
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing logical codon reassignment – Expanding the chemistry in biology†
Anaëlle
Dumas‡
,
Lukas
Lercher‡
,
Christopher D.
Spicer‡
and
Benjamin G.
Davis
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: Ben.Davis@chem.ox.ac.uk
First published on 14th July 2014
Abstract
Over the last decade, the ability to genetically encode unnatural amino acids (UAAs) has evolved rapidly. The programmed incorporation of UAAs into recombinant proteins relies on the reassignment or suppression of canonical codons with an amino-acyl tRNA synthetase/tRNA (aaRS/tRNA) pair, selective for the UAA of choice. In order to achieve selective incorporation, the aaRS should be selective for the designed tRNA and UAA over the endogenous amino acids and tRNAs. Enhanced selectivity has been achieved by transferring an aaRS/tRNA pair from another kingdom to the organism of interest, and subsequent aaRS evolution to acquire enhanced selectivity for the desired UAA. Today, over 150 non-canonical amino acids have been incorporated using such methods. This enables the introduction of a large variety of structures into proteins, in organisms ranging from prokaryote, yeast and mammalian cells lines to whole animals, enabling the study of protein function at a level that could not previously be achieved. While most research to date has focused on the suppression of ‘non-sense’ codons, recent developments are beginning to open up the possibility of quadruplet codon decoding and the more selective reassignment of sense codons, offering a potentially powerful tool for incorporating multiple amino acids. Here, we aim to provide a focused review of methods for UAA incorporation with an emphasis in particular on the different tRNA synthetase/tRNA pairs exploited or developed, focusing upon the different UAA structures that have been incorporated and the logic behind the design and future creation of such systems. Our hope is that this will help rationalize the design of systems for incorporation of unexplored unnatural amino acids, as well as novel applications for those already known.
Introduction
While DNA and RNA make up the essential genetic information needed to create a living cell,1 it is the proteins that they code for that are the ‘workhorses’ of the cell, playing a key role in virtually every biological process and structure.2 The vast diversity of proteins is even more striking, given that they are typically made of just 20 natural amino acid building blocks, with an even more limited set of chemical functionalities. While our understanding of cellular biology has undoubtedly expanded vastly over the past century, more questions that cannot be answered via the techniques currently available to researchers emerge with each new discovery. New tools are required to investigate and manipulate proteins in both an intra and extra-cellular context.3 The ability to incorporate unnatural amino acids (UAAs) into proteins offers the potential to study, alter, or even improve upon protein activity and function through molecular ingenuity, both in vitro and in vivo.4 Such UAAs may modulate enzyme activity, offer unique reactive handles for further modification, act as spectral or imaging probes, or simply offer a novel functional structure or natural post-translational modification (PTM) or mimic (Fig. 1).5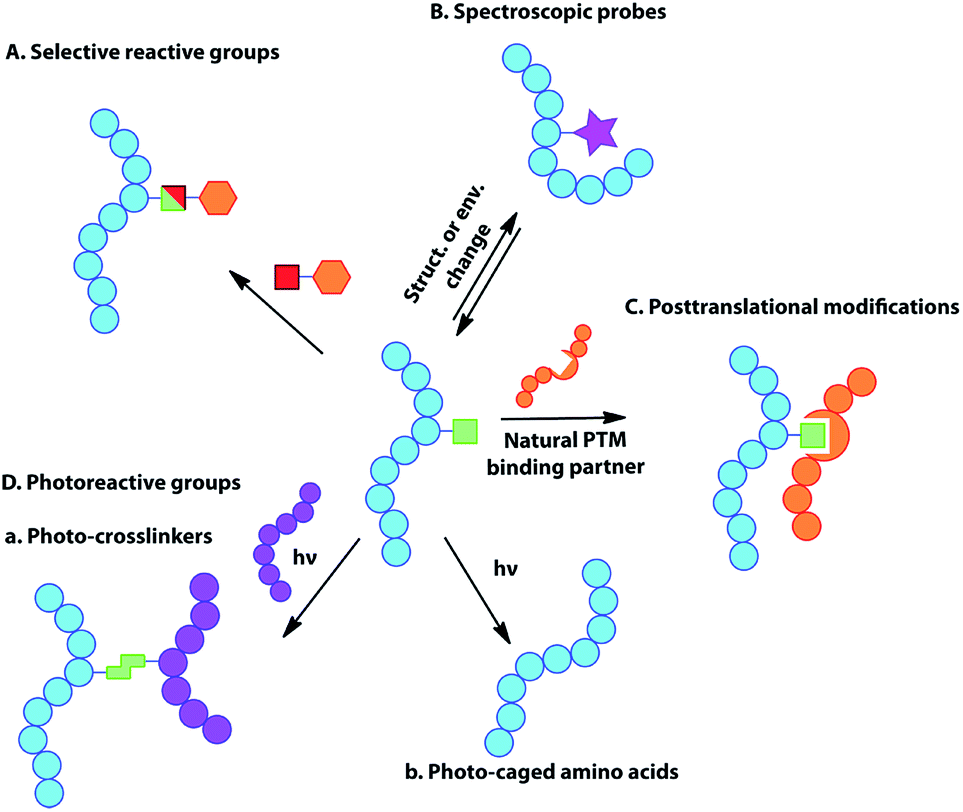 | ||
| Fig. 1 Schematic representation of the major uses of genetically encoded UAA side chains, including selectively reactive groups, spectroscopic probes, natural post-translational modifications or mimics and photoreactive-groups (photo-crosslinkers and photo-caged amino acids). | ||
Many techniques have emerged for incorporating UAAs into proteins in a variety of organisms over the past few decades.6 These include expansion of the genetic code by codon reassignment and suppression, of both sense and non-sense codons7 (see Table 1). Codon reassignment can allow the site-specific installation of an UAA into a protein of choice. In general, this technique relies on the exploitation or development of a selective amino-acyl tRNA synthetase/tRNA (aaRS/tRNA) pair, specific for the UAA of choice and the codon to be reassigned. Greater flexibility comes from an aaRS/tRNA pair that does not cross-react with the existing host cell’s pool of synthetases, tRNAs and translational machinery, but that is recognised by the host ribosome; this mutually compatible (or ‘orthogonal’) pair results in incorporation of the UAA. The most commonly used systems have relied upon the reassignment of ‘non-sense’ codons, as codons that are not used for amino acids by the endogenous systems, particularly (but not exclusively) the amber-stop codon, TAG. This has resulted in the incorporation of a diverse range of UAAs not only in prokaryote7a,8 and eukaryote7b,9 cell culture, but more recently in multicellular organisms and even animals.10 Other systems based on the reassignment of the ochre and opal stop codons,11 in addition to the use of quadruplet codons have also been reported (see later section).12 It is now also possible to consider their combined use for the incorporation of multiple distinct UAAs into a single protein of choice.11 Of course, competition with the host cell termination machinery at stop codons offers a significant drawback to ‘non-sense’ suppression, commonly resulting in lower yields and truncated protein products. The use of ‘sense’ codon reassignment offers a potential solution to this problem since it is a system with inherent translational efficiency. While such technical hurdles still exist, the field of UAA incorporation is still only in its infancy and the potential power of diverse, near unlimited chemical functionality in biology offers a truly exciting prospect (see ‘Outlook’ section).
|
In this review, we will focus on the site-specific incorporation of unnatural amino acids via codon reassignment. We aim not to describe the theory and techniques behind this technology, as this has been excellently reviewed elsewhere. Though a brief historical perspective will be given, the reader is directed to reviews by Liu,5 Young4 and Davis13 for a more detailed description. Rather, we hope to provide an overview of the structures and functionalities that have been incorporated, based on the aaRS/tRNA system that has been utilised (and hence the codons that have been reassigned), to provide a reference for those who wish to incorporate novel UAAs or use those that have already been described. We also provide a comprehensive table of all the UAAs incorporated to date along with details of their applications to date (see Table 1), as well as details of the mutations required to generate the requisite aaRS (see the ESI†). In particular, we wish to highlight the unique chemistries of the available UAAs, to aid researchers in developing novel applications for a field that has the potential to revolutionize the biological sciences in a way that site-directed mutagenesis did before it,14 perhaps more so.
Applications
The genetic encoding of UAAs via codon reassignment offers the advantage of site-specificity, to yield homogenously modified proteins substituted with a wide variety of otherwise hard to introduce functional groups. Since incorporation uses the host cell's own translational machinery, the modified proteins offer the advantage of being applicable to the study of intracellular biological processes in the very cellular system that produced them. To date, over 150 different UAAs have been incorporated into proteins. These have been used as tools for a number of applications including the study of protein–protein interactions, protein localization, enzymatic activity and cellular signalling, while also providing handles for protein imaging and spectroscopy. The applications of genetically-encoded UAAs have been extensively reviewed elsewhere,13,15 but a summary of the major uses is briefly given here.Selectively reactive groups (Fig. 1A)
The introduction of uniquely reactive, non-natural chemical groups provides a powerful tool for site-selective labelling and alteration of proteins, through the use of complementarily selective reactions. To date, a large number of biocompatible and selective (so-called ‘bioorthogonal’) reactions have been developed, including Cu(I)-catalyzed or “copper-free” alkyne–azide triazole-forming reactions, the Staudinger ligation, inverse-electron-demand Diels–Alder (IEDDA) reactions between tetrazines and strained alkenes, “photo-click” chemistry between tetrazoles and alkenes, or metal-mediated processes such as olefin metathesis and Suzuki–Miyaura or Sonogashira cross-couplings (for recent reviews, see ref. 16). The genetic encoding of most of the corresponding reaction partners into proteins provides chemical ‘tags’ that may be addressed selectively for site-specific labelling.17 Such reactive handles are particularly useful tools for the preparation of protein conjugates with unprecedented control over structure and homogeneity.In addition to advantageous applications in the development of protein therapeutics, site-specific protein labelling has also played a significant role in investigating a number of biological pathways (for reviews see ref. 15b, 16a, 16c and 18). Such chemoselective reactions are particularly important in enabling “tag-and-modify” approaches for the selective conjugation of proteins with entities that cannot otherwise be directly encoded, such as large posttranslational modifications (or their mimics), spectroscopic probes or other biomolecular moieties.17a
Spectroscopic probes (Fig. 1B)
The introduction of spectroscopic probes into proteins can provide useful information regarding protein structure, conformation, localization and intermolecular interactions, allowing the dissection of many complex biological processes. These spectroscopic probes, inserted directly into the protein backbone, may act as reporters of change in chemical environment occurring at the amino acid residue level. Examples of such spectroscopically-active amino acid side chains include fluorophores for fluorescent spectroscopy, spin labels for NMR and EPR, heavy atoms for X-ray crystallography or amino acids displaying unique infrared absorption properties (for reviews see ref. 19). Unfortunately, the genetic encoding of many spectroscopic probes is often limited by their complex structure and large size and it may therefore be preferable to introduce them via chemo-selective modification at a genetically-encoded chemical handle (‘tag’) (see previous section).Post-translational modifications or mimics (Fig. 1C)
