
Palladium(II) complexes containing ONO tridentate hydrazone for Suzuki–Miyaura coupling of aryl chlorides in aqueous-organic media†
Vignesh
Arumugam
a,
Werner
Kaminsky
b,
Nattamai S. P.
Bhuvanesh
c and
Dharmaraj
Nallasamy
*a
aInorganic & Nanomaterials Research Laboratory, Department of Chemistry, Bharathiar University, Coimbatore 641 046, India. E-mail: dharmaraj@buc.edu.in; Fax: +91 422 2422387; Tel: +91 422 2428316
bDepartment of Chemistry, University of Washington, Seattle, Washington 98195, USA
cDepartment of Chemistry, Texas A & M University, College Station, TX 77843, USA
First published on 3rd July 2015
Abstract
Facile synthesis of three new palladium(II) complexes bearing heterocyclic hydrazone ligands are presented along with their structural characterization using IR, 1H and 13C NMR spectra. Molecular structures of the complexes determined by single-crystal XRD revealed a distorted square-planar geometry around the metal ion to which the hydrazone was attached in a tridentate fashion. Catalytic activity of these complexes tested towards the Suzuki–Miyaura cross coupling reaction of substituted aryl boronic acids with aryl chlorides in a water–toluene system (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10%) without using any promoting additives or phase transfer agents, proved that they are highly active with 0.01 mol% loading under optimized conditions to afford 99% yield of the coupled product. Effects of temperature, solvent and base on the cross-coupling reaction were carried out as well. These complexes showed significant catalytic activity up to five cycles.
:10%) without using any promoting additives or phase transfer agents, proved that they are highly active with 0.01 mol% loading under optimized conditions to afford 99% yield of the coupled product. Effects of temperature, solvent and base on the cross-coupling reaction were carried out as well. These complexes showed significant catalytic activity up to five cycles.
Introduction
Transition metal-catalyzed C–C and C–X (X = heteroatom) bond forming reactions are a supreme tool in synthetic organic chemistry. The formation of C–C bond was achieved by Kumada, Heck, Negishi, Suzuki–Miyaura, Hiyama, Sonogashira and Stille coupling reactions.1 Among those, the palladium catalyzed Suzuki–Miyaura cross-coupling (SMC) reaction has triggered considerable enthusiasm in the synthetic chemistry community. SMC stands out as the most capable and helpful methodology for the synthesis of biphenyl derivative segments of numerous compounds such as pharmaceuticals, herbicides, natural products, polymers, organic electroluminescence materials and ligands.2 It is widely utilized for C–C coupling reactions due to low toxicity of boronic acids, stability at ambient conditions and the facile removal of boron-containing side products. SMC reaction permits the utilization of organic solvents and inorganic bases, endures numerous other functional groups, unaffected by steric hindrance of the substrates and it's suitable for modern procedures.3 The importance of palladium catalyzed cross-coupling reaction has been highlighted by the 2010 Nobel Prize in chemistry awarded to Professors Heck, Negishi and Suzuki.4 Though wealth of information are available on both homogeneous and heterogeneous SMC reactions of aryl bromides or iodides with palladium catalysts,5 relatively less is published with aryl chlorides as substrate due to the difficulty in the activation of C–Cl bond compared to C–Br or C–I bonds.6Over a longer period, large number of palladium complexes bearing carbene, imine, oxime, phosphorus and other Schiff base ligands has been successfully employed for cross-coupling reactions.7 In general, hydrazone is a class of Schiff base ligand that exhibits a wide range of analytical and biological applications.8 Their transition metal complexes are well exploited as anticancer drugs and show great promise as chemotherapeutic agents.9 In the recent years, hydrazone based palladium complexes emerge as a powerful catalytic system for the construction of C–C bond.10
Pincer type palladium(II) complexes are widely utilized as a catalyst in coupling reactions.11 The ONO tridentate heterocyclic hydrazone Pd(II) complexes may show similar behavior as their coordination mode is similar to that of pincer type complexes.11 In our group, we are currently investigating the synthesis and biological applications of hydrazone based transition metal complexes.9,12 These complexes have shown promising biological activity and as part of our continuing study into the versatility of hydrazone metal complexes,12 we now turn our attention to evaluate some hydrazone containing palladium complexes as catalysts for the Suzuki–Miyaura coupling reaction.
Herein, we report the synthesis and structural characterization of three new palladium complexes bearing heterocyclic hydrazone ligands (furoichydrazone (H2L1), thiophenehydrazone (H2L2) and nicotinichydrazone (H2L3)) and their catalytic activity towards SMC reaction of challenging aryl chlorides13 with substituted aryl boronic acids in water–toluene media. To the best of our insight, utilizing palladium(II) complexes containing heterocyclic hydrazone ligand as catalysts for SMC reaction of aryl chlorides has not been published earlier.
Results and discussion
Reactions of palladium precursor [PdCl2(PPh3)2] with the heterocyclic hydrazones (H2L1, H2L2 and H2L3) yielded complexes of the type [Pd(L)(PPh3)] as depicted in Scheme 1. Based on 1H, 13C NMR and single-crystal XRD data, we observed that the hydrazone ligand is coordinated to the palladium ion in a tridentate fashion by replacing both the chloride ions and a molecule of triphenylphosphine from the starting precursor.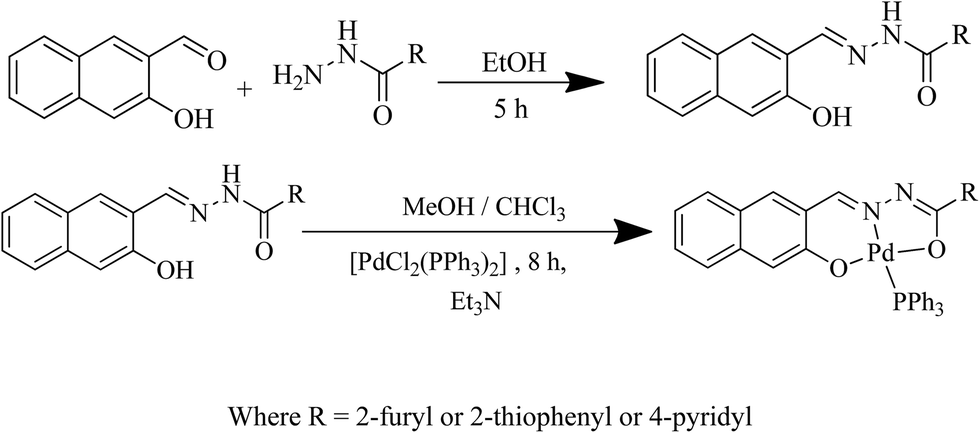 | ||
| Scheme 1 Synthetic route of ligands and their corresponding palladium complexes. | ||
