
The mechanism and kinetic studies on oxidation reaction of acetofenate initiated by HOx, NO3, O3, and Cl radicals†
Lingyan Kanga,
Chenxi Zhangb and
Xiaomin Sun*a
aEnvironment Research Institute, Shandong University, Jinan 250100, P. R. China. E-mail: sxmwch@sdu.edu.cn
bDepartment of Resource and Environment, Binzhou University, Binzhou 256600, PR China
First published on 5th November 2015
Abstract
Acetofenate (AF) is a widely used pesticide. The mechanism of HOx, NO3, O3, and Cl initiated oxidation reactions of AF was investigated with density functional theory. For each of the OH, NO3, and Cl, both addition and hydrogen abstraction were investigated, for each of the HO2 radicals and O3, addition reactions were investigated. The cycloaddition reactions of O3 were considered, including the exploration of isomerization. Based on the potential energy surface, the rate constants were calculated with the transition state theory method over a temperature range of 200–400 K and fitted with the Arrhenius formulas. The rate constants of the AF reaction with OH, HO2, NO3, O3, and Cl, are 4.04 × 10−13, 7.02 × 10−33, 6.93 × 10−20, 1.45 × 10−25, and 5.07 × 10−12 cm3 per molecule per s at 298.15 K, respectively. The OH-initiated reactions are dominant according to the branching ratio of reaction constants. The atmospheric lifetime of the reaction species was estimated according to rate constants.
Introduction
Acetofenate (AF), also known as benzethazet and plifenate, is used worldwide to control pests.1 As a commercial organochlorine pesticide, AF was developed as an analogue of DDT since the 1970s.2 Due to its low toxicity, selective insecticidal activity, and high efficacy, AF is considered a good alternative to combat pests, especially health pests.3,4 However, its popularity has led to the direct release of pesticides into the environment for a long time.5In the troposphere, radicals have great effects on the removal or transformation of organic compounds through controlling the oxidative capacity of the atmosphere.6 Once released into the atmosphere, AF could be oxidized by nucleophilic active species, HOx radicals, NO3 radical, O3, Cl atom, and other species, and generate a series of oxidation products.7–11
The HOx radicals (OH and HO2 radical), whose photochemical lifetimes are very short, are responsible for the majority of oxidation processes in the troposphere and control concentrations of many species.12,13 The degradation pathway of AF initiated by OH is important for the related chemistry features of OH, such as the inherent reactivity. However, the formation of OH mainly takes place in daytime through photolysis, and its concentration decreases rapidly after sunset.8,9,14 As for HO2, the principal sources are of photolysis and oxidation of HCHO, and the tropospheric abundances are about 100 times higher than that of OH during the daytime in clean air.15,16 On the whole, reactions with HOx radicals mainly occur during the day.
It is worth mentioning that not all atmospheric chemistry is initiated in the daytime. In the troposphere, the NO3 radical is thought to be the main night-time oxidant.17,18 During the daytime, the concentration of NO3 is very low. However, in the night-time atmosphere NO3 radicals were measured at typical concentrations of 10–100 parts per trillion.19 Clearly, the reactions of AF with NO3 pathway during nighttime cannot be ignored. In addition, in polluted environment, NO3 radical is also an important oxidant and may contribute to the removal of AF.20,21
Ozone is another powerful oxidant that cannot be ignored in atmospheric environment. Its strong oxidizing power and participation in the production of other radicals make ozone a key role in the oxidation of organic matters during both day and night.22,23 Most notably, the ozone arises from a variety of sources, and the concentration of O3 (7.0 × 1011 molecule per cm3) is much higher than that of OH radical (9.7 × 105 molecule per cm3).24,25
Moreover, in the marine atmosphere where the concentration of Cl atoms can reach a peak value of 105 molecule per cm3, the Cl atom-initiated chemistry can be an important removal process for AF.26 As for H-abstraction reactions, the rate constant of Cl atom is 103 times more active than that of OH radical.27 Thus, Cl atom also should be considered for the oxidation of AF in the atmosphere.
In this work, quantum chemistry method was applied to reveal the reaction mechanisms of AF. The initial reactions of HOx radicals, NO3 radical, O3, and Cl atom with AF were investigated, and thermodynamic parameters are included too. Based on quantum chemistry information, the rate constants are calculated using the transition state theory method. Arrhenius equations of the rate constants with a temperature range of 200–400 K are fitted, and the lifetimes of the reaction species in troposphere are estimated according to the rate constants.
Computational methods
Mechanism study
It is well known that the density functional theory (DFT) is a calculation tool to deal with atmospheric chemical reaction.28 In previous studies DFT have successfully applied to medium or even larger scale molecular system.29,30 The MPWB1K method is a hybrid DFT model with performance for thermochemistry, thermochemical dynamics, weak interactions, and hydrogen bonding.31 In this paper, all the work is performed using the Gaussian 03 programs and SGI workstation.32 The geometrical parameters of reactants, transition states (TS), intermediates (IM), and products were fully optimized at the MPWB1K/6-31+G(d,p) level. The vibrational frequencies have been calculated at the same level in order to determine the nature of stationary points. The single point energy is calculated at the basis sets 6-311+G(3df,2p).Kinetic calculation
The transition state theory (TST) is a model based on the interaction potential between reactants and products with a statistical representation of the dynamics to determine rate constants.33 It has been widely employed to discuss of rate processes in previous explorations. In this paper, the rate constants calculation of all elementary reactions have been carried out with the method over a suitable temperature range. Taking a bimolecular reaction as example:| A + BC → AB + C | (1) |
The rate constant is calculated using the following equation:
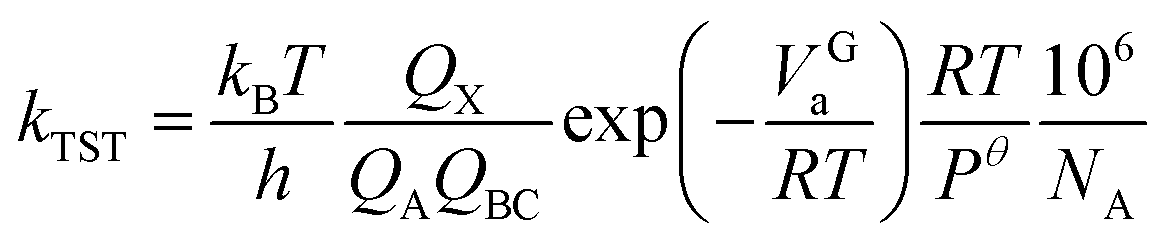 | (2) |
The definition of each parameter can be referred to literature.34 The numerical method for linear equation is used to fit the Arrhenius formula of rate constants with temperature.