During their life-time, most proteins in vivo undergo some form of chemical or enzymatic modification after translation, such as phosphorylation, sulfation, nitration, glycosylation, methylation, acetylation, ubiquitination or lipidation of the amino acid side chains. Such modifications are important in modulating protein function, interactions with binding partners, signal transduction and as a trigger for a number of cellular events. However, the elucidation of their exact roles and functions remains challenging, partly due to the difficulty in preparing homogenously-modified proteins, especially if the modified amino acid is present in multiple copies in the protein of interest. The genetic encoding of post-translational modifications or their structural mimics enables the selective and site-specific introduction of the modified residue in the protein. This provides homogenously-modified products that are highly valuable in elucidating the biological role of the respective modification. For example, lysine residues are known to undergo various modifications responsible for the regulation of a number of biological processes, notably in the context of histones.20 The development of selective PylRS/tRNA pairs has readily enabled the genetic encoding of lysine residues bearing natural modifications (due in part to the substructure resemblance of Pyl to Lys), providing valuable tools for the investigation of the ‘histone code’. Other areas such as the study of protein sulfation and phosphorylation, through the incorporation of sulfotyrosine21 and phosphoserine22 respectively, and oxidative damage23 have also been aided by codon reassignment.Photoreactive groups (Fig. 1D)
A number of photoreactive amino acids have been encoded in proteins. These can be broadly classified into two groups:Photo-crosslinkers
Photo-crosslinkers are able to rapidly form covalent bonds to organic molecules in their close proximity at the time of irradiation. Photo-crosslinking amino acids are therefore powerful tools for mapping biomolecular interactions in vivo and have been used extensively to probe protein–protein or nucleic acid–protein interactions and ligand binding. One major advantage offered by genetically-encoded photo-crosslinkers is their ability to provide positional and therefore structural information on protein complexes in their natural environment. The formation of a covalent bond between the two interacting partners allows the study of weak, transient or pH-dependent interactions that may be lost in non-covalent methods.Photo-caged amino acids
When site-specifically incorporated into a protein, the protection of a particular amino acid with a photo-labile protecting group (photo-cage) can temporarily block a specific protein function. The interactions of this amino acid with its environment and therefore protein function can then be restored by light irradiation. This enables rapid and specific protein activation inside living cells in order to facilitate the study of the biological consequences.Miscellaneous
The incorporation of UAAs can be exploited to generate novel protein properties. A few examples involve the introduction of redox-active, metal-chelating or stabilizing/destabilizing cores based on the introduction of large hydrophobic or aromatic scaffolds. As a result of such incorporations, stimulation of potent immune responses has been reported but one can envisage diverse applications including, e.g., the creation of de novo metalloenzymes.Non-sense suppression with orthogonal aaRS/tRNA pairs for genetic encoding of unnatural amino acids
Of the 64 naturally-occurring triplet codons, there are 3 which lack a corresponding tRNA capable of adding an amino acid to a growing peptide chain.§ These ‘stop’- or ‘non-sense’-codons, TAG (amber), TAA (ochre) and TGA (opal) instead result in termination of the translation process via the recruitment of release factors.24 As such, they are ideal candidates for re-assignment via the design of a suitable suppressor-tRNA, particularly the amber codon TAG, the rarest of the 3 codons in E. coli.25 Re-assignment effectively leads to an incorporation system that is in direct competition with translational termination by the host machinery. This was first achieved through the use of a chemically acylated TAG-specific tRNA by Noren et al. in a cell free system and subsequently in Xenopus oocytes by Nowak et al.26 Whilst allowing the site-specific incorporation of a large number of UAAs, the need to microinject the chemically-acylated tRNA limits the applicability to large cells such as Xenopus oocytes and leads to very small quantities of protein.27 In order to achieve general applicability in cells, it is necessary to identify an orthogonal aaRS/tRNA pair that can be uniquely acylated in the organism of choice by an engineered aaRS.28 Liu et al. reported the first use of an encoded suppressor-tRNA for incorporation of a natural glutamine amino acid in vivo.29 However, it was not until the discovery of a yeast aaRS/tRNA pair from S. cerevisiae that was completely orthogonal to the translational machinery of E. coli, that the prospect of incorporating an unnatural amino acid by stop-codon suppression in vivo became achievable.30The idea of transferring an aaRS/tRNA pair from another kingdom into the organism of interest would provide the basis for many of the subsequent discoveries in the field, since recognition sequences are often species specific. Indeed, the first incorporation of a UAA in vivo via codon reassignment was achieved by Wang et al. in E. coli, with an orthogonal TyrRS/tRNACUA pair from the archaebacteria Methanococcus jannaschii.7ap-Methoxyphenylalanine (1) was shown to be incorporated site-specifically into a model protein, with excellent selectivity and fidelity in response to the amber stop codon. This ‘Mj-Tyr’ aaRS/tRNA pair has since gone on to be widely used for the incorporation of a wide-range of UAAs, with the development of efficient +ve/−ve selection protocols allowing the rapid screening of UAA specificities from large libraries of mutants (usually 107–108).31
The first example of UAA codon reassignment in eukaryotic cells was demonstrated by Sakamoto et al. utilising an E. coli TyrRS/B. stearothermophilus tRNACUA pair to incorporate 3-iodotyrosine (4), albeit with low fidelity.9a Chin et al. subsequently demonstrated the use of an E. coli TyrRS/tRNACUA pair for efficient-suppression with a number of UAAs in S. cerevisiae. Again, an efficient selection system allowed selectivity screening, but efficiency was limited by the slow growth rate of yeast compared to bacteria.7b In this system, the rational design of eukaryotic promoter sequences was essential for effective incorporation.
More recently, the pyrrolysine (Pyl) pairs of Methanosarcina barkeri and Methanosarcina mazei have emerged as leading candidates in the quest to find a broader aaRS/tRNA pair.32 Unlike other pairs which have engineered codon-specificities, pyrrolysine aaRS/tRNACUA pairs occur naturally in some methanogenic archaea as amber-suppressors. These pairs have been shown to be selective in both prokaryotic and eukaryotic systems, with both wild-type (wt) and mutant synthetases being tolerant of a wide range of UAA structures, allowing their use in a diverse range of labelling and functional studies. Indeed, more recently the group of Chin have demonstrated that an ‘Mb-Pyl’ pair can be used for amber suppression in multicellular animals in an impressive demonstration of the power of stop-codon reassignment.10
While suppression of the amber-codon has been most widely investigated, the development of pairs for the suppression of both ochre and opal-codons have also been reported. While more prevalent in the genome, potentially leading to increased toxicity with suppression, their use may allow the incorporation of multiple distinct UAAs into the same protein, as achieved by the groups of Liu and Schultz.11,33 A further development has seen the decoding of quadruplet codons and orthogonal ribosomes, theoretically offering a large number of new unassigned codons and the possibility to develop a fully orthogonal genetic code in the cell (see ‘Quadruplet codon suppression’ section).12 However, to date this approach is still limited by availability of mutually orthogonal aaRS/tRNANNNN pairs.
Research in the field of codon reassignment has mostly focussed on the application of four main aaRS/tRNA pairs, the TyrRS/tRNA of M. jannaschii (Mj-Tyr), the E. coli TyrRS and LeuRS/tRNA pairs (Ec-Leu, Ec-Tyr), and the pyrrolysine aaRS/tRNA pair of methanogenic archaebacteria (Mb- or Mm-Pyl). While overlap certainly exists, in general each provides complementary UAA selectivity in different organisms (Fig. 2). Each will be discussed in turn, with a brief description of their identification and development as a non-sense-suppressor, followed by a focus on the varied UAA structures that have been incorporated.
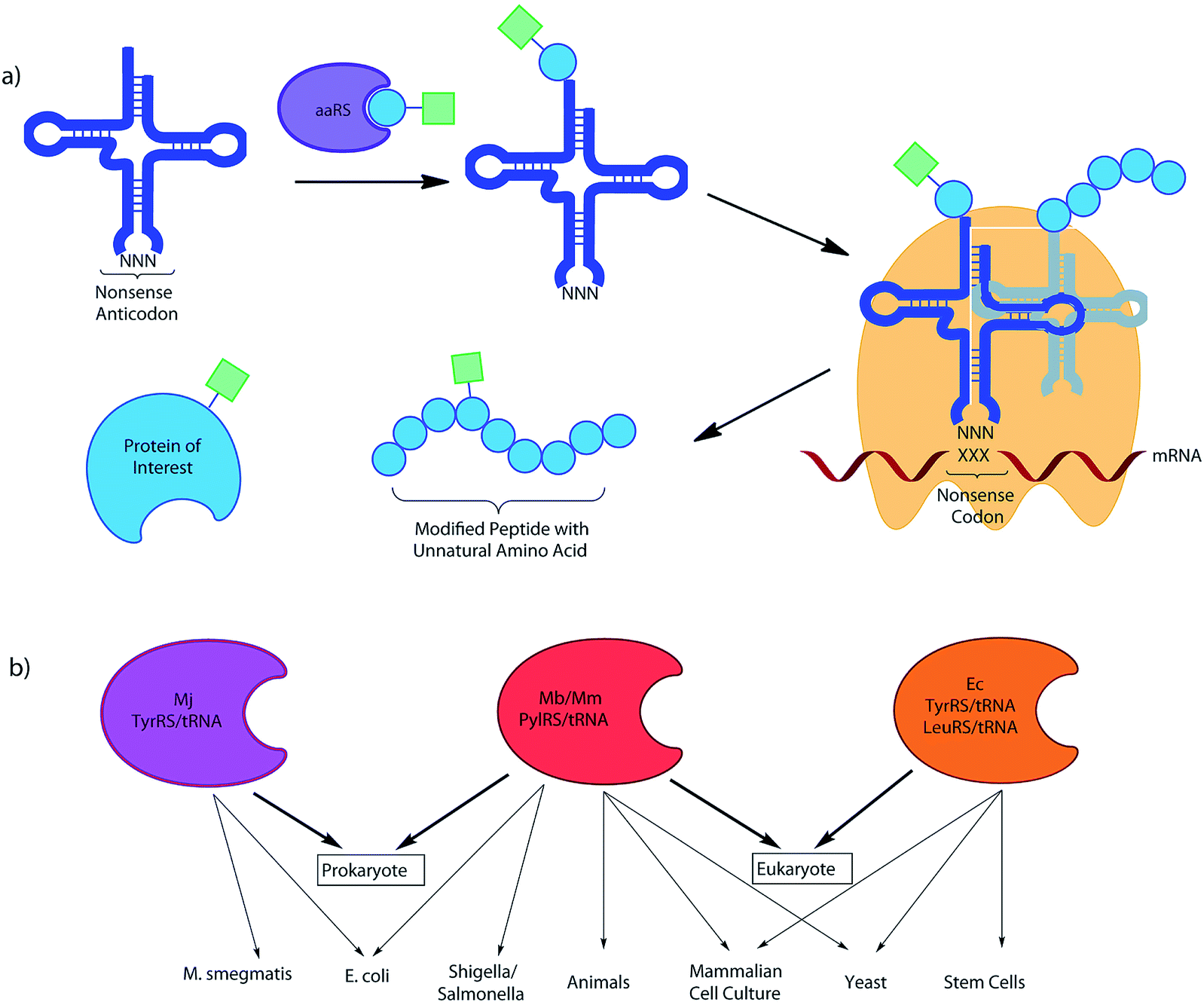 | ||
| Fig. 2 (a) An UAA is charged onto a tRNA with the required non-sense anticodon by an orthogonal aaRS. This tRNA then recognises its corresponding mRNA non-sense codon in the ribosome, leading to incorporation of the UAA into the protein of interest, where it may act as a ‘tag’ for further modification, as a spectroscopic probe, or as a PTM mimic. (b) The organism orthogonality of the 4 most commonly used systems for codon reassignment (M. jann TyrRS/tRNA, M. bar and M. maz PylRS/tRNA and E. coli Tyr/LeuRS/tRNA). | ||
TyrosylRS
Many early examples of incorporated UAAs contained reactive groups for bioconjugation at the para-position of the phenyl ring for undertaking subsequent chemical protein modification. For example, reactive allyl (2),36,37 propargyl (11)38 and alkynyl groups (41)39 have been incorporated to install unsaturated carbon–carbon bonds into proteins, while aryl halide (44 and 45),40 azide (6),41 aniline (7),36,40a ketone (10),42 diketone (16)43 and boronic acid (26)44 functionalities have all been used to provide reactive handles for further modification.45 The incorporation of the aniline based p-aminophenylalanine (7) is particularly noteworthy as it has been demonstrated that the amino acid can be biosynthesised in E. coli by hijacking the cellular machinery for the synthesis of aromatic amino acids, effectively generating a bacterium with a 21 amino acid genetic code.46 More recently, reactive handles for undertaking the inverse-electron demand Diels–Alder (tetrazine, 54),47 ‘photo-click’ (tetrazole, 55)48 and azo coupling (2-naphthol, 134)49 reactions have been incorporated using this pair. Photocrosslinking amino acids have also been extensively used to probe protein–protein interactions and ligand binding. Benzophenone 5 has been most widely incorporated due to its chemical stability,50 though arylazide (6)41 and diazirine (20)51 UAAs have also been incorporated.
