
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective construction of C-chiral allylic sulfilimines via the iridium-catalyzed allylic amination with S,S-diphenylsulfilimine: asymmetric synthesis of primary allylic amines†
Rebecca L.
Grange
a,
Elizabeth A.
Clizbe
b,
Emma J.
Counsell
b and
P. Andrew
Evans
*a
aQueen's University, Department of Chemistry, 90 Bader Lane, Kingston, ON K7L 3N6, Canada. E-mail: andrew.evans@chem.queensu.ca
bThe University of Liverpool, Department of Chemistry, Crown Street, Liverpool, L69 7ZD, UK
First published on 8th September 2014
Abstract
We have devised a highly regio- and enantioselective iridium-catalyzed allylic amination reaction with the sulfur-stabilized aza-ylide, S,S-diphenylsulfilimine. This process provides a robust and scalable method for the construction of aryl-, alkyl- and alkenyl-substituted C-chiral allylic sulfilimines, which are important functional groups for organic synthesis. Additionally, the combination of the allylic amination with an in situ deprotection of the sulfilimine constitutes a convenient one-pot protocol for the construction of chiral nonracemic primary allylic amines.
Introduction
Sulfur-stabilized aza-ylides, as exemplified by sulfilimines, provide an interesting class of molecules that display unique and diverse reactivity.1 For example, the ylide character of the nitrogen–sulfur group provides an ambidentate species that can either undergo nucleophilic or electrophilic aziridination of electron-deficient and electron-rich olefins, respectively.2,3 Sulfilimines also participate in cycloaddition reactions, which provides valuable opportunities for target directed synthesis.1 Notwithstanding the distinctive synthetic attributes of the sulfilimine, it represents a rather intriguing functional group in so much that it can be stereogenic at both carbon and sulfur. Although there are a number of convenient methods for the construction of enantioenriched S-chiral sulfilimines, the construction of C-chiral sulfilimines has not been forthcoming. Furthermore, sulfilimines provide important synthetic targets, as exemplified by their incorporation in pesticides and photographic recording materials.4 Additionally, a sulfilimine was recently implicated in the stabilization of collagen IV networks in the form of a critical cross-link between a hydroxylysine and methionine residue, which further underscores the growing significance of this unusual structural motif in organic chemistry.5In a program directed towards the utilization of charge separated nucleophiles in the metal-catalyzed allylic substitution reaction;6 we recently demonstrated the merit of pyridinium ylides, which provide a new class of air-stable and non-basic nitrogen nucleophiles (Scheme 1A).7 In contrast, the sulfur-stabilized derivative, which would permit the construction of the aforementioned C-chiral allylic sulfilimines, is a particularly poor nucleophile for the rhodium-catalyzed reaction due to the combination of field and resonance stabilization.8,9 Hence, we envisioned that the iridium-catalyzed reaction, which typically facilitates the alkylation of several weak nucleophiles,10 should overcome the poor reactivity and thereby provide exciting opportunities to illustrate the unique reactivity of the sulfilimine group. Herein, we now describe the first regio- and enantioselective iridium-catalyzed allylic amination11 of allylic carbonates and benzoates 1 with the commercially available nitrogen ylide S,S-diphenylsulfilimine12,13 for the construction of chiral non-racemic N-allylic sulfilimines 2, a hitherto unreported motif of considerable synthetic potential (Scheme 1B).