Results and discussion
OH-Initiated reaction
Under the atmospheric condition, the chemical reactions of AF can be initiated by the OH radical. Two possible reaction pathways are taken into consideration: OH radical addition and H atom abstraction. For convenience, the atom labels are marked in the structure of AF in Fig. 1. The reaction schemes with the potential barriers (ΔEb) and reaction heats (ΔEr) are depicted in Fig. 1, S1, and S2,† respectively.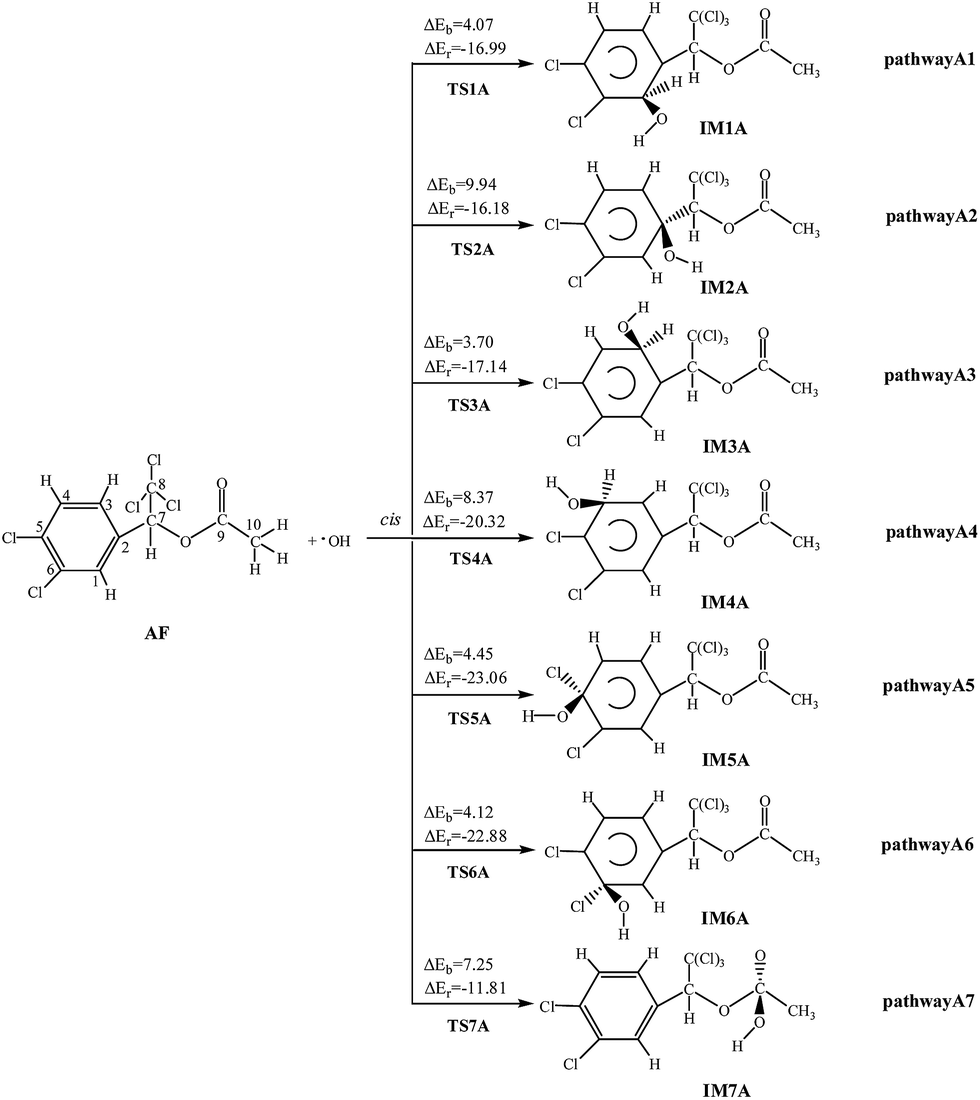 | ||
| Fig. 1 OH initiated addition pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
Because of its molecule asymmetry, AF has two kinds of different addition positions, that is, the same and opposite side of the benzene ring (the cis-addition and the trans-addition) relative to branched chain substituents. This paper mainly presents the cis-addition reactions. It is obvious that all of cis-addition channels can easily happen for their low potential barriers (3.70–9.94 kcal mol−1). As for trans-addition reactions, the channels are also easily happen, for the ΔEb values are less than 10.00 kcal mol−1. All these processes are exothermic (11.81–23.06 kcal mol−1), with less than 24.00 kcal mol−1 of energy released. Taken ΔEb values (<9.94 kcal mol−1) and ΔEr values (<23.06 kcal mol−1) into consideration, every OH addition reaction can occur easily. Among these pathways, the C3-cis-addition and C1-trans-addition are thermodynamically favorable since the barriers are low, 3.70 kcal mol−1 and 2.49 kcal mol−1.
Due to its nucleophilicity, OH radical can abstract the H atom from AF. There are five kinds of H atoms in AF molecule, suggesting that there are five possible pathways: three in the aromatic ring, one in the methylene group, and one in the methyl group. All these pathways can easily happen for their low potential barriers (0.59–9.10 kcal mol−1). Compared with other three exothermic (1.70–2.84 kcal mol−1) processes, pathway A11 (29.05 kcal mol−1) and A12 (17.62 kcal mol−1) are highly exothermic. Among the abstraction pathways, barriers are lower in the reaction of the H atom abstracted from the methylene group and methyl group than those from the aromatic ring.
HO2-Initiated reaction
