
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA catalytic asymmetric total synthesis of (−)-perophoramidine†
B. M.
Trost
*,
M.
Osipov
,
S.
Krüger
and
Y.
Zhang
Department of Chemistry, Stanford University, Stanford, CA 94305-5580, USA. E-mail: bmtrost@stanford.edu
First published on 30th July 2014
Abstract
We report a catalytic asymmetric total synthesis of the ascidian natural product perophoramidine. The synthesis employs a molybdenum-catalyzed asymmetric allylic alkylation of an oxindole nucleophile and a monosubstituted allylic electrophile as a key asymmetric step. The enantioenriched oxindole product from this transformation contains vicinal quaternary and tertiary stereocenters, and is obtained in high yield along with high levels of regio-, diastereo-, and enantioselectivity. To install the second quaternary stereocenter in the target, the route utilizes a novel regio- and diastereoselective allylation of a cyclic imino ether to deliver an allylated imino ether product in near quantitative yield and with complete regio- and diastereocontrol. Oxidative cleavage and reductive amination are used as final steps to access the natural product.
Introduction
In 2002, Ireland and coworkers reported the isolation and structural elucidation of a novel polycyclic alkaloid, (+)-perophoramidine (1) from the Philippine ascidian organism Perophora namei (Fig. 1).1 The structure of (+)-perophoramidine (1) was established using multidimensional NMR techniques, and the molecule was found to contain a densely functionalized hexacyclic structure containing two vicinal quaternary all-carbon stereocenters, two amidines, and several points of halogenation on the aromatic nuclei. The skeletal connectivity of perophoramidine (1) is related to the Penicillium derived communesin alkaloids, such as communesin B (2).2 Unlike perophoramidine (1), the communesin alkaloids contain a benzazepine ring and bear two aminal functionalities in place of two amidines. Additionally, the relative relationship of the vicinal quaternary stereocenters in perophoramidine (1) is trans while that of the communesins is cis. From a biological perspective, perophoramidine (1) displays cytotoxicity against the HCT116 colon carcinoma cell line with an IC50 of 60 μM. The combination of its complex, densely functionalized structure and cytotoxic properties make perophoramidine (1) an attractive target for asymmetric total synthesis.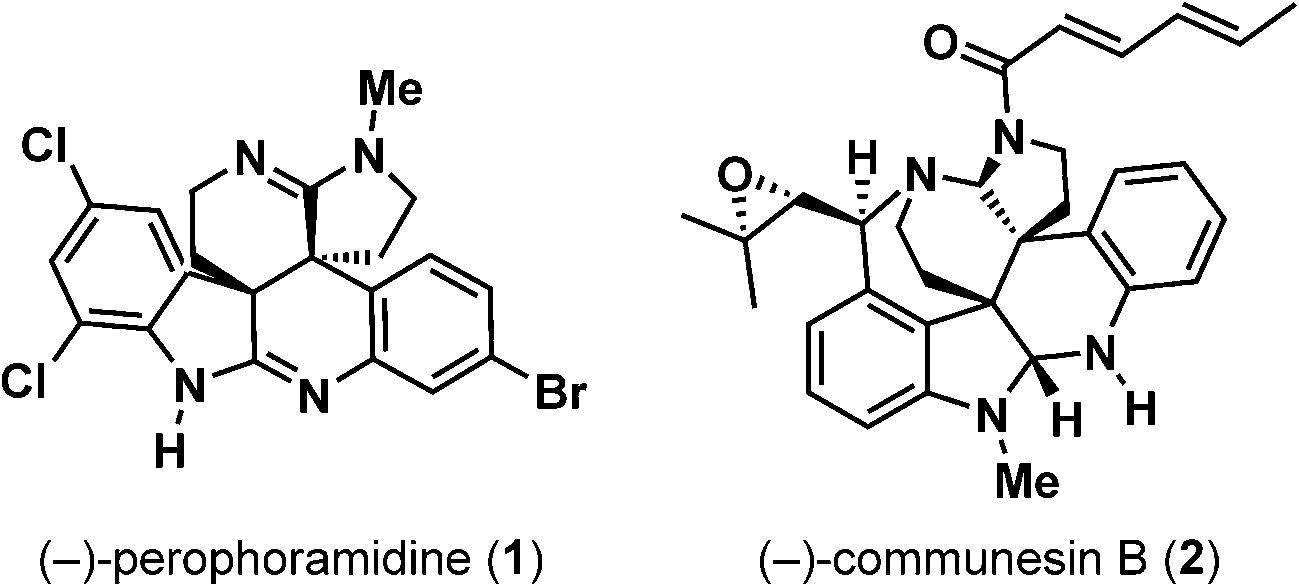 | ||
| Fig. 1 (−)-Perophoramidine (1) and (−)-communesin B (2). | ||
Several syntheses of perophoramidine (1) have been reported. In their synthesis of (±)-perophoramidine (1), Funk and coworkers constructed both quaternary stereocenters utilizing a [4 + 2]-cycloaddition between a 3-alkylindole and 3-bromooxindole, which proceeded through an ortho-aza-xylylene intermediate.3 Employing a chiral auxiliary-mediated hetero-Diels–Alder reaction as a key step, Qin et al. were able to access (+)-perophoramidine (1) in enantiopure form and also determined the absolute stereochemistry of the natural product.4 Most recently, Wang and coworkers described a catalytic asymmetric total synthesis of (+)-perophoramidine (1) using an alkylation reaction between an indole and a 3-bromooxindole, similar to that of Funk's.5 Performing the reaction under nickel-catalysis with chiral diamine ligands, the authors were able to install both vicinal quaternary stereocenters present in (+)-perophoramidine (1) in a diastereo- and enantioselective fashion. To date, all perophoramidine (1) syntheses have utilized a hetero-Diels–Alder-type strategy to access this natural product. Herein we describe a catalytic asymmetric total synthesis (−)–perophoramidine (1) based on a different strategy, which utilizes a molybdenum-catalyzed asymmetric allylic alkylation (Mo-AAA) as a key asymmetric step to create one quaternary stereocenter and an unusual regio- and diastereoselective allylation of an imino ether enolate, which constructs the other quaternary stereocenter in the target.