A number of para-substituted-Phe UAAs containing functional probes that do not require further modification for functionality have also been incorporated. For example, fluorescence quenching nitro (12),52 IR-53 and FRET-54 active cyano (17), NMR-active trifluoromethane (42)55 and photoisomerizable azobenzene (14)56 groups have all been incorporated. However, the use of the M. jann pair has not only been used to incorporate novel functionalities. The natural post-translationally modified AA sulfotyrosine (13) has been installed to study native sulfated proteins,21 while it has been suggested that p-carboxymethylphenylalanine (19) can act as a mimic of the natural modification phosphotyrosine.57
While most common, the substrate specificity of the M. jann pair does not have to be limited to para-substituted phenylalanine derivatives. Through suitable mutation of the TyrRS binding pocket a wider variety of aromatic substituents can be tolerated. For example, expansion of the pocket allows a number of meta-substituted tyrosine derivatives to be incorporated, including 3-aminotyrosine (18),58 used extensively as a probe of radical propagation;59 3-nitrotyrosine (36),23 a naturally occurring marker of photo-damage; 3,4-dihydroxyphenylalanine (39),60 a redox-active UAA and 3-iodotyrosine (4).61Meta-acetyl (51)62 and ortho-nitro (33) substituted phenylalanines have also been incorporated, the latter allowing the photo-induced cleavage of a protein backbone.63
The binding pocket has also been disrupted to allow the incorporation of alternative aromatic rings, including heteroaromatics. Fluorescent naphthyl (3),40a,64 hydroxyl (53)65 and methyl (52)65a-coumarins and hydroxylquinolines (24)66 can all be used as probes of unfolding and protein structure. Hydroxylquinoline 24 may alternatively act as an efficient chelator of zinc2+ ions,66 while copper2+ ions have been shown to bind to a bipyridine based UAA 22.67 The structure of the coumarin UAAs 52 and 53 are particularly intriguing, as unlike almost all other structures incorporated by the M. jann TyrRS/tRNACUA pair, the coumarinyl-UAAs contain an additional methylene group leading to a δ-, rather than γ-linked aromatic. This is a structure observed in only one other UAA incorporated via such a pair: phenylselenocysteine (48), although this contains a bulkier selenium atom at the γ-position which may go some way towards fulfilling the steric bulk required by the aaRS.68
Phenylselenocysteine is a member of another class of UAAs that can be incorporated utilising a M. jann pair, those that are used as a ‘protected’ or ‘latent’ precursor to an amino acid of interest. Phenylselenocysteine itself can be oxidatively eliminated to give the α,β-unsaturated amino acid dehydroalanine (Dha, a natural PTM in some peptide natural products, as well as a reactive handle for further chemical modification).68,69 ‘Protected’ tyrosine residues can also be installed, allowing for the selective activation of a protein under the requisite deprotection conditions. p-Boronophenylalanine (26) can be converted to either tyrosine or phenylalanine by oxidation or reduction respectively,44 and has thus been used as a fluorescent peroxynitrite sensor.70 While O-nitrobenzyltyrosine (43) can also be activated under photo de-caging conditions,71 a similar de-caging was used to generate fluorinated tyrosine analogues (46 and 47), important isosteric probes of tyrosine pKa and activity, that cannot be incorporated directly due to their cross-reactivity with the natural cell machinery.72
Initial efforts to rationally design M. jann TyrRS substrate specificities were based on the crystal structure of the homologous, but distinct, TyrRS of Bacillus stearothermophilus.73 Solving the crystal structure of the M. jann TyrRS allowed for an increased ability to screen for mutant synthetases and amino acid specificities (Fig. 3a). An observed high degree of structural plasticity goes some way to explaining the adaptability of this system.74 This crystal structure has even allowed the design of a pair for the introduction of an ester bond into protein backbones via an α-hydroxy acid 35. This was enabled through mutation around the amino binding region of the aaRS, although significant disruption of the cellular machinery was required to prevent metabolisation of the substrate.75
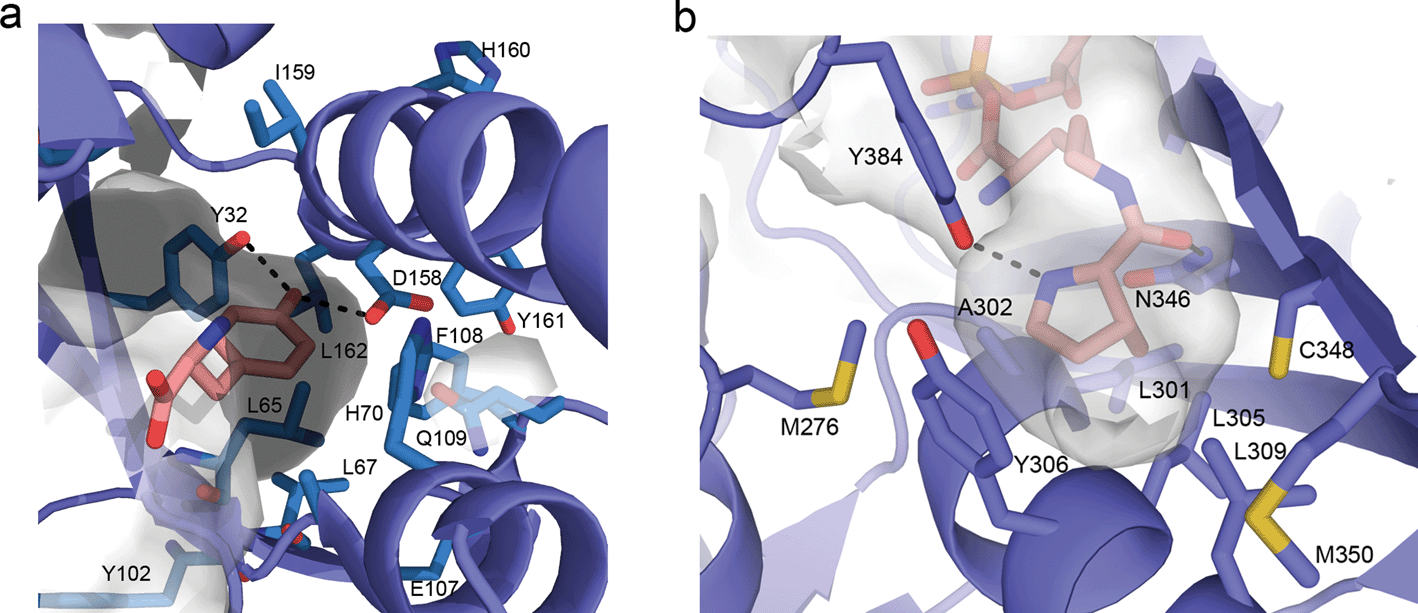 | ||
| Fig. 3 (a) Representation of the active site of the wt M. jann TyrRS (taken from PDB file 1j1u). Commonly mutated residues are drawn as sticks and labelled. Direct hydrogen bonding between the Tyrosine phenol and Y32 and D168 are indicated. (b) Active site of the wt M. mazei PylRS (taken from 2q7h). Hydrogen bonding between the side chain carbonyl oxygen of pyrrolysine and N346 and between the pyrroline nitrogen and Y384 are indicated. | ||
LeucylRS
Since the initial demonstrations of this pair for UAA incorporation, a variety of structurally diverse amino acids have been introduced. The fluorescent amino acid dansylalanine (58) has been incorporated in S. cerevisiae and also in human neural stem cells9b to act as a moderately effective environmental probe of unfolding.89 An improvement in sensitivity can be achieved utilising the prodan-based UAA Anap 73, incorporated using a LeuRS/tRNACUA pair mutant generated via a two-step strategy.90 First, a LeuRS mutant specific for 3-(naphthalen-2-ylamino)-2-aminopropanoic acid (74), an analogue of Anap lacking the ketone functionality, was selected for. A second mutant library was then constructed, yielding an Anap-specific LeuRS. This strategy of developing selectivity in steps allows gradual evolution to a desired aaRS via structurally similar derivatives, and has been used on a number of occasions when initial aaRS screening has failed to generate suitable mutants for an UAA of choice. In a further demonstration of LeuRS promiscuity, a ferrocene-containing UAA 49 has been incorporated with this pair, the first metallo-amino acid to be introduced into proteins biosynthetically.91
Often the structural space that can be incorporated with a single mutant aaRS is quite limited. The generation of promiscuous aaRSs, which can accept multiple different substrates greatly simplifies the introduction of new amino acids by negating the need for aaRS mutation. The LeuRS appears to be well suited to such applications given its tolerance for structural diversity. A promiscuous LeuRS mutant has been shown to be capable of charging different length methionine, cysteine and alkyl analogues, allowing the incorporation of a variety of structures using a single mutant (61–68).92 Similarly, through mutation of the active and the CP1 editing site of the aaRS, a number of alkene containing amino acids (60 and 69–72), of varied length and heteroatom substitution have been incorporated.93 Such alkene moieties could perhaps subsequently be used as selective ‘tags’ for chemical modification by olefin metathesis or thiol–ene reactions.
PyrrolysylRS
Early studies of the PylRS/tRNA pair utilized pyrrolysine mimics to map the biochemical activity of the aaRS, determining the structural features of the amino acid required for recognition. Additional motivation was provided not only by the potential biotechnological applications but by the difficulty in obtaining the amino acid pyrrolysine (necessary for further characterization of the PylRS mechanism but requiring a 16-step synthesis).95 Initial analogue studies showed that the PylRS/tRNACUA pair was able to efficiently incorporate a variety of amide or carbamate substituted lysines, such as 2-amino-6-((R)-tetrahydrofuran-2-carboxamido)hexanoic acid (2Thf-lys, 84),96N-ε-D-prolyl-L-lysine (81)95 and N-ε-cyclopentyloxycarbonyl-L-lysine (cyc, 82).95 It was also established that pyrrolysine incorporation was not mediated via a distinct mRNA structure,95 again in contrast to selenocysteine. The availability of the commercial pyrrolysine analogue cyc (82), enabled the structural characterization of a ternary PylRS–AMP-cyc complex (Fig. 3b).97 With this structural information in hand, directed evolution and rational design approaches to engineer PylRS specificity have became possible (see the following section for details).
The wild type PylRS has proven remarkably flexible in terms of substrate recognition, allowing the introduction of many useful functional groups, without the need for the generation of mutant synthetases. Typically, the unnatural amino acids present a specific functionality conjugated via a carbamate linker to the N-ε of lysine. The first examples of chemical handles/‘tags’ for bioconjugation labelling reactions were the introduction of alkyne (86) and azide (90) functionalities for CuAAC by Nguyen et al.98 Similarly, a Boc-protected lysine (77) was also incorporated, while Fekner et al.99 introduced an amide linked tetrahydrofuran moiety (85), which may mimic the pyrrolysine ring, bearing an alkyne to site-specifically label calmodulin (CaM). Selective modification of the pyrrolysine analogue by CuAAC and alkylation of a nearby cysteine with a FRET active dye pair, allowed the tracking of conformational changes in CaM by FRET measurements. The same group also reported the efficient incorporation of another alkyne-containing pyrrolysine analogue N6-(2-(R)-propargylglycyl)-lysine (105),100 which was used for the same purpose. In a follow-up, the authors replaced the lysine N-ε with an oxygen, substituting the ε-amide for a cleavable ester bond (106).101
In order to allow expansion of thioester-to-amide (so-called ‘native chemical ligation’, NCL) chemistry, cysteine (both D and L, 92) or a thiazolidine protected cysteine (104) conjugated to the lysine N-ε have been incorporated by amber suppression102 and used as chemical handles for protein ubiquitinylation via NCL,103 or fast ‘bioorthogonal’ labelling with 2-cyanobenzothiazoles.104 Although these amino acids are incorporated by the wt PylRS, the efficiency is relatively low and can be improved with mutant aaRSs.104 In order to allow IEDDA reactions for rapid protein labelling, a carbamate linked norbornene moiety (101 and 122) has recently been genetically-incorporated using both wt and mutant PylRSs.105 This has allowed rapid modification of target proteins with a tetrazine reaction partner, or by a [3 + 2]-dipolar cycloaddition with nitrile imines. This useful moiety has also been encoded in Drosophila cells.10a Small aliphatic diazirines (96), can also be incorporated using the wt PylRS/tRNA pair.106 This photocrosslinker was shown to covalently crosslink glutathione S-transferase (GST) dimers both in vivo and in vitro.