HO2-Initiated reaction schemes of AF embedded with the potential barriers and reaction heats are showed in Fig. 2 and S3,† respectively.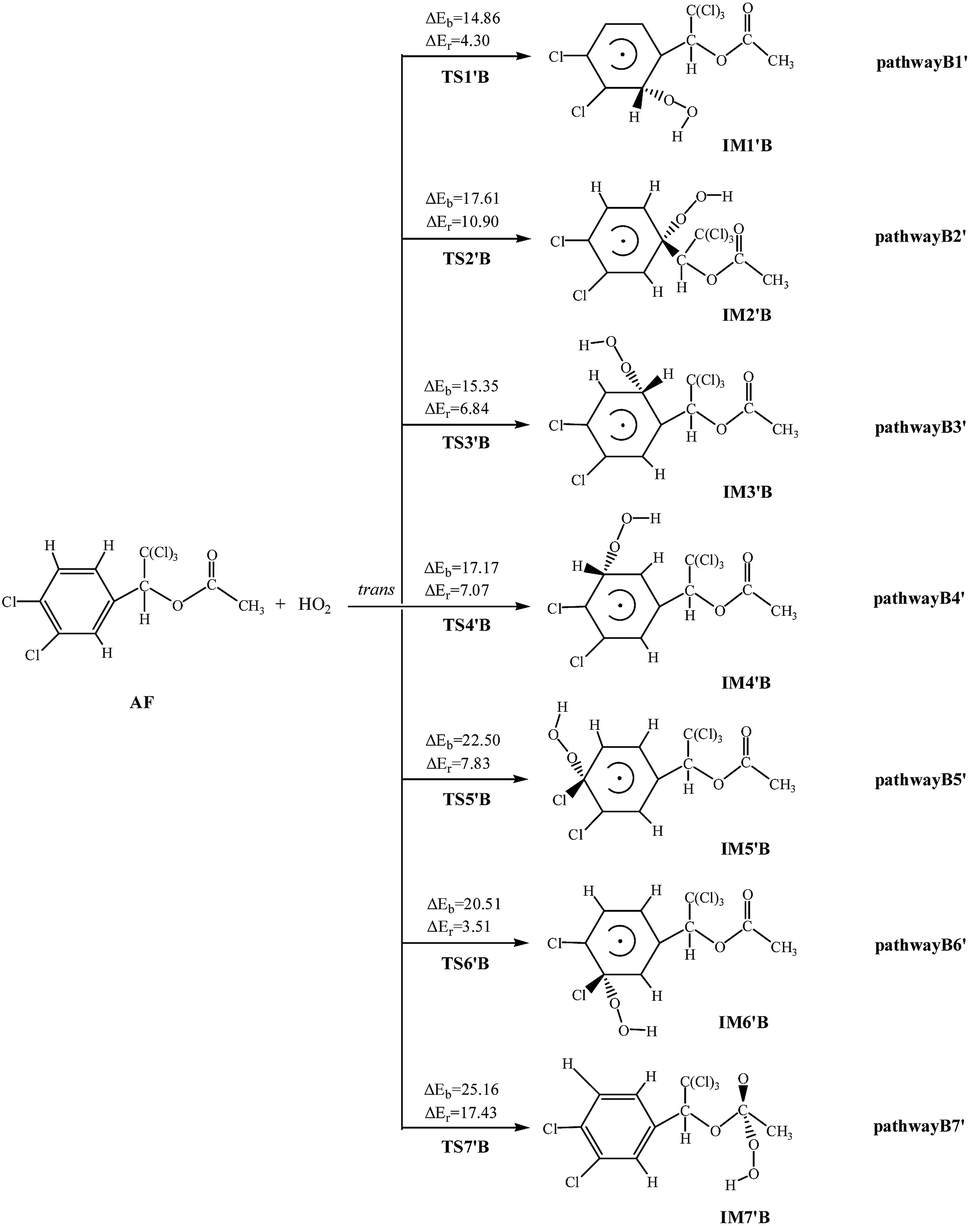 | ||
| Fig. 2 HO2 initiated pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
Unlike the OH radical, HO2 radical cannot abstract H atom from AF, and all channels are addition reactions. Clearly, HO2 radicals react with AF via TS to form products. All reactions are endothermic, and the ΔEr varies over the range of 3.51–17.43 kcal mol−1. The rate constant at 298.15 K is 7.02 × 10−33 cm3 per molecule per s, which is smaller than that of OH-initiated reaction.
NO3-Initiated reaction
NO3 radical can be added to the double bond and abstract H atom from AF which is similar to the reactions of AF and OH. The diagrams of NO3-initiated degradation mechanism of AF are illustrated in Fig. 3, 4, and S4,† respectively.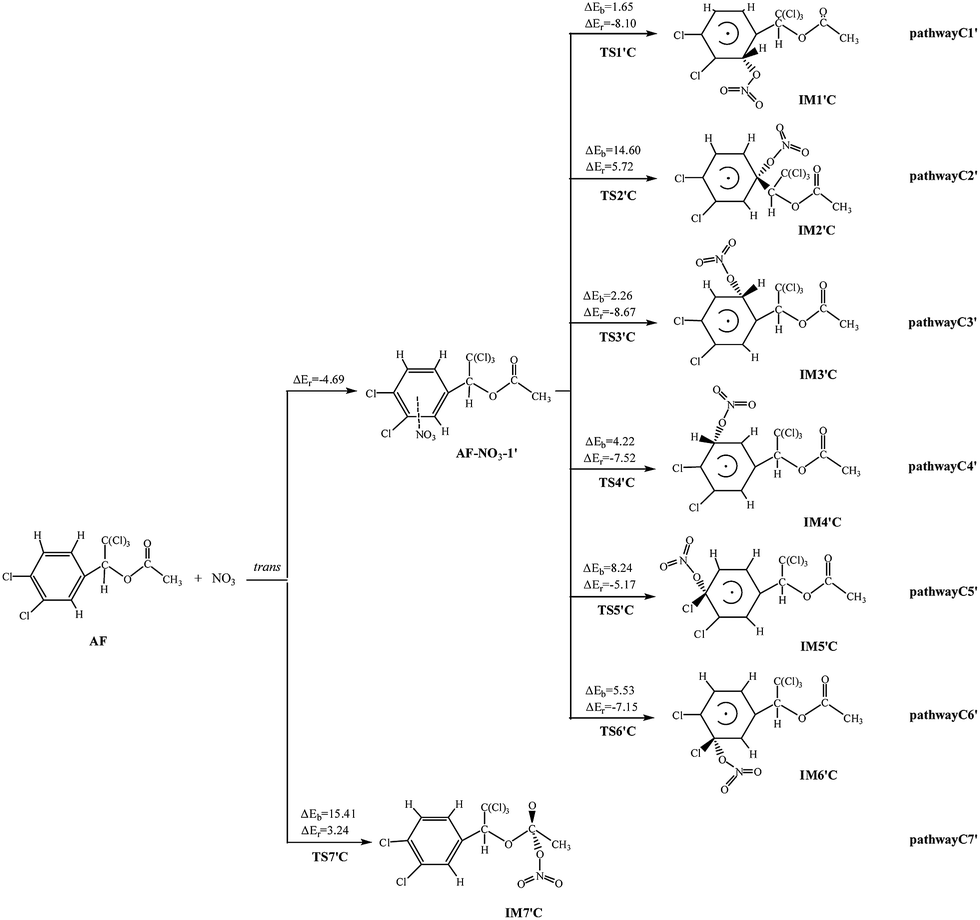 | ||
| Fig. 3 NO3 initiated addition pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
 | ||
| Fig. 4 NO3 initiated H atom abstraction pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
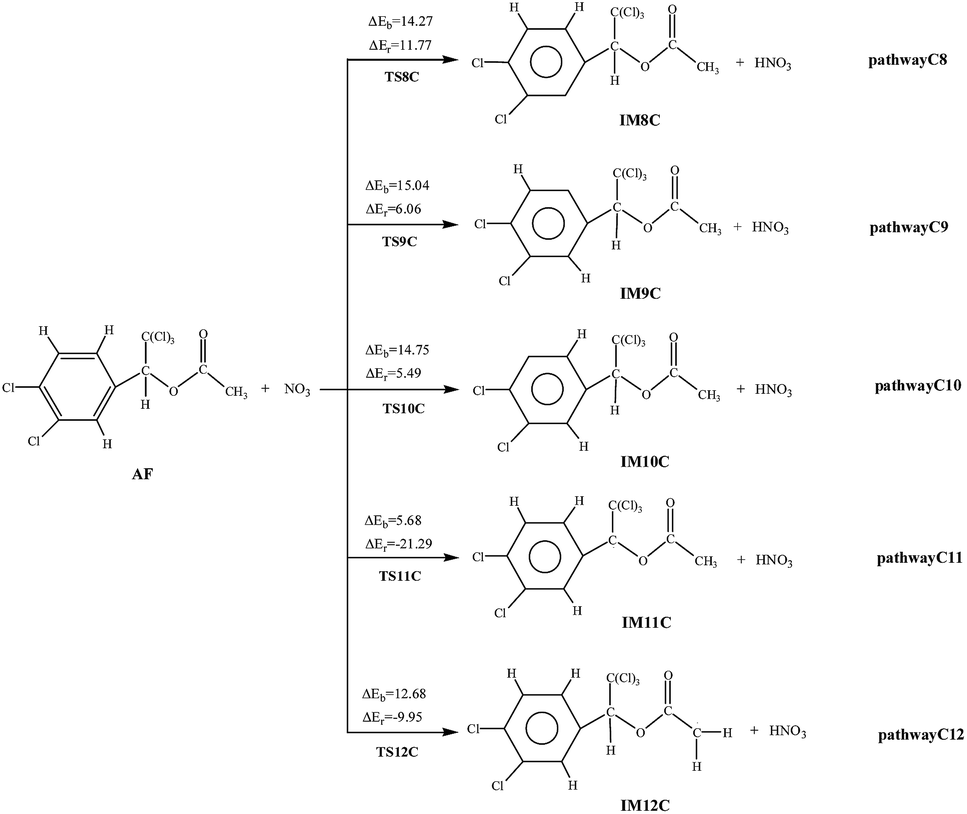
As for AF, NO3 can attract C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C or CO double bond to form two different kinds of NO3-adducts. It can be seen that in each of the pathways NO3 radicals are added to the benzene. There exists an intermediate AF–NO3, whose energy is lower than that of AF + NO3 by 5.89 kcal mol−1 of the cis-addition and by 4.69 kcal mol−1 of the trans-addition. Except the processes AF react with NO3 radical in the C2 and C7-trans-site are endothermic, the rest processes are all slightly exothermic with less than 8.67 kcal mol−1 of energy released. As shown from the cis-addition the calculated potential barriers and reaction heats, NO3 addition to C4 is the most favorable reaction with the lowest barrier (5.18 kcal mol−1) and releases the most heat (6.06 kcal mol−1). As for the trans-addition reactions, pathway C1′ and C3′ occur more early for the low potential barriers less than 2.26 kcal mol−1. Then followed by pathway C4′, the potential barriers is 4.22 kcal mol−1, giving out 7.52 kcal mol−1 of energy.