The FT-IR spectra of the ligands H2L1, H2L2 and H2L3 showed medium bands at 3442, 3399 and 3484 cm−1 respectively, due to the presence of O–H group. Similarly, a strong band observed at 3046, 3090 and 3054 cm−1 indicated the presence of N–H group in the free ligands. All the three ligands exhibited an intense C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O absorption at 1641, 1675 and 1613 cm−1 respectively. In the spectra of complexes 1, 2 and 3 absence of the strong band due to O–H indicated the deprotonation of naphthol oxygen and its coordination to palladium ion. The N–H and CO stretching vibrations are also not observed in the spectra of complexes and thus proved that the ligand experienced a tautomerization and consequently coordinated via the imidolate oxygen to palladium(II) ion. In the spectra of palladium complexes 1, 2 and 3 new bands were seen at 1526, 1577 and 1588 cm−1 due to the occurrence of CN–NC group in addition to the appearance of a new, sharp band at 1185, 1186 and 1182 cm−1 responsible for the C–O group. From the above discussed IR spectral features, it is clear that the hydrazones H2L1, H2L2 and H2L3 were coordinated to palladium(II) ion via the naphtholate oxygen, the azomethine nitrogen and imidolate oxygen in complexes 1, 2 and 3.14
O absorption at 1641, 1675 and 1613 cm−1 respectively. In the spectra of complexes 1, 2 and 3 absence of the strong band due to O–H indicated the deprotonation of naphthol oxygen and its coordination to palladium ion. The N–H and CO stretching vibrations are also not observed in the spectra of complexes and thus proved that the ligand experienced a tautomerization and consequently coordinated via the imidolate oxygen to palladium(II) ion. In the spectra of palladium complexes 1, 2 and 3 new bands were seen at 1526, 1577 and 1588 cm−1 due to the occurrence of CN–NC group in addition to the appearance of a new, sharp band at 1185, 1186 and 1182 cm−1 responsible for the C–O group. From the above discussed IR spectral features, it is clear that the hydrazones H2L1, H2L2 and H2L3 were coordinated to palladium(II) ion via the naphtholate oxygen, the azomethine nitrogen and imidolate oxygen in complexes 1, 2 and 3.14
In order to confirm the coordination of hydrazone ligand in the new palladium complexes, 1H & 13C NMR spectra of complexes 1, 2 and 3 were recorded. None of the complexes displayed any signal due to N–H proton revealed that an imidolate oxygen is coordinated to the palladium ion. Similarly, the non-existence of any resonance attributable to the O–H group of naphthalene ring revealed a deprotonation prior to its coordination to palladium ion in the complexes 1, 2 and 3. Sharp singlets found at δ 9.4, 8.5 and 8.9 ppm were assigned as due to that of azomethine proton of the coordinated hydrazone in the respective complexes. All the aromatic protons of coordinated triphenylphosphine and hydrazone of the palladium complexes (1, 2 and 3) showed their resonances in the region of δ 7.2–8.2 ppm. The 13C NMR spectra of all the Pd(II) complexes displayed signals at δ 164.2, 160.0 and 165.4 ppm respectively owing to the imidolate carbon (NC–O) involved in the coordination. The signal corresponding to the azomethine carbon of the hydrazone in complexes 1, 2 and 3 were shifted downfield and appeared at δ 163.1, 147.2 and 160.0 ppm respectively. The signals appeared in the region of δ 110.6–147.2 ppm were accounted to various aromatic carbons of the hydrazone as well as triphenylphosphine ligands of complexes 1, 2 and 3.15
The molecular structure of complexes 1, 2 and 3 were determined by single-crystal X-ray diffraction in order to identify the exact coordination mode of hydrazone ligands in the newly synthesized complexes. All the complexes tested here crystallize in a primitive monoclinic system, space group P21/n. The ORTEP diagrams (Fig. 1–3) reveal that the hydrazone ligand is coordinated to the palladium ion in a tridentate manner via the naphtholate oxygen, azomethine nitrogen and the deprotonated imidol, each forming five membered and six membered chelate rings. The Pd(II) ion adopted a distorted square-planar geometry satisfied by the hydrazone ligand as binegative tridentate ONO donor and the fourth site is occupied by a triphenylphosphine. The bond lengths and bite angles are very similar to those observed for other palladium(II) complexes.16 The bite angles (°) around Pd(II) ion in the complexes 1, 2 and 3 are N(1)–Pd(1)–O(1) = 93.86 (6), 93.21 (5) and 93.27 (10) respectively. Similarly, N(1)–Pd(1)–O(2) = 80.26 (6), 80.72 (5), 80.24 (10); O(1)–Pd(1)–O(2) = 173.64 (6), 173.89 (4), 173.50 (9) and N(1)–Pd(1)–P(1) = 178.52 (5), 175.52 (4), 178.83 (8) respectively (Table 1 in the ESI†).16 The Pd–N(1), Pd–O(1) and Pd(1)–O(2) bond length (Å) of the complexes 1, 2 and 3 are within the range reported for palladium complexes16 Pd(1)–N(1) = 1.9674 (16), 1.9638 (12), 1.9720 (3); Pd(1)–P(1) = 2.2956 (5), 2.2743 (5), 2.2996 (9) respectively. In all the complexes, the length of C(12)–O(2) bond is shorter when compared to C(3)–O(1) bond (Table 1 in the ESI†). The shortening of the above bond is due to the deprotonation of imidol group to coordinate with palladium ion in all the complexes.17 Selected bond angles and bond lengths are gathered in Table 1 in the ESI.† Details on the data collection and structure refinements are summarized in Table 2 in the ESI.†
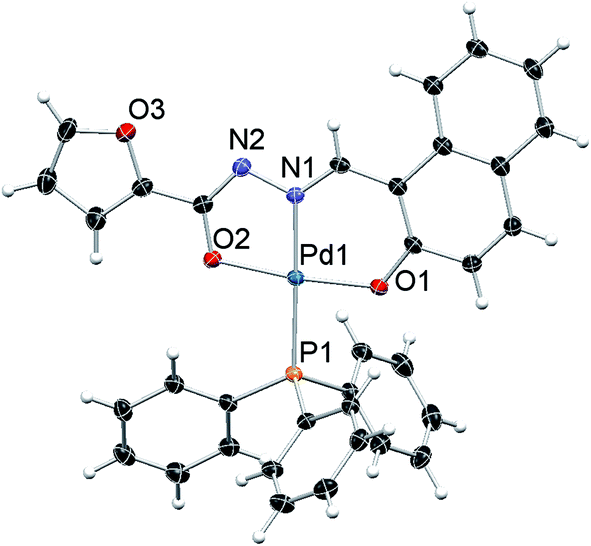 | ||
| Fig. 1 ORTEP diagram of complex 1; displacement parameters at their 50% probability level. | ||
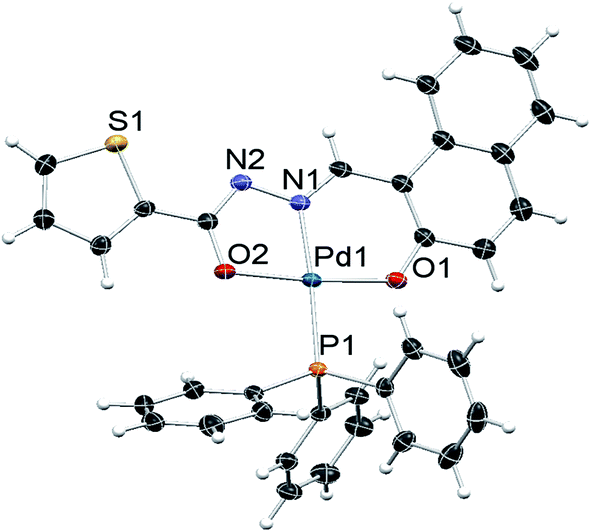 | ||
| Fig. 2 ORTEP diagram of complex 2; displacement parameters at their 50% probability level. | ||
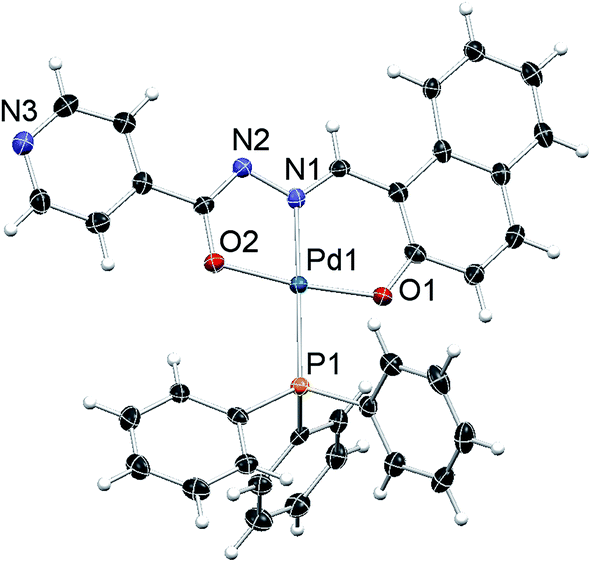 | ||
| Fig. 3 ORTEP diagram of complex 3; displacement parameters at their 50% probability level. | ||
Investigation on the catalytic activity of above synthesized Pd(II) complexes were performed against p-chlorobenzonitrile and 1-chloro-4-nitro-benzene with substituted aryl boronic acids in the presence of catalyst, inorganic base and solvent. Previous reports on the coupling reactions using p-chlorobenzonitrile and 1-chloro-4-nitro-benzene revealed that it was difficult to activate C–Cl bonds in them.6 Hence, we selected the challenging aryl chlorides13 namely, p-chlorobenzonitrile and 1-chloro-4-nitro-benzene for SMC with substituted aryl boronic acids in presence of the new palladium complexes as homogeneous catalysts.
