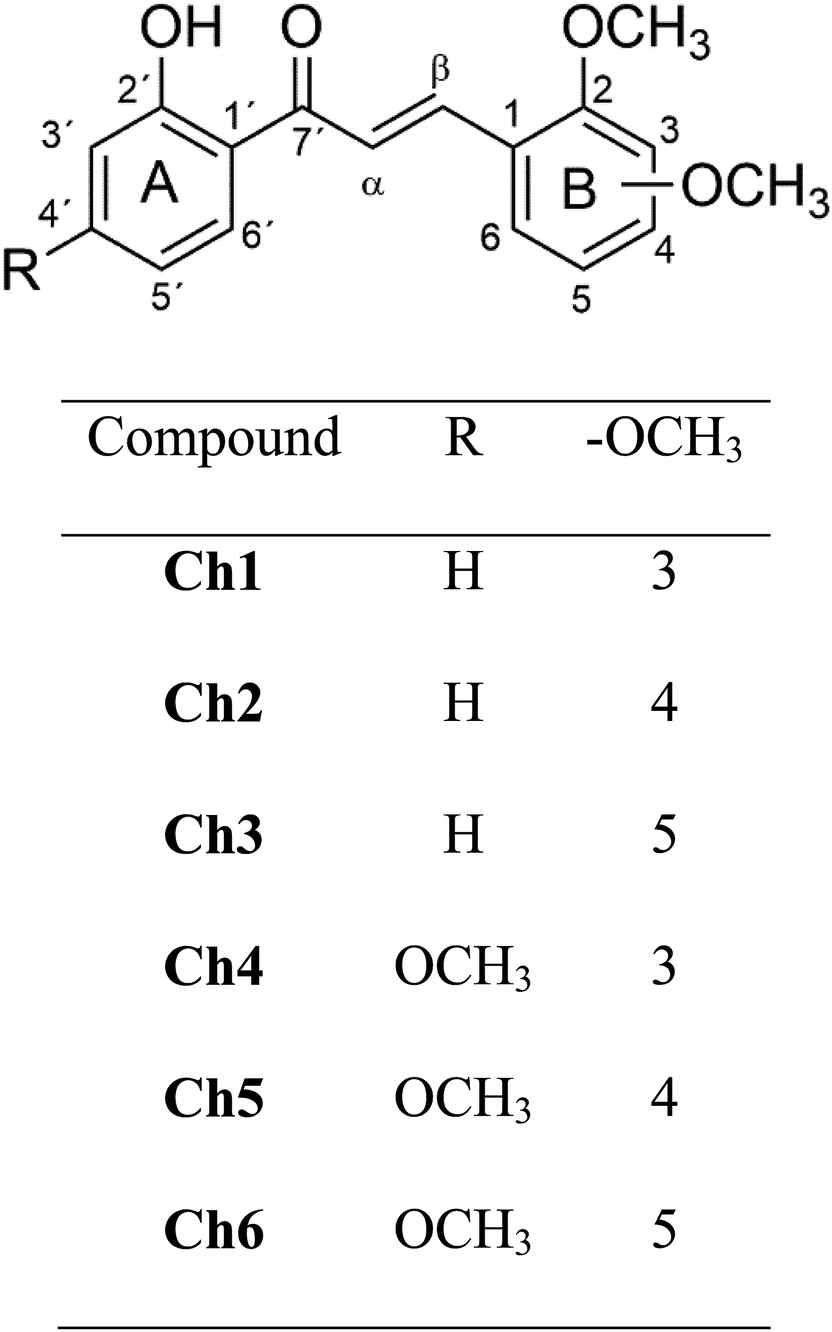 | ||
| Fig. 1 Compounds studied in this work. | ||
2. Experimental section
2.1. Synthesis
Chalcones were prepared following a previously described methodology,24 by adding, in a dropwise manner, a solution of the corresponding substituted benzaldehyde, (7.34 mmol in ethanol, 20 mL) to a stirred mixture of 4-methoxy-2-hydroxyacetophenone solution (7.34 mmol, in ethanol, 20 mL) and potassium hydroxide solution (2 g in 10 mL distilled water). The mixture was allowed to react overnight at room temperature. Then, it was diluted with distilled water (200 mL), neutralized with hydrochloric acid, and extracted four times with ethyl acetate (50 mL). The compounds were crystallized from ethanol. All these compounds have been described before.24,26 1H-NMR spectra were obtained for all compounds in DMSO and along with melting points were used to ensure the identity of the products. In addition, 1H-NMR spectra from Ch3 and Ch4 in CDCl3 were also obtained. 1H-NMR chemical shifts of H2 were used to experimentally assess the strength of IHB O2′–H2′⋯O7′.2.2 Electrochemical experiments
Cyclic Voltammetry (CV) experiments were carried out in DMSO with tetrabutylammonium hexafluorophosphate (TBFP) as supporting electrolyte using a totally automated BAS-100 voltammetric analyzer attached to a PC computer with the BAS 100-W software (v. 2.3), for total control of the experiments, data acquisition and treatment. All CV experiments were carried out with 1.0 mM solution of each chalcone. A hanging mercury drop electrode (HMDE) was used as working electrode (area 2.27 mm2). Platinum wire was used as auxiliary electrode and all potentials were measured against a non aqueous Ag/Ag+ CH Instrument 112 reference electrode. All the electrochemical experiments were obtained after purging the cell with N2 for ten minutes before each run, and the temperature was maintained at 25 ± 1 °C. Resistance was compensated before each experiment. To obtain comparable results within the complete series we choose this methodology, because it is the same previously used with Ch1–Ch3.232.3 Computational details
The calculations were carried out using the Gaussian 03![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 program package, running in a Microsystem cluster of blades. Geometries were optimized at DFT B3P86/6-31+G(d,p) level, using the conductor-like polarizable continuum model (C-PCM) approach to model the role of the solvent, DMSO. This methodology has been proved to be reliable in the study of redox properties of chalcones.28–30 No imaginary vibrational frequencies were found at the optimized geometries, which indicate that they are true minima of the potential energy surface. In solution, DMSO molecules can also act as hydrogen bond acceptors31,32 and thus they compete with the intramolecular hydrogen bond discussed here. It has been shown in previous works that even in this type of proton acceptor solvent, the intramolecular hydrogen bond remains.23,33
27 program package, running in a Microsystem cluster of blades. Geometries were optimized at DFT B3P86/6-31+G(d,p) level, using the conductor-like polarizable continuum model (C-PCM) approach to model the role of the solvent, DMSO. This methodology has been proved to be reliable in the study of redox properties of chalcones.28–30 No imaginary vibrational frequencies were found at the optimized geometries, which indicate that they are true minima of the potential energy surface. In solution, DMSO molecules can also act as hydrogen bond acceptors31,32 and thus they compete with the intramolecular hydrogen bond discussed here. It has been shown in previous works that even in this type of proton acceptor solvent, the intramolecular hydrogen bond remains.23,33
From frequency calculations, zero-point vibrational energies (ZPVE) were obtained and used to correct the calculated enthalpies. The IHB analysis were obtained using hyperconjugation stabilization energies within the framework of NBO analysis.34 The NBO method involves a population analysis, which distributes computed electron density into orbitals in the way chemist think, in terms of physical organic chemistry. The interaction between filled orbitals and empty antibonding orbitals represents the deviation of the molecule from the Lewis structure and can be used as a measurement of delocalization due to the presence of hydrogen bond interaction. These hyperconjugative interactions play an important role in hydrogen bonding. Donor–acceptor interaction energies (stabilization energy, ΔE(2)ij) can be calculated using second-order perturbation theory analysis.34 Natural resonance theory (NRT) calculations,35–37 available from the NBOPro6 software,38 have been used to determine the bond order and the fraction of covalency and ionicity of the O–H⋯O IHB.