C or CO double bond to form two different kinds of NO3-adducts. It can be seen that in each of the pathways NO3 radicals are added to the benzene. There exists an intermediate AF–NO3, whose energy is lower than that of AF + NO3 by 5.89 kcal mol−1 of the cis-addition and by 4.69 kcal mol−1 of the trans-addition. Except the processes AF react with NO3 radical in the C2 and C7-trans-site are endothermic, the rest processes are all slightly exothermic with less than 8.67 kcal mol−1 of energy released. As shown from the cis-addition the calculated potential barriers and reaction heats, NO3 addition to C4 is the most favorable reaction with the lowest barrier (5.18 kcal mol−1) and releases the most heat (6.06 kcal mol−1). As for the trans-addition reactions, pathway C1′ and C3′ occur more early for the low potential barriers less than 2.26 kcal mol−1. Then followed by pathway C4′, the potential barriers is 4.22 kcal mol−1, giving out 7.52 kcal mol−1 of energy.
The hydrogen on the AF can be abstracted by NO3 to produce nitric acid (HNO3). Noticeably, the pathway C11 and C12 are exothermic, while pathway C8, C9, and C10 are endothermic. The potential barrier of pathway C11 (5.68 kcal mol−1) is lower than that of other pathways. At the same time, pathway C11 (21.29 kcal mol−1) is more exothermic than pathway C12 (9.95 kcal mol−1). Thus, the pathway C11, i.e., NO3 abstracts H atom from the methylene group, occurs more easily. What's more, at 298.15 K the total rate constant of addition reactions (4.59 × 10−20 cm3 per molecule per s) is larger than that of abstraction reactions (2.34 × 10−20 cm3 per molecule per s) with NO3 at 298.15 K.
O3-Initiated reaction
The ozonolysis reactions are initiated by addition of O3 to the double bond of AF to produce primary ozonide. The reaction channels are provided in Fig. 5 and S5,† respectively.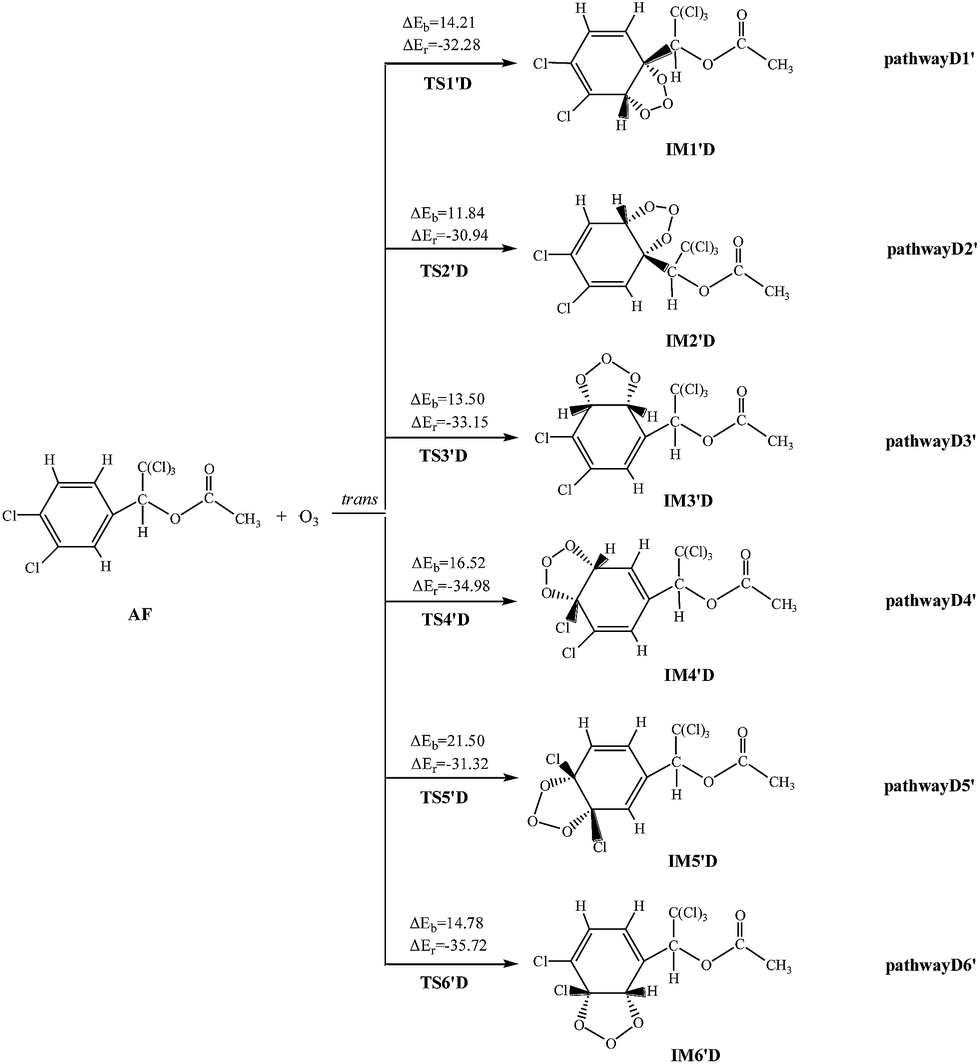 | ||
| Fig. 5 O3 initiated pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
The twelve elementary reactions all take place from AF with O3 via TS to form the ozonide. All processes are highly exothermic with released heat of 27.33–35.72 kcal mol−1, and the barrier varies over the range of 11.84–21.50 kcal mol−1. The smaller energy barrier comes from the trans-addition pathways that O3 addition to C2–C3 products IM2′D. The high reaction energy is retained as the internal energy of the adduct, the excited ozonide subsequently undergoes unimolecular decomposition due to highly exothermic.35 Then the C2–C3 and O–O of the added O3 bonds break, respectively. In consideration of the large system of reactants and IM2′D is obtained more easily, and only IM2′D is chosen to study the mechanism of isomerization. The reaction scheme is described as follows:
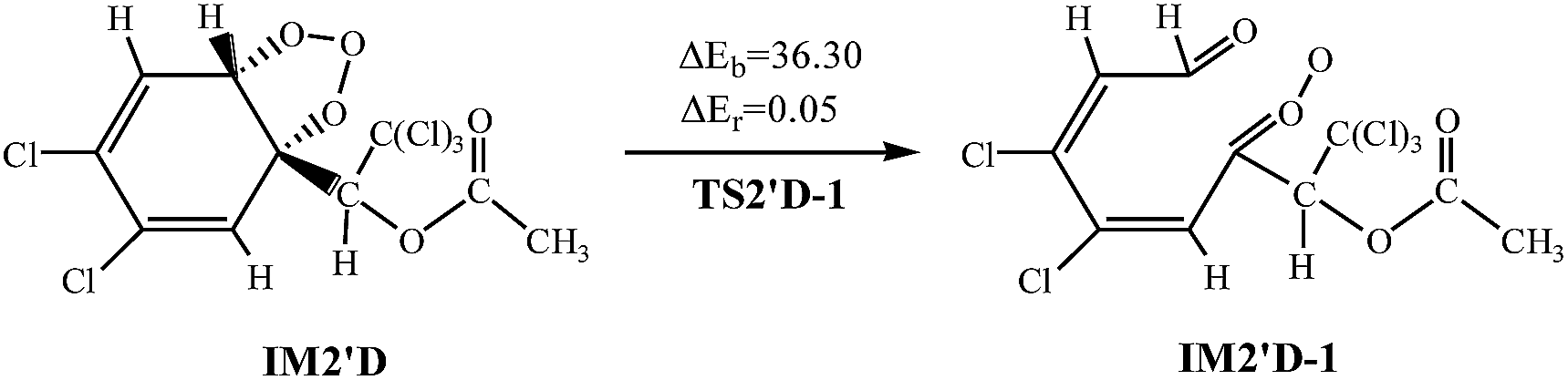
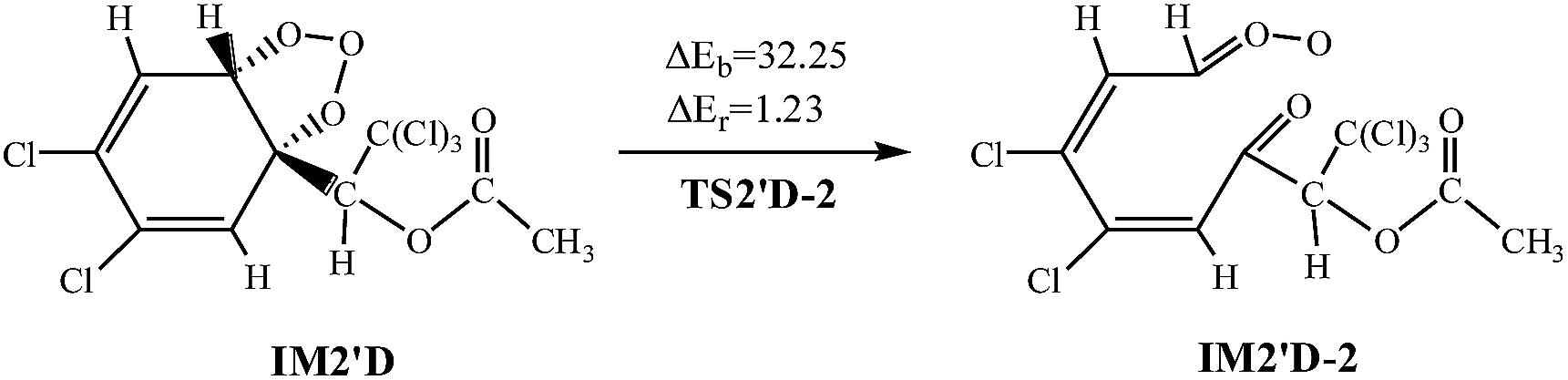
Calculations indicate that in the two processes there are apparent potential barriers of 36.30 kcal mol−1 and 32.25 kcal mol−1, absorbing 0.05 kcal mol−1 and 1.23 kcal mol−1 of heat.