Our investigations commenced with optimization of reaction conditions. For this purpose, p-chlorobenzonitrile (3 mmol) and phenylboronic acid (4 mmol) were used as model substrates in the presence of complex 2 as a catalyst (0.01 mol%). Various inorganic and organic bases listed in the Table 1 were surveyed identifying K2CO3 as a suitable base to increase yield (Table 1, entries 2–9). In our attempt to perform this coupling reaction in neat water, no progress was observed due to the fact that the catalyst is insoluble (Table 1, entry 1). Hence, we included some organic solvents to dissolve the catalyst in the reaction media. The impact of different polar and non-polar solvents on the yield of coupling reaction has been considered (Table 1, entries 10–17). Amongst the solvents examined, water-toluene mixture gave the best yield (Table 1, entry 11). Further, various ratios of water–toluene mixtures were analyzed, a ratio of 90:10% giving better yield (Table 1, entries 18–22). When the SMC reaction was carried out in room-temperature, the yield of coupled product was only 15% after 19 h. However, upon raising the temperature from 50 to 110 °C, maximum yield was obtained at 90 °C (94%) after 10 h followed by a decrease in the yield at 100 and 110 °C to 87 and 76%, respectively. Further heating to 120 °C had no influence on the quantity of the coupled product (Fig. 4).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yield (%) |
| 1 | Complex 2 | No base | H2O | No reaction |
| 2 | Complex 2 | No base | H2O/benzene | 12 |
| 3 | Complex 2 | KOH | H2O/benzene | 76 |
| 4 | Complex 2 | K2CO3 | H2O/benzene | 89 |
| 5 | Complex 2 | NaOH | H2O/benzene | 81 |
| 6 | Complex 2 | Na2CO3 | H2O/benzene | 83 |
| 7 | Complex 2 | CH3COONa | H2O/benzene | 40 |
| 8 | Complex 2 | Et3N | H2O/benzene | 25 |
| 9 | Complex 2 | Pyridine | H2O/benzene | 16 |
| 10 | Complex 2 | K2CO3 | H2O/benzene | 89 |
| 11 | Complex 2 | K2CO3 | H2O/toluene | 92 |
| 12 | Complex 2 | K2CO3 | H2O/CHCl3 | 65 |
| 13 | Complex 2 | K2CO3 | H2O/DMF | 63 |
| 14 | Complex 2 | K2CO3 | H2O/CH3CN | 60 |
| 15 | Complex 2 | K2CO3 | H2O/THF | 18 |
| 16 | Complex 2 | K2CO3 | H2O/DMSO | 35 |
| 17 | Complex 2 | K2CO3 | H2O/n-propanol | 22 |
| 18 | Complex 2 | K2CO3 | H2O/toluene (10%) | 94 |
| 19 | Complex 2 | K2CO3 | H2O/toluene (20%) | 92 |
| 20 | Complex 2 | K2CO3 | H2O/toluene (30%) | 85 |
| 21 | Complex 2 | K2CO3 | H2O/toluene (40%) | 81 |
| 22 | Complex 2 | K2CO3 | H2O/toluene (50%) | 76 |
| 23 | Complex 1 | K2CO3 | H2O/toluene (10%) | 98 |
| 24 | Complex 2 | K2CO3 | H2O/toluene (10%) | 92 |
| 25 | Complex 3 | K2CO3 | H2O/toluene (10%) | 89 |
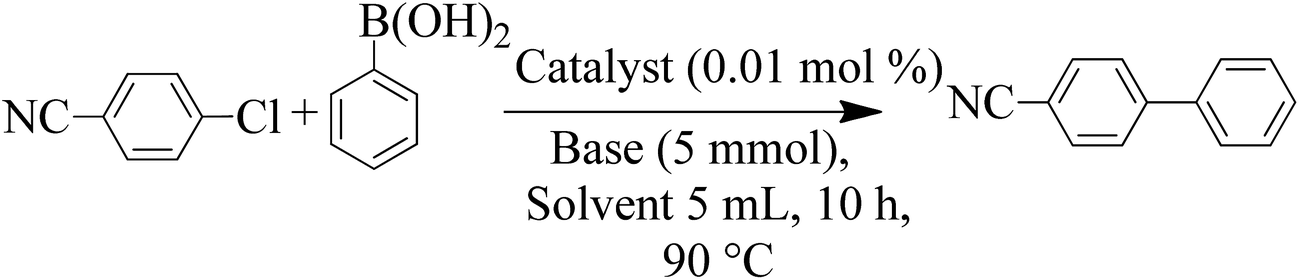
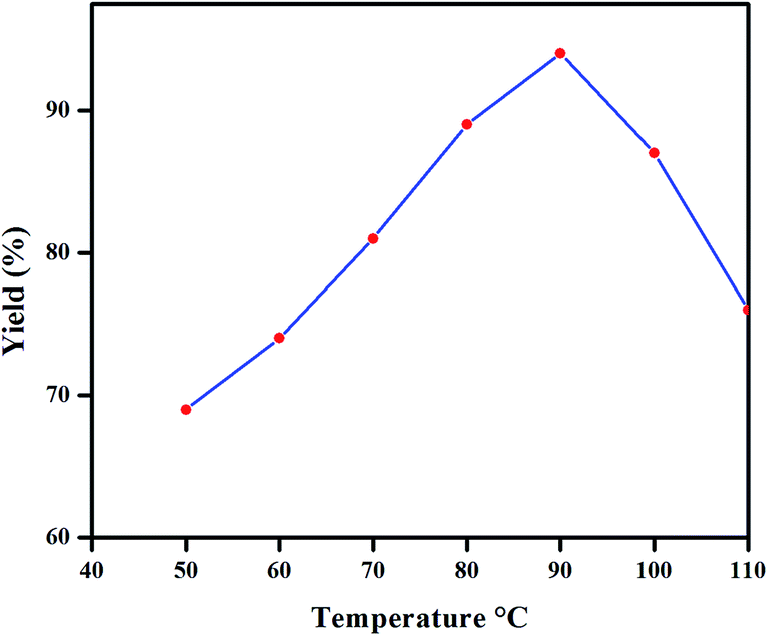 | ||
| Fig. 4 Effect of temperature on the SMC reaction. | ||
Among the catalysts (Table 1, entries 23–25), complex 1 showed superior catalytic activity towards SMC reaction of a series of aryl boronic acids with two different aryl chlorides. The higher catalytic potential of complex 1 is attributed to the presence of a furyl moiety at the terminal carbon of the coordinated hydrazone ligand.18a In general, phosphine ligands like triphenylphosphine, phosphanes, phosphane oxides bearing Pd complexes help to activate C–Cl bonds in electron-deficient and electron-rich aryl chlorides.18b
Low catalyst stacking tests (Table 2, entries 1–3) showed best execution utilizing 0.01 mol% of complex 1. Reducing catalyst stacking from 0.01–0.0001 mol% under optimized conditions resulted in low yield in accordance with previous reports.19 Utilizing 0.0001 mol% of catalyst increased turnover.