Metal-catalyzed asymmetric allylic alkylations have found widespread synthetic utility from their ability to construct multiple types of bonds, including C–C, C–N, C–O, and C–X bonds.6 While palladium catalyzed processes have been studied most extensively, these reactions typically lead to linear products when mono-substituted allylic electrophiles are employed.7 On the other hand, Mo-AAA reactions offer complementary regioselectivity, and lead to branched products with mono-substituted allylic electrophiles.8 While other transition metals also offer branched regioselectivity in allylic alkylation reactions,6 the low cost of molybdenum and simple structure of the ligands employed make the Mo-AAA an attractive asymmetric process to employ, especially on large scale. Previously, our group has demonstrated that 3-aryl- and 3-alkyloxindoles could serve as nucleophiles in the Mo-AAA.9 When mono-substituted allylic electrophiles were employed in this process, branched oxindole products were obtained with vicinal tertiary and quaternary stereocenters in high yield as well as with excellent levels of regio-, diastereo-, and enantioselectivity. With this powerful asymmetric methodology in hand, we planned to utilize our Mo-AAA methodology as a key asymmetric step to construct one of the two quaternary stereocenters in (−)-perophoramidine (1).
Results and discussion
Our retrosynthesis for (−)-perophoramidine (1) is outlined in Scheme 1. The title compound (1) could be accessed from allyl imino ether 3via oxidative cleavage of the olefin and a reductive amination/cyclization sequence. The allyl moiety of 3 would be installed using a regio- and diastereoselective allylation of pentacyclic imino ether 4. We realized that this transformation would be challenging, since little precedent for a regio- and diastereoselective allylation of an imino ether exists in the literature.10 Pentacyclic imino ether 4 could be formed in a straightforward manner from lactam 5, which would derive from dichlorinated tetracycle 6 by oxidative cleavage of the olefin, oxidation, and amidation. Dichlorinated tetracycle 6 would be accessed by chlorination of tetracycle 7 and azide formation by a Mitsunobu reaction. Tetracycle 7 could in turn be formed by reductive cyclization of enantioenriched oxindole 8, which would arise from the Mo-AAA reaction between two simple fragments: cinnamyl electrophile 9 and oxindole 10.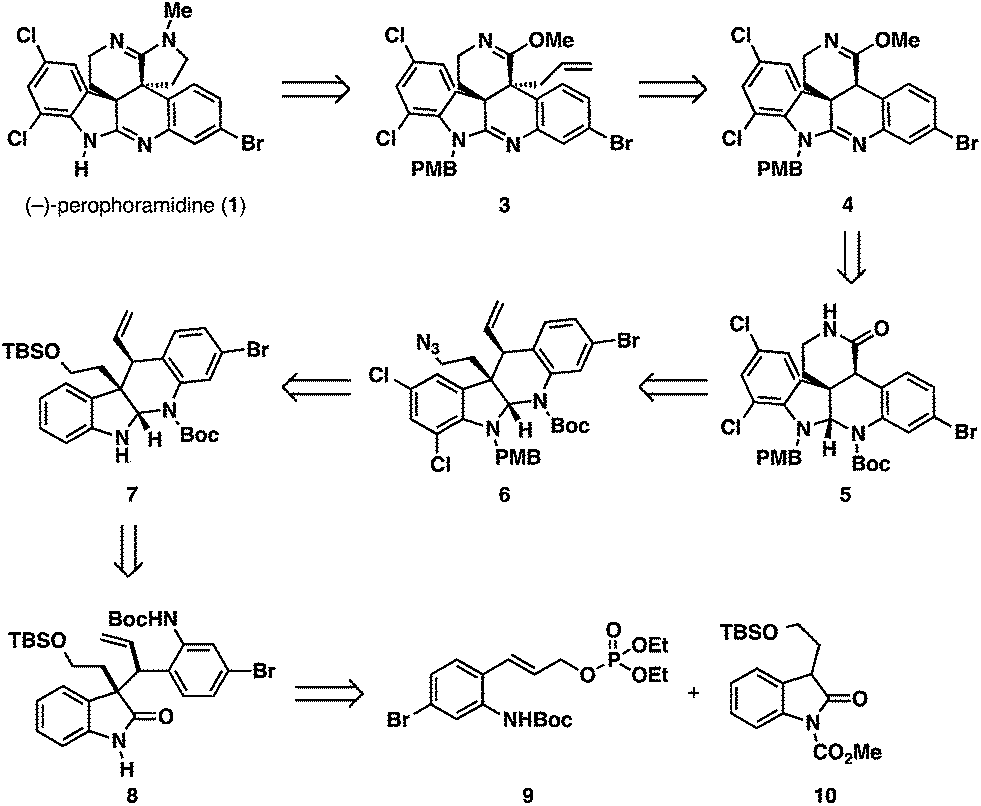 | ||
| Scheme 1 Retrosynthesis. | ||
Oxindole 10 was obtained from tryptophol using a known route.11 Allylic phosphate 9 was prepared from 4-bromo-2-fluorobenzaldehyde (11) in five steps detailed in Scheme 2. Nucleophilic aromatic substitution of aldehyde 11 with sodium azide provided an azidoaldehyde,12 which was treated under Horner–Wadsworth–Emmons olefination conditions to deliver azido ester 12. Reduction of the ester- and azido- functionalities with excess DIBAL-H provided the corresponding amino alcohol product, which was chemoselectively converted to a carbamate by treatment with Boc2O in aqueous dioxane. Allylic phosphate 9 was then accessed in 89% yield by treatment of the carbamate with diethylchlorophosphate.