Cl-Initiated reaction
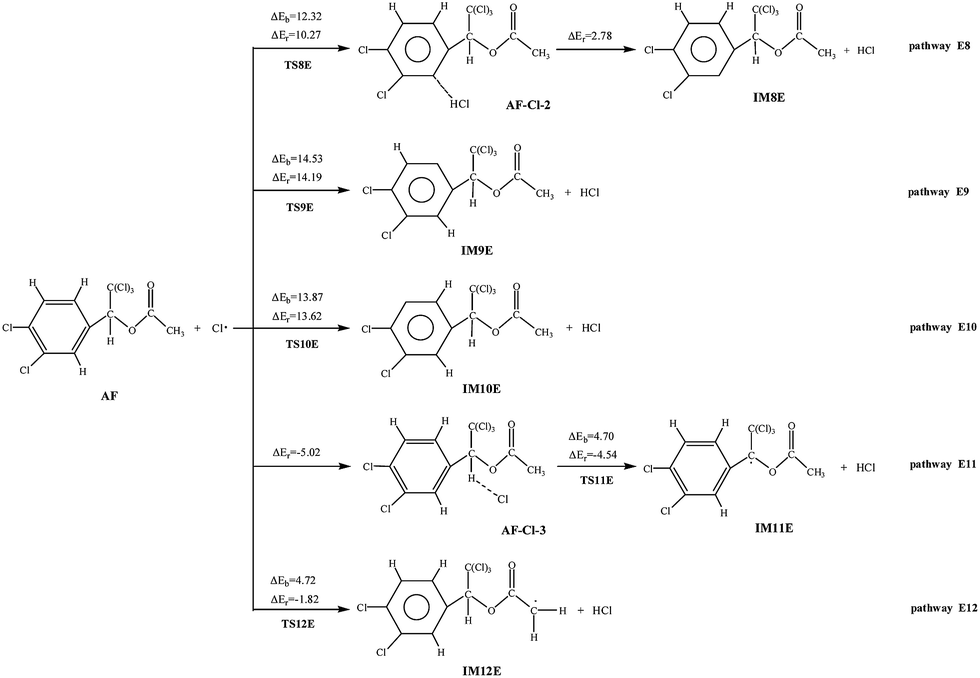
Similar to the above reaction pathways, the Cl atom can attack the double bonds of AF, and abstract hydrogen to generate hydrochloric acid (HCl). The probable pathways for the reactions are depicted in Fig. 6, 7 and S6,† respectively.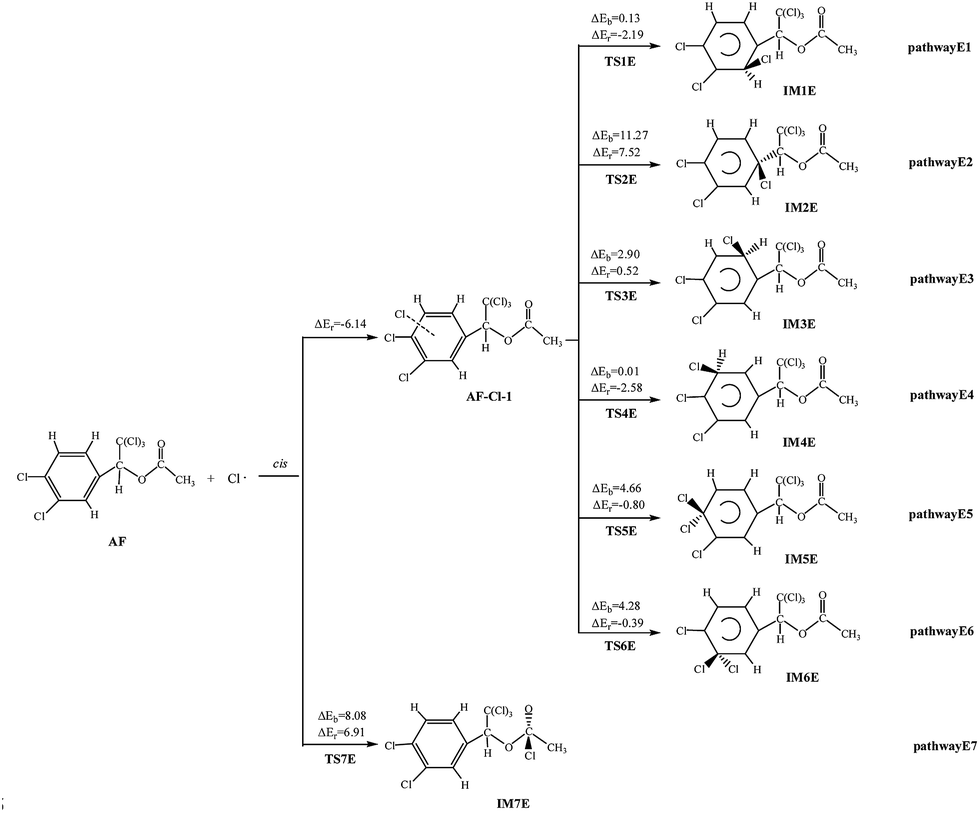 | ||
| Fig. 6 Cl initiated addition pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
 | ||
| Fig. 7 Cl initiated H atom abstraction pathways with the potential barriers ΔEb and reaction heats ΔEr (kcal mol−1). | ||
The cis-addition reactions are analysed in details. When Cl atom is added to the benzene ring to react with AF, they first form the intermediate AF–Cl-1, with the energy lower than that of AF + Cl by 6.14 kcal mol−1, and then generate IM via TS. The barriers of these processes vary over the range of 0.01–11.27 kcal mol−1. Among the six pathways, the barrierless process comes from pathway E4 (0.01 kcal mol−1), and the reaction is slightly exothermic, giving out 2.58 kcal mol−1 of energy while others are endothermic. When the Cl is added to CO double bond, this process without the formation of AF–Cl, so the process is endoergic, absorbing 6.91 kcal mol−1 of heat, and the potential barrier is 8.08 kcal mol−1.
The trans-addition reactions are similar with the cis-addition reactions. When Cl atom is added to the benzene ring, there also exists an intermediate AF–Cl-1′, and then generate IM via TS. The barriers of these processes vary over the range of 0.16–8.76 kcal mol−1. The process of Cl is added to CO double bond without the formation of AF–Cl, and it is endoergic, absorbing 8.24 kcal mol−1 of heat, and the potential barrier is 8.76 kcal mol−1. Among these reactions, pathway E1′, E3′, and E4′ could easily occur for low potential barriers (<1.18 kcal mol−1) and the reaction pathway E2′ is slightly endothermic, absorbing 1.39 kcal mol−1 of energy while others are exothermic.
The H atom abstraction pathways of AF with Cl atoms are all with positive barriers. The potential barriers of pathway E11 and E12 are lower than that of pathway E8, E9, and E10. Compared with other three endothermic (2.78–14.19 kcal mol−1) processes, pathway E11 (4.54 kcal mol−1) and E12 (1.82 kcal mol−1) are slight exothermic. In addition, the rate constants of pathway E11 and E12 are larger than those of other pathways. So taken the thermodynamic and kinetic into consideration, it is obvious that pathway E11 and E12 are favored in H abstraction pathways.
Subsequent reactions