The PylRS/tRNA pair has also been used for the incorporation of naturally occurring PTMs. Mono- and di-methyl-lysine have been incorporated via intermediate Boc-protected lysine derivatives (97 and 77 respectively) and subsequent chemical modification.107 This has been particularly useful for the study of histones, which are commonly modified at numerous lysine residues.
A detailed examination of the PylRS crystal structure reveals no significant interaction occurs with the lysine α-amine. This has been exploited by Kobayashi et al. to introduce the α-hydroxy acid of Boc-lysine 110 into proteins.108 This site-specific substitution of an amide for an ester bond in the protein backbone allows protein cleavage at this site by treatment with ammonium hydroxide (as previously also demonstrated with the M. jann incorporated hydroxyl acid 35 (ref. 75)).
The use of structural analogues has allowed the deduction of the necessary structural features required for efficient charging of an UAA to the aaRS. The most important is the presence of an amide or carbamate linkage at the N-ε. Here, both the planarity of this linkage and the hydrogen bonding capacity of the carbonyl oxygen appear important for recognition. In addition, the cyclic imine provides a further hydrogen bonding motif, which is recognized by the PylRS (Fig. 3b). While not essential, the presence of this hydrogen bond appears to significantly increase tRNA charging by the PylRS, as exemplified by the fact that 2-amino-6-((R)-tetrahydrofuran-2-carboxyamido)hexanoic acid (2-Thf-Lys, 84), which contains an oxygen in the same position as the imine nitrogen in pyrrolysine, is accepted as a substrate by PylRS, but 3- and 4-Thf-Lys variants with differently positioned endocyclic heteroatoms are not. Since the size of the active site is limiting in terms of the potential utility of the wt PylRS/tRNA pair, a number of groups have undertaken directed evolution or rational design to give mutants with altered specificity. Such mutants will be discussed in the next section.
In general, the mutants may be designed either rationally or by randomization of a number of active site residues of the PylRS. The randomized residues are either selected arbitrarily, or on the basis of the crystal structure of the M. mazei (Mm) PylRS in complex with pyrrolysine (PDB entry 2q7h).97,109 Since the binding pocket residues are well-conserved between the Mm-PylRS and M. barkeri (Mb)-PylRS,110 this model has been used for the engineering of both enzymes. The analogous Mb-PylRS and Mm-PylRS mutants indeed show similar efficiencies for the incorporation of the same substrate.111 As such, the two systems are often used arbitrarily based on the system available to the interested research group, and can in effect be used interchangeably.
The first example of a PylRS mutant for codon reassignment was reported by Chin and co-workers in 2008, when an Mb-PylRS mutant was evolved to incorporate the extensively studied natural lysine PTM N-ε-acetyllysine (78) in E. coli. The mutant Mb-PylRS possessed 6 mutations thought to partially plug the large hydrophobic cavity present around the pyrroline ring of the wt synthetase, while still accommodating the acetyl group.110 The same mutant was later shown by Liu and co-workers to successfully incorporate the corresponding alkyl analogue 2-amino-8-oxononanoic acid (118) in E. coli.112 Shortly after, Yokoyama and co-workers engineered a Mm-PylRS bearing 3 mutations at random sites and two in the pyrrolysine binding pocket, creating sufficient room to accommodate the large benzyloxy group of N-ε-benzyloxycarbonyl-L-lysine (76) and enable its incorporation into recombinant proteins in mammalian cells.111
Yokoyama and co-workers have shown that a single mutation of Mm-PylRS (Y384F), identified by random screening, significantly improves the amber suppression efficiency for BocLys (77) and AllocLys (107) compared to the wild type synthetase in E. coli. A second mutant, containing the structure-based mutation Y306A, in addition to Y384F, provided good yields for the incorporation of N-ε-benzyloxycarbonyl-L-lysine (76) and also enabled the large scale incorporation of N-ε-(O-azidobenzyloxycarbonyl)-L-lysine (80) in E. coli.113 The same Mm-PylRS mutant has been used for the incorporation of a variety of UAAs in E. coli including N-ε-[2-(furan-2-yl)ethoxy]carbonyl-lysine (140) for photo-activated protein–RNA cross-linking114 and Se-alkylselenocysteines (89).115 In addition, a number of tags designed for selective reactions have been incorporated, such as various norbornene (101 and 122),116 transcyclooctene (100)116,117 and cyclooctyne (99 and 102) derivatives118 in E. coli and mammalian cells. Small variations of this mutant, have enabled the incorporation of the two photo-crosslinking UAAs N-ε-(p-(nitrobenzyloxycarbonyl))-L-lysine (91) and N-ε-[((4-(3-(trifluoromethyl)-3H-diazirin-3-yl)-benzyl)oxy)carbonyl]-L-lysine (117) in E. coli and mammalian cells.119 Chin and co-workers have also demonstrated the incorporation of other selective reaction handles such as transcyclooctenes (100) and cyclooctynes (99) derivatives with Mb-PylRS mutants in E. coli and mammalian cells, for undertaking strain-promoted azide–alkyne cycloadditions and inverse-electron demand Diels–Alder reactions.120 A similar mutant was used for the incorporation of a five-membered 2,2,5,5-tetramethyl-pyrrolin-1-oxyl spin label moiety 139.121 The incorporation of the photocaged lysine derivative O-nitrobenzyl-oxycarbonyl-N-ε-L-lysine (91) was demonstrated by Schultz and co-workers with a Mm-Pyl mutant in E. coli and mammalian cells.122 Liu and co-workers later reported the encoding of a photocaged mono-methyllysine analogue 98 in E. coli. Irradiation of proteins containing this amino acid results in site-specifically incorporated methyl-lysine, for which no selective aaRS has been evolved to date.123N-ε-Acryloyl-L-lysine (115) has also been encoded and shown to undergo conjugation reactions in E. coli.124 This amino acid was also used to undertake ‘photo-click’ chemistry in E. coli, mammalian cells and even plants (A. thaliana).125 More recently, spiro-hexene 115 has been incorporated using the same pair to improve the rates of ‘photo-click’ reactions.126
Chin and co-workers have extended the technology further by engineering a PylRS/tRNACUA pair functional and orthogonal in yeast. Different Mb-PylRS mutants have been evolved for the incorporation of N-ε-acetyl-L-lysine (78), and its analogues trifluoroacetyl-L-lysine (93) and photocaged lysine derivative N-ε-[(1-(6-nitrobenzo[d][1,3]dioxol-5-yl)ethoxy)carbonyl]-L-lysine (94).127 The same group subsequently demonstrated the incorporation of photocaged-lysine 94 in mammalian cells with an Mb-PylRS mutant.128 A similar mutant enabled the incorporation of the photoreactive N-ε-(1-methylcycloprop-2-enecarboxamido)lysine (103) in E. coli and mammalian cells.129 The group also demonstrated the incorporation of a photocaged cysteine analogue 141 in E. coli and mammalian cells.130
Chen and co-workers originally identified an Mb-PylRS mutant for the incorporation of the reactive azide N-ε-(((1R,2R)-2-azidocyclopentyloxy)carbonyl)-L-lysine (87) in E. coli and mammalian cells.131 This mutant has subsequently been used for the incorporation of a variety of UAAs in E. coli such as α-hydroxy-BocLys (110)132 or chemical handles such as alkenylpyrrolysine (107–109)133 and alkynylpyrrolysine (86, 112 and 113) analogues.134 A related mutant was identified for the incorporation of the photo-crosslinker 3-(3-methyl-3H-diazirine-3-yl)-propaminocarbonyl-N-ε-L-lysine (96) in E. coli and mammalian cells135 and subsequently for the incorporation of aryl iodide (114) and alkynylpyrrolysine (113) analogues in the Gram-negative bacterial pathogens Shigella and Salmonella.8a Further single-site mutation enabled the incorporation of the newly identified natural histone PTM N-ε-crotonyl-L-lysine (121).136
One single mutation to the wild type Mb-PylRS, C313V enables significant improvement in the incorporation efficiency of the 1,2-aminothiols N-ε-D-cysteinyl-L-lysine and N-ε-L-cysteinyl-L-lysine (92) in E. coli.104 In the same paper, further evolution of the synthetase enabled the incorporation of N-ε-L-thiapropyl-L-lysine (104) through three additional mutations.104
Remarkably, PylRS can be evolved to incorporate unnatural amino acids bearing aromatic cores instead of the classical pyrrolysine –(CH2)4–NHR scaffold, while still remaining orthogonal towards endogenous amino acids, demonstrating the flexibility of the aaRS binding pocket towards substrate recognition. In a first example, Liu and co-workers demonstrated that the Mm-PylRS/tRNACUA pair can be evolved to preferentially tolerate L-phenylalanine (148), and its derivatives p-iodo- (45) and p-bromo-phenylalanine (44), whose side chain structures are drastically different from pyrrolysine.137 Strikingly, the same group reported a rationally designed Mm-PylRS-(N346A, C348A) mutant able to efficiently incorporate seven different O-substituted tyrosine derivatives (1, 2, 11, 31, 50 and 167)138 and twelve different meta-substituted phenylalanine analogues (146, 147 and 157–166)139 bearing diverse chemical functionalities in E. coli. This is particularly remarkable since meta-substituted phenylalanine derivatives are in general incorporated poorly by previously reported M. jann and E. coli TyrRS mutants. Notably, the same mutant was able to incorporate seven ortho-substituted phenylalanine derivatives (149–155)140 and thirteen meta-alkoxy- and meta-acyl-phenylalanines (135–138, 156 and 142),141 bringing the number of phenylalanyl based UAAs incorporated by this single mutant to almost 40. Similarly, Arbely et al. have reported the incorporation of photocaged o-nitrobenzyl-O-tyrosine 43 using a Mb-PylRS mutant in E. coli and mammalian cells.142 More recently, Schultz and co-workers reported the incorporation of a number of functionalised histidine analogues (129–133), further demonstrating the plasticity of the PylRS,143 while Wang's group used a mutant library generated by optimized saturation mutagenesis for the creation of mutants that incorporate conjugated aromatic rings (5 and 128).144
Miscellaneous aaRSs
The previously described aaRS/tRNA pairs have proven to be both robust and flexible, as illustrated by the multitude of structures that can be incorporated and the many applications in which they have been used. Interest remains though in the development of additional aaRS/tRNA pairs that are orthogonal in commonly-used model organisms. This could expand the structural space of incorporated amino acids and provide additional systems that can be combined for the incorporation of multiple amino acids (see following section). Towards this goal, a variety of novel pairs from different organisms have been explored. The Pyrococcus horikoshii glutamyl145 and lysyl,12S. cerevisiae aspartyl,146 glutaminyl,30 tyrosyl147 and tryptophanyl,148Methanobacterium thermoautotrophicum and Halobacterium sp. leucyl,149Pyrococcus horikoshii prolyl and Archaeoglobus fulgidus prolyl150 aaRS/tRNA pairs have all been reported to be orthogonal in E. coli. In yeast the E. coli glutaminyl,147b and in mammalian cells the Bacillus subtilis tryptophanyl151 aaRS/tRNA pairs have both also been shown to be orthogonal.UAA incorporation has been demonstrated utilising the P. horikoshii LysRS/tRNA pair that has also been used to incorporate homoglutamine (123) in response to a quadruplet codon (discussed further below). Meanwhile, the Methanosarcina acetivorans TyrRS/tRNA pair has been used to incorporate 3-azidotyrosine (75) and 3-iodotyrosine (4),152 while the Bacillus stearothermophilus TrpRS/tRNA pair allows the incorporation 5-hydroxytryptophan (124) into the foldon protein in mammalian cells. This amino acid possesses a distinctive absorption band at 310 nm and is also redox active, allowing for the oxidative cross-linking of a foldon protein dimer when a positive potential is applied. In E. coli a polyspecific S. cerevisiae yeast TrpRS/tRNA pair has also been used to incorporate a variety of tryptophan analogues (124–128) to replace the central tryptophan in the ECFP chromophore.148b This has a pronounced effect on the fluorescent properties of the protein, dependent on the choice of UAA. Also of particular note is the use of the M. jann Cys-tRNA and Methanococcus maripaludis phosphoserine-RS pair for the incorporation of the natural posttranslational modification phosphoserine (167).22
Many of the above mentioned pairs have not passed past the proof-of-orthogonality stage and have not yet been used to incorporate UAAs. Causes for this are certainly specific for each individual pair, but a lack of evolvability or inefficient suppression are commonly-encountered shortcomings. As such, the development of novel orthogonal aaRS/tRNA pairs still remains a relevant challenge.