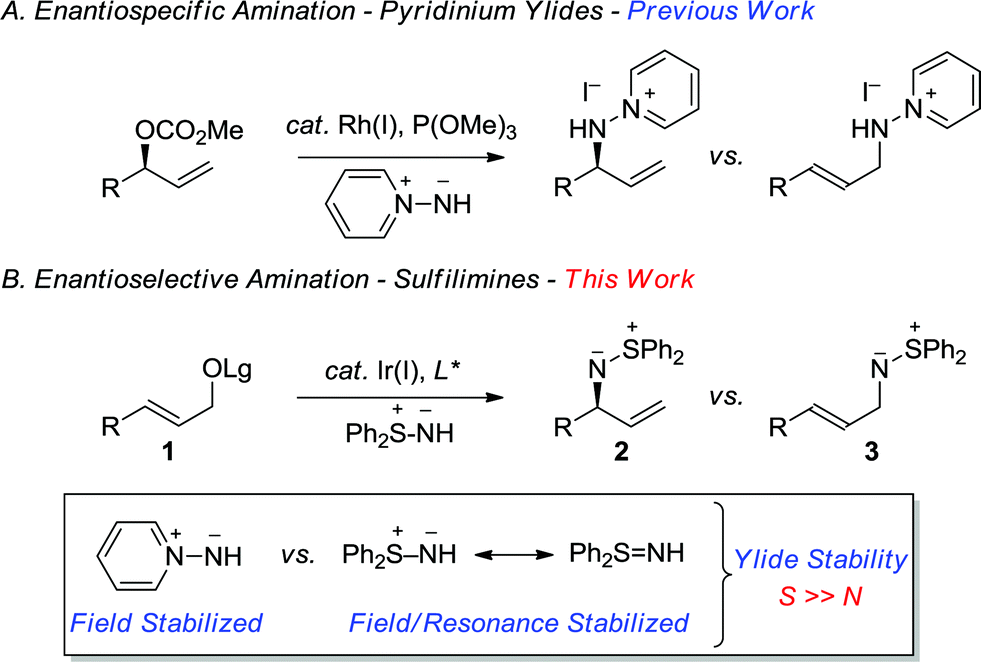 | ||
| Scheme 1 Enantiospecific and enantioselective metal-catalyzed allylic amination reactions with aza-ylides. | ||
Results and discussion
Table 1 outlines the preliminary studies on the development of the regio- and enantioselective iridium-catalyzed allylic amination with a sulfilimine nucleophile. Treatment of the cinnamyl carbonate 1a with S,S-diphenylsulfilimine and the chiral iridium complex derived from [Ir(cod)Cl]2 and the phosphoramidite ligand 4a14(Fig. 1) in dichloromethane at 35 °C furnished the sulfilimine 2a in 79% yield with excellent regio- and enantioselectivity (entry 1). Interestingly, the related phosphoramidite ligands 4b15 and 4c16 were significantly inferior to 4a in terms of both the efficiency and regioselectivity (Fig. 1). Further studies examined the feasibility of utilizing catalytic quantities of cesium carbonate as the exogenous base to improve the overall yield. Gratifyingly, the allylic amination reaction in the presence of catalytic cesium carbonate, furnished the allylic sulfilimine 2a with improved efficiency and comparable selectivity, making this an attractive synthetic protocol (entry 2).17|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Allylic alcohol derivative 1 | pKa (LgOH) | Cs2CO3 | Yield (%)b | 2/3c | ee (%)d,e | |
R ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) |
Lg |
||||||
| a All reactions were carried out on a 0.25 mmol reaction scale using 2 mol% of [Ir(cod)Cl]2, 4 mol% of 4a and 1.1 equivalents of S,S-diphenylsulfilimine in dichloromethane at 35 °C. b Isolated yields. c Regioselectivity was determined by 500 MHz 1H NMR on the crude reaction mixtures. d Enantioselectivity was determined by chiral HPLC analysis of the N-trifluoroacetamide or N-p-toluenesulfonamide derivative.19 e The absolute configurations of (S)-2a and (R)-2k were determined by conversion to either the N-trifluoroacetamide or the N-p-tosylsulfonamide derivative and comparison of the optical rotations with the reported values.19,20 | |||||||
| 1 | Ph | MeCO2 | 5.61 | — | 79 | ≥19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
96 |
| 2 | Ph | MeCO 2 | 5.61 | 0.25 | 87 |
≥19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
97 |
| 3 | n-Pr | “ | “ | 1.1 | 43 | 15:1 |
90 |
| 4 | “ | MeCO | 4.76 | “ | 18 | 14:1 |
99 |
| 5 | “ | PhCO | 4.20 | “ | 63 | ≥19:1 |
93 |
| 6 | n-Pr | 3-FC 6 H 4 CO | 3.87 | 1.1 | 91 |
≥19:1
|
95 |
| 7 | “ | “ | “ | 0.25 | 72 | ≥19:1 |
94 |
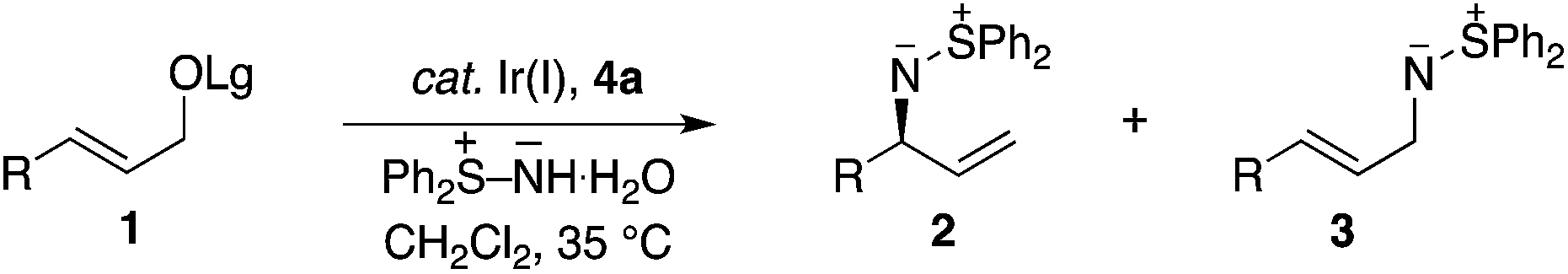