In general, the radicals formed from additions and abstractions could further be decomposed in the presence of O2 and NO/H2O in the atmosphere. Evidently, the addition reactions in which HO2, NO3, and Cl are added to C4-cis and C1-trans sites are the most favorable pathways with the lowest barriers in each group, which can further lose the H atom that connected with the C atom and degraded in the atmosphere. In the H atom abstraction pathways, different radicals obtain five similar intermediates. The barrierless and slight exothermic processes where H atom is abstracted from the methylene group and methyl group are more dominant. Some subsequent reactions for this part have been explored by our group.36Kinetic analysis
With TST method, the rate constants of OH, HO2, NO3, O3, and Cl initiated reactions of AF at a suitable temperature range of 200–400 K are listed in Tables S1A–S8A,† respectively. The Arrhenius equations i.e. k(T) = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/RT)37 for rate constants of element reactions are listed in Tables S1B–S5B,† respectively. The total Arrhenius equations and rate constants at 298.15 K are shown in Table 1. Obviously, most of the calculated rate constants can be fitted by Arrhenius equations. The correlation coefficient R2 is above 0.9. The branching ratios of addition and abstraction reactions among different initiated reaction from 200 to 400 K are shown in Fig. S7–S9.†
exp(−Ea/RT)37 for rate constants of element reactions are listed in Tables S1B–S5B,† respectively. The total Arrhenius equations and rate constants at 298.15 K are shown in Table 1. Obviously, most of the calculated rate constants can be fitted by Arrhenius equations. The correlation coefficient R2 is above 0.9. The branching ratios of addition and abstraction reactions among different initiated reaction from 200 to 400 K are shown in Fig. S7–S9.†
| Reactions | ktotal (T = 298.15) | Arrhenius formulas | R2 |
|---|---|---|---|
| AF + OH˙ → IMA | 4.04 × 10−13 | k = 7.71 × 10−12exp(−865.03/T) |
0.9941 |
| AF + HO2˙ → IMB | 7.02 × 10−33 | k = 2.91 × 10−15exp(−8133.1/T) |
0.9999 |
| AF + NO3 → IMC | 6.93 × 10−20 | k = 8.85 × 10−17exp(−2067.6/T) |
0.9799 |
| AF + O3 → IMD | 1.45 × 10−25 | k = 5.38 × 10−16exp(−6555.2/T) |
0.9999 |
| AF + Cl˙ → IME | 5.07 × 10−12 | k = 7.84 × 10−10exp(−947.77/T) |
0.9326 |
The rate constants at 298.15 K and 1 atom in the routes have been chosen for discussion. Take OH-initiated reaction for example. The individual rate constants for the addition pathway A1–A7 and A1′–A7′ are noted kA1–kA7 and as kA1′–kA7′ respectively, and rate constants for the H atom abstraction pathway 8–12 are noted as kA8–kA12 respectively. The rate constants of overall OH radical addition reaction and H atom abstraction are defined as kaddOH = kA1 + kA2 + kA3 + kA4 + kA5 + kA6 + kA7 + kA1′ + kA2′ + kA3′ + kA4′ + kA5′ + kA6′ + kA7′, and kabsOH = kA8 + kA9 + kA10 + kA11 + kA12, respectively. The overall rate constant for the AF with OH reaction is labeled as ktotalOH = kaddOH + kabsOH. At 298.15 K, the overall calculated rate constants for reactions of AF with OH, HO2, NO3, O3, and Cl, are 4.04 × 10−13, 7.02 × 10−33, 6.93 × 10−20, 1.45 × 10−25, and 5.07 × 10−12 cm3 per molecule per s.