Systems for incorporation of multiple amino acids
The wealth of UAAs that can be incorporated by codon reassignment already provides an important toolbox for biochemical studies of protein function. However, for more sophisticated purposes the incorporation of multiple functionalities into proteins would be desirable. For example, the incorporation of two different ‘tags’ for chemical modification would allow the addition of two complementary dyes for FRET measurements. Similarly, the incorporation of a photocrosslinker and a modifiable moiety, could allow highly specific capture and release, desirable for proteomic approaches to protein study. The in vivo incorporation of multiple unique amino acids is being pursued by a number of groups, currently only by two general approaches: the use of two distinct stop codons and the use of a stop codon in combination with a quadruplet codon (see below and Fig. 4). | ||
| Fig. 4 Incorporation of multiple UAAs into the same protein utilising the reassignment of 2 different non-sense codons. | ||
The first approach was initially realized by combining a para-azidophenylalanine specific M. jann TyrRS/tRNACUA and the wild type PylRS/tRNA pair.11 The authors first investigated whether the mRNA codon recognized by the Pyl tRNA could be changed from the naturally recognized UAG stop codon to UAA (ochre), UGA (opal) or a quadruplet UAGA codon. While the quadruplet codon significantly decreased protein yield, UAA and UGA actually resulted in higher protein expression when compared to the amber codon. An ochre specific PylRS/tRNAUUA pair was therefore combined with a M. jann TyrRS/tRNACUA pair to incorporate both azide (6) and alkyne (86) tags into GFP. The two orthogonal moieties were individually labelled with two different fluorescent dyes via a CuAAC reaction, resulting in the formation of a FRET pair. In a later report it was shown that by choosing appropriate conjugation chemistries, in this case strain-promoted azide–alkyne cycloaddition and hydrazone forming reactions, two different labels could be conjugated to a single protein in a one pot procedure.33b Subsequent detailed analysis of suppression efficiency of all three stop codons by different aaRS/tRNA variants, will allow further expansion and optimization of this approach.153 While optimization of the aaRS/tRNA-bearing plasmid can greatly improve expression yields for the incorporation of a single UAA.31c Similarly, Chatterjee et al. have presented an optimized system for the incorporation of two distinct UAAs.33a The constructed system allows the encoding of both distinct aaRS/tRNA pairs on a single plasmid, termed pUltraII, greatly simplifying incorporation.
Global suppression
While codon reassignment has the advantage of giving site specific incorporation of the desired UAA, the global replacement of a particular amino acid is another efficient way for the incorporation of single or multiple UAAs and is often sufficient to yield proteins with the desired biophysical property (e.g. enhanced stability or fluorescence). Global amino acid replacement commonly utilises auxotrophic strains, incapable of synthesizing a particular amino acid (or multiple amino acids if the strain is polyauxotrophic), to substitute residues with structurally similar counterparts. The most prevalent examples of replacement are the introduction of alkyne154 and azide155 functionalities through the substitution of methionine (Met) by azidohomoalanine (Aha) or homopropargylglycine (Hpg). These amino acids can then be used as reactive handles for further chemical modification, such as through CuAAC. The introduction of Aha is not limited to incorporation in auxotrophic strains of E. coli; in human cell culture Aha can be incorporated into proteins when supplied in the growth medium instead of Met. This incorporation in mammalian cells has been used to identify newly synthesized proteins with great temporal resolution, via pulse-chase labelling experiments.156 The need to incorporate UAAs that are structurally similar to endogenous amino acids157 is a limitation of this technique, yet still the functional space of proteins can be greatly expanded using codon suppression.158 The structural restraints can be partially alleviated by using engineered aaRSs that have relaxed specificity towards their natural substrate, increasing the number of UAAs that can be incorporated. A key advantage of global replacement is that multiple incorporations of the same, or up to three different UAAs, can be accomplished in the same protein while retaining good yields.159 Interestingly, these replacement experiments have generated proteins consisting of up to 10% of UAAs, which were shown to still retain activity.159a Combining UAA incorporation with directed evolution can further help to generate proteins with desired properties such as increased folding rates.160 The use of global replacement is in theory compatible with all codon reassignment methods making the combination of different UAA incorporation techniques an attractive future direction for further expanding protein function.Quadruplet codon suppression
The number of distinct UAAs that can be encoded by an organism is theoretically limited by the availability of non-coding triplet codons. Utilizing different codons within the same sequence with different register can also be viewed as a good strategy for increasing coding power. As a result, quadruplet codons have been used as an alternative for encoding UAAs. Early reports described the stoichiometric use of pre-aminoacylated, extended quadruplet anticodon tRNAs to incorporate UAAs in response to four-base codons in vitro or when microinjected in to Xenopus oocytes. These systems however require the synthesis of the pre-aminoacylated tRNA precursor, show low efficiency of incorporation and are not applicable to most cell types or larger scale expressions.161Here, we will focus on quadruplet codon-suppression involving orthogonal synthetase/tRNA pairs, able to selectively aminoacylate the tRNA and decode the quadruplet codons to incorporate UAAs in vivo. The first example of such a system was reported by Schultz and co-workers using a variant Pyrococcus horikoshii LysRS/tRNA pair in E. coli to incorporate L-homoglutamine (123) in response to the quadruplet codon AGGA. The orthogonality of this pair to the M. jann TyrRS/tRNACUA pair enabled the simultaneous incorporation of O-methyl-L-tyrosine (1) into recombinant myoglobin.12 However, this system proceeded with relatively low efficiency. Indeed, although the natural ribosome is capable of recognizing quadruplet anti-codon tRNAs, their decoding is relatively inefficient. This can be rationalized by a poor accommodation of the extended tRNA codon-binding region in the ribosome. At the same time, the natural ribosome is not evolved towards more efficient quadruplet decoding since this might lead to misreading and missynthesis of the proteome, toxicity and cell death.
To overcome this problem, further engineering of the cellular translational machinery has been achieved by Chin and co-workers, who have developed an orthogonal translation pathway within the cell. This has involved the creation of an orthogonal ribosome recognizing an alternative Shine–Dalgarno sequence, a short sequence upstream of the 5′-AUG translation initiation codon responsible for mRNA recognition by the ribosome in prokaryotes. This orthogonal ribosome was shown to be selectively directed to corresponding orthogonal mRNAs bearing such a Shine–Dalgarno sequence. The orthogonal mRNA is in turn not recognized by the endogenous ribosome.162 This orthogonal ribosome can be evolved towards a quadruplet code by mutating residues around the ribosome A site, responsible for the fidelity of triplet decoding. This ‘loosening’ enables efficient quadruplet codon decoding without affecting the translation of cellular mRNAs by natural ribosomes, and thus without toxic misreading of the proteome.163 In the presence of sufficient aminoacylated tRNA, the level of quadruplet decoding can approach that of triplet decoding by the natural ribosome.163
This orthogonal translation machinery was first used for quadruplet codon decoding using a system based on the M. jann TyrRS/tRNACUA pair. First, a M. jann tRNAUCCU was developed together with a synthetase able to identify p-azido-L-phenylalanine (6) and to recognize the quadruplet anticodon. Using this M. jann AzPheRS/tRNAUCCU variant, p-azido-L-phenylalanine (6) was incorporated in response to the AGGA codon in E. coli. The M. jann AzPheRS/tRNAUCCU pair, being orthogonal to the Mb-PylRS/tRNACUA pair, allowed simultaneous incorporation of p-azido-L-phenylalanine and an aliphatic alkyne (86) via quadruplet and amber codon suppression respectively in a single protein.163 Further evolving Pyl tRNA for quadruplet codon decoding has enabled optimized translation of quadruplet codons. Chin et al. recently reported the evolution of several PylRS/tRNAXXXX pairs, thereby enabling double quadruplet suppression in a single protein. In addition, using Mb PylRS/tRNAUACU and Mj TyrRS/tRNACUA pairs, two unique amino acids, norbornyl-lysine (122) and tetrazinyl-phenylalanine (54) were incorporated in the Ca2+-binding protein calmodulin with efficiencies of up to 20%. Elegantly using two orthogonal reactions involving a more activated tetrazine and a bicyclononyne probe, they achieved a one-pot incorporation of a FRET pair on the protein.164
In an alternative strategy, Schultz and co-workers proposed an engineered Pyl-tRNAUCCU to incorporate N-ε-(tert-butyloxy-carbonyl)-L-lysine (Boc-Lys, 77) in response to the AGGA codon in both bacterial and mammalian cells. The evolution of the tRNA improved aaRS recognition and ribosome affinity, and therefore enhanced the efficiency of quadruplet codon decoding by the natural ribosome, and without need for further engineering of the host cell translational machinery.165
Quadruplet decoding opens up interesting possibilities for the incorporation of multiple UAAs into proteins, by providing, theoretically, 256 additional blank codons. However, this approach is limited by the number of available aaRS/tRNA pairs orthogonal to both host cell machineries and to each other. Current efforts to further evolve new orthogonal synthetases and tRNAs de novo may soon lead to systems able to incorporate more than two different UAAs in a single protein.
Sense codon suppression
The reassignment of ‘non-sense’ codons has developed into a powerful tool for UAA incorporation, yet is still limited by competition with the host translational termination machinery and resultant low protein yields. As will be further discussed in the following section, one solution to this problem is by preventing termination as described by Johnson et al., deleting the ‘TAG’-specific release factor RF1, thus allowing efficient reassignment.166 However, the suppression of ‘native’-TAG codons may then subsequently begin to affect host fitness through the undesirable extension of peptide sequences.167 A further solution to this problem has been devised by Isaacs et al. Through the development of a technique known as hierarchical conjugative assembly genome engineering (CAGE), all 314 naturally occurring TAG codons in the genome of E. coli can be replaced with an alternative stop codon, thus minimising any deleterious effects of codon reassignment.168 It was subsequently shown that such engineering allows the deletion of RF1 and the complete reassignment of the TAG codon for UAA incorporation, even in strains for which RF1 knockout is usually lethal.169This ability to create an ‘amber-free’ system by completely replacing a codon throughout a genome highlights the potential of another powerful tool that we have covered only briefly thus far. The utilization of sense codons to incorporate UAAs has a long history170 and has been enhanced by the availability of associated auxotrophs to enable greater control (see ‘Global suppression’ section above). This is already an existing powerful form of sense codon utilization. Genome ‘editing’ now additionally opens the door to the reassignment of degenerate ‘sense’ codons. Since most amino acids are translated by multiple codons, those which are rarely used may be ideal candidates for such reassignment. Given that, in theory, up to 20 such codons may be replaced while maintaining a viable genome with no competition from the host termination machinery, such codons offer a potential technique for the efficient incorporation of multiple UAAs.171 However, significant challenges remain to be overcome in order to achieve such goals. The scarcity of ‘natural’ reassignments highlights the challenges that may be encountered due to the possibility of differing translational efficiencies associated with seemingly equal codons, or hidden effects on gene regulation. Indeed, in pioneering work by Lajoie et al. the removal of 13 rare codons from a number of essential genes proved to be widely tolerated by the host, yet a decrease in fitness in many instances demonstrated that ‘synonymous codons can be non-equivalent in unpredictable ways’.172
Another challenge is the misidentification of UAA-associated tRNAs by host synthetases. Krishnakumar et al. have shown that while the Pyl-tRNACUA is efficiently aminoacylated by the Pyl-aaRS, when suppression of the arginine codon CGG was attempted, using the altered anti-codon Pyl-tRNACCG, unexpected misacylation with arginine was observed as the major product.171 This can be attributed to the use of the tRNA-anticodon as a major recognition element for many aaRSs, leading to a need for further engineering than simply native-tRNA knockout for ‘sense’-codon reassignment. There may therefore be a need to focus on codons which do not rely on such recognition, or alternatively the addition of anti-determinants of recognition to minimise misacylation.173
Despite these potential difficulties, important early steps towards ‘sense’-codon reassignment have already been achieved by a number of groups. Bohlke and Budisa have shown that the rare isoleucine-codon AUA can be liberated from its natural translation pathway via tRNA knockout and subsequent replacement with a tRNA from an alternative species, albeit continuing to incorporate isoleucine.174 More recently, Bröcker et al. have demonstrated that the incorporation machinery of selenocysteine (the rarely used ‘21st amino acid’, see reviews by Böck et al.175 and Johansson et al.176 for details) can be modulated to recognise 58 of the 64 naturally occurring codons, in many cases completely outcompeting the endogenous tRNA, although other codons resulted in ambiguous translation.177 These reports demonstrate that while limitations still exist and UAAs are yet to be incorporated, ‘sense’-reassignment remains an exciting topic of research. In particular, while many rare codons or tRNA/aaRS systems may prove not to be of use, further research into the subtleties of tRNA recognition and the interactions of tRNA/aaRS with the host machineries may prove highly fruitful in allowing UAA incorporation with greatly improved efficiency.