Electron affinities and bond dissociation energies were calculated as follow (where H corresponds to enthalpies):39
| Adiabatic electron affinity (AEA) = H(optimized neutral) − H(optimized anion) |
| Vertical electron affinity (VEA) = H(optimized neutral) − H(anion at optimized neutral geometry) |
| Vertical detachment energy (VDE) = H(neutral at optimized anion geometry) − H(optimized anion) |
3. Results and discussion
3.1. Electrochemistry
Chalcones 4–6 were electrochemically reduced on Hg electrode in a non-aqueous medium containing DMSO as solvent and tetrabutylammonium hexafluorophosphate (TBFP) as supporting electrolyte, in the same way as it was previously reported for Ch1–Ch3.24 The cyclic voltammograms were obtained for all three compounds, showing a behavior for the redox process similar to that observed in Ch1–Ch324 (Fig. 2).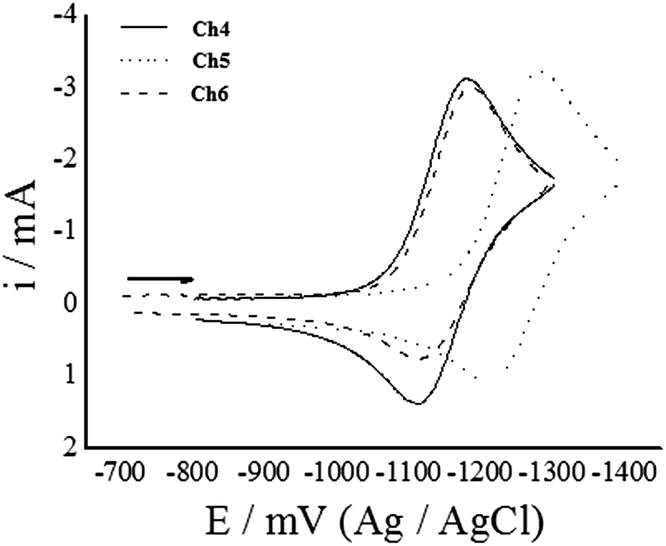 | ||
| Fig. 2 Cyclic voltammogram of 1 mM 2′-hydroxy chalcone derivatives in DMSO + 0.1 M TBPF at 1.0 V s−1. | ||
When the reduction potentials are compared (Table 1), it can be noticed that Ch5 needs more energy for reducing than Ch4 and Ch6. Similar behavior was reported for Ch1–Ch3,24 where Ch2 needs more energy than Ch1 and Ch3 to be reduced. A systematic decrease in the reduction potential is observed for Ch4–Ch6 when compared with their respective analogues Ch1–Ch3, indicating that the presence of the –OCH3 substituent, in para position with respect to the carbonyl on ring A, increases the energy of the molecular electronic state hindering the electron capture.
| Compound | −Epc (mV) | k2 × 10−3 (M−1 s−1) | t1/2 (s) |
|---|---|---|---|
| Ch1 | 1096 | 5.6 ± 0.30 | 0.18 |
| Ch2 | 1187 | 30.5 ± 0.30 | 0.03 |
| Ch3 | 1110 | 40.5 ± 0.27 | 0.03 |
| Ch4 | 1180 | 4.4 ± 0.30 | 0.21 |
| Ch5 | 1282 | 18.5 ± 0.26 | 0.05 |
| Ch6 | 1184 | 20.8 ± 0.14 | 0.04 |
Under the mentioned experimental conditions, the one electron reduction process produces very well resolved cyclic voltammograms due to the formation of the anion radicals. When the concentration of the species in solution increased, an increase in the magnitude of the current for the couple was observed (Fig. 3A). The stability of the anion radical depends mainly on the reaction media, and can be monitored by following the decay of the corresponding oxidation current, Ipa, subsequent to the electrochemical generation (return sweep). In effect, a suitable measurement of the stability of the radical anion is expressed by the current ratio parameter, Ipa/Ipc, which reveals the tendency of an electrochemically generated species, i.e. the anion radical, to undergo subsequent chemical reactions.40 The current ratio equals to one in the absence of further reactions but decreases if the radical subsequently reacts.
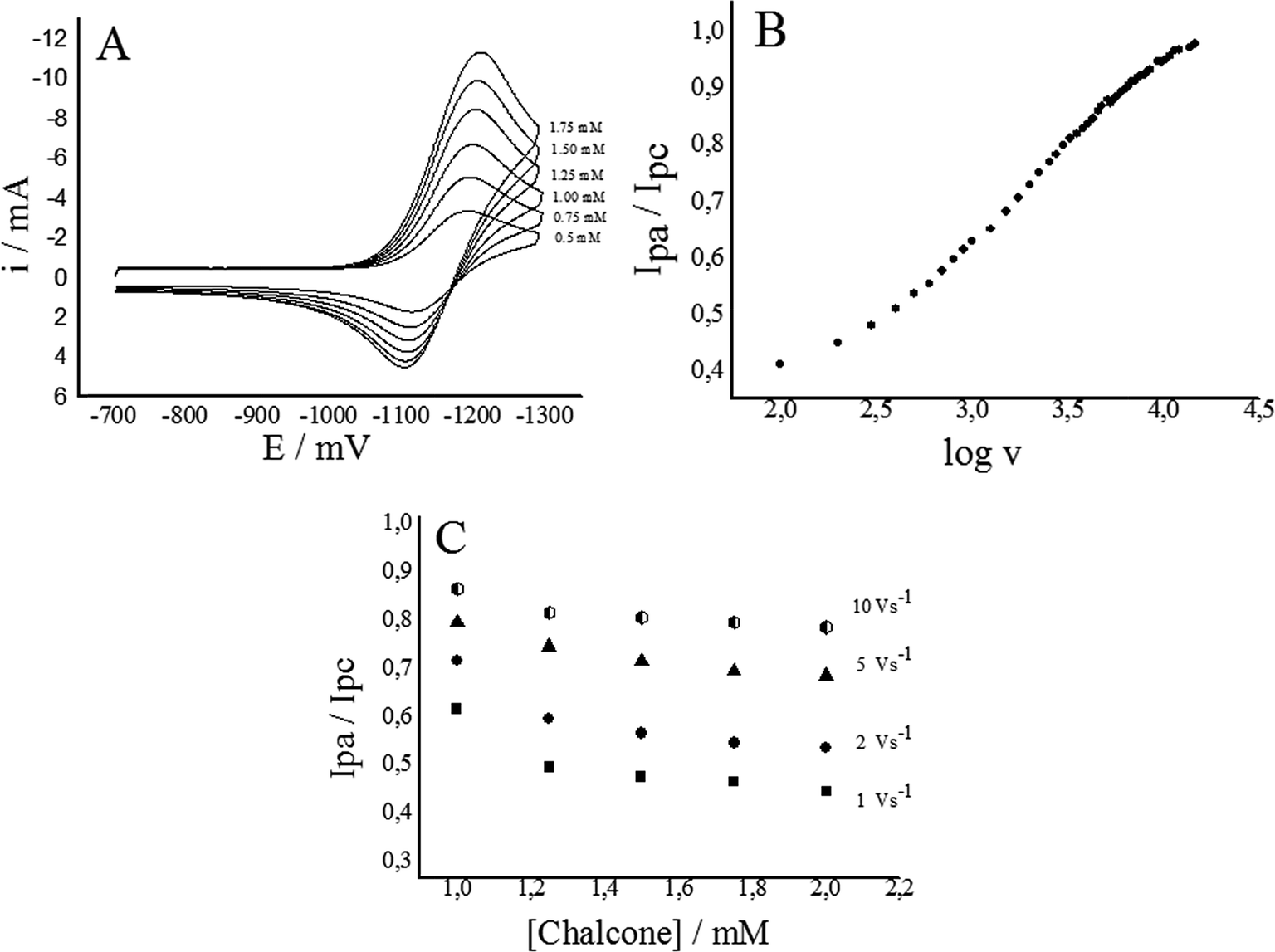 | ||
| Fig. 3 (A) Cyclic voltammogram for chalcone 4 at different concentrations, with a scan rate of 5 V s−1. (B) Dependence of the current ratio with the sweep rate for the one-electron redox couple of chalcone 4. (C) Current ratio dependence of chalcone 4 at different concentrations and different sweep rates. All experiments were performed in DMSO and 0.1 M TBPF. | ||
The calculation of the ratios Ipa/Ipc for the three species show that when the scan rate increases, it takes values close to 1 (Fig. 3B), but when the concentration increases, the ratio of currents decreases (Fig. 3C), indicative of a non-reversible process.