The degradation rate can be expressed by the formula r = kX[X][AF]. Once the concentrations of AF and X radical in atmosphere are determined the degradation rate can be calculated. The rate branching ratio can be presented by
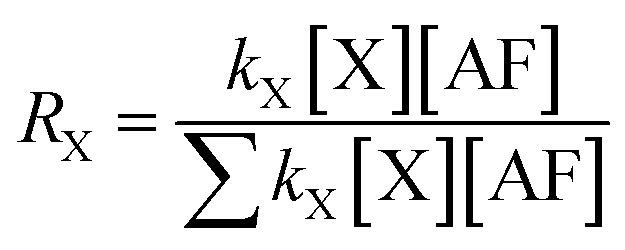
Atmospheric lifetime
To assess its impact on the environment, it is critical to know the atmospheric lifetime of AF. The atmospheric lifetimes (τ) of AF, with respect to reactions with OH, HO2, NO3, O3, and Cl, can be calculated and analyzed on the basis of the rate constants in the elementary reaction, using the formula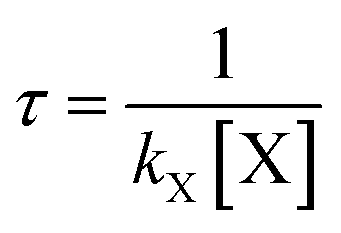 . In which [X] term is the concentration of radical and can be taken as a constant in the troposphere. The calculated lifetimes at the range of 200–400 K are listed in Tables S1C–S8C.† Overall, the higher the temperature is, the shorter the lifetime.
. In which [X] term is the concentration of radical and can be taken as a constant in the troposphere. The calculated lifetimes at the range of 200–400 K are listed in Tables S1C–S8C.† Overall, the higher the temperature is, the shorter the lifetime.
The total atmospheric lifetime can be calculated with the formula 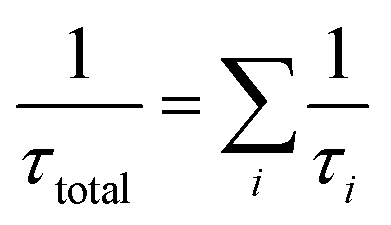 . Total atmospheric lifetimes of AF with different oxidants are listed in Table SD.† According to the results, the τtotal for all OH, HO2, NO3, O3, and Cl initiated oxidation reactions is 2.14 days. And the τ for the reaction of OH radical is around 2.18 days, and 135 days for Cl. Evidently, AF is more likely to be removed quickly by the reaction with ozone near their emission sources. However, both the concentration of radicals and the rate constant of reactions with radicals determine the atmospheric lifetime of AF. In some places, the abundant OH radicals may play an important role in the degradation of AF. At night or in seriously polluted regions, the 12 h averaged NO3 radical concentration is about 5 × 108 cm3 per molecule per s, the reaction with NO3 may contribute to controlling AF.40 Moreover, in some coastal areas where the concentration of Cl atoms can reach 1 × 105 molecule per cm3, the reaction of AF with Cl atom may contribute great portion to the removal of AF in the atmosphere.26
. Total atmospheric lifetimes of AF with different oxidants are listed in Table SD.† According to the results, the τtotal for all OH, HO2, NO3, O3, and Cl initiated oxidation reactions is 2.14 days. And the τ for the reaction of OH radical is around 2.18 days, and 135 days for Cl. Evidently, AF is more likely to be removed quickly by the reaction with ozone near their emission sources. However, both the concentration of radicals and the rate constant of reactions with radicals determine the atmospheric lifetime of AF. In some places, the abundant OH radicals may play an important role in the degradation of AF. At night or in seriously polluted regions, the 12 h averaged NO3 radical concentration is about 5 × 108 cm3 per molecule per s, the reaction with NO3 may contribute to controlling AF.40 Moreover, in some coastal areas where the concentration of Cl atoms can reach 1 × 105 molecule per cm3, the reaction of AF with Cl atom may contribute great portion to the removal of AF in the atmosphere.26
Conclusions
The degradation mechanism of AF initiated by OH radical, HO2 radical, NO3 radical, O3, and Cl atom were investigated at the MPWB1K/6-31+G(d,p) level. The kinetic calculations were carried out with TST method. Some valuable conclusions can be drawn as follows:The mechanism of OH, HO2, NO3, O3, and Cl initiated reactions of AF includes H abstraction pathways and the addition pathways.
The O3-adducts can undergo unimolecular decomposition, leading to C–C/C–O bond cleavage. For cis-addition HO2, NO3, and Cl added to C4 are the most favorable reaction pathways with the lowest barrier and release the more heat, while they added C1 are the most favorable reaction pathways among the trans-addition, the adducts can further leave the H atom connect with C4 or C1 and be degraded in the atmosphere.
At 298.15 K, the total rate constants of AF with OH, HO2, NO3, O3, and Cl, are 4.04 × 10−13, 7.02 × 10−33, 6.93 × 10−20, 1.45 × 10−25, and 5.07 × 10−12 cm3 per molecule per s, respectively. The rate branching ratio of OH initiated reactions are dominant. The HO2, NO3, O3 and Cl initiated reactions make little contribution to the reaction rate of AF from 200 to 400 K compared with the OH initiated reactions.
The τ for all OH, HO2, NO3, O3, and Cl initiated oxidation reactions is 2.14 days.
Acknowledgements
This work is supported by National Natural Science Foundation of China (21277082, 21337001), the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province (BS2012HZ009), Natural Science Foundation of Shandong Province (No. ZR2014BP012), Program for New Century Excellent Talents in University (NCET-13-0349), Marie Curle International Research Staff Exchange Scheme Fellowship within the 7th European Community Framework Program (No. 295132), Project for science and technology development of Shandong province (2014GSF117028), Beijing National Laboratory for Molecular Science (20140160), Strategic Priority Research Program (B) of the Chinese Academy of Sciences, Grant No. XDB05010200), and the Fundamental Research Funds of Shandong University (2015JC020).Notes and references
- M. Zhao, Y. Zhang, C. Wang, Z. Fu, W. Liu and J. Gan, Chem. Res. Toxicol., 2009, 22, 504–510 CrossRef CAS PubMed.