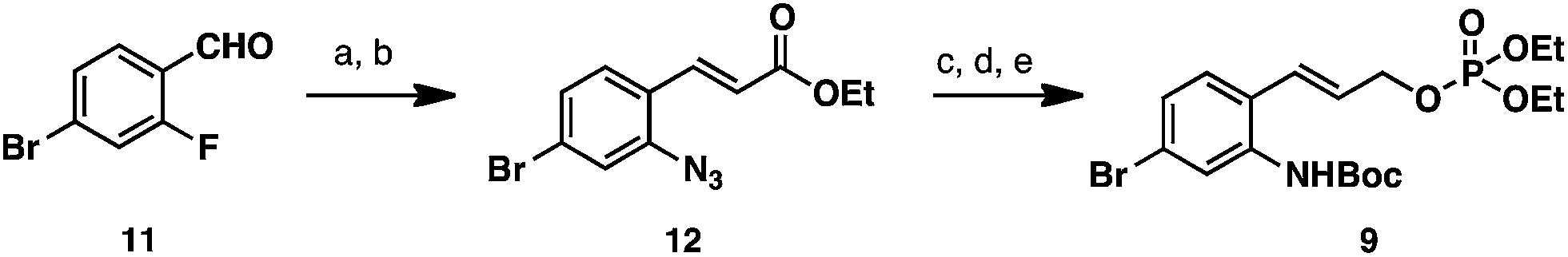 | ||
| Scheme 2 Synthesis of allylic phosphate 9. Reagents and conditions: (a) NaN3, DMSO, 50 °C, 83%; (b) NaH, THF, triethylphosphonoacetate, –78 °C, 86%; (c) DIBAL-H, THF, –78 °C, 99%. (d) Boc2O, Na2CO3, H2O/dioxane, 70 °C, 78%; (e) diethylchlorophosphate, DCM, pyridine, 0 °C, 76%. Boc = tert-butoxycarbonyl. | ||
With both the oxindole nucleophile 10 and allylic electrophile 9 in hand, the Mo-AAA was explored (Scheme 3). The coupling of oxindole 10 and allylic phosphate 9 could be affected employing 20 mol% Mo(CO)3(C7H8), 30 mol% (S,S)-L1, and NaHMDS as base in THF.13 These conditions provided the desired Mo-AAA product, which was treated with NaOH to hydrolyze the methyl carbamate to deliver enantioenriched oxindole 8 in 89% isolated yield, 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 branched to linear ratio, 8:1 dr, and 97% ee. It is noteworthy to mention that the Mo-AAA reaction could be easily conducted on >10 mmol scale to provide multigram quantities of enantioenriched oxindole 8, and that ligand (S,S)-L1 could be recovered at the end of the transformation.
:1 branched to linear ratio, 8:1 dr, and 97% ee. It is noteworthy to mention that the Mo-AAA reaction could be easily conducted on >10 mmol scale to provide multigram quantities of enantioenriched oxindole 8, and that ligand (S,S)-L1 could be recovered at the end of the transformation.
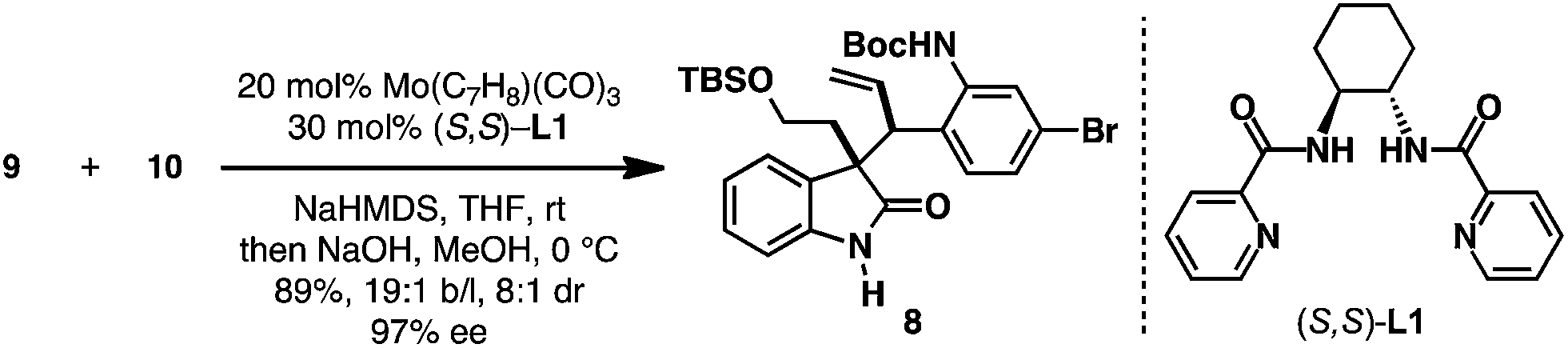 | ||
| Scheme 3 Mo-AAA. C7H8 = cycloheptatriene, HMDS = hexamethyldisilazide. Yield of mixture of isomers. Both b/l ratio and dr were determined by analysis of the 1H NMR spectrum of the crude reaction mixture. Enantiopurity determined by HPLC using a chiral stationary phase. | ||
Enantioenriched oxindole 8 was elaborated to tetracycle 7via reductive cyclization (Scheme 4). In order to undergo chemoselective reduction, the oxindole carbonyl group required electrophilic activation. For this, we chose to install a sterically encumbered carbamate at the oxindole nitrogen, which would favor nucleophilic attack on the oxindole carbonyl group but can be chemoselectively removed without affecting the pre-existing Boc group. We elected to utilize the Pd-labile dimethylallyl protecting group, which possesses the steric properties of a Boc group, but can be removed using Pd-catalysis instead of strong acids.14 Oxindole 8 was deprotonated with NaHMDS and was treated with nitrophenyl carbonate 13 facilitating the formation of protected oxindole 14. Reaction of this intermediate with LiEt3BH chemoselectively reduced the oxindole carbonyl group leading to an N,O-hemiaminal intermediate. Subsequent treatment with Pd(PPh3)4 and morpholine cleaved the dimethylallyl protecting group and spontaneously cyclized the N,N-aminal delivering tetracycle 7 in 75% yield over 2 steps.