3.2. DFT calculations
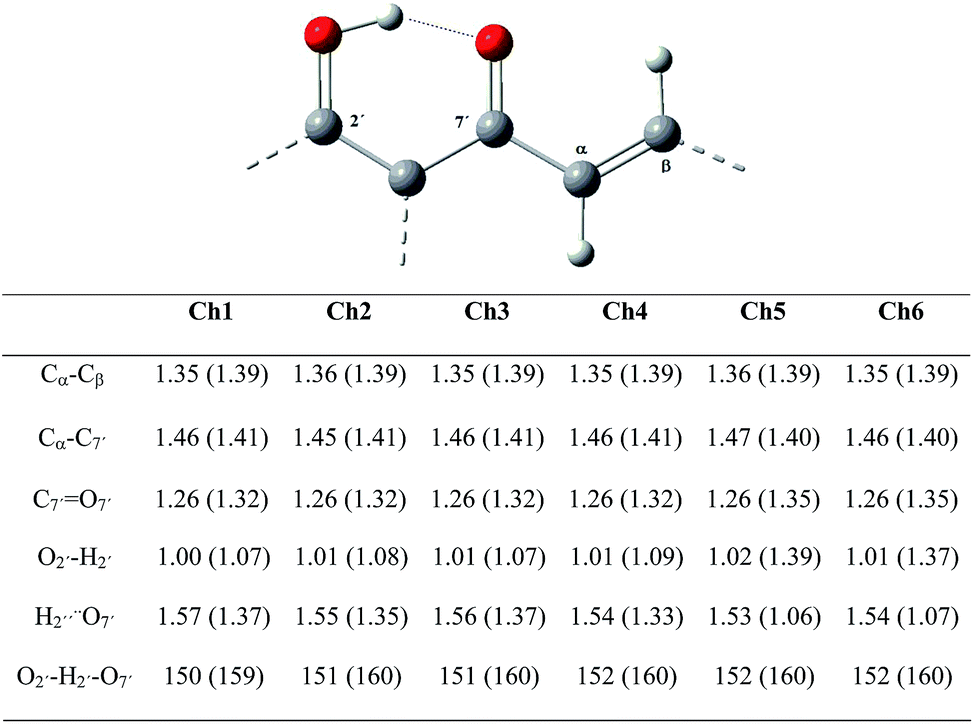 | ||
| Fig. 4 Main geometrical parameters for neutral and radical anion chalcones. | ||
| AEA | VEA | VDE | |
|---|---|---|---|
| a AEAs, VEAs and VDEs values in eV. | |||
| Ch1 | 3.78 | 3.59 | 4.03 |
| Ch2 | 3.67 | 3.49 | 3.88 |
| Ch3 | 3.78 | 3.60 | 4.01 |
| Ch4 | 3.74 | 3.48 | 3.95 |
| Ch5 | 3.61 | 3.38 | 3.92 |
| Ch6 | 3.73 | 3.49 | 4.07 |
As can be observed from the table attached to Fig. 4, most geometrical parameters do not show significant differences between the neutral and the anion radical species, for all chalcones. Nevertheless, the Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ– bond increases its length by 0.03–0.04 Å, while the Cα–C7′ bond shortens by 0.04–0.07 Å in the anion radicals. Carbonyl C7′O7′ also present a bond lengthening of 0.06–0.09 Å. The only notable geometrical differences between the neutral species and the anion radicals were found for parameters corresponding to the IHB, for instance, the distance O2′–H2′ lengthens by 0.06–0.08 Å for Ch1–Ch4 and notably by 0.37–0.36 Å for Ch5 and Ch6, while oppositely, the distance H2′⋯O7′, shortens by 0.20–0.21 Å for Ch1–Ch4 and 0.47 Å for both Ch5 and Ch6. In addition, the angle O2′–H2′–O7′ increased from 150–152° to 159–160° for all chalcones. These results indicate that the IHB and the double bond are mostly affected upon the electron attachment.
Cβ– bond increases its length by 0.03–0.04 Å, while the Cα–C7′ bond shortens by 0.04–0.07 Å in the anion radicals. Carbonyl C7′O7′ also present a bond lengthening of 0.06–0.09 Å. The only notable geometrical differences between the neutral species and the anion radicals were found for parameters corresponding to the IHB, for instance, the distance O2′–H2′ lengthens by 0.06–0.08 Å for Ch1–Ch4 and notably by 0.37–0.36 Å for Ch5 and Ch6, while oppositely, the distance H2′⋯O7′, shortens by 0.20–0.21 Å for Ch1–Ch4 and 0.47 Å for both Ch5 and Ch6. In addition, the angle O2′–H2′–O7′ increased from 150–152° to 159–160° for all chalcones. These results indicate that the IHB and the double bond are mostly affected upon the electron attachment.
Particularly interesting is that the presence of a para-methoxy substituent on position 4′ of ring A, along with the presence of a methoxy substituent on ring B at C4 or C5 in the anion radical, shortens the H2′′⋯O7 distance (lengthening the O2′–H2′ distance) to a point in which H1′⋯O7 could be consider a covalent bond, in particular for Ch5 and Ch6. However, the latter do not occur for Ch4, which exhibit the methoxy group in C3. Even though both, Ch4 and Ch6 present a methoxy substituent in ortho- and meta-position with regard to the double bond, only the ortho-methoxy group can deliver electron density to the C7′O7′ bond through conjugation with the Cα–Cβ double bond. In the case of Ch4, both methoxy groups are neighbors, which can affect the ability of the methoxy in C2 to remain in the ring plane and deliver electron density to the C7′O7′ bond, reducing the effect exhibited in Ch5 and Ch6. Further bond order analysis (vide infra) helps to rationalizes these results.