Outlook
Enormous strides have been made in the field of codon-reassignment in little over a decade, yet there remain a number of key challenges to be overcome. The vast majority of studies so far have focused on the incorporation of UAAs in single cell cultures. While this has proved a powerful tool for understanding a number of important cellular processes, a transition into multicellular organisms would be an important discovery for studying inter-cellular functions. To this extent a number of recent studies have begun to address the challenges of genetic code expansion in animals.Chin and co-workers, first demonstrated the incorporation of N-ε-(tert-butyloxy-carbonyl)-L-lysine (BocLys, 77) and N-ε-[(2-propynloxy)carbonyl]-L-lysine (85) in Caenorhabditis elegans using a wt Mm-PylRS/tRNACUA pair.10b In order to generate a worm containing the required genetic information, they used ‘biolistic’ bombardment to deliver an extrachromosomal array, requiring antibiotic-based selections to maintain the DNA constructs in the cell line. While undoubtedly an important discovery, suppression efficiencies were very low and observed only in a small subset of transformed worms. A similar technique was used for the expansion of the genetic code of Drosophila melongaster. Suppressions were undertaken in both fly embryos, and in specific tissues and cell subsets in adult flies.10a
In an alternative approach, Parrish et al. reported the dual encoding of O-methyltyrosine (1) and dansylalanine (58) with E. coli TyrRS/tRNA and LeuRS/tRNA pairs respectively, again in C. elegans. In this case however, they first generated stable transgenic worms with chromosomally integrated reporter and aaRS/tRNA genes, resulting in increased levels of suppression and genetic stability.178 Further optimisation of incorporation may have important implications in the study of embryonic development, neural processing and cancer biology that can only be addressed in an in vivo setting. Recently, Wang and co-workers transferred this technology into plants, using a MbPylRS mutant incorporating an acryllysine UAA 111 as a handle for ‘photo-click’ chemistry, into proteins. This was achieved by first constructing a vector containing the corresponding genes that could replicate in Agrobacterium, which could in turn deliver the genes into the plant host.125
Another significant challenge in the field is the improvement of suppression efficiency. Current non-sense techniques suffer from UAA incorporation, in effect, being in direct competition with protein truncation. As such, incorporation efficiency can be drastically reduced and in many cases fails entirely. Knock out of RF1, the release factor responsible for termination at UAG, eliminates the problem of competing termination.166 Since this protein is primarily responsible for decoding the amber stop codon, UAG, and causing translational termination, its deletion reduces competition and greatly increases incorporation efficiency. As a result, up to 10 stop codons can be suppressed in a single protein. Similar discoveries are now required in eukaryotes in order to improve the efficiency of UAA incorporation in these more challenging cell lines. An efficient codon reassignment will also likely greatly aid the incorporation of multiple amino acids into the same protein (see above) using multiple orthogonal RS/tRNA pairs.
While over 150 UAAs have been incorporated by codon reassignment, these are mainly based around the same few core structural motifs. There are many structures or functionalities that still cannot be incorporated, and as such there remains a need to discover additional orthogonal pairs that can be used. In particular, there are many natural PTMs that cannot be encoded, either in their natural forms, or as close structural or electronic mimics. Site-specific incorporation of such UAAs would greatly enhance our ability to study natural processes. The inability to create glycoproteins, for example, remains a glaring frustration.
For those wishing to incorporate a novel UAA, it is worth noting that while many aaRS/tRNA pairs have been developed for a specific amino acid of choice and have no observed cross-reactivity with natural amino acids, these may still have a broad substrate specificity, allowing for the incorporation of a number of related UAAs by a single aaRS.55a,179 As such, there may be no need to go through the complex process of developing a novel specific system for incorporation of a desired UAA in every case. Rather, it may be possible to utilise an aaRS designed for a structurally related UAA, at least as a starting point from which further manipulations can lead to increased selectivities.67,179b
Importantly, though, logistical barriers also exist. In order to facilitate further progress in the field of codon reassignment, it is not only important to explore new techniques, organisms, and orthogonal pairs. It is also vital that the requisite plasmids and constructs become more widely available, as at present gaining access to some variants can be limited and is often a slow and tedious process. Deposition, which is now common for many plasmids, seems to have been slowly embraced by the suppression community.
Finally, perhaps more ambition is needed? Only when more researchers begin to apply their ideas not just to the incorporation of novel UAAs, but to their applications and functions also, can the true potential of this powerful tool be realized. In the future, codon reassignment may have at least a similar impact on the biological sciences as site-directed mutagenesis did before it and given the potential for further selective chemical elaboration in an almost unlimited manner, perhaps even more so.
Notes and references
- F. Crick, Nature, 1970, 227, 561 CrossRef CAS
.
-
H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Molecular Cell Biology, W. H. Freeman, New York, 2000 Search PubMed
- A. B. Martin and P. G. Schultz, Trends Cell Biol., 1999, 9, M24 CrossRef CAS
- T. S. Young and P. G. Schultz, J. Biol. Chem., 2010, 285, 11039 CrossRef CAS PubMed
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413 CrossRef CAS PubMed
- A. J. de Graaf, M. Kooijman, W. E. Hennink and E. Mastrobattista, Bioconjugate Chem., 2009, 20, 1281 CrossRef CAS PubMed
-
(a) L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498 CrossRef CAS PubMed
-
(a) J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. Chen, J. Am. Chem. Soc., 2013, 135, 7330 CrossRef CAS PubMed
-
(a) K. Sakamoto, A. Hayashi, A. Sakamoto, D. Kiga, H. Nakayama, A. Soma, T. Kobayashi, M. Kitabatake, K. Takio, K. Saito, M. Shirouzu, I. Hirao and S. Yokoyama, Nucleic Acids Res., 2002, 30, 4692 CrossRef CAS PubMed
-
(a) A. Bianco, F. M. Townsley, S. Greiss, K. Lang and J. W. Chin, Nat. Chem. Biol., 2012, 8, 748 CrossRef CAS PubMed
- W. Wan, Y. Huang, Z. Wang, W. K. Russell, P.-J. Pai, D. H. Russell and W. R. Liu, Angew. Chem., Int. Ed., 2010, 49, 3211 CrossRef CAS PubMed
- J. C. Anderson, N. Wu, S. W. Santoro, V. Lakshman, D. S. King and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7566 CrossRef CAS PubMed
- L. Davis and J. Chin, Nat. Rev. Mol. Cell Biol., 2012, 13, 168 CAS
- P. E. Carrigan, P. Ballar and S. Tuzmen, Methods Mol. Biol., 2011, 700, 107 CAS
-
(a) H. Neumann, FEBS Lett., 2012, 586, 2057 CrossRef CAS PubMed
-
(a) M. Grammel and H. Hang, Nat. Chem. Biol., 2013, 9, 475 CrossRef CAS PubMed
-
(a) J. Chalker, G. Bernardes and B. Davis, Acc. Chem. Res., 2011, 44, 730 CrossRef CAS PubMed
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC
- H.-W. Ai, Anal. Bioanal. Chem., 2012, 403, 2089 CrossRef CAS PubMed
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381 CrossRef CAS PubMed
- C. Liu and P. Schultz, Nat. Biotechnol., 2006, 24, 1436 CrossRef CAS PubMed
- H. S. Park, M. J. Hohn, T. Umehara, L. T. Guo, E. M. Osborne, J. Benner, C. J. Noren, J. Rinehart and D. Söll, Science, 2011, 333, 1151 CrossRef CAS PubMed
- H. Neumann, J. L. Hazen, J. Weinstein, R. A. Mehl and J. W. Chin, J. Am. Chem. Soc., 2008, 130, 4028 CrossRef CAS PubMed
-
B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, 2008 Search PubMed
- C. Alff-Steinberger and R. Epstein, J. Theor. Biol., 1994, 168, 461 CrossRef CAS PubMed
-
(a) C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182 CAS
- D. Mendel, V. W. Cornish and P. G. Schultz, Annu. Rev. Biophys. Biomol. Struct., 1995, 24, 435 CrossRef CAS PubMed
- R. Furter, Protein Sci., 1998, 7, 419 CrossRef CAS PubMed
- D. R. Liu, T. J. Magliery, M. Pastrnak and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10092 CrossRef CAS
- D. R. Liu and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4780 CrossRef CAS
-
(a) Y. Ryu and P. G. Schultz, Nat. Methods, 2006, 3, 263 CrossRef CAS PubMed
- S. Blight, R. Larue, A. Mahapatra, D. Longstaff, E. Chang, G. Zhao, P. Kang, K. Green-Church, M. Chan and J. Krzycki, Nature, 2004, 431, 333 CrossRef CAS PubMed
-
(a) A. Chatterjee, S. B. Sun, J. L. Furman, H. Xiao and P. G. Schultz, Biochemistry, 2013, 52, 1828–1837 CrossRef CAS PubMed
- L. Wang, T. J. Magliery, D. R. Liu and P. G. Schultz, J. Am. Chem. Soc., 2000, 122, 5010 CrossRef CAS