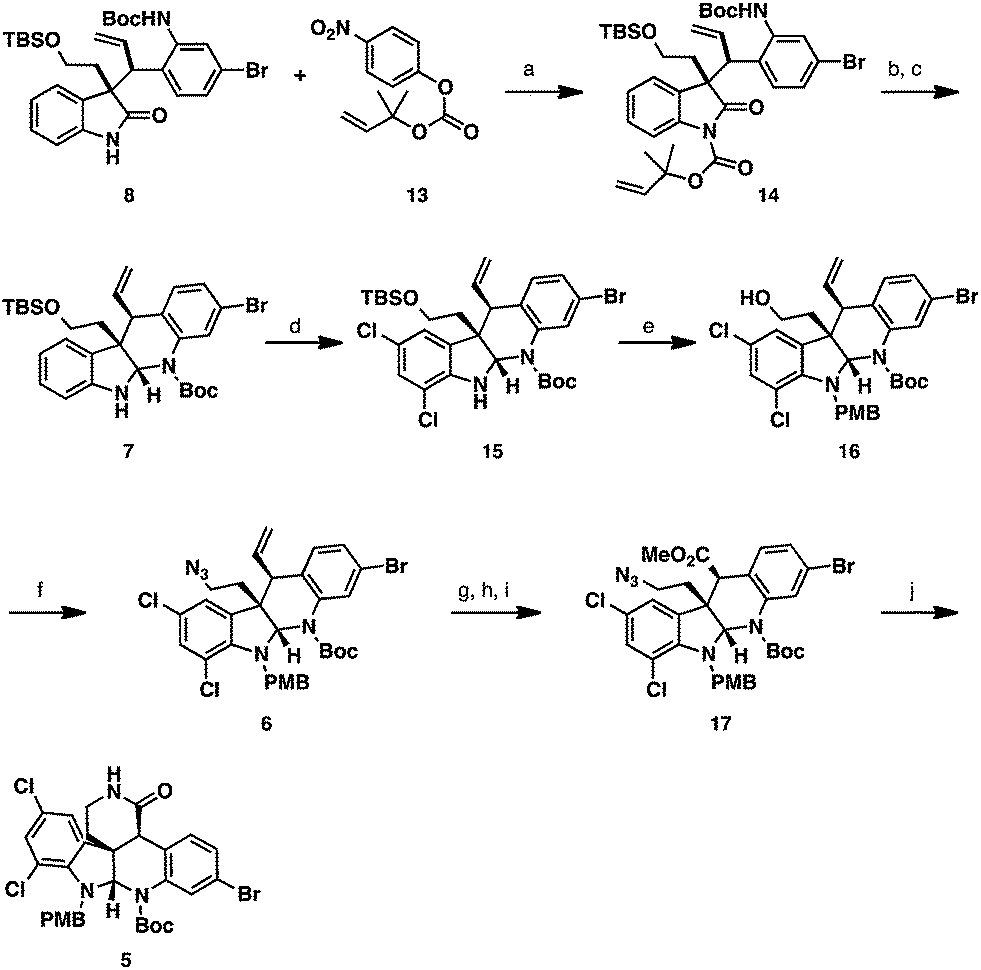 | ||
| Scheme 4 Synthesis of Lactam 5. Reagents and conditions: (a) NaHMDS, THF, 0 °C, then 13, 73%; (b) LiEtBH3, THF, –78 °C; (c) 10 mol% Pd(PPh3)4, morpholine, DCM, rt, 74% over 2 steps; (d) NCS, NaH2PO4, AcOH, rt, 88%; (e) NaHMDS, TBAI, PMB-Br, THF, 0 °C then CSA, MeOH; (f) HN3, PPh3, DEAD, THF, 0 °C, 82% over 2 steps; (g) OsO4, NaIO4, 2,6-lutidine, dioxane, rt; (h) NaClO2, NaH2PO4, tBuOH, H2O, rt; (i) TMSCHN2, MeOH, PhH, rt; (j) PMe3, THF, rt, 47% over 4 steps. NCS = N-chlorosuccinimide, TBAI = tetra-n-butylammonium iodide, PMB = p-methoxybenzyl, CSA = (±)-camphor-10-sulfonic acid, DEAD = diethyl azodicarboxylate. | ||
At this stage, the two aromatic nuclei of tetracycle 7 were electronically differentiated to allow for regioselective dichlorination of the more electron-rich indoline ring.15 This transformation was accomplished employing NCS as the chlorinating agent in AcOH as solvent. In order to obtain high yields reproducibly, the inclusion of NaH2PO4 as a weak base to remove HCl was found to be necessary. Under the optimized reaction conditions, dichloroaminal 15 could be furnished in 88% yield as a single product. To avoid undesired side reactions, the free N–H in dichloroaminal 15 was protected with a PMB group employing NaHMDS and PMBBr. The inclusion of catalytic TBAI to form PMBI in situ was found to be crucial in order to obtain high yields of the product. We discovered that the primary TBS group in the PMB-protected intermediate was very sensitive to Brønsted acid, such that variable quantities of the protiodesilylated primary alcohol product 16 were observed in the crude reaction mixture by proton NMR. Fortunately, working up the reaction mixture with CSA protiodesilylated the silyl ether quantitatively and delivered primary alcohol 16 in 99% yield. Conversion of the primary alcohol to an azide functionality was accomplished utilizing a Mitsunobu reaction with hydrazoic acid, which provided azide 6 in high yield. To access lactam 5, the olefin functionality had to be cleaved oxidatively. Attempts to utilize ozonolysis for this transformation led to decomposition of the starting material. This likely occurred due to the presence of the electron-rich aminal functionality, which could undergo competitive oxidation. Conversely, performing the oxidative cleavage employing modified Johnson–Lemieux conditions with osmium tetroxide and sodium periodate, readily provided the desired aldehyde product.16 Subsequent Pinnick oxidation of the aldehyde intermediate and methylation of the resulting carboxylic acid with TMSCHN2, delivered methyl ester 17. Treatment of this material with PMe3 under Staudinger conditions led to the formation of a primary amine intermediate that underwent spontaneous lactamization, providing lactam 5 in 47% yield over 4 steps.