Table 2 shows the values of AEA, VEA and VDE calculated for Ch1–Ch6. If the VEA is positive, the attachment of the electron to the molecule is energetically favorable, even before geometrical rearrangements occur, i.e. the molecule can attract the excess of charge density. On the other hand, a positive AEA indicates that the formed anion is stable in its final geometry, i.e. that once the electron is trapped by the molecule, it stays there long enough to rearrange and possibly to play a role in chemical reactions. When VDE is positive the energy of the neutral molecule is higher than their anion, and it is stable with respect to the vertical electron auto-detachment. VDE can be interpreted as the vertical ionization potential of the anion. If the nuclear configuration of the anion and the neutral molecule are similar, the values of VEA and VDE can be understood as lower and upper bounds of the AEA, as can be seen in Table 2. Only in the case that electron attachment generates a significant change in geometry, VDE could be lower than AEA.41
We found that all AEAs are positive, indicating a favorable tendency to form stable anion radicals in this series. For Ch1–Ch3 calculated AEAs correlate well with observed reduction potentials, with Ch2 showing the lowest potential and the lowest AEA. The same tendency is observed for Ch4–Ch6, where Ch5 also possesses the lowest potential and the lowest AEA. These results indicate that the presence of a methoxy substituent in C4 of ring B (Ch2 and Ch5) decreases the ability of chalcones to form a stable anion radical. When Ch1–Ch3 are compared with Ch4–Ch5, they present higher AEA and higher reduction potential. These results indicate that the presence of a methoxy group at C4′ of ring A decreases the ability of the molecule to form a stable anion radical. On the other hand, all VEAs are positive, which indicate that all molecules have favorable tendency to take an electron. The effects of substituents have a similar impact for VEAs than for AEAs, which indicate that the presence of a methoxy groups at C4′ of ring A and C4 of ring B decrease the ability of the molecule to take an electron and form a stable anion radical. Besides, all VDEs are positive, which indicates that electron auto-detachment from the anion radical is unlikely. In this case it is observed that the presence of a methoxy substituent at C4 of ring B (Ch2 and Ch5) increases the possibility of an electron auto-detachment from the anion radical, but does not affect in the same way to Ch4 and Ch6. Ch4 present a lower VDE than their analog Ch1, but Ch5 and Ch6 present a higher VDE than Ch2 and Ch3, which indicates that a methoxy substituent at C4 of ring B decreases the capability of suffering an electron auto-detachment in Ch4, but increases in Ch5 and Ch6.
The attachment of an extra electron to a neutral molecule leads to one of two different types of anions, valence-bound or dipole-bound anion.42 In the first case, the attached electron is strongly bound occupying a valence molecular orbital. In the second case, the attached electron is weakly bound, mainly by electrostatic charge–dipole interactions.43 Therefore, a valence-bound anion suffer a much notable effect on their structural parameters than a dipole-bound anion.44,45 In our cases, we found that the differences between AEAs and VEAs is in all cases 0.2–0.3 eV. These differences indicate that the radical anions formed in all molecules should be valence-bound anions, since a difference energy of ∼0.3 eV is close to the expected amount for relaxation energy of a valence-bound anion.39,46 With the purpose to study the distribution of the excess electron density of the anion radicals, the singly occupied molecular orbitals (SOMOs) of the stable and vertically attached electron were obtained (see Fig. 1 and 2 of ESI†). This approach also helps to distinguish between valence-bound and dipole-bound anions, given that SOMO of a dipole-bound anion is centered in a limited part of the molecule, while SOMO of a valence-bound anion is distributed throughout the whole molecule.6,39,46 All anion radicals display valence-bound character, confirming the above statement about the difference between AEAs and VEAs.