| Entry | Catalyst (mol%) | Yield (%) | TON |
|---|---|---|---|
| 1 | 0.01 | 98 | 9800 |
| 2 | 0.001 | 90 | 90000 |
| 3 | 0.0001 | 71 | 710000 |
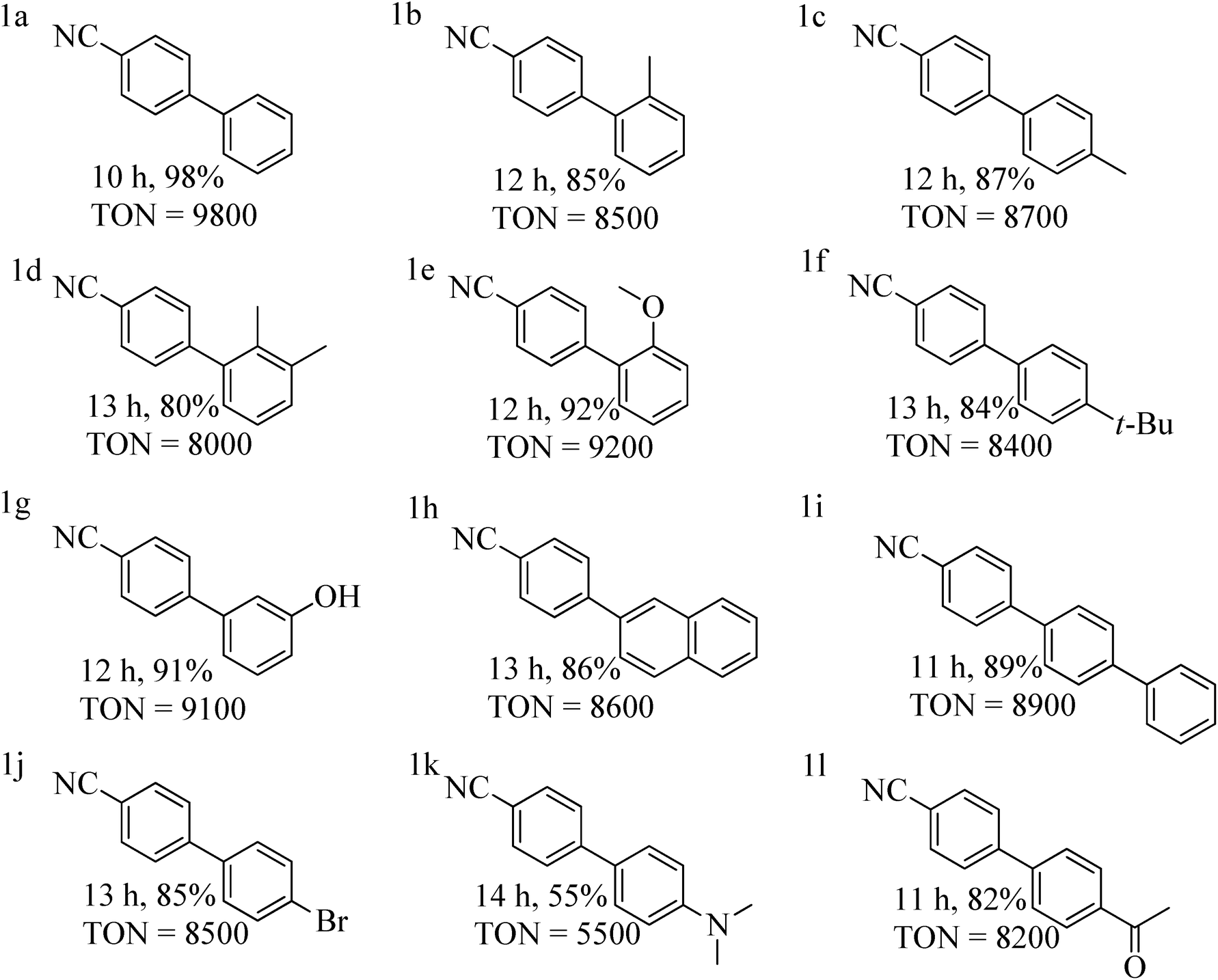
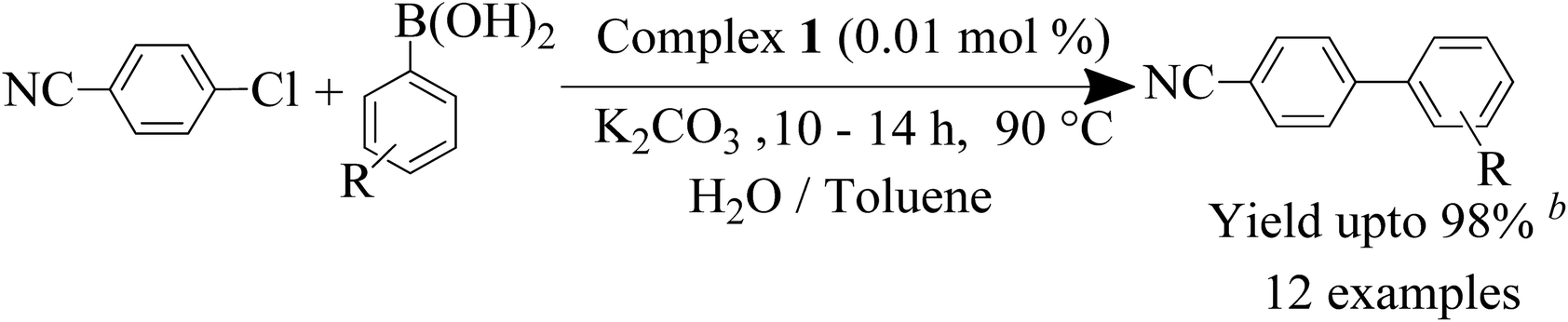
With the optimized reaction conditions in hand, we examined the scope of the SMC reaction of p-chlorobenzonitrile with diverse aryl boronic acids with catalyst 1 (0.01 mol%) (Table 3). Here, p-chlorobenzonitrile featuring a moderately deactivating nitrile group, is an ideal coupling partner for SMC reaction, while the other coupling partner aryl boronic acid bears both electron donating and withdrawing groups. Aryl boronic acids having deactivating groups such as –Br, –COCH3, produce good yields within 10–14 h (Table 3, entries 1j and 1l). In the case of aryl boronic acids with activating groups such as –CH3, –OCH3, t-(CH3)3C–, –OH and –NR2, a yield of 55–92% were achieved (Table 3, entries 1c, 1e, 1f, 1g and 1k). Remarkably, sterically demanding aryl boronic acids were also smoothly converted to the corresponding products in 85–90% yield (Table 3, entries 1c, 1h and 1i). Gratifyingly, the coupling reaction was also operative for aryl boronic acid with high steric hindrances at a 80–85% yield (Table 3, entries 1b, 1d and 1e).