Having established the optimal procedure for the aryl derivatives, we sought to examine aliphatic substituted electrophiles, which are generally more challenging substrates for the iridium-catalyzed allylic substitution reaction.6f In this context, the application of similar reaction conditions to the propyl substituted allylic carbonate 1 led to poor yield with both catalytic and stoichiometric base (entry 3), which prompted the examination of alternative leaving groups. Although the acetate leaving-group was inferior to the carbonate (entry 3 vs. 4), the benzoate provided significant improvement in yield and regioselectivity (entry 5). Additional modifications to the stereoelectronics of the benzoate identified the 3-fluorobenzoate as the optimal leaving group (3-FC6H4CO > PhCO > MeOCO > MeCO) in terms of selectivity and efficiency (entry 6).18 Furthermore, this reaction can also be conducted with catalytic base, albeit with slightly diminished yield (entry 7). Hence, the scope of the reaction was examined using stoichiometric cesium carbonate.
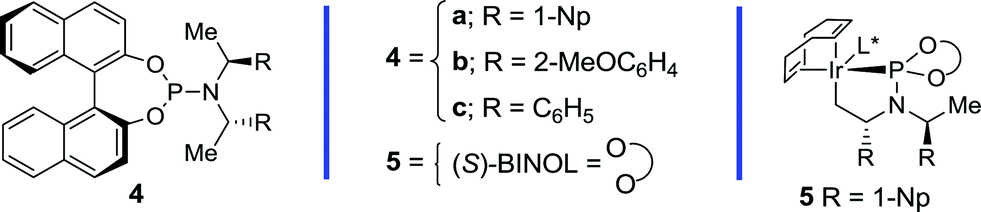 | ||
| Fig. 1 Chiral phosphoramidite ligands and the cyclometallated iridium complex utilized for the enantioselective allylic amination. | ||
Table 2 outlines the application of the optimized reaction conditions (Table 1, entries 2 and 6) to aryl-, alkyl- and alkenyl-substituted allylic carbonates and benzoates.‡ Interestingly, the reaction is tolerant of a wide array of electron-rich and electron-poor cinnamyl alcohol derivatives, in which 2-, 3- and 4-substituted aromatic allylic carbonates afford excellent yields and regioselectivities (entries 1–10), albeit with a slightly diminished selectivity for the chloro- and bromo-derivatives (entries 7 and 8). In addition, excellent enantioselectivities were observed in all cases with the exception of the 2-substituted variant (entry 2), which often provides lower selectivity in related iridium-catalyzed allylic substitution reactions.6f The 2-naphthyl derivative also provides excellent regio- and enantioselectivity for this process (entry 10), thereby further illustrating the scope with aryl derivatives. Gratifyingly, the application of the optimized reaction conditions to the more challenging alkyl and alkenyl derivatives provided exquisite regio- and enantioselectivity. For instance, the linear and branched alkyl derivatives provide excellent yields and selectivities under these conditions (entries 11–17), albeit the α-branched derivatives required pre-activation of the catalyst (entries 14–16) with n-propylamine to provide 5 (Fig. 1).10a Additional studies demonstrated that tert-butyldimethylsilyl