Correlations between the experimental reduction potentials (−Epc) and calculated AEAs and VEAs are presented in Fig. 5a and b respectively. This kind of correlations have been made for different kind of compounds47–49 and an interpretation of the results can be carried out from the work of Parker.50 We found that the correlation between VEAs and experimental reduction potentials (R2 = 0.98) was better than the correlation with AEAs (R2 = 0.88). The above result suggests that the reduction potential depends more on the ability of taking an extra electron than on the ability to form a stable anion radical. The slopes in both cases are close to one, which indicate the absence of secondary effects such as differential solvation and relaxation of the resulting anions. The results suggest that the different methoxy substitution patterns of these chalcones do not modify such secondary effects which remain fairly constant, regardless the electron affinities.
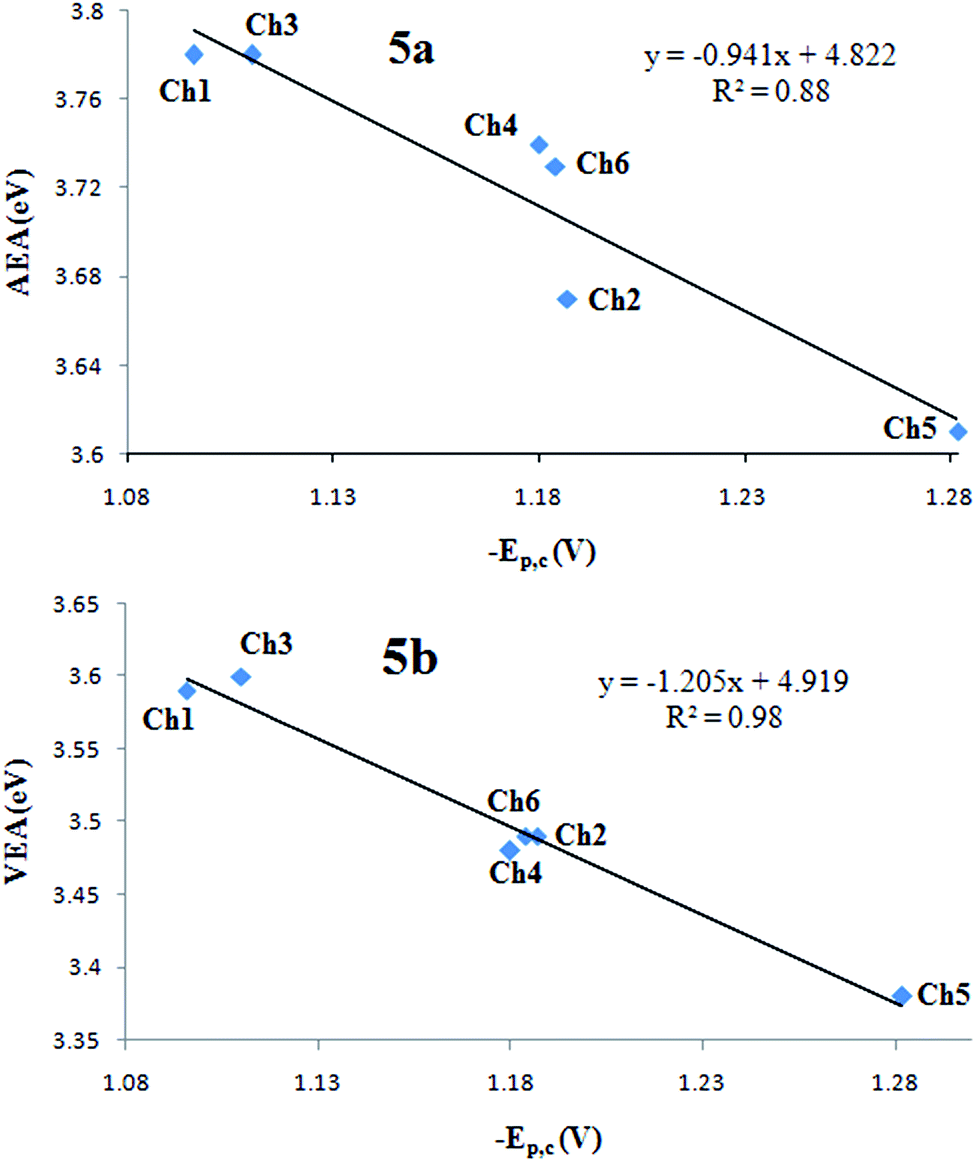 | ||
| Fig. 5 Correlation graphics of experimental reduction potentials vs. theoretical AEAs (a) and VEAs (b). | ||
These results support the reliability of our calculations and are in agreement with previously reported works, which states that the energetics of the reduction process should be controlled by the first single-electron uptake.48–51
| Molecule | δH2′ DMSO-d6 (CDCl3) | Φi | Φj | ΔE(2)ij | εj − εi/au | Fij/au |
|---|---|---|---|---|---|---|
| Ch1 | 12.40 | LP1O7′ | σ*O2′–H2′ | 4.82 | 1.54 | 0.078 |
| LP2O7′ | σ*O2′–H2′ | 39.12 | 1.18 | 0.194 | ||
| Ch2 | 12.82 | LP1O7′ | σ*O2′–H2′ | 4.90 | 1.53 | 0.078 |
| LP2O7′ | σ*O2′–H2′ | 42.82 | 1.18 | 0.203 | ||
| Ch3 | 12.62 | LP1O7′ | σ*O2′–H2′ | 4.87 | 1.54 | 0.078 |
| LP2O7′ | σ*O2′–H2′ | 40.13 | 1.18 | 0.197 | ||
| Ch4 | 13.42 | LP1O7′ | σ*O2′–H2′ | 4.88 | 1.52 | 0.078 |
| (13.43) | LP2O7′ | σ*O2′–H2′ | 44.75 | 1.17 | 0.207 | |
| Ch5 | 13.67 | LP1O7′ | σ*O2′–H2′ | 4.93 | 1.51 | 0.078 |
| (13.68) | LP2O7′ | σ*O2′–H2′ | 48.06 | 1.17 | 0.214 | |
| Ch6 | 13.51 | LP1O7′ | σ*O2′–H2′ | 4.89 | 1.52 | 0.078 |
| LP2O7′ | σ*O2′–H2′ | 45.52 | 1.17 | 0.209 |
Few quantitative relationships of NBO second-order stabilization energy with other characteristic parameters of the hydrogen bond have been reported. Senthilkumar et al.53 showed a quantitative correlation between NBO energies and calculated hydrogen bond distances at B3LYP and MP2 level, in formic acid dimers. Also, recently we found good correlations among the experimental 1H-NMR chemical shift of O–H,52 electrochemical oxidation potentials54 and NBO stabilization energy for the interaction O–H⋯O–C, in a series of hydroquinones possessing IHB. As far as we know, there are no other reports in the literature about a quantitative correlation between experimental IHB parameters and calculated NBO energies. Fig. 6a shows a good correlation between the 1H-NMR chemical shift of H2′ and the ΔE(2)ij for IHB. Having shown that this stabilization energy is a good parameter for the IHB strength in these chalcones, we study their correlation with the experimental reduction potentials (−Epc) and the results are displayed in Fig. 6b. As observed from this figure, there exist a good correlation between the strength of the IHB and the experimental reduction potentials (R2 = 0.90). This correlation allow us to establish that, in this series of chalcone derivatives, the IHB strength is a very important factor to determine their capability to attach an extra electron, a remarkable observation.