|
|
|---|
| a Reaction conditions: p-chlorobenzonitrile (3.0 mmol), aryl boronic acid (4.0 mmol), 0.01 mol% of complex 1, K2CO3 (5.0 mmol), H2O (4.5 mL) and toluene (0.5 mL) stirred for 10–14 h at 90 °C. b Isolated yield after column chromatography, TON = turnover number = ratio of moles of product formed to moles of catalyst used. |
|
Encouraged by these results, we next performed the SMC reaction of 1-chloro-4-nitro-benzene as one of the partners instead of p-chlorobenzonitrile considering the fact that the former aryl chloride is also a challenging substrate possessing a strongly deactivating –NO2 group at para position.13 Reaction conditions were very similar to those optimized for p-chlorobenzonitrile except that the nitro substituted aryl chloride was reactive enough to couple with aryl boronic acids at room-temperature within 4–7 min. Coupling with aryl boronic acids containing both activating and deactivating groups were proceeded smoothly (Table 4 entries 2a–2l).
|
|
|---|
| a Reaction conditions: 1-chloro-4-nitro-benzene (3.0 mmol), aryl boronic acid (4.0 mmol), 0.01 mol% of complex 1, K2CO3 (5.0 mmol), H2O (4.5 mL) and toluene (0.5 mL) stirred for 4–7 min at room-temperature. b Isolated yield after column chromatography, TON = turnover number = ratio of moles of product formed to moles of catalyst used. |
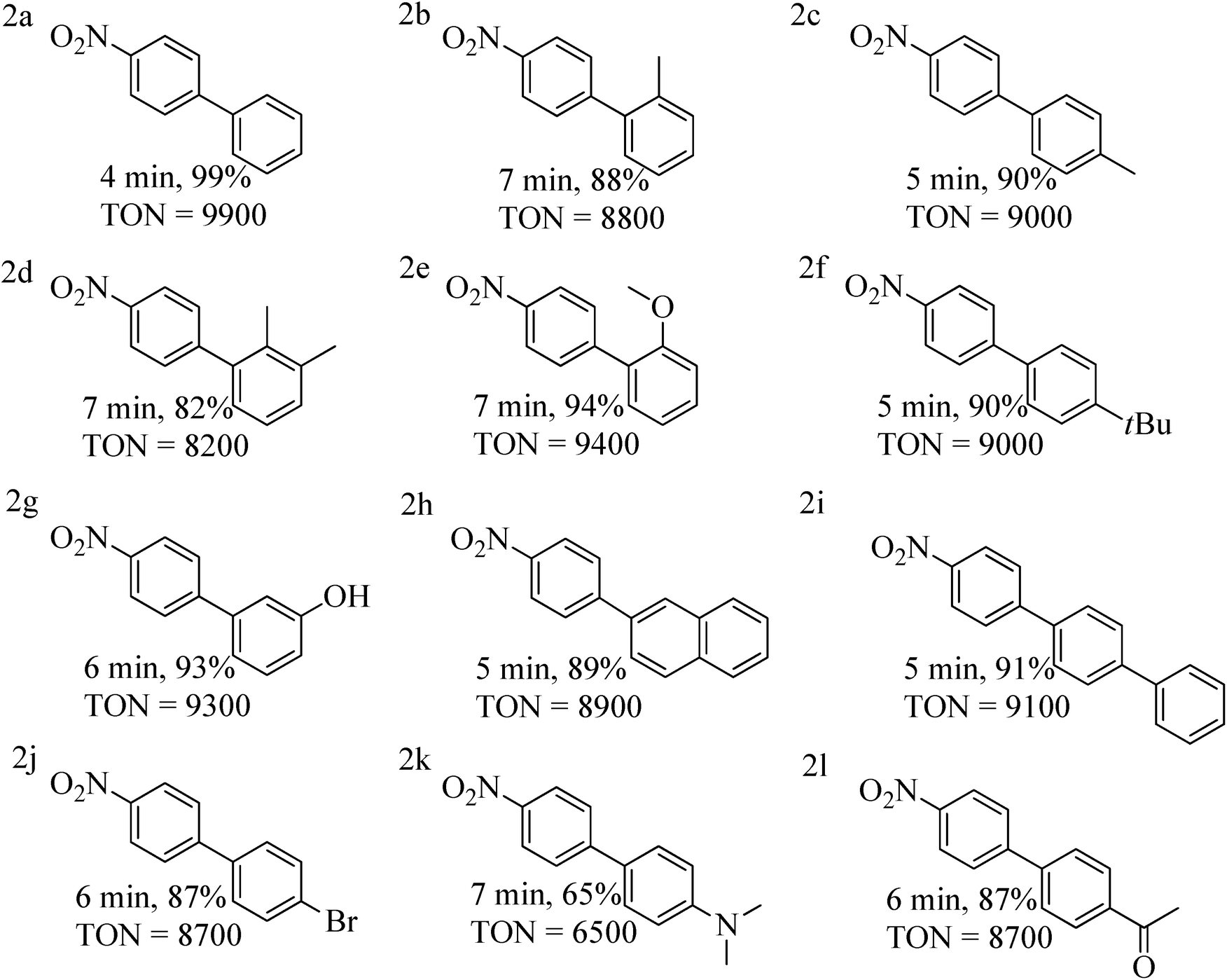
|
Mechanism for the SMC reaction
Generally, pincer type ligand can increase the electron density at palladium(II) centre and facilitate the generation of Pd(IV) species. Therefore, Pd(IV) intermediates are considered as reactive intermediates in the mechanism of the SMC reaction of aryl halides at higher temperature.11,20a Advantageously, Pd(II)/Pd(IV) cycle helps facile reductive elimination from Pd(IV) intermediate. However, Pd(IV) intermediates are unstable and hence, it can't be isolated.20a Based on these facts and previous reports,20b,c a possible mechanism for SMC reaction was proposed in Scheme 2. The active species, palladium(II) hydrazone complex underwent the oxidative addition of p-chlorobenzonitrile to give an intermediate II. Addition of base to an