and benzyl protected hydroxymethyl and ethyl derivatives are also well tolerated (entries 18–21). Furthermore, the tethered N-Boc and N-Cbz derivatives also provide suitable substrates for this process to afford differentially protected diamines (entries 22–23). Finally, the chloro- and alkenyl-substituted allylic benzoates afford excellent selectivity (entry 24–25), in which the latter also requires catalyst pre-activation similar to the α-branched derivatives. Overall, this study highlights the synthetic versatility of the allylic amination reaction with a sulfilimine nucleophile, which encompasses an array of aryl-, alkyl- and alkenyl-substituted allylic alcohol derivatives.
| Entry | Allylic alcohol derivative 1 | Method | Yieldb (%) | b/l2/3c | eed (%) | ||
|---|---|---|---|---|---|---|---|
| Lg |
R |
||||||
| a All reactions were carried out on a 0.25 mmol reaction scale using 2 mol% of [Ir(cod)Cl]2, 4 mol% of 4a and 1.1 equivalents of S,S-diphenylsulfilimine in dichloromethane at 35 °C. Method A: 0.25 equiv. Cs2CO3; Method B: 1.1 equiv. Cs2CO3; Method C: 1.1 equiv. Cs2CO3 and catalyst pre-activated with n-PrNH2.10a b Isolated yields. c Regioselectivity was determined by 500 MHz 1H NMR on the crude reaction mixtures. d Enantioselectivity was determined by chiral HPLC analysis of the N-trifluoroacetamides (entries 1–10 and 20–21) and the N-p-tosylsulfonamides (entries 11–19 and 22–25). e The absolute configurations were determined by conversion to the known N-p-tosylsulfonamide and comparison of the optical rotations, which are consistent with the Hartwig model.19,20 | |||||||
| 1 | MeOCO– | Ph | a | A | 87 | ≥19:1 |
98e |
| 2 | “ | 2-MeOC6H4 | b | A | 87 | ≥19:1 |
81 |
| 3 | “ | 3-MeOC6H4 | c | A | 85 | ≥19:1 |
94 |
| 4 | “ | 4-MeOC6H4 | d | A | 80 | ≥19:1 |
95 |
| 5 | “ | 4-MeC6H4 | e | A | 90 | ≥19:1 |
93 |
| 6 | “ | 4-FC6H4 | f | A | 88 | ≥19:1 |
96 |
| 7 | “ | 4-ClC6H4 | g | A | 78 | 17:1 |
96 |
| 8 | “ | 4-BrC6H4 | h | A | 81 | 18:1 |
91 |
| 9 | “ | 3,5-(MeO)2C6H3 | i | A | 81 | ≥19:1 |
94 |
| 10 | “ | 2-Naphthyl | j | A | 89 | ≥19:1 |
96 |
| 11 | 3-FC6H4CO– | n-Pr | k | B | 91 | ≥19:1 |
95e |
| 12 | “ | Me | l | B | 91 | ≥19:1 |
95e |
| 13 | “ | Ph(CH2)2 | m | B | 86 | ≥19:1 |
91 |
| 14 | “ | i-Pr | n | C | 82 | ≥19:1 |
95e |
| 15 | “ | c-Pen | o | C | 84 | ≥19:1 |
97 |
| 16 | “ | c-Hex | p | C | 74 | ≥19:1 |
97 |
| 17 | “ | i-Pen | q | B | 86 | ≥19:1 |
94 |
| 18 | “ | TBSOCH2 | r | C | 78 | 17:1 |
95 |
| 19 | “ | TBSO(CH2)2 | s | C | 77 | 14:1 |
93 |
| 20 | “ | BnOCH2 | t | B | 78 | 16:1 |
92 |
| 21 | “ | BnO(CH2)2 | u | B | 71 | 17:1 |
90 |