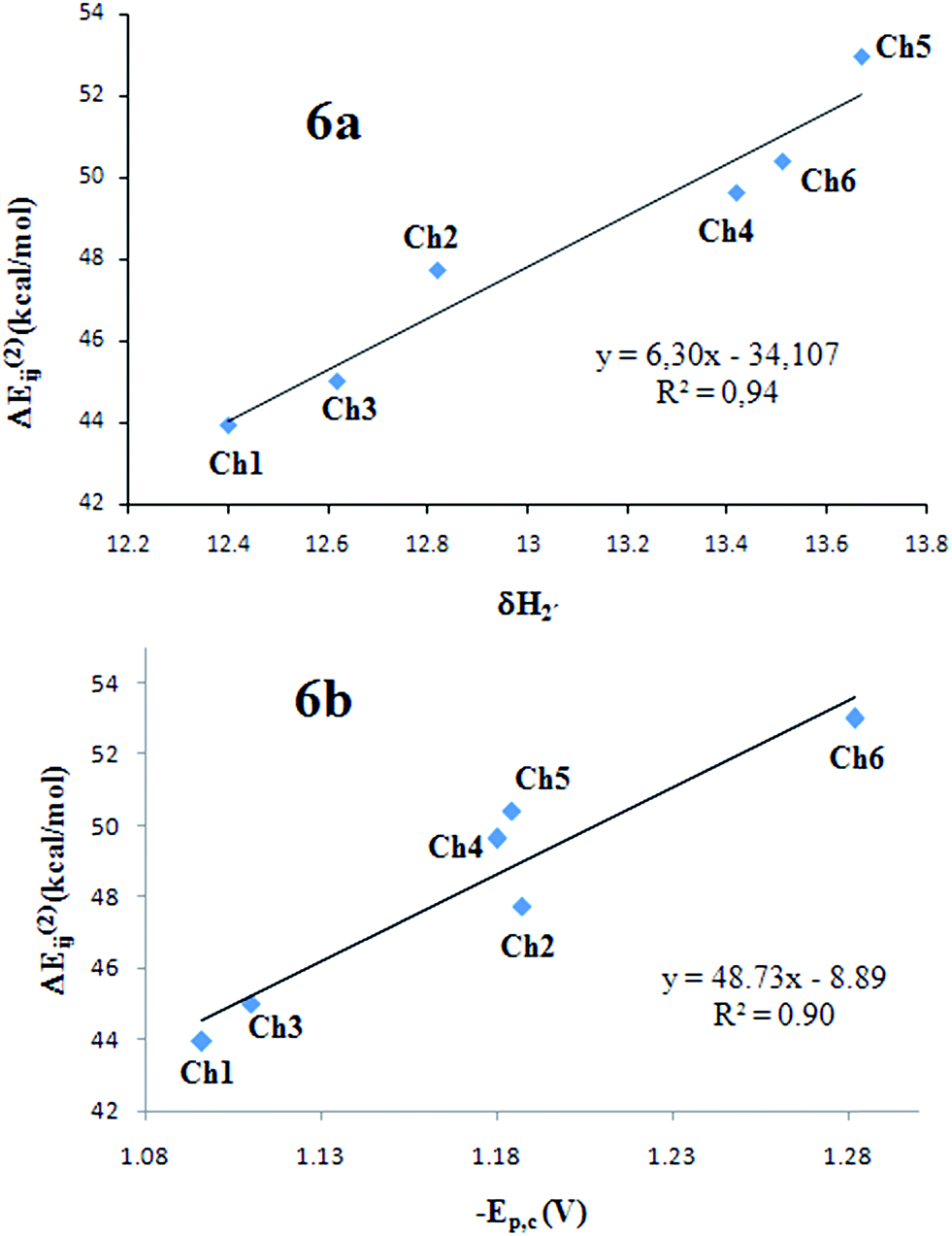 | ||
| Fig. 6 Correlations between experimental chemical shift for δH2′ in DMSO-d6 (a) and reduction potentials (b) vs. NBO stabilization energies for LP → σ* interactions in IHB O7′⋯H2–O2′. | ||
NBO method, besides determining the second-order stabilization energies, also allow us to carry out a bond order analysis and to establish the covalency and ionicity of the bonds, based in the natural resonance theory (NRT, for details see references).35–37,55 We focused on calculating the bond order and the degree of covalency and ionicity for the bonds involved in the O2′–H2′⋯O7′ IHB, both, in the neutral and the anion radical species. There were three resonance structures considered by the NBO programme, third resonance structures were neglected from all the analysis, since they represented less than 2% in all cases. Fig. 7 presents the resonance structures 1 and 2, along with the main data from NRT calculations. Resonance structure 1 was always the major contributor to the neutral species (∼95%) and has been consistently major for Ch1 to Ch3 than Ch4 to Ch6. The latter observation is supported by the small but consistent bond order differences for O–H in Ch1 to Ch3 when compared with Ch4 to Ch6 (see table attached to Fig. 7). The O–H bond for all chalcones has a low covalent character, around 40%. On the other hand, as can be expected, the bond orders for H2′⋯O7′ are lower than for O2′–H2′, being higher for Ch4 to Ch6 than for Ch1 to Ch3. The ionic character dominates this bond in all cases (96–97%).
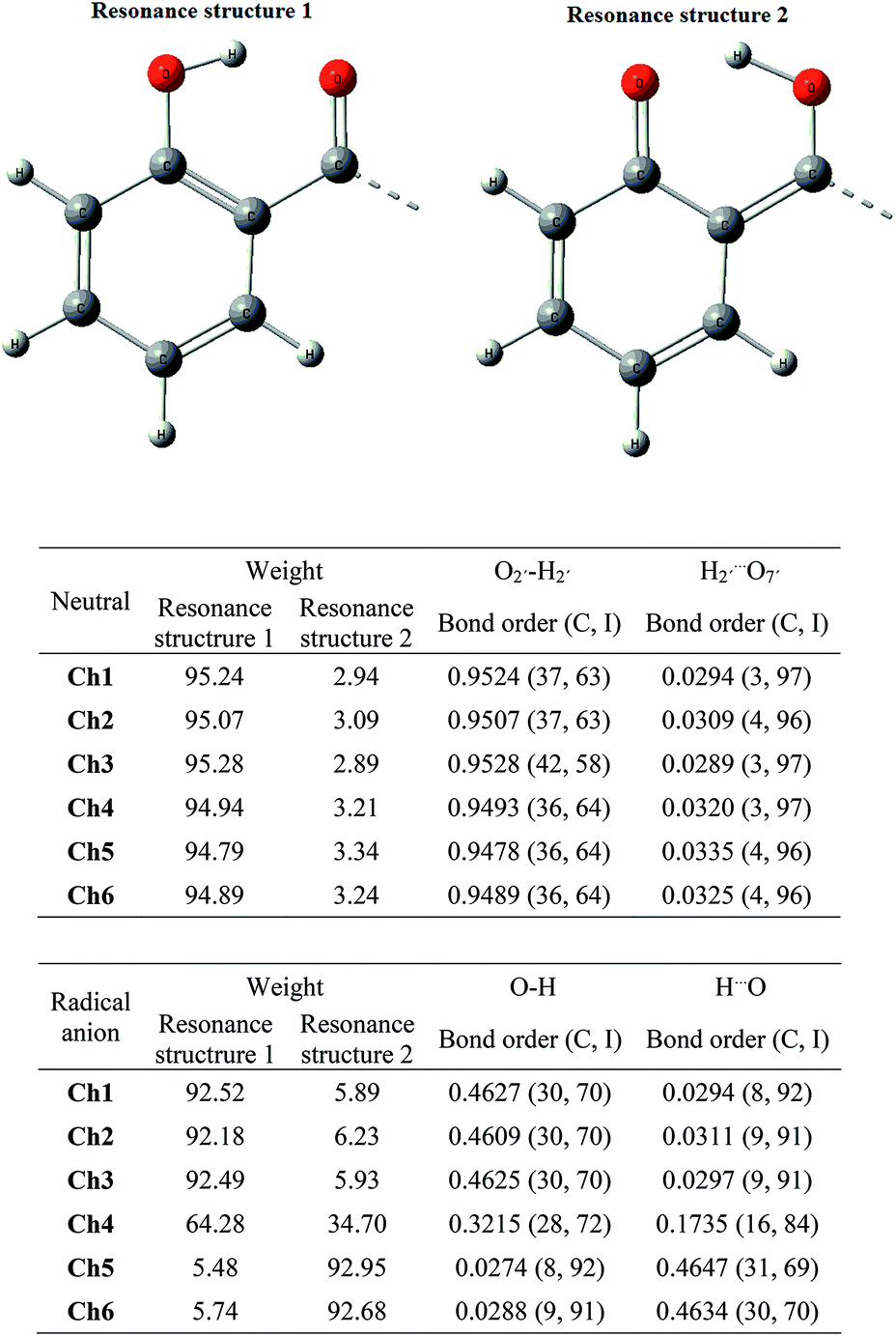 | ||
| Fig. 7 Resonance structures and main data from NRT calculation. | ||
When the results for the anion radicals are examined, a very different behaviour is observed between Ch1–Ch3 and Ch4–Ch6. Weights of resonance structures for Ch1 to Ch3 are very similar to those that have the neutral species, ∼92% for resonance structure 1 and ∼6% for resonance structure 2. Natural bond orders for O2′–H2′ of Ch1–Ch3 are ∼0.46, with a 30% of covalency and 70% of ionicity, while H2′⋯O7′ present a natural bond order around 0.0300 with ∼91% of ionicity for all three cases. On the other hand, for Ch4 the resonance structure 2 takes a much greater weight than for Ch1 to Ch3, reaching ∼34%, while for Ch4 and Ch5 the resonance structure 2 becomes the dominant one, with a 93% for both. Natural bond order and covalency of O2′–H2′ for Ch4 decreases slightly, compared with Ch1 to Ch3, to values of 0.3214 and 28% respectively. However, both natural bond order and covalency notably decrease to ∼0.0280 and 8% respectively for Ch5 and Ch6. Moreover, H2′⋯O2′ bond in Ch4 presents a notable increases in their natural bond order and even more for Ch5 and Ch6. The above was accompanied by a decrease in ionic character and an increase in covalent character, being more important for Ch5 and Ch6. Taking together, these results evidence that the presence of the methoxy substituents on ring B do not significantly affect the IHB in the radical anion when compared with the neutral molecule. Besides, the presence of methoxy substituent in C4′ of ring A has a high impact, modifying the IHB of the anion radical compared with the neutral molecule, being more different for Ch4 than for Ch5 and Ch6. The later suggests that the difference described above in the values of VDEs between Ch4 and Ch5–Ch6 is closely related to their difference in the radical anion IHB structure. There is a switching of the minimum in the potential energy surface along the IHB coordinate.