intermediate II triggered a metathetic process to generate an intermediate III. Transmetallation of phenylboronic acid to an intermediate III produced an intermediate IV which on reductive elimination gave the desired cyanobiphenyls followed by the regeneration of Pd(II) active species. The mechanism for the SMC reaction involving 1-chloro-4-nitro-benzene and aryl boronic acids is very similar to the one depicted in Scheme 2.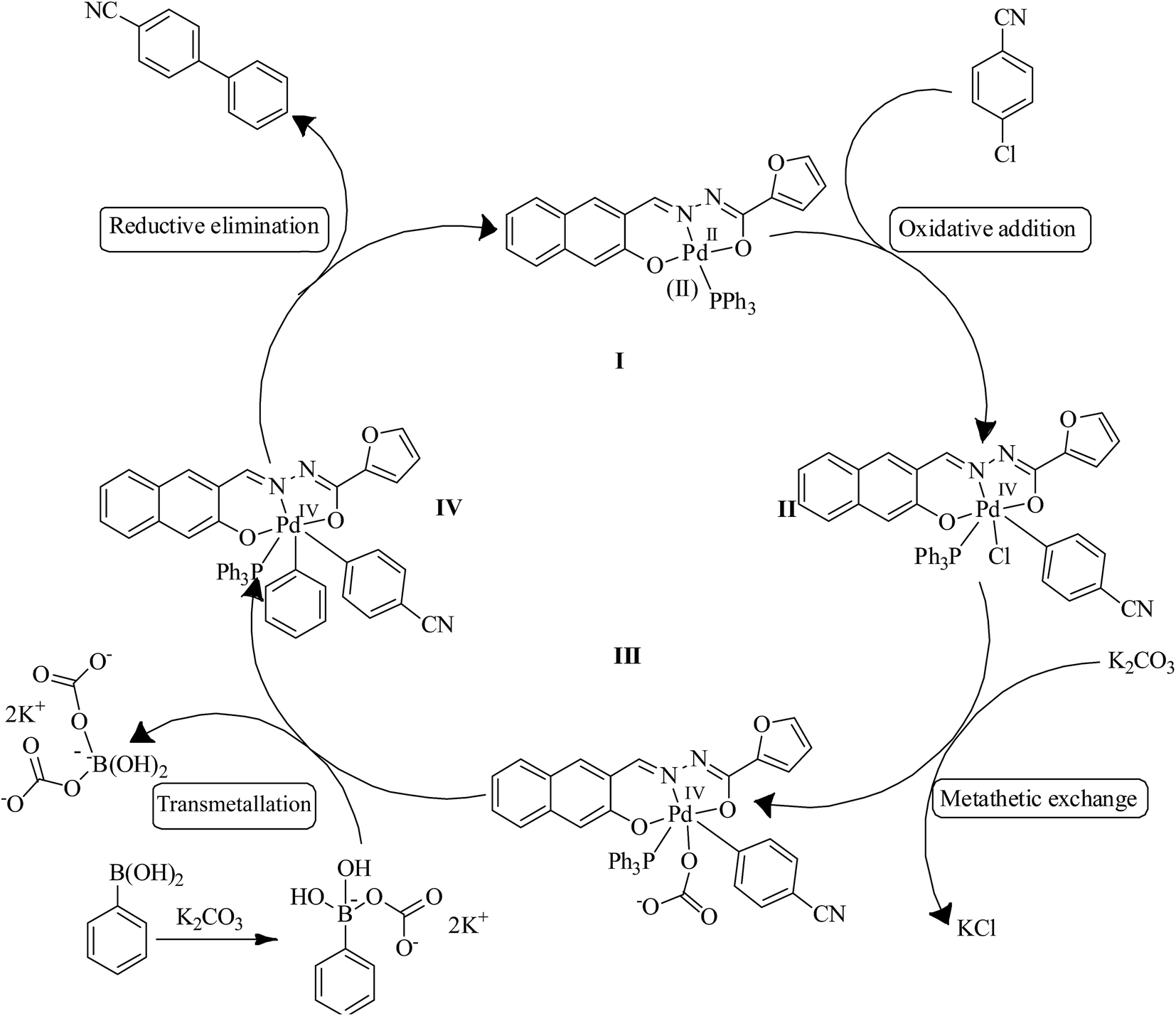 | ||
| Scheme 2 Plausible mechanism for SMC reaction of p-chlorobenzonitrile with phenylboronic acid. | ||
Reusability of catalyst
Recovery and reusability of the catalyst was studied in a reaction that involved p-chlorobenzonitrile (3.0 mmol), phenylboronic acid (4 mmol), K2CO3 (5 mmol) and catalyst (0.01 mol%) in water–toluene (4.5–0.5 mL) at 90 °C. After completion of the reaction, the mixture was extracted with ethyl acetate to separate the catalyst and the product by column chromatography. The recovered catalyst was dried and utilized for the next cycle under the same reaction conditions. Though a marginal decrease in the catalytic activity of the selected complex 1 was observed upon moving from the first to fifth cycle, there occurred a significant loss in its performance after the fifth cycle and yielded only 45% of the coupled product in the sixth cycle (Fig. 5). The identity of the catalyst after the reaction was confirmed by comparing its physical data (melting point and Rf value) with the original complex.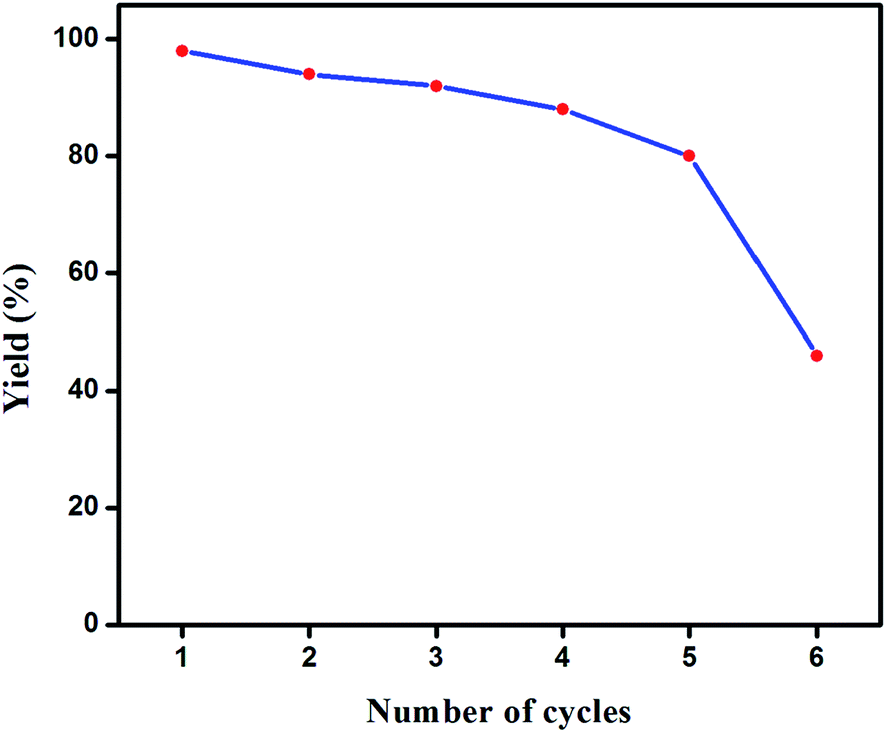 | ||
| Fig. 5 Reusability of catalyst 1. | ||
H. M. Lee et al. reported recently21 that Zwitter ionic phosphine complexes were highly efficient at room-temperature for SMC reactions between sterically hindered aryl chloride and aryl boronic acids in an aqueous media. 94% of products were reported for the coupling reaction between p-chlorobenzonitrile and phenylboronic acid during 7 h. However, in the present report, we achieved 98% of the coupled product from the same substrates in 10 h by utilizing the newly synthesized palladium(II)-hydrazone complex 1 as a catalyst with comparably low catalyst loading.