To oxidize the aminal functionality to the amidine present in the natural product, the Boc group had to be removed first (Scheme 5). Attempts to cleave this group under Brønsted or Lewis acidic conditions led to the loss of both Boc and PMB groups, or decomposition of the starting materials. Previous work by Rawal and Cava showed that Boc groups were readily removed from pyrroles and indoles under thermal conditions.17 Analogously, we were able to cleave the Boc group present in lactam 5 by heating the substrate to 170 °C under high vacuum. In the same pot, the N–H aminal intermediate could be dissolved in a DCM/AcOH mixture and treated with PhI(OAc)2 to deliver the amidine 18 in 67% yield. To set the stage for the diastereoselective imino ether allylation, amidine 18 was converted to the requisite imino ether intermediate 4. This was accomplished in 71% yield employing Meerwein's reagent in the presence of NaHCO3 as an acid scavenger to afford the O-alkylation product with complete chemoselectivity.
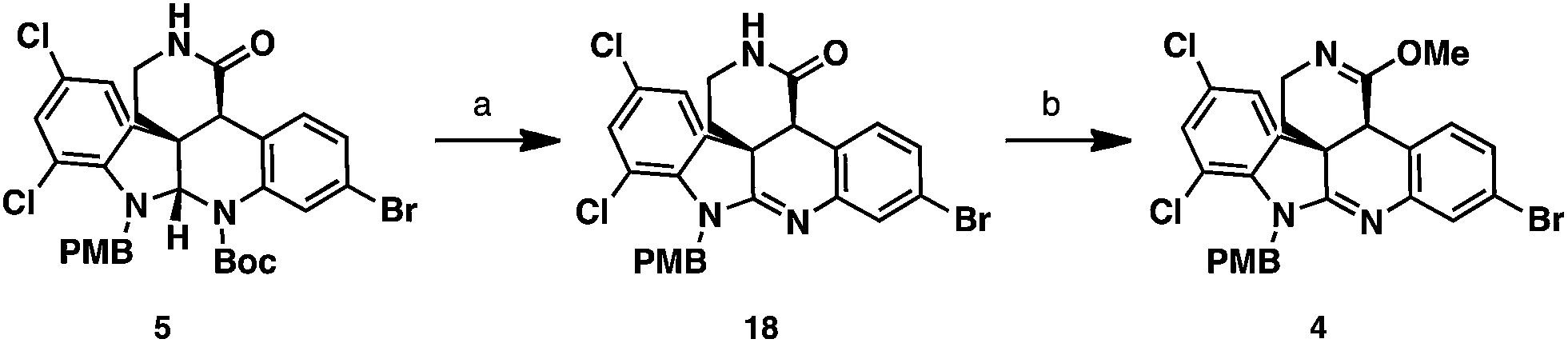 | ||
| Scheme 5 Synthesis of imino ether 4. Reagents and conditions: (a) 170 °C, 0.1 mmHg, neat, then PhI(OAc)2, DCM, AcOH, 0 °C to rt, 65%; (b) Me3OBF4, NaHCO3, DCM, 0 °C, 71%. | ||
With imino ether 4 in hand, allylation of this substrate was explored using allyl iodide as an electrophile (Table 1). Employing LDA in THF under conditions developed by our group for the chemoselective C-allylation of imino ethers led to complete consumption of the starting material and provided C- and N-allylation adducts, 3 and 19 as a 1:10 mixture (entry 1).18 Attempts to thermally rearrange the N-allyl ketene aminal 19 to allyl imino ether 3 at 150 °C via [3,3] sigmatropic rearrangement was unsuccessful. On the other hand changing the base from LDA to KHMDS led to a dramatic change in the regiochemical course of the transformation. Under these conditions, the C- and N-allylation products, 3 and 19 products were obtained in a 3:1 ratio favoring C-allyl imino ether product 3 (entry 2). While studying this transformation, it was noted that the ratio of 3:19 eroded to ∼2:1 when imino ether 4 was treated with KHMDS and the reaction mixture was allowed to stir for 30 min at 0 °C prior to the addition of allyl iodide (entry 3). We postulated that the 3:1 ratio of 3:19 could be improved further by the slow addition of KHMDS solution to a mixture of imino ether 4 and electrophile, rather than allowing the deprotonated intermediate to equilibrate. To our delight, addition of KHMDS to a solution of imino ether 4 and allyl iodide over 10 min led to exclusive formation of C-allyl imino ether 3 in 95% isolated yield (entry 4).