3.3 Implications with the biological activities of 2′-hydroxychalones
The results of this work provide insights about the effect of methoxy substituents on the IHB properties of Ch1–Ch6 and their ability to take an electron and generate a stable anion radical. As we mentioned in the introduction, previous works have shown that this chalcones can inhibit the proliferation of different cancer cells lines,14,25,26 where the presence of methoxy substituents seems to play a critical role. It has been proposed that some of the anti-proliferative activities of these compounds is related to their capability of inducing mitochondrial mediated apoptosis.25 The ability of chalcones to generate radical species could impact, for example, in their capability to modify the mitochondrial membrane potential, inhibit the electron chain or affect their ability to act as uncoupler of the oxidative phosphorylation.13,15Our results suggest that if it is necessary to increase the ability of the chalcone to form an anion radical and their persistency in time, the presence of the methoxy substituent in C4 of ring B should be avoided; their presence do not significantly affect the IHB in the radical anion. On the other hand, the presence of a methoxy substituent in C4′ has opposite impact in neutral and radical anion species. For the neutral specie, it decreases the ability to form a radical anion, but once it is formed, it presents lower possibility to return back to the neutral form. The above can have a high impact on the IHB, making it weaker for the anion radical than for the neutral species.
Our results also suggest that in addition to the predictability of the AEAs and VEAs to determine the reduction potential of these 2′-hydroxychalcone derivatives, the strength of the IHB is also an acceptable parameter to predict this important experimental data, which has been widely related to different biological activities. Nevertheless, this relationships needs to be tested for a more extensive series of chalcones to establish their actual reliability.
4. Conclusions
Cyclic voltammetry is a very useful technique to investigate the formation and stability of anion radicals from chalcone derivatives. We found that the presence of a –OCH3 substituent in para position with respect to the carbonyl on ring A, decreases the energy of the electronic state of the anion radical when compared with derivatives without this substituent. This is closely related with the energy of the molecular orbital where the extra electron is going to be placed. The calculation of the ratios Ipa/Ipc for Ch4–Ch6 shows that, as expected, the formed radicals appears to be stable at low concentration, but when the concentration increases they experience subsequent reactions.DFT results show that the IHB O2′–H2′⋯O7′ and the double bond CαCβ are the most affected regions upon the electron attachment process. From the analysis of the AEAs, VEAs and VDEs, we found that all chalcones can take an electron and form a stable radical anion. Besides the auto-detachment process is very unlikely for all. The presence of methoxy groups in C4 of ring B and in C4′ of ring A decreases the ability to take an electron and form a stable radical anion. On the other hand, the presence of a methoxy group in C4 of ring B, increases the ability to experience an electron auto-detachment by the radical anion, in all cases. However, the presence of a methoxy group at C4′ of ring A has a different affect on Ch4 that on Ch5 and Ch6. For Ch4 the presence of a methoxy decreases the trend to experience an electron auto-detachment once the anion radical is formed, but for Ch5 and Ch6 this trend increases.
VEAs exhibit a better correlation than AEAs, with experimental reduction potentials which suggests that it is more influenced by the capability of taking the extra electron than by forming a stable anion radical. The slopes of the correlations, close to one, strongly suggests the absence of secondary effects, such as differential solvation and relaxation of the resulting anions. These indicate that the different methoxy substituents patterns of these chalcones does not modify such secondary effects, which remain fairly constant, regardless the electro affinity values.
Analyses of the second order stabilization NBO energies (ΔE(2)ij) for the IHBs, allows us to quantify the strength of the donor–acceptor orbital interaction. A comparison with 1H-NMR data shows that NBO energy is a good parameter to measure the strength of the IHB. Stabilization energies for the IHBs are pointing in the same direction that reduction potentials, and they present a good semi-quantitative correlation (R2 = 0.90), which indicates that the strength of IHB has a significant influence in the reduction potential of these chalcones.
Natural bond order analysis shows that the presence of a methoxy substituent on ring B does not significantly affect the IHB in the radical anion, when compared with the neutral species. On the other hand, the presence of a methoxy substituent at C4′ of ring A has a significant impact weakening the IHB of the radical anion when compared with the neutral species.
Our results highlight the possibility of establishing direct links between molecular structure details, such as the position of substituents on the aromatic rings and the presence of intramolecular hydrogen bonds, with the electrochemical behavior of methoxylated 2-hydroxychalcones. We hope this work drives the search for similar correlations in a broader spectrum of molecules engaged in IHB, and can be useful in the design of new chalcones with potential pharmacological activity.
Acknowledgements
We are grateful to FONDECYT grants 1140753, FONDECYT/POSTDOCTORADO 3140286 (M.M-C) and ACT grant 1107.References
- Z. V. Todres, Organic Ion Radicals, Marcel Decker, New York, 2003 Search PubMed
.