| 22 | “ | BocNH(CH2)5 | v | B | 82 | ≥19:1 |
93 |
| 23 | “ | CbzNH(CH2)5 | w | B | 82 | ≥19:1 |
92 |
| 24 | “ | Cl(CH2)5 | x | B | 82 | ≥19:1 |
92 |
| 25 | “ | (E)-MeCHCH |
y | C | 72 | ≥19:1 |
95 |
Scheme 2 illustrates the synthetic utility of the iridium-catalyzed allylic amination with S,S-diphenylsulfilimine, which represents a novel ammonia equivalent.21–23 The development of ammonia equivalents remains an important area of investigation, since chiral nonracemic primary allylic amines are versatile synthons for target directed synthesis. Recent landmark reports on the ability to utilize ammonia to directly prepare the primary allylic amine represents an important advance in this area. Nevertheless, this approach has limitations, which provides the impetus for further developments. For instance, the allylic amination with ammonia requires a large excess of the nucleophile (100 fold) and a specialized catalyst to reduce dialkylation. Although this process affords exquisite selectivity, the yields of the primary amine are generally modest. Alternatively, the allylic amination with sulfamic acid, which forms ammonia through in situ fragmentation, provides modest yields and enantioselectivities with simple alkyl substrates, thereby making this less attractive for synthetic applications. Consequently, we envisioned that the allylic amination with S,S-diphenylsulfilimine could be combined with a simple deprotection to provide a practical and scalable route to this important functional group.21 For example, the allylic amination of 1k can be accomplished on a gram-scale to afford the enantiomerically enriched sulfilimine 2k in 81% yield with excellent selectivity. Interestingly, the cleavage of the N–S bond could also be accomplished on a similar scale to afford the allylic amine hydrochloride salt 6 in 94% yield (99% cee) (Scheme 2A).24 Alternatively, enantiomerically enriched primary allylic amines can be accessed directly using the one-pot process outlined in Scheme 2B. Treatment of the primary allylic carbonate 1w under the optimal reaction conditions, followed by the in situ cleavage of the N–S bond of the sulfilimine, furnished the mono-protected diamine 7 in 76% overall yield (b/l ≥19:1, 91% ee), thereby illustrating the synthetic utility of the sulfilimine.
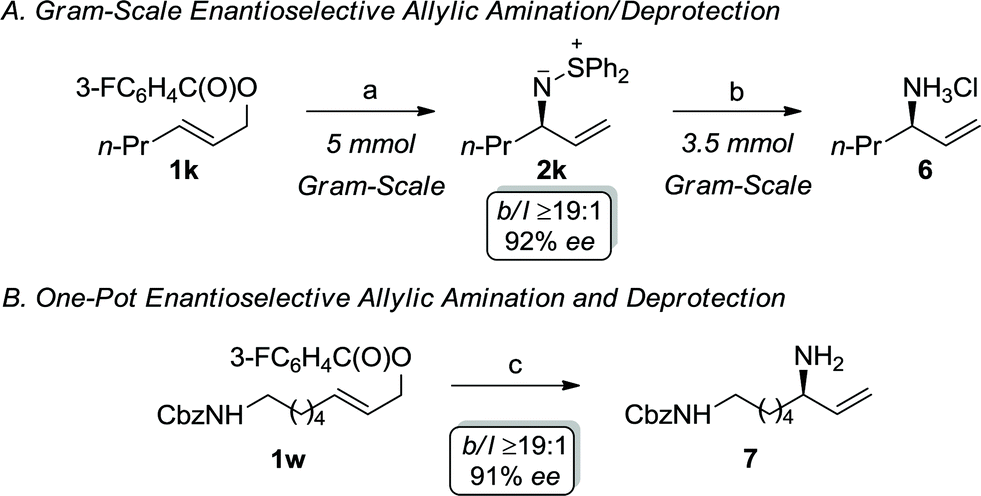 | ||
| Scheme 2 Gram-scale allylic amination/deprotection and one-pot allylic amination/deprotection: (a) cat. [Ir(cod)Cl]2, 4a, Ph2S+NH−, Cs2CO3, CH2Cl2, 35 °C, 81%; (b) aqueous 10M HCl, CH3CN, RT, 94%; c) cat. [Ir(cod)Cl]2, 4a, Ph2S+NH−, Cs2CO3, CH2Cl2, 35 °C; then aqueous 10M HCl, CH3CN, RT, 76%. | ||