|
|
||
|---|---|---|
| Entry | Conditions | Result |
| 1 | LDA, THF, 0 °C then allyl iodide | >95% conversion, 3:19, 1:10 |
| 2 | KHMDS, THF, 5 min, 0 °C then allyl iodide | >95% conversion, 3:19, 3:1 |
| 3 | KHMDS, THF, 30 min, 0 °C then allyl iodide | >95% conversion, 3:19, 2:1 |
| 4 | Allyl iodide THF, add KHMDS over 10 min | 98% Isolated yield, 3:19, >20:1 |
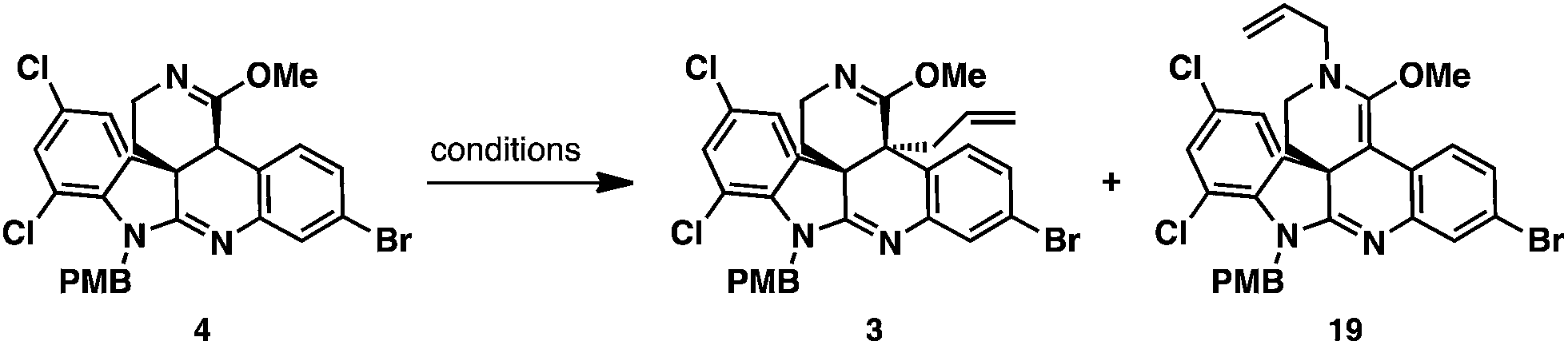
To rationalize the regiochemical outcome of the allylation reaction, we propose that the imino ether, like other enolates,19 forms an aggregate when deprotonated with strong base. The aggregated enolate form leads to the undesired N-allyl ketene aminal product 19, while the monomeric form leads to the desired C-allyl imino ether 3. Slow addition of base to a solution of nucleophile and electrophile reduces the concentration of imino ether enolate and the likelihood of aggregation. Conversely, addition of base to the imino ether and delayed addition of the electrophile favors the formation of aggregates. Furthermore, lithium favors the formation of aggregates to a greater extent than potassium. To rationalize the diastereoselectivity for the transformation, we propose that allylation occurs from the more accessible bottom face of the molecule such that steric interactions with the aminoethylene chain of the adjacent quaternary stereocenter are avoided.
With a method to access allyl imino ether 3, oxidative cleavage of the allyl moiety was explored. Attempts to utilize ozonolysis for the transformation led to decomposition of the starting material. This likely occurred due to the oxidative lability of the electron-rich amidine present in the molecule. We postulated that oxidative decomposition could be circumvented if the basic amidine in allyl imino ether 3 would be “protected” via protonation. Indeed, treatment of allyl imino ether 3 with CSA and subsequent introduction of ozone followed by reductive basic workup smoothly delivered the desired aldehyde product 20.
To install the final nitrogen atom present in the natural product, aldehyde 20 was converted to amine 21 using a reductive amination. Attempts to condense methylamine with the aldehyde 20 and subsequent treatment of the imine intermediate with reducing agents provided the desired amine product 21 in trace quantities. Instead, the primary alcohol product 22 resulting from the reduction of the aldehyde was observed as the major product. This indicated that the condensation process between aldehyde 20 and methylamine was not occurring readily. Addition of dehydrating agents to the reaction mixture to favor imine formation, such as molecular sieves or MgSO4 did not improve the ratio of 21 to 22. However, employing Ti(OiPr)4 as a Lewis acid to catalyze imine formation under conditions developed by Bhattacharyya20 and subsequent addition of NaBH4 at 0 °C led to formation of the desired amine 21 as the only product, with no observable quantity of the undesired primary alcohol product 22. It is noteworthy to mention that under these reaction conditions no reduction of the imino ether functionality was observed (Scheme 6).