- V. V. Roznyatovskiy, D. M. Gardner, S. W. Eaton and M. R. Wasielewski, Org. Lett., 2014, 16, 696–699 CrossRef CAS PubMed
- B. Chen, D. A. Hrovat, S. H. M. Deng, J. Zhang, X. B. Wang and W. T. Borden, J. Am. Chem. Soc., 2014, 136, 3589–3596 CrossRef CAS PubMed
- P. A. Frey, A. D. Hegeman and G. H. Reed, Chem. Rev., 2006, 106, 3302–3316 CrossRef CAS PubMed
- M. Salamone, G. A. DiLabio and M. Bietti, J. Org. Chem., 2012, 77, 10479–10487 CrossRef CAS PubMed
- X. G. Bao, J. Wang, J. D. Gu and J. Leszczynski, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5658–5663 CrossRef CAS PubMed
- A. Kumar and M. D. Sevilla, ChemPhysChem, 2009, 10, 1426–1430 CrossRef CAS PubMed
- J. D. Gu, Y. M. Xie and H. F. Schaefer, J. Am. Chem. Soc., 2006, 128, 1250–1252 CrossRef CAS PubMed
- J. L. G. Ruano, J. Aleman, A. Parra and L. Marzo, Eur. J. Org. Chem., 2014, 2014, 1577–1588 CrossRef PubMed
- X. L. Zheng, L. Yang, W. Y. Du, A. S. Ding and H. Guo, Chem.–Asian J., 2014, 9, 439–442 CrossRef CAS PubMed
- I. Thome, C. Besson, T. Kleine and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 7509–7513 CrossRef CAS PubMed
- S. M. Kuo, Crit. Rev. Oncog., 1997, 8, 47–69 CrossRef CAS
- O. Sabzevari, G. Galati, M. Y. Moridani, A. Siraki and P. J. O'Brien, Chem.–Biol. Interact., 2004, 148, 57–67 CrossRef CAS PubMed
- G. Valdameri, C. Gauthier, R. Terreux, R. Kachadourian, B. J. Day, S. M. B. Winnischofer, M. E. M. Rocha, V. Frachet, X. Ronot, A. Di Pietro and A. Boumendjel, J. Med. Chem., 2012, 55, 3193–3200 CrossRef CAS PubMed
- S. Y. Kim, I. S. Lee and A. Moon, Chem.–Biol. Interact., 2013, 203, 565–572 CrossRef CAS PubMed
- Y. Roh, H. Y. Jang, V. Lynch, N. L. Bauld and M. J. Krische, Org. Lett., 2002, 4, 611–613 CrossRef CAS PubMed
- H. Cong, D. Ledbetter, G. T. Rowe, J. P. Caradonna and J. A. Porco, J. Am. Chem. Soc., 2008, 130, 9214–9215 CrossRef CAS PubMed
- F. Ferrari, F. Delle Monache, A. I. Suarez and R. S. Compagnone, Fitoterapia, 2000, 71, 213–215 CrossRef CAS
- E. M. Stocking and R. M. Williams, Angew. Chem., Int. Ed., 2003, 42, 3078–3115 CrossRef CAS PubMed
- C. M. Brito, D. Pinto, A. M. S. Silva, A. M. G. Silva, A. C. Tome and J. A. S. Cavaleiro, Eur. J. Org. Chem., 2006, 2558–2569 CrossRef CAS PubMed
- C. Qi, H. Cong, K. J. Cahill, P. Muller, R. P. Johnson and J. A. Porco, Angew. Chem., Int. Ed., 2013, 52, 8345–8348 CrossRef CAS PubMed
- L. D. Hicks, A. J. Fry and V. C. Kurzweil, Electrochim. Acta, 2004, 50, 1039–1047 CrossRef CAS PubMed
- N. Cotelle, P. Hapiot, J. Pinson, C. Rolando and H. Vezin, J. Phys. Chem. B, 2005, 109, 23720–23729 CrossRef CAS PubMed
- P. Quintana-Espinoza, C. Yanez, C. A. Escobar, D. Sicker, R. Araya-Maturana and J. A. Squella, Electroanalysis, 2006, 18, 521–525 CrossRef CAS PubMed
- C. Echeverria, J. Francisco Santibanez, O. Donoso-Tauda, C. A. Escobar and R. Ramirez-Tagle, Int. J. Mol. Sci., 2009, 10, 221–231 CrossRef CAS PubMed
- A. Boumendjel, J. Boccard, P. A. Carrupt, E. Nicolle, M. Blanc, A. Geze, L. Choisnard, D. Wouessidjewe, E. L. Matera and C. Dumontet, J. Med. Chem., 2008, 51, 2307–2310 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin and J. C. Burant, et al., Gaussian 03, Revision E. 01, Inc., Wallingford CT, 2004 Search PubMed
- D. Kozlowski, P. Trouillas, C. Calliste, P. Marsal, R. Lazzaroni and J. L. Duroux, J. Phys. Chem. A, 2007, 111, 1138–1145 CrossRef CAS PubMed
- E. Anouar, S. A. A. Shah, N. B. Hassan, N. El Moussaoui, R. Ahmad, M. Zulkefeli and J. F. F. Weber, Molecules, 2014, 19, 3489–3507 CrossRef PubMed
- Y. Z. Rong, Z. W. Wang and B. Zhao, Food Biophys., 2013, 8, 250–255 CrossRef
- M. J. Bausch, R. Gostowski, C. Guadalupefasano, D. Selmarten, A. Vaughn and L. H. Wang, J. Org. Chem., 1991, 56, 7191–7193 CrossRef CAS
- F. G. Bordwell, R. J. McCallum and W. N. Olmstead, J. Org. Chem., 1984, 49, 1424–1427 CrossRef CAS
- Y. Fu, R. Liu, L. Liu and Q. X. Guo, J. Phys. Org. Chem., 2004, 17, 282–288 CrossRef CAS PubMed
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 610–627 CrossRef CAS
- E. D. Glendening, J. K. Badenhoop and F. Weinhold, J. Comput. Chem., 1998, 19, 628–646 CrossRef CAS
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013, http://nbo6.chem.wisc.edu/ Search PubMed
- H. N. Liu, L. A. Walker and R. J. Doerksen, Chem. Res. Toxicol., 2011, 24, 1476–1485 CrossRef CAS PubMed
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS
- T. J. VanHuis, J. M. Galbraith and H. F. Schaefer, Mol. Phys., 1996, 89, 607–631 CrossRef CAS PubMed
- J. Simoms and K. Jordan, Chem. Rev., 1987, 87, 535–555 CrossRef
- D. Svozil, P. Jungwirth and Z. Havlas, Collect. Czech. Chem. Commun., 2004, 69, 1395–1428 CrossRef CAS
- C. Desfrancois, H. AbdoulCarime and J. P. Schermann, Int.
J. Mod. Phys. B, 1996, 10, 1339–1395 CrossRef CAS
- M. Gutowski and P. Skurski, J. Phys. Chem. B, 1997, 101, 9143–9146 CrossRef CAS
- X. F. Li, Z. L. Cai and M. D. Sevilla, J. Phys. Chem. A, 2002, 106, 1596–1603 CrossRef CAS
- Y. A. Getmanenko, C. Risko, P. Tongwa, E. G. Kim, H. Li, B. Sandhu, T. Timofeeva, J. L. Bredas and S. R. Marder, J. Org. Chem., 2011, 76, 2660–2671 CrossRef CAS PubMed
- N. Florini, M. Michelazzi, F. Parenti, A. Mucci, M. Sola, C. Baratti, V. De Renzi, K. Daasbjerg, S. U. Pedersen and C. Fontanesi, J. Electroanal. Chem., 2013, 710, 41–47 CrossRef CAS PubMed
- K. L. Phillips, S. I. Sandler and P. C. Chiu, J. Comput. Chem., 2011, 32, 226–239 CrossRef CAS PubMed
- V. D. Parker, J. Am. Chem. Soc., 1976, 98, 2942–2946 CrossRef
- A. Alparone and V. Librando, Monatsh. Chem., 2012, 143, 1123–1132 CrossRef CAS
- M. Martínez-Cifuentes, B. E. Weiss-López, L. S. Santos and R. Araya-Maturana, Molecules, 2014, 19, 9354–9368 CrossRef PubMed
- L. Senthilkumar, T. K. Ghanty, S. K. Ghosh and P. Kolandaivel, J. Phys. Chem. A, 2006, 110, 12623–12628 CrossRef CAS PubMed
- R. Salazar, J. Vidal, M. Martínez-Cifuentes, R. Araya-Maturana and O. Ramírez-Rodríguez, New J. Chem., 2015, 39, 1237–1246 RSC
- A. Shahi and E. Arunan, Phys. Chem. Chem. Phys., 2014, 16, 22935–22952 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries, SOMO's and energies for all molecules. See DOI: 10.1039/c5ra10140a |
| This journal is © The Royal Society of Chemistry 2015 |