Conclusion
This study presented the synthesis and spectral characterization of three new palladium(II) complexes bearing heterocyclic hydrazone ligands. X-ray diffraction data of all these complexes confirmed a distorted square-planar geometry around palladium(II) ion satisfied by ONO tridentate coordination of hydrazone ligands and a triphenylphosphine as a fourth ligand. One of the new complexes acted as a fabulous catalyst for Suzuki–Miyaura coupling of p-chlorobenzonitrile or 1-chloro-4-nitro-benzene with substituted aryl boronic acids in water–toluene media. Extension of the scope of this catalyst to aryl boronic acids possessing a range of functional groups attested that a smooth coupling occurred in all the cases under optimized conditions without the formation of either homocoupled products of boronic acids or coupling at the hydroxyl group present in boronic acid. Hence, we believe that this methodology involving a robust palladium based homogeneous catalyst towards the Suzuki–Miyaura coupling offers a novel route to synthesis a series of substituted biaryl segments.Experimental procedure
Reagents and materials
All reagent grade chemicals were used without further purification unless otherwise specifically mentioned. Solvents were purified and dried according to standard procedures.22 Elemental analysis (C, H, N and S) were performed on a Vario EL III Elemental analyzer instrument. IR spectra (4000–400 cm−1) were recorded on a Nicolet Avatar Model FT-IR spectrophotometer. Melting points were determined with a Lab India instrument.1H and 13C NMR spectra were recorded in deuterated CHCl3 as solvent on BRUKER 400 and 100 MHZ instruments, respectively.Synthesis of the ligands
Pincer type heterocyclic hydrazone ligands (H2L) were synthesized by condensing equimolar amounts of o-hydroxy naphthaldehyde with diverse hydrazides such as furoic acid hydrazide (H2L1), thiophenecarboxylic acid hydrazide (H2L2) and nicotinic acid hydrazide (H2L3) in ethanol according to a literature method (Scheme 1).9b The reaction mixture was then refluxed on a water bath for 5 h and poured into crushed ice. The corresponding solid hydrazone formed was filtered, washed several times with distilled water and recrystallized from ethanol with 85–90% yield. The purity of the ligands were checked by various analytical techniques and is in accordance with literature reports.9bSynthesis of palladium complexes [Pd(L1)(PPh3)] (1)
A solution of [PdCl2(PPh3)2] (0.100 g, 143 mM) in chloroform (20 cm3) was added drop wise to the hot methanolic (15 cm3) solution of ligand H2L1 (0.40 g, 143 mM) and two drops of triethylamine were added. The reaction mixture was refluxed for 8 h and thereafter kept at room-temperature for crystallization. Red colored crystal needles suitable for X-ray studies were obtained on slow evaporation of the reaction mixture over of 45 days. Yield: 85%, mp: 241–243 °C. Elemental analysis calculated for (%): C34H25N2O3PPd: C, 63.12; H, 3.89; N, 4.33. Found (%) C, 62.98; H, 3.44; N, 4.01. UV-visible (solvent: DMSO, nm): 319, 327, 383 and 425. Selected IR bands (KBr, ν in cm−1): 1605 (imidolate NC–O), 1526 (CN) and 1185 (naphtholate C–O). 1H NMR (CDCl3, δ ppm) 9.42 (s, 1H), 8.45 (d, J = 8 Hz, 4H), 8.29 (dd, J = 1.2, 1.2 Hz, 3H), 8.01–8.03 (m, 3H), 7.97 (d, J = 1.2 Hz, 5H), 7.84 (t, J = 7.8 Hz, 3H), 7.49 (t, J = 7.6 Hz, 2H), 7.36–7.40 (m, 4H). 13C NMR (CDCl3, δ ppm) 164.2, 146.4, 136.5, 129.9, 128.7, 127.4, 127.3, 126.8, 125.7, 119.9, 118.5, 115.0, 114.8.
Synthesis of [Pd(L2)(PPh3)] (2)
Complex 2 was prepared by the same procedure as that described for complex 1 with H2L2 (0.42 g, 0.143 mM) and [PdCl2(PPh3)2] (0.100 g, 0.143 mM). Red colored crystals of the product were obtained on slow evaporation of the reaction mixture over the period of two months. The crystals obtained were suitable for X-ray diffraction. Yield: 80%, mp: 261–264 °C. Elemental analysis calculated for (%): C34H25N2O2PPdS: C, 61.59; H, 3.80; N, 4.23; S, 4.84. Found (%) C, 61.02; H, 3.34; N, 4.05; S, 4.26. UV-visible (solvent: DMSO, nm): 266, 328, 376 and 444. IR bands (KBr, ν in cm−1): 1776 (imidolate NC–O), 1588 (CN) and 1186 (naphtholate C–O). 1H NMR (CDCl3, δ ppm) 8.57 (s, 1H), 8.14 (s, 6H), 8.02–8.08 (m, 7H), 7.99 (d, J = 7.2 Hz, 4H), 7.95 (d, J = 7.2 Hz, 2H), 7.65 (d, J = 4 Hz, 2H), 7.40–7.56 (m, 3H). 13C NMR (CDCl3, δ ppm) 160.0, 147.2, 135.9, 136.5, 129.6, 129.0, 128.6, 120.4, 118.1, 116.7, 114.4, 110.6.
Synthesis of [Pd(L3)(PPh3)] (3)
Complex 3 was prepared using the same procedure as described for 1 by the reaction of H2L3 (0.42 g, 0.143 mM) and [PdCl2(PPh3)2] (0.100 g, 0.143 mM). After the completion of reaction the solute was kept at room-temperature. Needle shape orange crystals of X-ray diffraction quality were obtained after 50 days. Yield: 79%, mp: 254–257 °C. Elemental analysis calculated for (%): C35H26N3O2PPd: C, 63.89; H, 3.98; N, 6.39. Found (%) C, 63.15; H, 3.22; N, 6.16. UV-visible (solvent: DMSO, nm): 308, 340, 370 and 420. IR bands (KBr, ν in cm−1): 1693 (imidolate NC–O), 1577 (CN), 1182 (naphtholate C–O) and 501(CN in ring). 1H NMR (CDCl3, δ ppm) 8.92 (s, 1H), 8.21 (s, 2H), 8.01–8.04 (t, J = 8.4 Hz, 2H), 7.84 (d, J = 5.6 Hz, 8H), 7.77 (d, J = 2.8 Hz, 3H), 7.55 (t, J = 6.4 Hz, 4H), 7.46 (t, J = 3.6 Hz, 3H), 7.23–7.29 (m, 3H). 13C NMR (CDCl3, δ ppm) 165.4, 160.0, 148.23, 135.9, 131.5, 129.9, 129.7, 128.7, 121.4, 117.7, 115.4, 112.8.
Single-crystal X-ray diffraction studies
Catalysis
Acknowledgements
One of the authors Mr Vignesh Arumugam thank the University Grants Commission (UGC), New Delhi, India for the award of Junior Research Fellowship under UGC-BSR RFSMS to carry out this work. We thank Dr B. R. Venkatraman, Associate Professor, Department of Chemistry, Periyar E.V.R. College, Tiruchirappalli-23, India, for his useful scientific discussions on this work and Prof. Dr Ramasubbu Jeyaraman Science Foundation (RJSF), Chennai, India, for extending NMR facility.References
- (a) K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS; (b) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (c) A. O. King, N. Okukado and E. Negishi, J. Chem. Soc., Chem. Commun., 1977, 19, 683 RSC; (d) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (e) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS; (f) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 50, 4467 CrossRef; (g) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef.