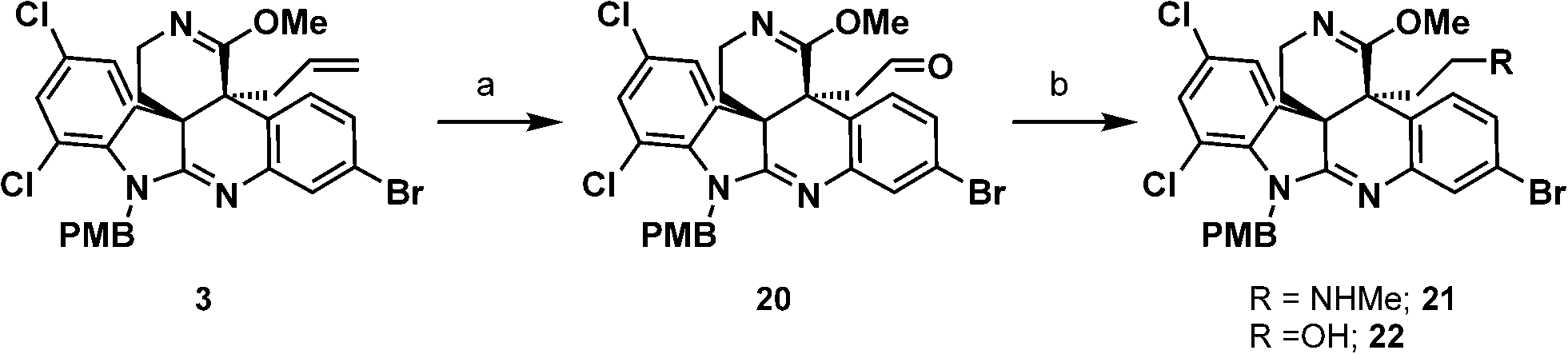 | ||
| Scheme 6 Synthesis of amine 21. (a) CSA, –78 °C, DCM, then O3, then MeOH, Me2S, NaHCO3, to rt; (b) Ti(OiPr)4, MeNH2, EtOH, rt, then NaBH4, 0 °C, 51% over 2 steps. | ||
In the final stages of the synthesis, we were left with the tasks of cyclizing the amine into the pendant imino ether to afford amidine 23 and removal of the PMB protecting group to access the natural product (1). Treatment of the amine 21 with Brønsted acids did not facilitate amidine formation but led to acid-mediated hydrolysis of the imino ether functionality to a secondary lactam. On the other hand, thermal cyclization of the amidine proved quite amenable to our system, and heating amine 21 at 120 °C in either toluene (sealed tube) or anisole as solvents furnished PMB-amidine 23. To complete the total synthesis of (−)-perophoramidine (1), the PMB group needed to be removed. Attempts to utilize ceric ammonium nitrate to cleave this group oxidatively led to decomposition of the starting material. However, PMB cleavage could be performed under strongly acidic conditions; treatment of PMB-amidine 23 with TFA in the presence of anisole at 90 °C afforded (−)-perophoramidine (1). Since both the amidine cyclization and PMB cleavage steps were performed in anisole, we were curious if the two steps could be coupled into a one-pot process. Indeed, heating amine 21 in anisole at 120 °C to cyclize the amidine and then subsequent introduction of TFA at 90 °C to cleave the PMB group delivered (−)-perophoramidine (1) in 62% yield for the single operation (Scheme 7).
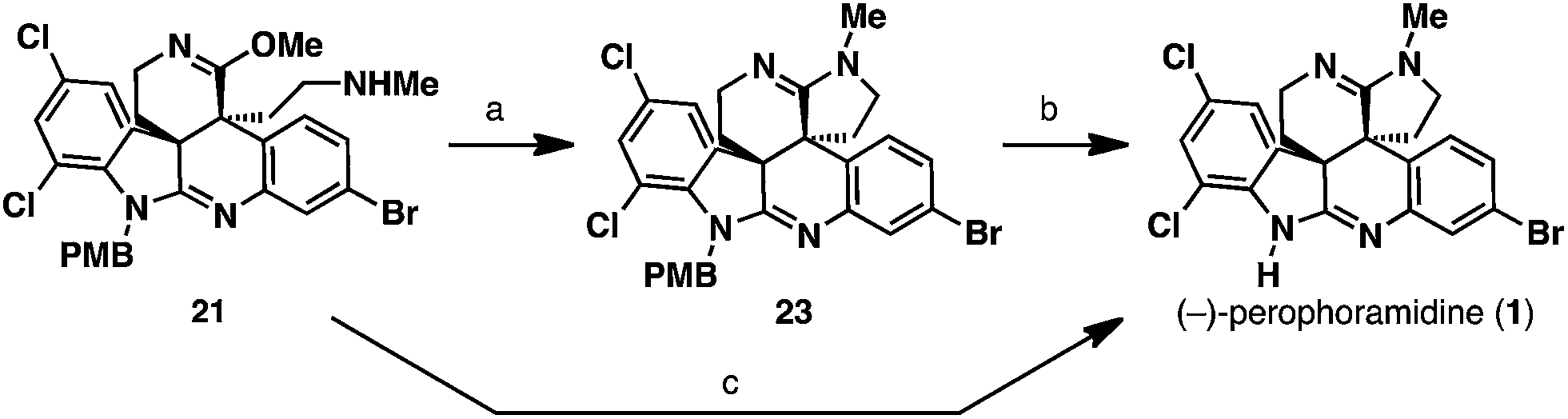 | ||
| Scheme 7 Completion of the total synthesis. (a) Anisole, 120 °C, 100% conversion; (b) TFA, anisole, 90 °C, 100% conversion; (c) Anisole, 120 °C, then 90 °C, TFA, 62%. | ||
Conclusions
In summary, we have developed a catalytic asymmetric total synthesis of the alkaloid natural product (−)-perophoramidine (1). The route utilizes a regio- diastereo- and enantioselective Mo-AAA to construct one of the two vicinal quaternary carbon stereocenters present in the target. The second quaternary carbon stereocenter is constructed employing a regio- and diastereoselective allylation of an imino ether anion, which shows an unprecedented dependence of regioselectivity on the nature of the metal cation. With the potassium salt the reaction proceeds with complete regio- and diastereoselectivity for the desired product. Manipulation of the allyl moiety via oxidative cleavage, reductive amination, cyclization and protecting group cleavage are used to complete the synthesis. The strategy permits structural flexibility particularly at the quaternary stereocenters for analog synthesis.Acknowledgements
We thank the NSF (CHE-1145236) and the NIH (GM 033049) for their generous support of our programs. M.O. thanks The John Stauffer Memorial Fellowship and Stanford Graduate Fellowship for financial support. S.K. thanks the ISAP program of DAAD. We thank Johnson-Matthey for their generous gifts of palladium salts.Notes and references
- S. M. Verbitski, C. L. Mayne, R. A. Davis, G. P. Concepcion and C. M. Ireland, J. Org. Chem., 2002, 67, 7124 CrossRef CAS PubMed.