- H. Yang, B. Xue, P. Yu, S. Zhou and W. Liu, Chemosphere, 2010, 80, 652–659 CrossRef CAS PubMed.
- Y. Zhu, Z. Wang and B. Li, Encyclopedia of agricultural chemicals, 2006 Search PubMed.
- L. Li, F. Hu, C. Wang and X. Wang, J. Environ. Sci., 2010, 22, 1980–1986 CrossRef CAS.
- F. Chen, Q. Zhang, C. Wang, Y. Lu and M. Zhao, Reprod. Toxicol., 2012, 33, 53–59 CrossRef CAS PubMed.
- P. S. Monks, Chem. Soc. Rev., 2005, 34, 376–395 RSC.
- R. Atkinson, Atmos. Environ., 2000, 34, 2063–2101 CrossRef CAS.
- H. M. Walker, D. Stone, T. Ingham, S. Vaughan, B. Bandy, M. Cain, R. L. Jones, O. J. Kennedy, M. McLeod, B. Ouyang, J. Pyle, S. Bauguitte, B. Bandy, G. Forster, M. J. Evans, J. F. Hamilton, J. R. Hopkins, J. D. Lee, A. C. Lewis, R. T. Lidster, S. Punjabi, W. T. Morgan and D. E. Heard, Atmos. Chem. Phys. Discuss., 2015, 15, 2997–3061 Search PubMed.
- S. S. Brown and J. Stutz, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- E. M. Knipping and D. Dabdub, Environ. Sci. Technol., 2003, 37, 275–284 CrossRef CAS PubMed.
- X. Qu, Q. Zhang and W. Wang, Chem. Phys. Lett., 2006, 426, 13–19 CrossRef CAS.
- X. Ren, H. Harder, M. Martinez, R. Lesher, A. Oliger, J. B. Simpas, W. H. Brune, J. J. Schwab, K. L. Demerjian, Y. He, X. Zhou and H. Gao, Atmos. Environ., 2003, 37, 3639–3651 CrossRef CAS.
- A. Hofzumahaus, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C. Chang, H. Fuchs, F. Holland, K. Kita, Y. Kondo, X. Li, S. Lou, M. Shao, L. Zeng, A. Wahner and Y. Zhang, Science, 2009, 324, 1702–1704 CrossRef CAS PubMed.
- S. E. Paulson and J. J. Orlando, Geophys. Res. Lett., 1996, 23, 3727–3730 CrossRef CAS.
- K. D. Lu, F. Rohrer, F. Holland, H. Fuchs, B. Bohn, T. Brauers, C. C. Chang, R. Häseler, M. Hu, K. Kita, Y. Kondo, X. Li, S. R. Lou, S. Nehr, M. Shao, L. M. Zeng, A. Wahner, Y. H. Zhang and A. Hofzumahaus, Atmos. Chem. Phys., 2012, 12, 1541–1569 CAS.
- D. E. Heard and M. J. Pilling, Chem. Rev., 2003, 103, 5163–5198 CrossRef CAS PubMed.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosa-Mas, J. Hjorth, G. L. Bras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, Atmos. Environ., Part A, 1991, 25, 1–203 CrossRef.
- A. M. Winer, R. Atkinson and J. N. Pitts, Science, 1984, 224, 156–159 CAS.
- U. F. Platt, A. M. Winer, H. W. Biermann, R. Atkinson and J. N. Pitts, Environ. Sci. Technol., 1984, 18, 365–369 CrossRef CAS PubMed.
- U. Platt, D. Perner, A. M. Winer, G. W. Harris and J. N. Pitts, Geophys. Res. Lett., 1980, 7, 89–92 CrossRef CAS.
- A. Geyer, B. Alicke, R. Ackermann, M. Martinez, H. Harder, W. Brune, P. D. Carlo, E. Williams, T. Jobson, S. Hall, R. Shetter and J. Stutz, J. Geophys. Res., 2003, 108, 4368 CrossRef.
- A. E. Perring, S. E. Pusede and R. C. Cohen, Chem. Rev., 2013, 113, 5848–5870 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Atmospheric chemistry, Fundamentals and experimental techniques, 1986 Search PubMed.
- J. A. Logan, J. Geophys. Res., 1985, 90, 10463–10482 CrossRef.
- S. J. Oltmans, A. S. Lefohn, D. Shadwick, J. M. Harris, H. E. Scheel, I. Galbally, D. W. Tarasick, B. J. Johnson, E. G. Brunke, H. Claude, G. Zeng, S. Nichol, F. Schmidlin, J. Davies, E. Cuevas, A. Redondas, H. Naoe, T. Nakano and T. Kawasato, Atmos. Environ., 2013, 67, 331–351 CrossRef CAS.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Nature, 1998, 394, 353–356 CrossRef CAS.
- H. B. Singh, A. N. Thakur, Y. E. Chen and M. Kanakidou, Geophys. Res. Lett., 1996, 23, 1529–1532 CrossRef CAS.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- A. K. Chakrabartty, B. K. Mishra, D. Bhattacharjee and R. Chandra, Mol. Phys., 2013, 111, 860–867 CrossRef CAS.
- R. C. Deka and B. K. Mishra, Chem. Phys. Lett., 2014, 595, 43–47 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908–6918 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Milla, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, A. D. Daniels, O. Farkas, A. D. Rabuck, K. Raghavachari and J. V. Ortiz, Gaussian 03, revision B. 05, Gaussian, Inc., Pittsburgh, PA, 2003, p. 12478 Search PubMed.
- Y. Chu, B. Han, H. Fang, A. Zheng and F. Deng, Microporous Mesoporous Mater., 2012, 151, 241–249 CrossRef CAS.
- P. R. Barreto, A. F. Vilela and R. Gargano, J. Mol. Struct.: THEOCHEM, 2003, 639, 167–176 CrossRef CAS.
- J. Bai, X. Sun, C. Zhang, Y. Zhao and C. Gong, Chemosphere, 2013, 92, 933–940 CrossRef CAS PubMed.
- L. Kang, X. Sun, C. Zhang, X. Zhang and J. Chen, Atmos. Environ., 2015, 103, 357–364 CrossRef CAS.
- K. J. Laidler, J. Chem. Educ., 1984, 61, 494–498 CrossRef CAS.
- M. Vrekoussis, M. Kanakidou, N. Mihalopoulos, P. J. Crutzen, J. Lelieveld, D. Perner, H. Berresheim and E. Baboukas, Atmos. Chem. Phys., 2004, 4, 169–182 CrossRef CAS.
- O. W. Wingenter, D. R. Blake, N. J. Blake, B. C. Sive, F. S. Rowland, E. Atlas and F. Flocke, J. Geophys. Res., 1999, 104, 21819–21828 CrossRef CAS.
- Y. Shu and R. Atkinson, J. Geophys. Res., 1995, 100, 7275–7281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11453e |
| This journal is © The Royal Society of Chemistry 2015 |