- B. A. Steer and P. Schimmel, J. Biol. Chem., 1999, 274, 35601 CrossRef CAS PubMed
- S. W. Santoro, L. Wang, B. Herberich, D. S. King and P. G. Schultz, Nat. Biotechnol., 2002, 20, 1044 CrossRef CAS PubMed
- Z. Zhang, L. Wang, A. Brock and P. G. Schultz, Angew. Chem., Int. Ed., 2002, 41, 2840 CrossRef CAS
- A. Deiters and P. G. Schultz, Bioorg. Med. Chem. Lett., 2005, 15, 1521 CrossRef CAS PubMed
- S. J. Miyake-Stoner, A. M. Miller, J. T. Hammill, J. C. Peeler, K. R. Hess, R. A. Mehl and S. H. Brewer, Biochemistry, 2009, 48, 5953 CrossRef CAS PubMed
-
(a) L. Wang, J. Xie, A. A. Deniz and P. G. Schultz, J. Org. Chem., 2003, 68, 174 CrossRef CAS PubMed
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026 CrossRef CAS PubMed
- L. Wang, Z. Zhang, A. Brock and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 56 CrossRef CAS PubMed
- H. Zeng, J. Xie and P. G. Schultz, Bioorg. Med. Chem. Lett., 2006, 16, 5356 CrossRef CAS PubMed
- E. Brustad, M. L. Bushey, J. W. Lee, D. Groff, W. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 8220 CrossRef CAS PubMed
-
(a) E. M. Brustad, E. A. Lemke, P. G. Schultz and A. A. Deniz, J. Am. Chem. Soc., 2008, 130, 17664 CrossRef CAS PubMed
- R. A. Mehl, J. C. Anderson, S. W. Santoro, L. Wang, A. B. Martin, D. S. King, D. M. Horn and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 935 CrossRef CAS PubMed
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898 CrossRef CAS PubMed
- J. Wang, W. Zhang, W. Song, Y. Wang, Z. Yu, J. Li, M. Wu, L. Wang, J. Zang and Q. Lin, J. Am. Chem. Soc., 2010, 132, 14812 CrossRef CAS PubMed
- S. Chen and M.-L. Tsao, Bioconjugate Chem., 2013, 24, 1645–1649 CrossRef CAS PubMed
-
(a) J. W. Chin, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11020 CrossRef CAS PubMed
- E. M. Tippmann, W. Liu, D. Summerer, A. V. Mack and P. G. Schultz, ChemBioChem, 2007, 8, 2210 CrossRef CAS PubMed
- M.-L. Tsao, D. Summerer, Y. Ryu and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 4572 CrossRef CAS PubMed
- K. C. Schultz, L. Supekova, Y. Ryu, J. Xie, R. Perera and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 13984 CrossRef CAS PubMed
- H. Taskent-Sezgin, J. Chung, V. Patsalo, S. J. Miyake-Stoner, A. M. Miller, S. H. Brewer, R. A. Mehl, D. F. Green, D. P. Raleigh and I. Carrico, Biochemistry, 2009, 48, 9040 CrossRef CAS PubMed
-
(a) S. E. Cellitti, D. H. Jones, L. Lagpacan, X. Hao, Q. Zhang, H. Hu, S. M. Brittain, A. Brinker, J. Caldwell, B. Bursulaya, G. Spraggon, A. Brock, Y. Ryu, T. Uno, P. G. Schultz and B. H. Geierstanger, J. Am. Chem. Soc., 2008, 130, 9268 CrossRef CAS PubMed
- M. Bose, D. Groff, J. Xie, E. Brustad and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 388 CrossRef CAS PubMed
- J. Xie, L. Supekova and P. G. Schultz, ACS Chem. Biol., 2007, 2, 474 CrossRef CAS PubMed
- M. R. Seyedsayamdost, J. Xie, C. T. Y. Chan, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2007, 129, 15060 CrossRef CAS PubMed
- M. R. Seyedsayamdost and J. Stubbe, Methods Enzymol., 2009, 462, 45 CAS
- L. Alfonta, Z. Zhang, S. Uryu, J. A. Loo and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 14662 CrossRef CAS PubMed
- K. Sakamoto, K. Murayama, K. Oki, F. Iraha, M. Kato-Murayama, M. Takahashi, K. Ohtake, T. Kobayashi, S. Kuramitsu, M. Shirouzu and S. Yokoyama, Structure, 2009, 17, 335 CrossRef CAS PubMed
- Z. Zhang, B. A. C. Smith, L. Wang, A. Brock, C. Cho and P. G. Schultz, Biochemistry, 2003, 42, 6735 CrossRef CAS PubMed
- F. B. Peters, A. Brock, J. Wang and P. G. Schultz, Chem. Biol., 2009, 16, 148 CrossRef CAS PubMed
- L. Wang, A. Brock and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 1836 CrossRef CAS PubMed
-
(a) I. N. Ugwumba, K. Ozawa, Z.-Q. Xu, F. Ely, J.-L. Foo, A. J. Herlt, C. Coppin, S. Brown, M. C. Taylor, D. L. Ollis, L. N. Mander, G. Schenk, N. E. Dixon, G. Otting, J. G. Oakeshott and C. J. Jackson, J. Am. Chem. Soc., 2011, 133, 326 CrossRef CAS PubMed
- H. S. Lee, G. Spraggon, P. G. Schultz and F. Wang, J. Am. Chem. Soc., 2009, 131, 2481 CrossRef CAS PubMed
- J. Xie, W. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 9239 CrossRef CAS PubMed
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849 CrossRef CAS PubMed
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630 CrossRef CAS PubMed
- Z. J. Chen, W. Ren, Q. E. Wright and H. W. Ai, J. Am. Chem. Soc., 2013, 135, 14940 CrossRef CAS PubMed
- A. Deiters, D. Groff, Y. Ryu, J. Xie and P. G. Schultz, Angew. Chem., Int. Ed., 2006, 45, 2728 CrossRef CAS PubMed
- B. J. Wilkins, S. Marionni, D. D. Young, J. Liu, Y. Wang, M. L. D. Salvo, A. Deiters and T. A. Cropp, Biochemistry, 2010, 49, 1557 CrossRef CAS PubMed
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498 CrossRef CAS PubMed
-
(a) T. Kobayashi, O. Nureki, R. Ishitani, A. Yaremchuk, M. Tukalo, S. Cusack, K. Sakamoto and S. Yokoyama, Nat. Struct. Biol., 2003, 10, 425 CrossRef CAS PubMed
- J. Guo, J. Wang, J. C. Anderson and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 722 CrossRef CAS PubMed
-
(a) H. Edwards and P. Schimmel, Mol. Cell. Biol., 1990, 10, 1633 CAS
- K. Oki, K. Sakamoto, T. Kobayashi, H. M. Sasaki and S. Yokoyama, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13298 CrossRef CAS PubMed
-
(a) N. Hino, A. Hayashi, K. Sakamoto and S. Yokoyama, Nat. Protoc., 2006, 1, 2957 CrossRef CAS PubMed
- W. Liu, A. Brock, S. Chen, S. Chen and P. G. Schultz, Nat. Methods, 2007, 4, 239 CrossRef CAS PubMed
-
(a) S. Ye, T. Huber, R. Vogel and T. P. Sakmar, Nat. Chem. Biol., 2009, 5, 397 CrossRef CAS PubMed
- A. Deiters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 11782 CrossRef CAS PubMed
-
(a) S. Chen, P. G. Schultz and A. Brock, J. Mol. Biol., 2007, 371, 112 CrossRef CAS PubMed
- W. Wang, J. K. Takimoto, G. V. Louie, T. J. Baiga, J. P. Noel, K.-F. Lee, P. a. Slesinger and L. Wang, Nat. Neurosci., 2007, 10, 1063 CrossRef CAS PubMed
- F. Iraha, K. Oki, T. Kobayashi, S. Ohno, T. Yokogawa, K. Nishikawa, S. Yokoyama and K. Sakamoto, Nucleic Acids Res., 2010, 38, 3682 CrossRef CAS PubMed
- A. Soma and H. Himeno, Nucleic Acids Res., 1998, 26, 4374 CrossRef CAS PubMed
- H. Asahara, H. Himeno, K. Tamura, T. Hasegawa, K. Watanabe and M. Shimizu, J. Mol. Biol., 1993, 231, 219 CrossRef CAS PubMed
- S. Cusack, A. Yaremchuk and M. Tukalo, EMBO J., 2000, 19, 2351 CrossRef CAS PubMed
- N. Wu, A. Deiters, T. A. Cropp, D. King and P. G. Schultz, J. Am. Chem. Soc., 2004, 126, 14306 CrossRef CAS PubMed
- D. Summerer, S. Chen, N. Wu, A. Deiters, J. W. Chin and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9785 CrossRef CAS PubMed
- H. S. Lee, J. Guo, E. A. Lemke, R. D. Dimla and P. G. Schultz, J. Am. Chem. Soc., 2009, 131, 12921 CrossRef CAS PubMed
- E. M. Tippmann and P. G. Schultz, Tetrahedron, 2007, 63, 6182 CrossRef CAS PubMed
- E. Brustad, M. L. Bushey, A. Brock, J. Chittuluru and P. G. Schultz, Bioorg. Med. Chem. Lett., 2008, 18, 6004 CrossRef CAS PubMed
- H.-W. Ai, W. Shen, E. Brustad and P. G. Schultz, Angew. Chem., Int. Ed., 2010, 49, 935 CrossRef CAS PubMed
-
(a) B. Hao, W. Gong, T. K. Ferguson, C. M. James, J. A. Krzycki and M. K. Chan, Science, 2002, 296, 1462 CrossRef CAS PubMed
- C. R. Polycarpo, S. Herring, A. Bérubé, J. L. Wood, D. Söll and A. Ambrogelly, FEBS Lett., 2006, 580, 6695 CrossRef CAS PubMed
- W.-T. Li, A. Mahapatra, D. G. Longstaff, J. Bechtel, G. Zhao, P. T. Kang, M. K. Chan and J. A. Krzycki, J. Mol. Biol., 2009, 385, 1156 CrossRef CAS PubMed
- J. M. Kavran, S. Gundliapalli, P. O'Donoghue, M. Englert, D. Soell and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11268 CrossRef CAS PubMed
- D. Nguyen, H. Lusic, H. Neumann, P. Kapadnis, A. Deiters and J. Chin, J. Am. Chem. Soc., 2009, 131, 8720 CrossRef CAS PubMed
- T. Fekner, X. Li, M. M. Lee and M. K. Chan, Angew. Chem., Int. Ed., 2009, 48, 1633 CrossRef CAS PubMed
- X. Li, T. Fekner and M. K. Chan, Chem.–Asian J., 2010, 5, 1765 CrossRef CAS PubMed
- M. M. Lee, T. Fekner, T.-H. Tang, L. Wang, A. H.-Y. Chan, P.-H. Hsu, S. W. Au and M. K. Chan, ChemBioChem, 2013, 14, 805 CrossRef CAS PubMed
- X. Li, T. Fekner, J. J. Ottesen and M. K. Chan, Angew. Chem., Int. Ed., 2009, 48, 9184 CrossRef CAS PubMed
- T. Fekner, X. Li and M. K. Chan, ChemBioChem, 2011, 12, 21–33 CrossRef CAS PubMed
- D. Nguyen, T. Elliott, M. Holt, T. Muir and J. Chin, J. Am. Chem. Soc., 2011, 133, 11418 CrossRef CAS PubMed
-
(a) K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298 CrossRef CAS PubMed
- C. Chou, R. Uprety, L. Davis, J. W. Chin and A. Deiters, Chem. Sci., 2011, 2, 480–483 RSC
-
(a) D. P. Nguyen, M. M. G. Alai, S. Virdee and J. W. Chin, Chem. Biol., 2010, 17, 1072 CrossRef CAS PubMed
- T. Kobayashi, T. Yanagisawa, K. Sakamoto and S. Yokoyama, J. Mol. Biol., 2009, 385, 1352 CrossRef CAS PubMed
-
(a) T. Yanagisawa, R. Ishii, R. Fukunaga, O. Nureki and S. Yokoyama, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 1031 CrossRef CAS PubMed
- H. Neumann, S. Peak-Chew and J. Chin, Nat. Chem. Biol., 2008, 4, 232 CrossRef CAS PubMed
- T. Mukai, T. Kobayashi, N. Hino, T. Yanagisawa, K. Sakamoto and S. Yokoyama, Biochem. Biophys. Res. Commun., 2008, 371, 818 CrossRef CAS PubMed
- Y. Huang, W. Wan, W. Russell, P.-J. Pai, Z. Wang, D. Russell and W. Liu, Bioorg. Med. Chem. Lett., 2010, 20, 878 CrossRef CAS PubMed