- (a) S. P. Stanforth, Tetrahedron, 1998, 54, 263 CrossRef CAS; (b) A. Suzuki and Y. Yamamoto, Chem. Lett., 2011, 40, 894 CrossRef CAS; (c) A. Fihri, M. Bouhrara, B. Nekoueishahraki, J. M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC; (d) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC; (e) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633 CrossRef CAS; (f) M. Kertesz, C. H. Choi and S. Yang, Chem. Rev., 2005, 105, 3448 CrossRef CAS PubMed; (g) F. A. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef; (h) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS PubMed; (i) A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. Elshof and G. Rothenberg, Chem.–Eur. J., 2007, 13, 6908 CrossRef CAS PubMed; (j) S. Kaye, J. M. Fox, F. A. Hicks and S. L. Buchwald, Adv. Synth. Catal., 2001, 343, 789 CrossRef CAS; (k) J. D. Sellars and P. G. Steel, Chem. Soc. Rev., 2011, 40, 5170 RSC.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed.
- (a) E. Sindhuja, R. Ramesh and Y. Li, Dalton Trans., 2012, 5351 RSC; (b) S. Kumar, G. K. Rao, A. Kumar, M. P. Singh and A. K. Singh, Dalton Trans., 2013, 16939 RSC; (c) X. Li, X. Zhao, J. Zhang and Y. Zhao, Chem. Commun., 2013, 49, 10004 RSC; (d) L. Liu, Y. Dong and N. Tang, Green Chem., 2014, 16, 2185 RSC; (e) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed.
- (a) F. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC; (b) M. J. Jin and D. H. Lee, Angew. Chem., Int. Ed., 2010, 122, 1137 CrossRef; (c) Y. Tsuji and T. Fujihara, Inorg. Chem., 2007, 46, 1895 CrossRef CAS PubMed; (d) V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047 CrossRef CAS.
- (a) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195 CrossRef CAS PubMed; (b) D. Domin, D. B. Garagorri, K. Mereiter and K. Kirchner, Organometallics, 2005, 24, 3957 CrossRef CAS; (c) A. Schnyder, A. F. Indolese, M. Studer and H. U. Blaser, Angew. Chem., Int. Ed., 2002, 41, 3668 CrossRef CAS; (d) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- (a) M. Katual and G. Dutt, Talanta, 1975, 22, 151 CrossRef PubMed; (b) B. Singh, R. Shrivastava and K. K. Narang, Synth. React. Inorg. Met.-Org. Chem., 2000, 30, 1175 CrossRef CAS.
- (a) P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2012, 4423 RSC; (b) P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2011, 9690 RSC; (c) P. Sathyadevi, P. Krishnamoorthy, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Metallomics, 2012, 4, 498 CrossRef CAS PubMed; (d) P. Krishnamoorthy, P. Sathyadevi, P. T. Muthiah and N. Dharmaraj, RSC Adv., 2012, 2, 12190 RSC.
- (a) T. Mino, Y. Shirae, M. Sakamoto and T. Fujita, J. Org. Chem., 2005, 70, 2191 CrossRef CAS PubMed; (b) K. Watanabe, T. Mino, T. Abe, T. Kogure and M. Sakamoto, J. Org. Chem., 2014, 79, 6695 CrossRef CAS PubMed; (c) T. Mino, K. Kajiwara, Y. Shirae, M. Sakamoto and T. Fujita, Synlett, 2008, 2711 Search PubMed; (d) T. Mino, T. Koizumi, S. Suzuki, K. Hirai, K. Kajiwara, M. Sakamoto and T. Fujita, Eur. J. Org. Chem., 2012, 678 CrossRef CAS; (e) T. Mino, Y. Shirae, Y. Sasai, M. Sakamoto and T. Fujita, J. Org. Chem., 2006, 71, 6834 CrossRef CAS PubMed; (f) T. Mino, Y. Shirae, T. Saito, Y. Sasai, M. Sakamoto and T. Fujita, J. Org. Chem., 2006, 71, 9499 CrossRef CAS PubMed.
- H. Zhang and A. Lei, Dalton Trans., 2011, 8745 RSC.
- (a) P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley, N. S. P. Bhuvanesh and N. Dharmaraj, Dalton Trans., 2012, 6842 RSC; (b) P. Sathyadevi, P. Krishnamoorthy, N. S. P. Bhuvanesh, P. Kalaiselvi, V. Vijaya Padma and N. Dharmaraj, Eur. J. Med. Chem., 2012, 55, 420 CrossRef CAS PubMed; (c) P. Sathyadevi, P. Krishnamoorthy, E. Jayanthi, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Inorg. Chim. Acta, 2012, 384, 83 CrossRef CAS; (d) P. Sathyadevi, P. Krishnamoorthy, K. Thanigaimani, P. T. Muthiah and N. Dharmaraj, Polyhedron, 2012, 31, 294 CrossRef CAS; (e) P. Krishnamoorthy, P. Sathyadevi, K. Senthilkumar, P. T. Muthiah, R. Ramesh and N. Dharmaraj, Inorg. Chem. Commun., 2011, 14, 1318 CrossRef CAS; (f) P. Krishnamoorthy, P. Sathyadevi, R. R. Butorac, A. H. Cowley and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376 CrossRef CAS PubMed.
- A. Fihri, D. Luart, C. Len, A. Solhy, C. Chevrin and V. Polshettiwar, Dalton Trans., 2011, 3116 RSC.
- R. N. Prabhu and R. Ramesh, J. Organomet. Chem., 2012, 718, 43 CrossRef CAS.
- A. A. Ibrahim, H. Khaledi and H. M. Ali, Polyhedron, 2014, 81, 457 CrossRef CAS.
- S. Das and S. Pal, J. Organomet. Chem., 2006, 691, 2575 CrossRef CAS.
- A. R. B. Rao and S. Pal, J. Organomet. Chem., 2012, 701, 62 CrossRef CAS.
- (a) T. Mahamoa, M. M. Mogorosi, J. R. Moss, S. F. Mapolie, J. C. Slootweg, K. Lammertsma and G. S. Smith, J. Organomet. Chem., 2012, 703, 34 CrossRef; (b) L. Botella and C. Najera, Angew. Chem., Int. Ed., 2002, 41, 179 CrossRef CAS.
- (a) M. T. Reetz and J. G. Vries, Chem. Commun., 2004, 14, 1559 RSC; (b) A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. Elshof and G. Rothenberg, Chem.–Eur. J., 2007, 13, 6908 CrossRef CAS PubMed.
- (a) J. L. Bolliger, O. Blacque and C. M. Frech, Chem.–Eur. J., 2008, 14, 7969 CrossRef CAS PubMed; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (c) A. J. J. Lennox and G. C. L. Jones, Chem. Soc. Rev., 2014, 43, 412 RSC.
- J. Y. Lee, D. Ghosh, J. Lee, S. S. Wu, C. H. Hu, S. D. Liu and H. M. Lee, Organometallics, 2014, 33, 6481 CrossRef CAS.
- A. I. Vogel, Text Book of Practical Organic Chemistry, Longman, London, 5th edn, 1989 Search PubMed.
- A. Altomare, C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- A. Altomare, G. L. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- S. Mackay, C. Edwards, A. Henderson, C. Gilmore, N. Stewart, K. Shankland and D. A. MaXus, A computer program for the solution and refinement of crystal structures from diffraction data, University of Glasgow, Scotland, 1997 Search PubMed.
- D. Waasmaier and A. Kirfel, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 416 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1005675, 852732 and 1017037. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra10973f |
| This journal is © The Royal Society of Chemistry 2015 |