- (a) P. Siengalewicz, T. Gaich and J. Mulzer, Angew. Chem., 2008, 120, 8290 CrossRef; (b) Z. Zuo and D. Ma, Isr. J. Chem., 2011, 51, 434 CrossRef CAS.
- J. R. Fuchs and R. L. Funk, J. Am. Chem. Soc., 2004, 126, 5068 CrossRef CAS PubMed.
- H. Wu, F. Xue, X. Xiao and Y. Qin, J. Am. Chem. Soc., 2010, 132, 14052 CrossRef CAS PubMed.
- H. Zhang, L. Hong, H. Kang and R. Wang, J. Am. Chem. Soc., 2013, 135, 14098 CrossRef CAS PubMed.
- U. Kazmaier, Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, in Topics in Organometallic Chemistry, Springer-Verlag Berlin Heidelberg, 2011, vol. 38 Search PubMed.
- (a) B. M. Trost, Acc. Chem. Res., 1996, 29, 355 CrossRef CAS; (b) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (c) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 4545 CrossRef CAS; (d) B. M. Trost, Chem. Pharm. Bull., 2002, 50, 1–14 CrossRef CAS; (e) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (f) B. M. Trost, J. Org. Chem., 2004, 69, 5813 CrossRef CAS PubMed; (g) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427 RSC; (h) B. M. Trost, Org. Process Res. Dev., 2012, 16, 185 CrossRef CAS PubMed.
- For recent reviews see: (a) O. Belda and C. Moberg, Acc. Chem. Res., 2004, 37, 159 CrossRef CAS PubMed; (b) C. Moberg, Top. Organomet. Chem., 2012, 38, 209 CrossRef.
- (a) B. M. Trost and Y. Zhang, J. Am. Chem. Soc., 2006, 128, 4590 CrossRef CAS PubMed; (b) B. M. Trost and Y. Zhang, J. Am. Chem. Soc., 2007, 129, 14548 CrossRef CAS PubMed; (c) B. M. Trost and Y. Zhang, Chem.–Eur. J., 2010, 16, 296 CrossRef CAS PubMed; (d) B. M. Trost and Y. Zhang, Chem.–Eur. J., 2011, 17, 2916 CrossRef CAS PubMed.
- (a) U. Groth, L. Richter, U. Schoellkopf and J. Zindel, Liebigs Ann. Chem., 1992, 11, 1179 CrossRef; (b) P. J. M. Taylor, S. D. Bull and P. C. Andrews, Synlett, 2006, 9, 1347 Search PubMed; (c) The bis-lactim alkylation method developed by Schollkopf is closely related and allows for the formation of quaternary amino acids. For a review, see U. Schollkopf, Pure Appl. Chem., 1983, 55, 1799 CrossRef.
- B. M. Trost, Y. Zhang and T. Zhang, J. Org. Chem., 2009, 74, 5115 CrossRef CAS PubMed.
- J. Hu, Y. Cheng, Y. Yang and Y. Rao, Chem. Commun., 2011, 47, 10133 RSC.
- When 10 mol% catalyst loadings were employed, b/l ratios and diastereoselectivities were less reproducible. Typically, these were around a 9:1 b/l ratio and 2.5 dr.
- J.-K. Lee, Y.-W. Suh, M. C. Kung, C. M. Downing and H. H. Kung, Tetrahedron Lett., 2007, 48, 4919 CrossRef CAS PubMed.
- Funk and co-workers have demonstrated that dichlorination of a similar intermediate could be performed with NCS in AcOH. See: ref. 3.
- W. Yu, Y. Mei, Y. Kang, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217 CrossRef CAS PubMed.
- V. H. Rawal and M. P. Cava, Tetrahedron Lett., 1985, 26, 6141 CrossRef CAS.
- B. M. Trost and R. A. Kunz, J. Org. Chem., 1974, 39, 2475 CrossRef CAS.
- L. T. Tomasevich and D. B. Collum, J. Am. Chem. Soc., 2014, 136, 9710 CrossRef CAS PubMed.
- K. A. Neidigh, M. A. Avery, J. S. Williamson and S. Bhattacharyya, J. Chem. Soc., Perkin Trans. 1, 1998, 1, 2527 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, spectroscopic data, and copies of 1H and 13C spectra. See DOI: 10.1039/c4sc01826e |
| This journal is © The Royal Society of Chemistry 2015 |