- T. Yanagisawa, R. Ishii, R. Fukunaga, T. Kobayashi, K. Sakamoto and S. Yokoyama, Chem. Biol., 2008, 15, 1187 CrossRef CAS PubMed
- M. J. Schmidt and D. Summerer, Angew. Chem., Int. Ed., 2013, 52, 4690 CrossRef CAS PubMed
- Z. Wang, Y.-S. Wang, P.-J. Pai, W. Russell, D. Russell and W. Liu, Biochemistry, 2012, 51, 5232 CrossRef CAS PubMed
- T. Plass, S. Milles, C. Koehler, J. Szymański, R. Mueller, M. Wiessler, C. Schultz and E. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166 CrossRef CAS PubMed
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878 CrossRef CAS PubMed
- A. Borrmann, S. Milles, T. Plass, J. Dommerholt, J. M. Verkade, M. Wiessler, C. Schultz, J. van Hest, F. van Delft and E. Lemke, ChemBioChem, 2012, 13, 2094 CrossRef CAS PubMed
- T. Yanagisawa, N. Hino, F. Iraha, T. Mukai, K. Sakamoto and S. Yokoyama, Mol. BioSyst., 2012, 8, 1131 RSC
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. Cox, M. Blackman, J. Fox and J. Chin, J. Am. Chem. Soc., 2012, 134, 10317 CrossRef CAS PubMed
- M. J. Schmidt, J. Borbas, M. Drescher and D. Summerer, J. Am. Chem. Soc., 2014, 136, 1238 CrossRef CAS PubMed
- P. Chen, D. Groff, J. Guo, W. Ou, S. Cellitti, B. Geierstanger and P. Schultz, Angew. Chem., Int. Ed., 2009, 48, 4052 CrossRef CAS PubMed
- Y.-S. Wang, B. Wu, Z. Wang, Y. Huang, W. Wan, W. Russell, P.-J. Pai, Y. Moe, D. Russell and W. Liu, Mol. BioSyst., 2010, 6, 1557 RSC
- Y. J. Lee, B. Wu, J. E. Raymond, Y. Zeng, X. Q. Fang, K. L. Wooley and W. R. S. Liu, ACS Chem. Biol., 2013, 8, 1664 CrossRef CAS PubMed
- F. H. Li, H. Zhang, Y. Sun, Y. C. Pan, J. Z. Zhou and J. Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 9700 CrossRef CAS PubMed
- Z. Yu and Q. Lin, J. Am. Chem. Soc., 2014, 136, 4153 CrossRef CAS PubMed
- S. Hancock, R. Uprety, A. Deiters and J. Chin, J. Am. Chem. Soc., 2010, 132, 14819 CrossRef CAS PubMed
-
(a) A. Gautier, D. Nguyen, H. Lusic, W. An, A. Deiters and J. Chin, J. Am. Chem. Soc., 2010, 132, 4086 CrossRef CAS PubMed
- Z. Yu, Y. Pan, Z. Wang, J. Wang and Q. Lin, Angew. Chem., Int. Ed., 2012, 51, 10600 CrossRef CAS PubMed
- D. P. Nguyen, M. Mahesh, S. J. Elsasser, S. M. Hancock, C. Uttamapinant and J. W. Chin, J. Am. Chem. Soc., 2014, 136, 2240 CrossRef CAS PubMed
- Z. Hao, Y. Song, S. Lin, M. Yang, Y. Liang, J. Wang and P. Chen, Chem. Commun., 2011, 47, 4502 RSC
- Y.-M. Li, M.-Y. Yang, Y.-C. Huang, Y.-T. Li, P. Chen and L. Liu, ACS Chem. Biol., 2012, 7, 1015 CrossRef CAS PubMed
- L. Yiming, Y. Maiyun, H. Yichao, S. Xiaoda, L. Lei and R. C. Peng, Chem. Sci., 2012, 3, 2766–2770 RSC
- Y. Li, M. Pan, Y. Li, Y. Huang and Q. Guo, Org. Biomol. Chem., 2013, 11, 2624 CAS
- M. Zhang, S. Lin, X. Song, J. Liu, Y. Fu, X. Ge, X. Fu, Z. Chang and P. Chen, Nat. Chem. Biol., 2011, 7, 671 CrossRef CAS PubMed
- C. H. Kim, M. Kang, H. J. Kim, A. Chatterjee and P. G. Schultz, Angew. Chem., Int. Ed., 2012, 51, 7246 CrossRef CAS PubMed
- Y.-S. Wang, W. Russell, Z. Wang, W. Wan, L. Dodd, P.-J. Pai, D. Russell and W. Liu, Mol. BioSyst., 2011, 7, 714 RSC
- Y. S. Wang, X. Q. Fang, A. L. Wallace, B. Wu and W. S. R. Liu, J. Am. Chem. Soc., 2012, 134, 2950 CrossRef CAS PubMed
- Y.-S. Wang, X. Fang, H.-Y. Chen, B. Wu, Z. Wang, C. Hilty and W. Liu, ACS Chem. Biol., 2013, 8, 405 CrossRef CAS PubMed
- J. M. Tharp, Y.-S. Wang, Y.-J. Lee, Y. Yang and W. R. Liu, ACS Chem. Biol., 2014, 9, 884–890 CrossRef CAS PubMed
-
(a) A. Tuley, Y. S. Wang, X. Q. Fang, Y. Kurra, Y. H. Rezenom and W. R. Liu, Chem. Commun., 2014, 50, 2673 RSC
- E. Arbely, J. Torres-Kolbus, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 11912 CrossRef CAS PubMed
-
(a) H. Xiao, F. B. Peters, P.-Y. Yang, S. Reed, J. R. Chittuluru and P. G. Schultz, ACS Chem. Biol., 2014, 9, 1092–1096 CrossRef CAS PubMed
- V. K. Lacey, G. V. Louie, J. P. Noel and L. Wang, ChemBioChem, 2013, 14, 2100 CrossRef CAS PubMed
- S. W. Santoro, J. C. Anderson, V. Lakshman and P. G. Schultz, Nucleic Acids Res., 2003, 31, 6700 CrossRef CAS PubMed
- M. Pastrnak, T. J. Magliery and P. G. Schultz, Helv. Chim. Acta, 2000, 83, 2277 CrossRef CAS
-
(a) S. Ohno, T. Yokogawa, I. Fujii, H. Asahara, H. Inokuchi and K. Nishikawa, J. Biochem., 1998, 124, 1065 CrossRef CAS
-
(a) R. A. Hughes and A. D. Ellington, Nucleic Acids Res., 2010, 6813–6830 CrossRef CAS PubMed
- J. C. Anderson and P. G. Schultz, Biochemistry, 2003, 42, 9598 CrossRef CAS PubMed
- A. Chatterjee, H. Xiao and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14841 CrossRef CAS PubMed
- Z. Zhang, L. Alfonta, F. Tian, B. Bursulaya, S. Uryu, D. S. King and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8882 CrossRef CAS PubMed
- A. Ikeda-Boku, S. Ohno, Y. Hibino, T. Yokogawa, N. Hayashi and K. Nishikawa, J. Biochem., 2013, 153, 317 CrossRef CAS PubMed
- K. A. Odoi, Y. Huang, Y. H. Rezenom and W. R. Liu, PLoS One, 2013, 8, e57035 CAS
- J. C. M. van Hest, K. L. Kiick and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 1282 CrossRef CAS
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19 CrossRef CAS PubMed
-
(a) A. J. M. Howden, V. Geoghegan, K. Katsch, G. Efstathiou, B. Bhushan, O. Boutureira, B. Thomas, D. C. Trudgian, B. M. Kessler, D. C. Dieterich, B. G. Davis and O. Acuto, Nat. Methods, 2013, 10, 343 CrossRef CAS PubMed
-
(a) N. Budisa, Angew. Chem., Int. Ed., 2004, 43, 6426 CrossRef CAS PubMed
-
(a) S. Lepthien, M. G. Hoesl, L. Merkel and N. Budisa, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16095 CrossRef CAS PubMed
-
(a) L. Merkel, M. Schauer, G. Antranikian and N. Budisa, ChemBioChem, 2010, 11, 1505 CrossRef CAS PubMed
- T. H. Yoo, A. J. Link and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13887 CrossRef CAS PubMed
- K. H. Wang, W. H. Schmied and J. W. Chin, Angew. Chem., Int. Ed., 2012, 51, 2288 CrossRef CAS PubMed
- K. H. Wang, H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Biotechnol., 2007, 25, 770 CrossRef CAS PubMed
- H. Neumann, K. Wang, L. Davis, M. Garcia-Alai and J. Chin, Nature, 2010, 464, 441 CrossRef CAS PubMed
- K. Wang, A. Sachdeva, D. J. Cox, N. W. Wilf, K. Lang, S. Wallace, R. A. Mehl and J. W. Chin, Nat. Chem., 2014, 6, 393 CrossRef CAS PubMed
- W. Niu, P. Schultz and J. Guo, ACS Chem. Biol., 2013, 8, 1640–1645 CrossRef CAS PubMed
-
(a) B. F. Johnson, J. Xu, K. Jeffrey, Z. Xiang and L. Wang, Nat. Chem. Biol., 2011, 7, 779 CrossRef PubMed
- T. Mukai, A. Hayashi, F. Iraha, A. Sato, K. Ohtake, S. Yokoyama and K. Sakamoto, Nucleic Acids Res., 2010, 38, 8188 CrossRef CAS PubMed
- F. J. Isaacs, P. A. Carr, H. H. Wang, M. J. Lajoie, B. Sterling, L. Kraal, A. C. Tolonen, T. a. Gianoulis, D. B. Goodman, N. B. Reppas, C. J. Emig, D. Bang, S. J. Hwang, M. C. Jewett, J. M. Jacobson and G. M. Church, Science, 2011, 333, 348 CrossRef CAS PubMed
- M. J. Lajoie, A. J. Rovner, D. B. Goodman, H.-R. Aerni, A. D. Haimovich, G. Kuznetsov, J. A. Mercer, H. H. Wang, P. A. Carr, J. A. Mosberg, N. Rohland, P. G. Schultz, J. M. Jacobson, J. Rinehart, G. M. Church and F. J. Isaacs, Science, 2013, 342, 357 CrossRef CAS PubMed
-
(a) A. R. Fersht and C. Dingwall, Biochemistry, 1979, 18, 1250 CrossRef CAS
- R. Krishnakumar, L. Prat, H.-R. Aerni, J. Ling, C. Merryman, J. I. Glass, J. Rinehart and D. Söll, ChemBioChem, 2013, 14, 1967 CrossRef CAS PubMed
- M. J. Lajoie, S. Kosuri, J. A. Mosberg, C. J. Gregg, D. Zhang and G. M. Church, Science, 2013, 342, 361 CrossRef CAS PubMed
-
(a) R. Krishnakumar and J. Ling, FEBS Lett., 2014, 588, 383 CrossRef CAS PubMed
- N. Bohlke and N. Budisa, FEMS Microbiol. Lett., 2014, 351, 133 CrossRef CAS PubMed
- A. Böck, K. Forchhammer, J. Heider, W. Leinfelder, G. Sawers, B. Veprek and F. Zinoni, Mol. Microbiol., 1991, 5, 515 CrossRef PubMed
- L. Johansson, G. Gafvelin and E. S. J. Arnér, Biochim. Biophys. Acta, 2005, 1726, 1 CrossRef CAS PubMed
- M. J. Bröcker, J. M. L. Ho, G. M. Church, D. Söll and P. O'Donoghue, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef
- A. Parrish, X. She, Z. Xiang, I. Coin, Z. Shen, S. Briggs, A. Dillin and L. Wang, ACS Chem. Biol., 2012, 7, 1292 CrossRef CAS PubMed
-
(a) J. C. Jackson, S. P. Duffy, K. R. Hess and R. A. Mehl, J. Am. Chem. Soc., 2006, 128, 11124 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: A comprehensive table of the UAAs incorporated to date (also summarized in Table 1), their reported/potential uses, and the required mutations in the aaRS to allow their uses. See DOI: 10.1039/c4sc01534g |
| ‡ These authors contributed jointly. |
| § A brief note on nomenclature: Throughout this review, we will often refer to different amino acyl synthetase–tRNA pairs in shorthand. This will be comprised of the abbreviated name of the organism from which the pair is derived (e.g. M. jann), the 3 letter code for the natural amino acid substrate from which it is derived (e.g. Tyr), which will proceed the synthetase (RS), followed by the tRNA with its anticodon given in subscript (e.g. CUA). So for example, the tyrosyl synthetase–tRNA pair for amber suppression from the archaebacteria Methanococcus jannaschii would become M. jann TyrRS/tRNACUA. Colloquially, this may also be referred to as Mj-Tyr. |
| This journal is © The Royal Society of Chemistry 2015 |