Conclusions
In conclusion, we have devised the first highly regio- and enantioselective iridium-catalyzed allylic amination reaction with the sulfur-stabilized aza-ylide, S,S-diphenylsulfilimine, which is directly applicable to aryl-, alkyl- and alkenyl-substituted sulfilimines. Additionally, the sulfilimine provides a convenient and commercially available ammonia equivalent, which is readily cleaved with acid to afford the enantiomerically enriched primary amine hydrochloride salt. Furthermore, the ability to conduct this sequence either on a gram-scale or in one-pot, permits the direct formation of the primary allylic amine to illustrate the utility of this process for challenging synthetic applications. Overall, this work demonstrates that aza-ylides are a valuable class of nucleophiles for enantioselective metal-catalyzed allylic substitution reactions.25Acknowledgements
We sincerely thank the National Sciences and Engineering Research Council (NSERC) for a Discovery Grant and Queen's University for generous financial support. NSERC is also thanked for supporting a Canada Research Chair (PAE). Additionally, we acknowledge the Royal Society for a Wolfson Research Merit Award (PAE) and the University of Liverpool for a Postgraduate Research Studentship (EAC).Notes and references
- (a) I. V. Koval, Sulfur Rep., 1993, 14, 149 CrossRef CAS; (b) P. C. Taylor, Sulfur Rep., 1999, 21, 241 CrossRef CAS.
- For leading examples, see: (a) D. A. Clark, F. De Riccardis and K. C. Nicolaou, Tetrahedron, 1994, 50, 11391 CrossRef CAS; (b) B. M. Trost and T. Zhang, Chem.–Eur. J., 2011, 17, 3630 CrossRef CAS PubMed.
- For leading examples, see: (a) Y. Hayashi and D. Swern, J. Am. Chem. Soc., 1973, 95, 5205 CrossRef CAS; (b) V. Desikan, Y. Liu, J. P. Toscano and W. S. Jenks, J. Org. Chem., 2007, 72, 6848 CrossRef CAS PubMed; (c) T. Nakahodo, M. Okada, H. Morita, T. Yoshimura, M. O. Ishitsuka, T. Tsuchiya, Y. Maeda, H. Fujihara, T. Akasaka, X. Gao and S. Nagase, Angew. Chem., Int. Ed., 2008, 47, 1298 CrossRef CAS PubMed.
- (a) H. H. Credner, Ger. Patent, 3215834, 1983; (b) D. T. Manning, T. T. Wu and M. Pilato, Eur Patent, 839809, 1997.
- (a) R. Vanacore, A.-J. L. Ham, M. Voehler, C. R. Sanders, T. P. Conrads, T. D. Veenstra, K. B. Sharpless, P. E. Dawson and B. G. Hudson, Science, 2009, 325, 1230 CrossRef CAS PubMed; (b) G. Bhave, C. F. Cummings, R. M. Vanacore, C. Kumagai-Cresse, I. A. Ero-Tolliver, M. Rafi, J.-S. Kang, V. Pedchenko, L. I. Fessler, J. H. Fessler and B. G. Hudson, Nat. Chem. Biol., 2012, 8, 784 CrossRef CAS PubMed.
- For recent reviews on the metal-catalyzed allylic substitution, see: (a) D. K. Leahy and P. A. Evans, in Modern Rhodium Catalyzed Reactions, ed. P. A. Evans, Wiley-VCH, Weinheim, 2005; ch. 10, p. 191 Search PubMed; (b) G. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675 RSC; (c) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (d) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427 RSC; (e) M. L. Crawley, in Science of Synthesis, ed. J. G. De Vries, G. A. Molander and P. A. Evans, Thieme, Stuttgart, 2011, vol. 3, p. 403 Search PubMed; (f) J. F. Hartwig and M. J. Pouy, Top. Organomet. Chem., 2011, 34, 169 CrossRef CAS; (g) W.-B. Liu, J.-B. Xia and S.-L. You, Top. Organomet. Chem., 2012, 38, 155 CrossRef; (h) J.-M. Begouin, J. E. M. N. Klein, D. Weickmann and B. Plietker, Top. Organomet. Chem., 2012, 38, 269 CrossRef; (i) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147 RSC.
- P. A. Evans and E. A. Clizbe, J. Am. Chem. Soc., 2009, 131, 8722 CrossRef CAS PubMed.
- The rhodium-catalyzed allylic amination with S,S-diphenylsulfilimine provides the allylic sulfilimine in poor yield (8%).
- For examples of the stereospecific intermolecular rhodium-catalyzed allylic amination reactions, see: (a) P. A. Evans, J. E. Robinson and J. D. Nelson, J. Am. Chem. Soc., 1999, 121, 6761 CrossRef CAS; (b) P. A. Evans, J. Qin, J. E. Robinson and B. Bazin, Angew. Chem., Int. Ed., 2007, 46, 7417 CrossRef CAS PubMed.
- For leading examples, see: (a) C. Shu, A. Leitner and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4797 CrossRef CAS PubMed; (b) T. Graening and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 17192 CrossRef CAS PubMed; (c) D. J. Weix and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7720 CrossRef CAS PubMed.
- For selected examples of enantioselective iridium-catalyzed allylic substitution reactions that provide protected or functionalized allylic amines, see: (a) T. Ohmura and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 15164 CrossRef CAS PubMed; (b) H. Miyabe, A. Matsumura, K. Moriyama and Y. Takemoto, Org. Lett., 2004, 6, 4631 CrossRef CAS PubMed; (c) T. Nemoto, T. Sakamoto, T. Matsumoto and Y. Hamada, Tetrahedron Lett., 2006, 47, 8737 CrossRef CAS; (d) S. Spiess, C. Welter, G. Franck, J.-P. Taquet and G. Helmchen, Angew. Chem., Int. Ed., 2008, 47, 7652 CrossRef CAS PubMed; (e) Y. Ichikawa, S.-I. Yamamoto, H. Kotsuki and K. Nakano, Synlett, 2009, 2281 CrossRef CAS; (f) K.-Y. Ye, H. He, W.-B. Liu, L.-X. Dai, G. Helmchen and S.-L. You, J. Am. Chem. Soc., 2011, 133, 19006 CrossRef CAS PubMed; (g) W.-B. Liu, X. Zhang, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 5183 CrossRef CAS PubMed; (h) A. Sharma and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 17983 CrossRef CAS PubMed; (i) K.-Y. Ye, L.-X. Dai and S.-L. You, Chem.–Eur. J., 2014, 20, 3040 CrossRef CAS PubMed and pertinent references cited therein.
- Sulfilimines have a coordinate covalent (dative) single bond with strong polarization towards the nitrogen, thereby making them sufficiently nucleophilic to promote amination, see: F. Pichierri, Chem. Phys. Lett., 2010, 487, 315 CrossRef CAS.
- S,S-Diphenylsulfilimine is stable at ambient temperature, whereas many dialkyl sulfilimines decompose above –30 °C, see: R. Appel and W. Büchner, Chem. Ber., 1962, 95, 855 CrossRef CAS.
- R. Naasz, L. A. Arnold, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 927 CrossRef CAS.
- K. Tissot-Croset, D. Polet and A. Alexakis, Angew. Chem., Int. Ed., 2004, 43, 2426 CrossRef CAS PubMed.
- N. Sewald and V. Wendisch, Tetrahedron: Asymmetry, 1998, 9, 1341 CrossRef CAS.
- Enhanced reactivity of sulfilimines in the presence of base is well precedented, see: R. P. Claridge, R. W. Millar, J. P. B. Sandall and C. Thompson, Tetrahedron, 1999, 55, 10243 CrossRef CAS.
- Interestingly, the 3-fluorobenzoate leaving group provided sub-optimal results with an aryl-substituted electrophile: the reaction of 1 (R = Ph, LG = 3-FC6H4CO) furnished 2a in 59% yield and with 97% enantiomeric excess.
- For the conversion of sulfilimines to N-trifluoroacetamides or N-tosylsulfonamides, see: J. Drabowicz, P. Łyzwa and M. Mikołajczyk, Synthesis, 1981, 890 CrossRef CAS.
- S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228 CrossRef CAS PubMed.
- For recent examples of the regio- and enantioselective iridium-catalyzed allylic substitution using ammonia and ammonia equivalent pro-nucleophiles, see: (a) M. J. Pouy, L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11312 CrossRef CAS PubMed; (b) R. Weihofen, A. Dahnz, O. Tverskoy and G. Helmchen, Chem. Commun., 2005, 3541 RSC; (c) R. Weihofen, O. Tverskoy and G. Helmchen, Angew. Chem., Int. Ed., 2006, 45, 5546 CrossRef CAS PubMed; (d) M. J. Pouy, A. Leitner, D. J. Weix, S. Ueno and J. F. Hartwig, Org. Lett., 2007, 9, 3949 CrossRef CAS PubMed; (e) D. J. Weix, D. Marković, M. Ueda and J. F. Hartwig, Org. Lett., 2009, 11, 2944 CrossRef CAS PubMed.
- For enantio-convergent iridium-catalyzed allylic substitution using in-situ generated ammonia as the pro-nucleophile, see: M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470 CrossRef CAS PubMed.
- S,S-Diphenylsulfilimine has also been utilized as an ammonia equivalent in a 1,6-conjugate addition.2b.
- H. J. Shine and K. Kim, Tetrahedron Lett., 1974, 16, 99 CrossRef.
- Enantiomerically enriched allylic sulfilimines readily undergo a novel metathesis reaction to afford chiral-nonracemic allylic isocyanates, see: R. L. Grange and P. A. Evans, J. Am. Chem. Soc., 2014, 136, 11870 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc01317d |
| ‡ Representative procedure: the phosphoramidite ligand 4a (128 mg, 0.20 mmol) and [Ir(cod)Cl]2 (67 mg, 0.10 mmol) were dissolved in anhydrous dichloromethane (7.5 mL) and stirred at room temperature for ca. 15 minutes under an atmosphere of argon resulting in a homogeneous orange solution. A suspension of S,S-diphenylsulfilimine monohydrate (1.21 g, 5.5 mmol) and cesium carbonate (1.72 g, 5.3 mmol) in anhydrous DCM (2.5 mL) was heated in a 35 °C oil bath for ca. 15 minutes. The catalyst was added via syringe to the nucleophile, followed by the addition of the allylic benzoate 1k (1.11 g, 5.0 mmol) using a tared 5 mL gas-tight syringe. The resulting reaction mixture was stirred in a 35 °C oil bath for ca. 20 hours and then concentrated in vacuo to afford the crude product. Purification by flash chromatography (SiO2, gradient elution with ethyl acetate/hexanes 70:30 followed by ethyl acetate/hexanes/triethylamine 50:50:0.5) afforded the allylic sulfilimine2k (1.15 g, 81%, 92% ee) as a colorless oil. |
| This journal is © The Royal Society of Chemistry 2015 |